")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
p53 Mediates GnRH Secretion via Lin28/let-7 System in GT1-7 Cells
Authors Chen T , Wu H, Chen X, Xie R, Wang F, Sun H, Chen L
Received 1 September 2020
Accepted for publication 17 November 2020
Published 1 December 2020 Volume 2020:13 Pages 4681—4688
DOI https://doi.org/10.2147/DMSO.S279901
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Ting Chen, Haiying Wu, Xiuli Chen, Rongrong Xie, Fengyun Wang, Hui Sun, Linqi Chen
Department of Endocrinology, Genetics and Metabolism, Children’s Hospital of Soochow University, Suzhou 215000 Jiangsu, People’s Republic of China
Correspondence: Ting Chen
Department of Endocrinology, Genetics, and Metabolism, Children’s Hospital of Soochow University, Suzhou, Jiangsu, People’s Republic of China
Tel +86-512-8069-8322
Email [email protected]
Study Objective: The well-known tumor suppressor transcriptional factor p53 has been proposed to be one of the central hubs of a functionally related and hierarchically arranged gene network coordinating pubertal timing. Our previous studies revealed that p53 is involved in the metabolic control of puberty. The current study aimed to investigate the underlying signaling pathway, through which p53 mediated the metabolic control of puberty.
Design, Setting, Participants, Interventions, and Main Outcome Measures: We engineered the expression of p53 and/or Lin28a in GT1-7 cells to investigate the interaction between p53 and Lin28/let-7 system, and their impact on GnRH secretion.
Results: Overexpression of p53 stimulated, while inhibition of p53 by pifithrin-α significantly suppressed the GnRH secretion and GPR54 expression levels in response to kisspeptin stimulation in GT1-7 cells. Furthermore, overexpressed p53 suppressed Lin28a and c-Myc expression levels and increased let-7 expression levels in GT1-7 cell lines. On the other hand, inhibition of p53 by pifithrin-α upregulated Lin28a and c-Myc levels and downregulated let-7 expression levels. Moreover, Lin28a overexpression counteracted the effect of p53 overexpression in p53 and Lin28a co-overexpression cells, whose GnRH secretion and GPR54 expression levels were not different from controls. Meanwhile, Lin28a suppression counteracted the effect of pifithrin-α, and the GnRH secretion and GPR54 expression levels are not different from controls in p53 and Lin28a co-suppression cells.
Conclusion: These data suggest that p53 is a central mediator of GnRH secretion in hypothalamus, and this effect is at least partly through the Lin28/let-7 pathway.
Keywords: pubertal timing, p53, Lin28/let-7 system, gonadotropin-releasing hormone
Introduction
The timing of pubertal onset is affected by complex interactions among genetic, nutritional, environmental, as well as socioeconomic factors.1,2 Compelling evidence has proved very prominent regulatory roles of nutritional and metabolic signals on the timing of pubertal onset.3 The secular trends in early pubertal maturation coincide with surging prevalence of overweight and obesity have raised discussion for a possible causal link between greater body fat mass and advanced pubertal age, especially in girls.4 This association of obesity and advanced puberty has been further supported by observations from animal studies.5–7 In the previous study, we also observed advanced vaginal opening (VO) in high-fat diet (HFD) rodents.8
The pivotal event indicating the onset of puberty is the re-emergence of pulsatile gonadotropin-releasing hormone (GnRH) secretion from the GnRH neuron. The pubertal activation of GnRH neuron is regulated by both trans-synaptic inputs and glial inputs.9 Components of the trans-synaptic network mainly include the KNDy neurons releasing both Kisspeptin and neurokinin B (NKB),10 GABAergic neurons, opiatergic neurons, and glutamatergic neurons.11 All these different, yet partially overlapping, sub-systems contribute to the neuroendocrine mechanism controlling the pubertal onset. More importantly, a host of functionally related and hierarchically arranged gene network has been proposed to coordinate all the neuronal and glial inputs in the initiation of pubertal process.9 This genetic network has been supposed to consist of five central hubs: CDP/CUTL1, MAF, p53, YY1, and USF2, all of which control the network at the transcriptional level.9,12 Among these major hubs, MAF, p53, and YY1 have been reported to be involved in obesity and/or metabolic conditions.13–16
Although the mechanisms underlying the metabolic control of puberty remain to be fully elucidated, there have been multiple studies addressing this issue. Kisspeptins, a family of structurally related peptides, have been recognized as a pivotal neuroendocrine regulator of GnRH neurons.17,18 Accumulating evidence has revealed that kisspeptin neurons convey metabolic information to the control center of puberty onset.19 The well-known adipose hormone leptin, levels of which are proportional to fat mass, has been implicated to regulate GnRH neuron via indirect mechanisms.20 A leptin-kisspeptin-GnRH pathway has been proposed based on the fact that exogenous leptin can induce Kiss1 mRNA expression in the ARC of leptin-deficient mice.21 Moreover, leptin is also supposed to control puberty onset indirectly through mTOR (mammalian target of rapamycin) and its downstream effectors.17,22 Besides, Sirtuin 1 (SIRT1), a fuel-sensing deacetylase, has recently been reported to mediate obesity and nutrient-dependent perturbation to timing of puberty onset via epigenetic control of Kiss1 expression.23
p53, a well-known tumor suppressor protein and one of the central hubs in the gene network controlling pubertal onset, has long been regarded as a regulator of metabolism. Human studies have shown the P27R polymorphism of p53 predisposes to obesity and metabolic dysfunction.15 Animals and in vitro studies also revealed the roles of p53 in diet-induced obesity.16 In the previous study, we revealed that HFD mice have higher expression of p53 in hypothalamus than mice fed with normal chow. More importantly, in HFD mice, hypothalamus-specific overexpression of p53 can make VO much earlier, while inhibition of p53 expression relatively delayed VO.8
Lin28/let-7 axis has been proposed as a subordinate node of the gene network controlling puberty onset.9 In this study, we hypothesized that the impact of p53 was via Lin28/let-7 axis. To test this hypothesis, we manipulated the expression of p53 and Lin28a in GT1-7 cells, which are murine hypothalamic GnRH neuronal cells, and explored the interaction between p53 and Lin28/let-7 axis, as well as their impact on kisspeptin-stimulated GnRH secretion function in these cells.
Materials and Methods
Ethical Approval
All procedures performed in the present study were in accordance with the ethical standards of the ethical committee of Children’s Hospital of Soochow University. The ethical committee of Children’s Hospital of Soochow University approved this study and the use of GT1-7 cell lines kindly provided by Shanghai Ruijin Hospital.
Lentiviral Vectors, Transfection, and Expression
For mouse p53 overexpression, the lentiviral vector (CL1128_PDS159-MUS-p53) was designed and constructed as previously described.8 The p53 was prepared via mouse cDNA library using RT-PCR. Then, we subcloned the p53 cDNA sequence into Nhe I/ASC I restriction enzyme site between the CMV promoter and the IRES-GGFPa1 sequence of the lentiviral expression vector, PDS159_pL6.3-CMV-GFPa1-IRES-MCS (Novobio, Shanghai, China). For high titer lentiviruses collection, recombinant lentiviruses were produced by transient transfection in 293T cells. Infectious particles were harvested at 48 h after transfection, filtered through 0.45-μm-pore cellulose acetate filters, concentrated by ultracentrifugation (50,000g for 2h), redissolved in 1 mL sterile DMEM, aliquoted, and stored at −80°C. For mouse Lin28a overexpression and knockdown, the lentiviral vectors CL1126_ PDS159-MUS-lin28a and CL1127_ PDS19-MUS- SH-lin28a, respectively, were designed and constructed.
Cell Transfection
GT1-7 cells (2×105, passage number 7) were inoculated in each well of 96-well plates, and transfection was then conducted by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). GT1-7 cells were infected with different lentiviruses described above. The effect of Lin28a knockdown, as well as Lin28a and p53 overexpression were validated by measuring their mRNA levels by qRT-PCR. Cell lysates were harvested for further qRT-PCR and Western blot (WB) analysis.
GnRH Secretion Studies
Cells were treated with Metastin (45–54) amide (kisspeptin-10, M2816, Sigma-aldrich, France) at final concentrations of 100nM for 1h. RIA for GnRH was performed by the RIA kit (Phoenix pharmaceuticals, RK-040-02) according to the manufacturer’s instructions. Protein content was consistently equivalent between wells (≤10% variation) (P0012, Beyotime Biotechnology, China). GnRH content in 200μL of media from each treated plate was harvested for quantification of GnRH secretion. The GnRH concentration in each media was also assayed by Western blot as described below.
qRT-PCR
We used qRT-PCR to detect the mRNA levels in the in vitro experiments, with mus β-actin mRNA expression level as the internal control. Total RNA from GT1-7 cells was isolated using TRIzol reagent (Invitrogen). The qRT-PCR was performed by a CFX96TM Real-Time System (Bio-Rad) using fluorescent SYBR Green technology (CS7561, Invitrogen). Each qPCR contained 10μL chamQ SYBR QPCR master Mix (Q311-02, vazyme), and a final primer concentration of 200 nM. The melting curve analysis was used to verify the specificity of the amplification products. The primer sequences were all summarized in Supplementary Table 1.
Western Blot
The cellular protein lysates were obtained from the GT1-7 cells. The expression levels of β-actin, GAPDH and tubulin were used as the internal controls. In brief, protein (40μg each) from cell lysates were subjected to gel electrophoresis on SDS-polyacrylamide gel, and separated proteins were transferred onto nitrocellulose membranes and probed with rabbit antiserum against c-Myc (ab32072, abcam), Lin28a (ab46020, abcam), Kiss1 (ab19028, abcam), p53 (#32,532, cell signaling technology), p21 (ab188224, abcam), GRP54 (ab100896, abcam), β-actin (bs-0061R, Bioss, Beijing, China), and GAPDH (bsm-0978M, Bioss, Beijing, China). Then, the goat anti-rabbit secondary antibody (BV-S8008, Biovol Biotech, Shanghai, China) was used to incubate the membranes and an enhanced chemiluminescence Western blotting substrate kit (Pierce Rockford, IL, USA) was used to detect the signals.
Statistical Analysis
Statistical analyses were assessed by SPSS 22.0. Differences among groups were analyzed by one-way ANOVA, followed by post hoc Tukey’s test. Each experiment was repeated at least three times and data are expressed as mean ± SD. The P<0.05 was considered significant.
Results
Overexpression of p53 Suppressed Lin28a Expression in GT1-7 Cell Lines
The effect of overexpression of p53 on gene expression of Lin28/let-7 pathway was measured by qRT-PCR and Western blot (Figure 1). We found that GT1-7 cells with p53 overexpression had significantly lower levels of Lin28a and c-Myc (Figure 1A–D). Besides, p53 overexpression significantly elevated the let-7a levels, while Lin28a overexpression decreased the let-7a levels in GT1-7 cells (Figure 1E). Moreover, the levels of Lin28a and c-Myc were not different from controls in p53 and Lin28a co-overexpressed cells (Figure 1A–D).
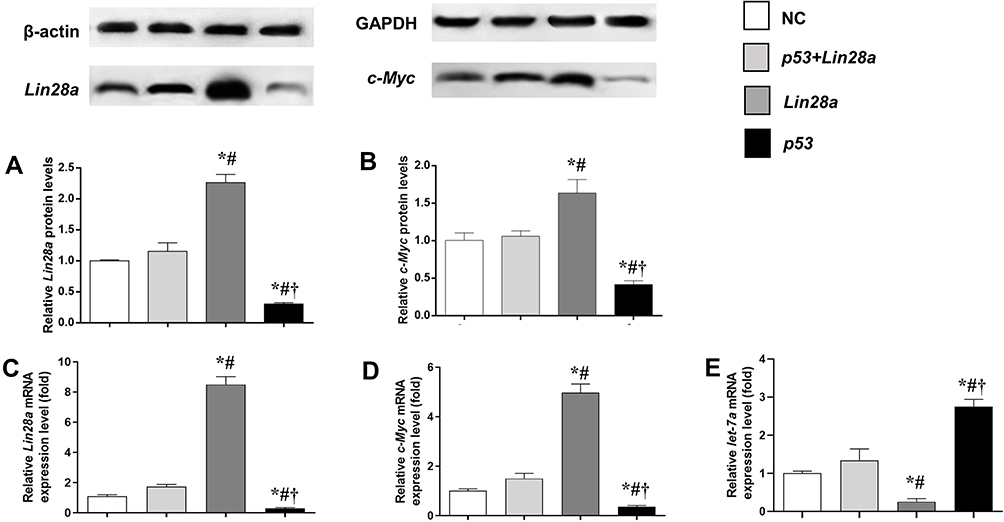 |
Figure 1 The protein and mRNA expression levels of components of Lin28/let-7 axis in GT1-7 cells with p53 and/or Lin28a overexpression. (A, B) are the relative protein levels of Lin28a and c-Myc, respectively. (C–E) are the relative mRNA levels of Lin28a, c-Myc, and let-7a, respectively. Results are expressed as mean±SD; *p<0.05 vs control; #p<0.05 vs cells with both p53 and Lin28a overexpression; †p<0.05 vs cells with Lin28a overexpression. |
Inhibition of p53 Elevated Lin28a Expression in GT1-7 Cell Lines
The gene expression of Lin28/let-7 pathway was measured in GT1-7 cells with p53 inhibition via qRT-PCR and Western blot (Figure 2). Pifithrin-α was used to suppress the expression of p53. We found that the expression levels of Lin28a and c-Myc significantly increased after p53 suppression (Figure 2A–D). Additionally, sh-Lin28a significantly downregulated Lin28a and c-Myc expressions in GT1-7 cells (Figure 2A–D). Besides, pifithrin-α suppressed but sh-Lin28a elevated the levels of let-7a (Figure 2E). The use of pifithrin-α in GT1-7 cells together with sh-Lin28a make the expressions of Lin28a and c-Myc not different from normal controls (Figure 2A–D).
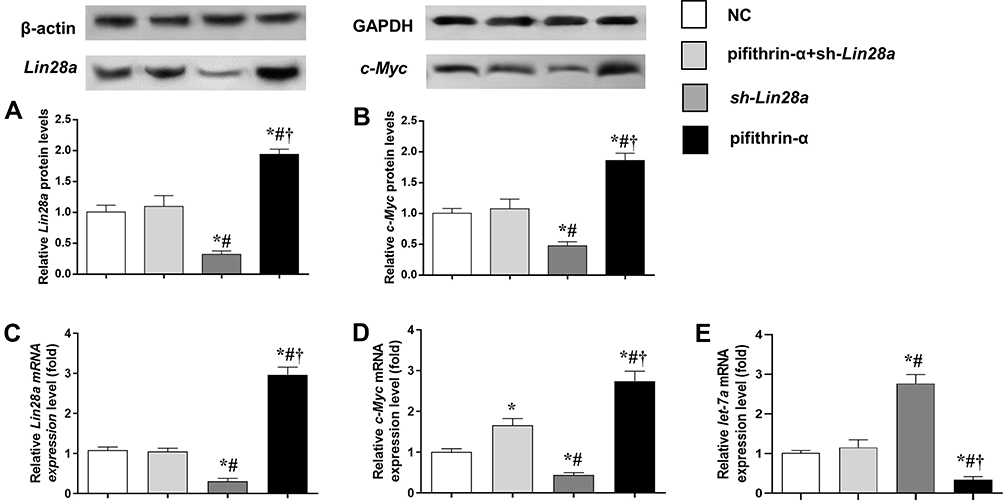 |
Figure 2 The protein and mRNA expression levels of components of Lin28/let-7 axis in GT1-7 cells with p53 and/or Lin28a inhibition. (A, B) are the relative protein levels of Lin28a and c-Myc, respectively. (C–E) are the relative mRNA levels of Lin28a, c-Myc, and let-7a, respectively. Results are expressed as mean±SD; *p<0.05 vs control; #p<0.05 vs cells with both p53 and Lin28a inhibition; †p<0.05 vs cells with Lin28a suppression. |
GnRH Secretion and GPR54 Expression Levels in Response to Kisspeptin Stimulation in GT1-7 Cells
The mRNA and protein expression levels of the GPR54 gene, as well as GnRH concentrations in the culture media were assessed in different GT1-7 cell groups (Figures 3 and 4). After kisspeptin stimulation, significantly higher GnRH concentration was observed in culture media of p53 overexpression cells (Figure 3C), while inhibition of p53 by pifithrin-α significantly suppressed the GnRH secretion of GT1-7 cells (Figure 4C). Upregulation of Lin28a significantly suppressed (Figure 3C), while downregulation of Lin28a significantly elevated the GnRH secretion of GT1-7 cells (Figure 4C). Co-overexpression or co-down-expression of Lin28a with p53 resulted in similar GnRH secretion levels as controls (Figures 3C and 4C). The trend of GPR54 expression levels of GT1-7 cells was similar as the GnRH concentrations in culture media (Figures 3A and B and 4A and B).
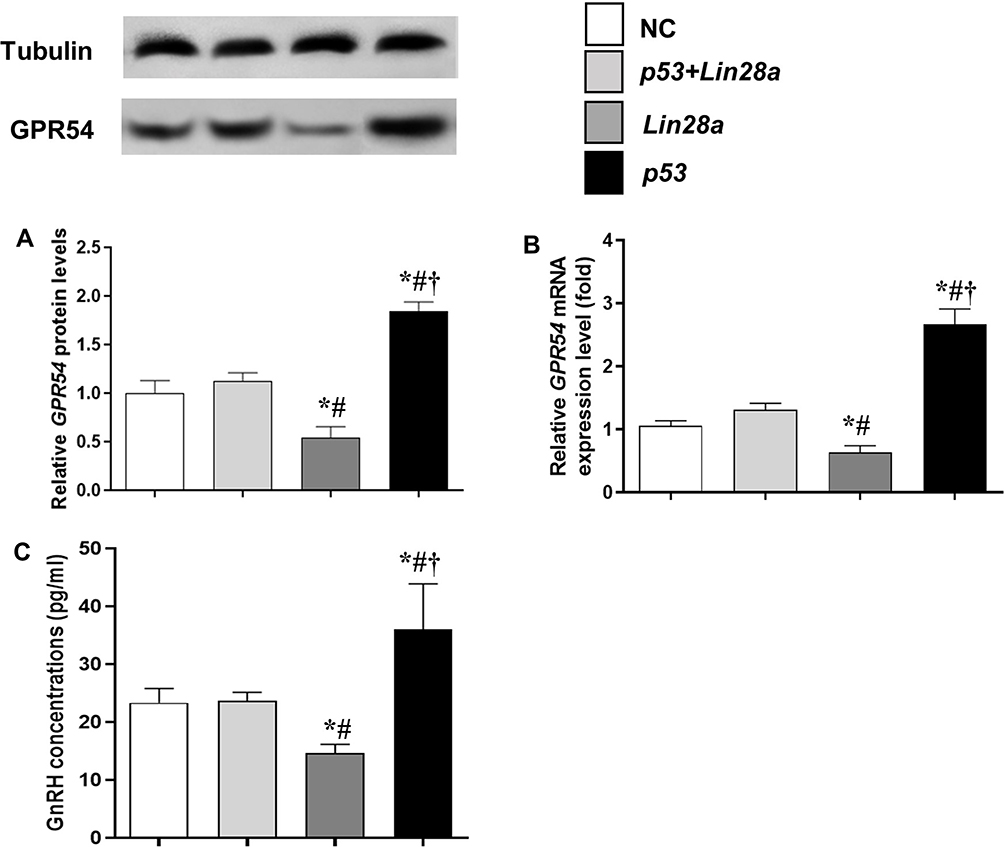 |
Figure 3 The protein and mRNA expression levels of GPR54 in GT1-7 cells with p53 and/or Lin28a overexpression after kisspeptin stimulation. (A, B) are the relative protein and mRNA levels of GPR54, respectively. (C) Is the GnRH concentrations in the cell culture media. Results are expressed as mean±SD; *p<0.05 vs control; #p<0.05 vs cells with both p53 and Lin28a overexpression; †p<0.05 vs cells with Lin28a suppression. |
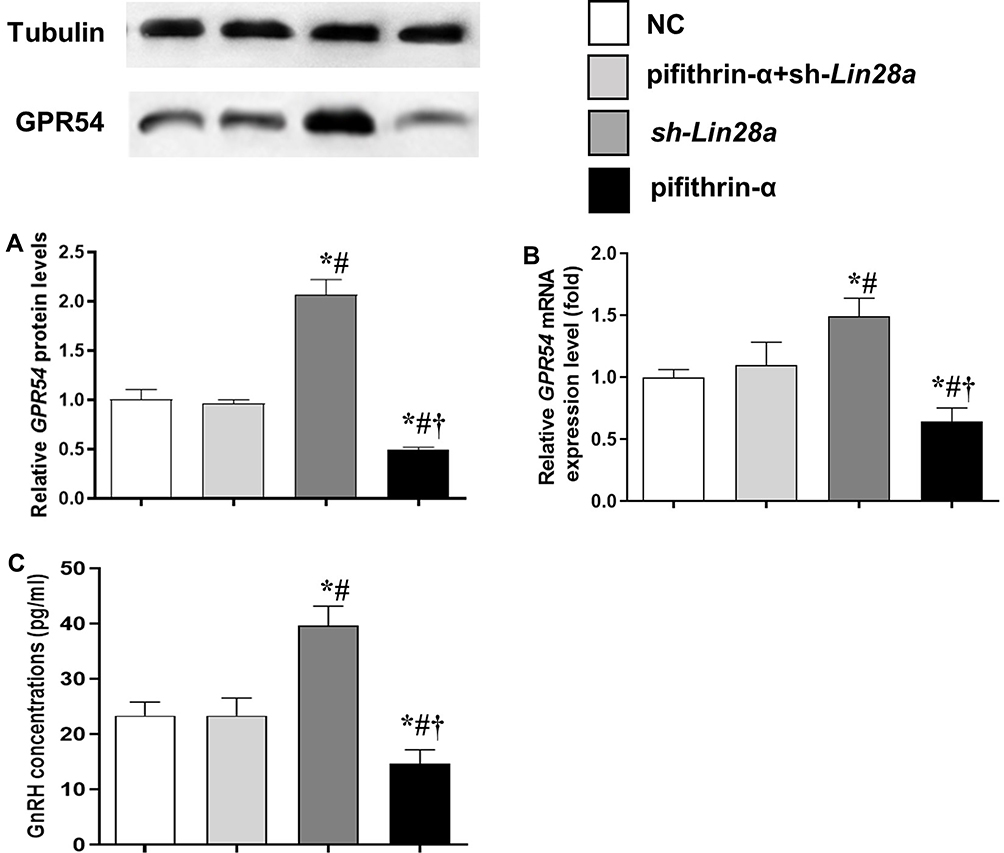 |
Figure 4 The protein and mRNA expression levels of GPR54 in GT1-7 cells with p53 and/or Lin28a inhibition after kisspeptin stimulation. (A, B) are the relative protein and mRNA levels of GPR54, respectively. (C) Is the GnRH concentrations in the cell culture media. Results are expressed as mean±SD; *p<0.05 vs control; #p<0.05 vs cells with both p53 and Lin28a inhibition; †p<0.05 vs cells with Lin28a suppression. |
Discussion
Although epidemiological studies have demonstrated the pivotal roles of metabolic and nutritional factors in the pubertal onset, the underlying mechanisms of such relationships have not been fully elucidated.1,2 p53, a well-known tumor suppressor protein with metabolic regulating function, has been predicted to be one of the central hubs of a genetic network controlling pubertal onset.15,16 Our previous study revealed that hypothalamus-specific overexpression of p53 could induce earlier vaginal opening (VO) in high-fat diet mice, while inhibition of p53 expression delayed VO.8 In the present study, we further showed that overexpression of p53 stimulated, while inhibition of p53 significantly suppressed GnRH secretion and GPR54 expression levels in response to kisspeptin stimulation of GT1-7 cells. Moreover, the puberty-controlling function of p53 is at least partly through the Lin28/let-7 axis.
The GnRH neurons are regulated by a network of excitatory and inhibitory afferents, and the highest level of this intra-network is transcriptionally controlled.9 Among the five central transcriptional hubs, MAF, p53, and YY1 have been shown participating in obesity and/or metabolic conditions.13–16 The extensively expressed transcription factor c-MAF is crucial for developmental and cellular differentiation processes, especially in adipose tissue,24 pancreas,25 and immune system.26 It has been proposed as a risk loci of early-onset and morbid adult obesity in the European population by a genome-wide association study (GWAS).13 The key transcription factor Yin Yang 1 (YY1), which is mainly involved in cell proliferation and differentiation, has been revealed mediating hepatic lipogenesis and glucogenesis in animal models.14,27 More importantly, among all the three central transcriptional hubs, the roles of p53 in metabolism and obesity are best established.15,28 Compelling evidence suggests that p53 in not only related to obesity but also engaged in the glucose homeostasis, insulin resistance, and the development of diabetes.29 High-calorie diet can upregulate endothelial p53 expression, while inhibition of endothelial p53 expression improves dietary metabolic abnormalities.28 In the previous study, we showed that hypothalamic p53 expression was involved in the metabolic control of puberty in HFD mice.8 In the present study, we engineered the expressions of p53 in GT1-7 cells and found that overexpression of p53 stimulated GnRH secretion and GPR54 expression levels in response to kisspeptin stimulation in GT1-7 cells, which further supported the role of p53 in pubertal regulation.
Previous studies on metabolic control of puberty onset have implicated the regulation effect of leptin, SIRT1, and mTOR through kisspeptin, a gatekeeper of puberty.17,20–22 Leptin is a known regulator of p53 expression in multiple tissues.30,31 SIRT1 modulates p53 transcriptional-dependent function via regulating p53 acetylation.32,33 Prior studies have revealed the function of p53 in the regulation of IGF-1/AKT/mTOR pathway.34 Therefore, we suggest that p53 might be a central mediator of leptin, SIRT1, and mTOR pathways in the metabolic control of puberty onset.
The heterochronic genes, Lin28a and Lin28b, were first identified in the nematode C. elegans, regulating the timing of larval development.35 Lin28a and Lin28b are RNA-binding proteins that have been shown to selectively repress the expression of microRNAs (miRNAs), including those belonging to the let-7 family.36,37 They can bind to the terminal loops of precursors of miRNAs in the let-7 family, inhibiting their maturation.38 Besides, Lin28a and Lin28b can derepress c-Myc expression by suppressing mature let-7 synthesis, while c-Myc reversely activates both Lin28a and Lin28b expressions.39,40 The Lin28/let-7 axis has been established as a regulator of puberty control.9 Lin28b was suggested to have potential impact on pubertal regulation based on GWASs.41,42 Lin28a was established as a negative regulator of puberty since mice with Lin28a overexpression had delayed puberty.43 Moreover, apparent decrease of Lin28a, Lin28b, and c-Myc mRNA levels has been observed in the hypothalamus of both male and female rats before/around puberty.40 Additionally, Lin28/let-7 axis is a well-known central regulator of metabolism.44 Hypothalamic ventromedial Lin28a expression positively correlated with energy balance.45 Therefore, Lin28/let-7 axis might as well play a role in the metabolic control of puberty onset.
As central nodes of the gene networks of puberty control, we hypothesized that Lin28/let-7 axis might be regulated by the central hub p53 to mediate the metabolic control of puberty onset. In HFD mice, we showed that the effect of p53 on pubertal regulation might be via Lin28/let-7 axis. In this study, we further explored this observation in GT1-7 cells, and confirmed that Lin28/let-7 axis can modulate GnRH secretion of GnRH neurons, and this effect was regulated by p53.
In conclusion, the transcriptional factor p53 is a central hub, while Lin28/let-7 axis is a subordinate node of the gene network controlling puberty. In GnRH neuron, p53 interacts actively with Lin28/let-7 axis to regulate the GnRH secretion.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (project code 81700793), a Suzhou Personnel Planning Project (project code GSWS2019051), and a Suzhou Science and Technology Development Project (SS202064) awarded to Dr Ting Chen.
Disclosure
The authors declared no potential conflicts of interest with respect to the research, authorship, and publication of this article.
References
1. Gajdos ZKZ, Hirschhorn JN, Palmert MR. What controls the timing of puberty? An update on progress from genetic investigation. Curr Opin Endocrinol Diabetes Obes. 2009;16(1):16–24. doi:10.1097/MED.0b013e328320253c
2. Macedo DB, Brito VN, Latronico AC. New causes of central precocious puberty: the role of genetic factors. Neuroendocrinology. 2014;100(1):1–8. doi:10.1159/000366282
3. Kaplowitz PB. Link between body fat and the timing of puberty. Pediatrics. 2008;121(Supplement 3):S208–S217. doi:10.1542/peds.2007-1813F
4. Aksglaede L, Juul A, Olsen LW, et al. Age at puberty and the emerging obesity epidemic. PLoS One. 2014;63(12):e8450. doi:10.1371/journal.pone.0008450
5. Brill DS, Moenter SM. Androgen receptor antagonism and an insulin sensitizer block the advancement of vaginal opening by high-fat diet in mice. Biol Reprod. 2009;81(6):1093–1098. doi:10.1095/biolreprod.109.079301
6. Feng Li X, Lin YS, Kinsey-Jones JS, et al. High-fat diet increases LH pulse frequency and kisspeptin-neurokinin B expression in puberty-advanced female rats. Endocrinology. 2012;153(9):4422–4431. doi:10.1210/en.2012-1223
7. Ullah R, Su Y, Shen Y, et al. Postnatal feeding with high-fat diet induces obesity and precocious puberty in C57BL/6J mouse pups: a novel model of obesity and puberty. Front Med. 2012;153(2):4422–4431. doi:10.1007/s11684-017-0530-y
8. Chen T, Chen C, Wu H, et al. Overexpression of p53 accelerates puberty in high-fat diet–fed mice through Lin28/let-7 system. Exp Biol Med. 2020:1535370220961320. doi10.1177/1535370220961320
9. Ojeda SR, Dubay C, Lomniczi A, et al. Gene networks and the neuroendocrine regulation of puberty. Mol Cell Endocrinol. 2010;324(1–2):3–11. doi:10.1016/J.MCE.2009.12.003
10. Kauffman AS, Clifton DK, Steiner RA. Emerging ideas about kisspeptin–GPR54 signaling in the neuroendocrine regulation of reproduction. Trends Neurosci. 2007;30(10):504–511. doi:10.1016/j.tins.2007.08.001
11. Terasawa EI, Fernandez DL. Neurobiological mechanisms of the onset of puberty in primates. Endocr Rev. 2001;22:111–151.
12. Roth CL, Mastronardi C, Lomniczi A, et al. Expression of a tumor-related gene network increases in the mammalian hypothalamus at the time of female puberty. Endocrinology. 2007;148(11):5147–5161. doi:10.1210/en.2007-0634
13. Meyre D, Delplanque J, Chèvre J-C, et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat Genet. 2009;41(2):157. doi:10.1038/ng.301
14. Lu Y, Ma Z, Zhang Z, et al. Yin Yang 1 promotes hepatic steatosis through repression of farnesoid X receptor in obese mice. Gut. 2014;63(1):170LP– 178. doi:10.1136/gutjnl-2012-303150
15. Kung C-P, Leu -J-J, Basu S, et al. The P72R polymorphism of p53 predisposes to obesity and metabolic dysfunction. Cell Rep. 2016;14(10):2413–2425. doi:10.1016/J.CELREP.2016.02.037
16. Molchadsky A, Ezra O, Amendola PG, et al. p53 is required for brown adipogenic differentiation and has a protective role against diet-induced obesity. Cell Death Differ. 2013;20(5):774–783. doi:10.1038/cdd.2013.9
17. Roa J, Tena-Sempere M. Energy balance and puberty onset: emerging role of central mTOR signaling. Trends Endocrinol Metab. 2010;21(9):519–528. doi:10.1016/j.tem.2010.05.003
18. Oakley AE, Clifton DK, Steiner RA. Kisspeptin signaling in the brain. Endocr Rev. 2009;30:713–743.
19. Castellano JM, Roa J, Luque RM, et al. KiSS-1/kisspeptins and the metabolic control of reproduction: physiologic roles and putative physiopathological implications. Peptides. 2009;30(1):139–145. doi:10.1016/j.peptides.2008.06.007
20. Quennell JH, Mulligan AC, Tups A, et al. Leptin indirectly regulates gonadotropin-releasing hormone neuronal function. Endocrinology. 2009;150(6):2805–2812. doi:10.1210/en.2008-1693
21. Kalamatianos T, Grimshaw SE, Poorun R, et al. Fasting reduces KiSS-1 expression in the anteroventral periventricular nucleus (AVPV): effects of fasting on the expression of KiSS-1 and neuropeptide Y in the AVPV or arcuate nucleus of female rats. J Neuroendocrinol. 2008;20(9):1089–1097. doi:10.1111/j.1365-2826.2008.01757.x
22. Roa J, Garcia-Galiano D, Varela L, et al. The mammalian target of rapamycin as novel central regulator of puberty onset via modulation of hypothalamic Kiss1 system. Endocrinology. 2009;150(11):5016–5026. doi:10.1210/en.2009-0096
23. Vazquez MJ, Toro CA, Castellano JM, et al. SIRT1 mediates obesity- and nutrient-dependent perturbation of pubertal timing by epigenetically controlling Kiss1 expression. Nat Commun. 2018;9(1):4194. doi:10.1038/s41467-018-06459-9
24. Serria MS, Ikeda H, Omoteyama K, et al. Regulation and differential expression of the c-MAF gene in differentiating cultured cells. Biochem Biophys Res Commun. 2003;310(2):318–326. doi:10.1016/j.bbrc.2003.08.144
25. Tsuchiya M, Taniguchi S, Yasuda K, et al. Potential roles of large mafs in cell lineages and developing pancreas. Pancreas. 2006;32(4):408–416. doi:10.1097/01.mpa.0000220867.64787.99
26. Agnello D, Lankford CSR, Bream J, et al. Cytokines and transcription factors that regulate T helper cell differentiation: new players and new insights. J Clin Immunol. 2003;23:147–161. doi:10.1023/A:1023381027062
27. Lai C-Y, Lin C-Y, Hsu -C-C, et al. Liver-directed microRNA-7a depletion induces nonalcoholic fatty liver disease by stabilizing YY1-mediated lipogenic pathways in zebrafish. Biochim Biophys Acta. 2018;1863:844–856.
28. Yokoyama M, Okada S, Nakagomi A, et al. Inhibition of endothelial p53 improves metabolic abnormalities related to dietary obesity. Cell Rep. 2014;7(5):1691–1703. doi:10.1016/j.celrep.2014.04.046
29. Kung C-P, Murphy ME. The role of the p53 tumor suppressor in metabolism and diabetes. J Endocrinol. 2016;231(2):R61–R75. doi:10.1530/JOE-16-0324
30. Zwezdaryk K, Sullivan D, Saifudeen Z. The p53/adipose-tissue/cancer nexus. Front Endocrinol (Lausanne). 2018;9:457. doi:10.3389/fendo.2018.00457
31. Zahid H, Subbaramaiah K, Iyengar NM, et al. Leptin regulation of the p53-HIF1α/PKM2-aromatase axis in breast adipose stromal cells: a novel mechanism for the obesity–breast cancer link. Int J Obes. 2018;42(4):711–720. doi:10.1038/ijo.2017.273
32. Liu X, Wang D, Zhao Y, et al. Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proc Natl Acad Sci. 2011;108:1925–1930.
33. Gonfloni S, Iannizzotto V, Maiani E, et al. P53 and Sirt1: routes of metabolism and genome stability. Biochem Pharmacol. 2014;92(1):149–156. doi:10.1016/j.bcp.2014.08.034
34. Feng Z, Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010;20(7):427–434. doi:10.1016/j.tcb.2010.03.004
35. Pasquinelli AE, Reinhart BJ, Slack F, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408(6808):86–89. doi:10.1038/35040556
36. Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science (80-). 2008;320(5872):97–100. doi:10.1126/science.1154040
37. Thornton JE, Gregory RI. How does Lin28 let-7 control development and disease? Trends Cell Biol. 2012;22(9):474–482. doi:10.1016/j.tcb.2012.06.001
38. Viswanathan SR, Daley GQ. Lin28: a MicroRNA regulator with a macro role. Cell. 2010;140(4):445–449. doi:10.1016/j.cell.2010.02.007
39. Chang T-C, Zeitels LR, Hwang H-W, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci. 2007;148(9):5147–5161. doi:10.1073/pnas.0808300106
40. Sangiao-Alvarellos S, Manfredi-Lozano M, Ruiz-Pino F, et al. Changes in hypothalamic expression of the Lin28/let-7 system and related microRNAs during postnatal maturation and after experimental manipulations of puberty. Endocrinology. 2013;154(2):942–955. doi:10.1210/en.2012-2006
41. Ong KK, Elks CE, Li S, et al. Genetic variation in LIN28B is associated with the timing of puberty. Nat Genet. 2009;41(6):729. doi:10.1038/ng.382
42. Perry JRB, Stolk L, Franceschini N, et al. Meta-analysis of genome-wide association data identifies two loci influencing age at menarche. Nat Genet. 2009;41(6):648. doi:10.1038/ng.386
43. Zhu H, Shah S, Shyh-Chang N, et al. Lin28a transgenic mice manifest size and puberty phenotypes identified in human genetic association studies. Nat Genet. 2010;42(7):626. doi:10.1038/ng.593
44. Zhu H, Ng SC, Segr AV, et al. The Lin28/let-7 axis regulates glucose metabolism. Cell. 2011;147:81–94.
45. Kim JD, Toda C, Ramírez CM, et al. Hypothalamic ventromedial Lin28a enhances glucose metabolism in diet-induced obesity. Diabetes. 2017;66(8):2102–2111. doi:10.2337/db16-1558
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.