")
Back to Archived Journals » Research and Reports in Biochemistry » Volume 5
p21-activated kinase family: promising new drug targets
Received 25 February 2015
Accepted for publication 1 April 2015
Published 14 May 2015 Volume 2015:5 Pages 119—128
DOI https://doi.org/10.2147/RRBC.S57278
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Nikolay Dokholyan
Nhi Huynh, Hong He
Department of Surgery, University of Melbourne, Austin Health, Melbourne, VIC, Australia
Abstract: The p21-activated kinase (PAK) family of serine/threonine protein kinases are downstream effectors of the Rho family of GTPases. PAKs are frequently upregulated in human diseases, including various cancers, and their overexpression correlates with disease progression. Current research findings have validated important roles for PAKs in cell proliferation, survival, gene transcription, transformation, and cytoskeletal remodeling. PAKs are shown to act as a converging node for many signaling pathways that regulate these cellular processes. Therefore, PAKs have emerged as attractive targets for treatment of disease. This review discusses the physiological and pathological roles of PAKs, validation of PAKs as new promising drug targets, and current challenges and advances in the development of PAK-targeted anticancer therapy, with a focus on PAKs and human cancers.
Keywords: p21-activated kinase, cancer, inhibitor
Introduction
The first p21-activated kinase, PAK1, was discovered in 1994 as a Cdc42/Rac-interacting protein.1 Cdc42 and Rac proteins are members the Rho family of GTPases, which are known regulators of cell proliferation and motility.2 Thereafter, a significant amount of work has revealed the connection of PAKs to a plethora of cellular processes, including proliferation, survival, motility, gene transcription, and oncogenic transformation.3,4 Activation of PAKs has been shown to drive oncogenic signaling in cells and contribute to the progression of cancer. This review summarizes the role of PAKs in tumorigenesis and the current status of PAK1-targeted drug development.
Functional characteristics and molecular mechanisms of action
Structure and activation mechanisms of PAKs
Six mammalian PAKs are classified into two groups based on their sequence, structural homology, and activation mechanism, ie, group I (PAK1–3) and group II (PAK4–6). All PAKs have a conserved C-terminal kinase domain and an N-terminal regulatory domain.5 Group I PAKs are approximately 70% homologous in their sequence but with over 90% homology in their kinase domains.6 The N-terminal regulatory domain contains a p21-protein binding domain that overlaps with an autoinhibitory domain (AID, Figure 1). Group I PAKs normally exist as inactive homodimers, in which the AID of one PAK molecule interacts with the kinase domain of another. Binding of Cdc42/Rac to the p21-protein binding domain disrupts this interaction and initiates conformational changes to trigger autophosphorylation of the activation loop and several C-terminal serine residues, which are critical for full kinase activity.1,7 Unlike group I, the kinase domains of group II PAKs are constitutively phosphorylated. Binding to Cdc42/Rac does not result in activation but rather affects subcellular localization.8 It was believed that group II PAKs do not possess an AID and are constitutively catalytically active, until recent work revealed the presence of AID-like domains in group II PAKs.9–11 PAK5 was first identified to contain an AID and its kinase activation was found to be stimulated by Cdc42 binding similar to group I PAKs.9 This domain, however, was absent in PAK4 and PAK6, leading to the assumption of distinct regulation within group II PAKs. Recent studies suggest that the AID-like domain interacts with and inhibits the kinase domain. Activation does not rely on autophosphorylation of the activation loop, but rather occurs readily once this interaction is disrupted, permitting assumption of active conformation.10,11
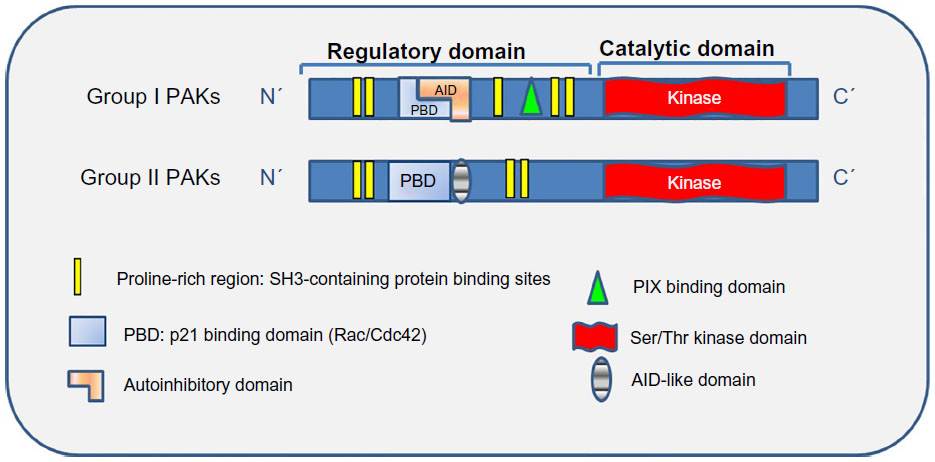 | Figure 1 Structure of PAKs. |
Biological functions of PAKs
PAKs play important roles in many cellular processes, including cell proliferation, motility, and survival. PAKs stimulate cell proliferation by activating the mitogen-activated protein kinase signaling pathway.12,13 Two components of this pathway, c-Raf and MEK1, have been reported to be direct substrates for group I PAKs.14–16 PAKs also promote cell proliferation via the Wnt signaling pathway. PAK1 associates and phosphorylates β-catenin to stabilize and facilitate its nuclear translocation, thus controlling transcriptional activity.17,18 In addition, PAKs promote cell cycle progression by stimulating cyclin D1 expression, a key regulator of G1 cell cycle progression.19 PAK1 has also been shown to localize to centrosomes and to phosphorylate and activate centrosomal kinase Aurora A during mitosis.20 PAKs regulate actin-cytoskeleton rearrangement during cell motility, division, and migration; these are processes that, when deregulated, can be exploited by cancer cells during metastatic and invasive disease progression. PAKs participate in cytoskeletal remodeling by phosphorylating many substrates that control different aspects of cytoskeletal dynamics, such as LIM kinase,21 p41-ARC,22 filamin A,22 and myosin light chain kinase.23 PAK1 phosphorylates c-Raf, causing its relocalization from the cell membrane to the mitochondria, where it binds to Bcl-2 to replace Bad, thereby relieving Bcl-2 from the inhibitory complex to stimulate cell survival signals.24 PAK1 and PAK5 can also phosphorylate Bad directly and dissociate it from Bcl-2,25 thus enhancing cell survival.
PAKs in disease: aberrant PAK function in cancer
PAKs have been shown to affect three main areas of human health, ie, cancer, brain function, and viral infection.26 However, much interest and studies accentuate the understanding of their roles in cancer. This review focuses on the functions of PAKs in different aspects of cancer.
Upregulation of PAKs in cancer
Abnormalities in expression and activity of PAKs are often reported in human cancers. Multiple PAK isoforms have been shown to contribute to tumorigenesis by stimulating signaling pathways controlling cell proliferation, survival, motility, angiogenesis, anchorage-independent growth, and epithelial–mesenchymal transition,27 processes that are conceptualized to constitute the hallmarks of cancer. PAKs are not found to be frequently mutated in cancers; yet, overexpression and/or gene amplification of PAKs are common.28,29 This generally results in elevated mRNA and protein levels, with subsequent accumulation of phosphorylated PAK and presumably increased PAK activity. In particular, PAK1 and PAK4 genes are located on chromosomal regions that are frequently amplified in cancer, and are most strongly associated with cancer compared with other PAK members.
Aberrant function of PAKs, particularly PAK1, is well documented in breast cancer. PAK1 gene amplification at 11q13 is reported as prevalent in luminal breast cancer.30 The frequency of PAK1 amplification is 17% in the tumor panel examined and well correlated with mRNA expression. Immunohistochemistry analysis of breast tumor tissues of different stages revealed that PAK1 expression was upregulated and positively correlated with disease progression30 as well as recurrence rate and mortality.31,32 PAK1 small interfering RNA induces robust cell apoptosis associated with caspase activation and attenuated phosphorylation of MEK1 and ERK1/2. Amplification of PAK1 is also reported in ovarian cancer, and more importantly, elevated PAK1 levels are used as an independent prognostic predictor of poor survival in ovarian cancer.33 Overexpression of PAK1 is observed in over 70% of colorectal cancers (CRCs),34 where its expression increases with disease progression from adenoma to carcinoma, with significant increases in invasive and metastatic CRC.35 In addition, expression of PAK4 and PAK5 is also elevated in CRC.36 PAK4 gene amplification at 19q13.2 has been shown in 22% and 11% of pancreatic cancers and CRCs, respectively.37,38 Increased expression of PAK4 is positively correlated with disease aggressiveness and a poor prognosis.39
Loss-of-function mutations of the tumor suppressor genes NF1 and NF2 predispose mutation carriers to development of the dominantly inherited autosomal disease neurofibromatosis type 1 or type 2 (NF1 and NF2), respectively, which is characterized by formation of tumors of the central and peripheral nervous systems. PAK1 promotes the malignant growth of both NF1 and NF2 through different mechanisms. Neurofibromin, encoded by NF1, is a GTPase-activating protein that stimulates Ras GTPase activity, leading to its inactivation and downstream inhibition of PAK1 through the effector pathways. Merlin, the tumor suppressor gene encoded by NF2, directly interacts with the Cdc42/Rac binding domain of PAK1 to prevent its activation.40,41 Merlin is a negative regulator of PAK1 and exerts growth suppressive activity. PAK1 can phosphorylate Merlin on S518, causing conformational changes and consequently disrupting interaction with PAK1, liberating PAK1 for activation.41 Although mutations of PAKs are not frequently described in cancer, a gain-of-function point mutation of PAK5 was recently reported in 5%–10% of lung cancers.42 Inhibition of PAK5 by small interfering RNA decreased cell survival and ERK activity.42 Hyperactivated PAK functions can also result from mutations in upstream regulators such as Ras/Rac. The discovery of the role of PAKs in carcinogenesis warrants the potential of targeting PAKs in cancer therapy.
PAK signaling in cancer
Many human cancers are driven by the oncogenic protein Ras, with more than 30% showing somatic gain-of-function mutations of this oncogene.43 Over 90% of pancreatic adenocarcinomas44 and 30%–50% of CRCs45,46 carry Ras mutations. Of the three human Ras isoforms, NRas, HRas, and KRas, KRas mutations comprise 86%47 of all Ras mutations and occur in 17%–25% of all cancers.46 KRas mutations are probably best described in CRC, and are clinically associated with a poorer prognosis, increased tumor aggressiveness, and treatment resistance.45,46,48 Mutations of Kras cause deregulated cell growth, migration, apoptosis, and differentiation via activation of its downstream effectors. The two canonical Ras effectors are the Raf serine/threonine protein kinases and the phosphoinositide 3-kinases (PI3Ks). The development of anti-Ras therapy has not proved successful due to the complex nature of Ras signaling. The next most sensible approach therefore has been focused on targeting the two mentioned Ras effector signaling pathways. Over 30 and 50 inhibitors targeting the RAF/MEK/MAPK/ERK and the PI3K/AKT cascade, respectively, are currently under evaluation.28 However, the efficacy of targeting components in these two pathways is compromised by the cross-talk and inhibitory effects that exist between the two pathways, and ablating signal from one pathway may result in compensatory signal in the other. Consequently, combination targeting of both pathways may prove helpful in anti-Ras therapy.
PAK1, acting downstream of Ras, can enhance the RAF/MEK/ERK signaling pathway and increase cell proliferation by phosphorylating Raf1 (S338)49 and MEK1 (S298).50 PAK3 can also phosphorylate Raf1 on S338 to enhance its activation both in vitro and in vivo.15 PAK1 regulates the PI3K/AKT pathway in a kinase-independent manner by facilitating the PDK1-mediated recruitment of AKT to the plasma membrane where it is activated.51 We have recently reported that PAK1 is required for proliferation, survival, migration, and vascular endothelial growth factor secretion of CRC cells harboring mutations in Ras, PI3K, and Apc13 and that PAK1 knockdown inhibited these cellular processes by inactivation of ERK and AKT.13 Moreover, PAK1 knockdown suppressed growth and metastasis of CRC cell lines in xenograft and liver metastasis models in mice.17 These findings are supported by the work of Li et al showing that PAK1 regulates CRC cell metastasis via ERK-dependent phosphorylation of FAK in both cell lines and clinical samples.52 Inhibition of PAK1, both genetically and pharmacologically, causes inhibition of ERK and AKT activity, and tumor regression in a KRas-driven skin cancer model.53 These results imply that instead of combination therapies targeting both RAF/MEK/ERK and PI3K/AKT pathways, targeting PAK1 alone could be an alternative approach in cancer treatment, specifically Kras-driven cancers.
However, there was a report that knockdown of PAK1 or PAK4 inhibited the proliferation of mutant KRas colon cancer cells via RAF/MEK/ERK-independent and/or PI3K/AKT-independent pathway(s),54 suggesting an alternative signaling pathway is involved. Indeed, the interactions of PAKs with the Wnt pathway have also been reported by several studies.17,34,55,56 In breast cancer and CRC cell lines, PAK1 associates with and phosphorylates β-catenin on S663 and S675,34,56 and promotes its nuclear translocation and subsequent transcriptional upregulation of T-cell factor-responsive genes including myc and cyclin D1, a key driver of cell cycle progression. Consistent with this, we have reported that PAK1 binds to β-catenin in CRC cells and that PAK1 knockdown inhibited β-catenin expression and β-catenin/TCF4 transcriptional activity.17 Taken together, PAK1 facilitates cross-talk between the Ras effector pathways and the Wnt signaling pathway, which is important in tumorigenesis, and thus becomes an appealing target in cancer therapy (Figure 2).
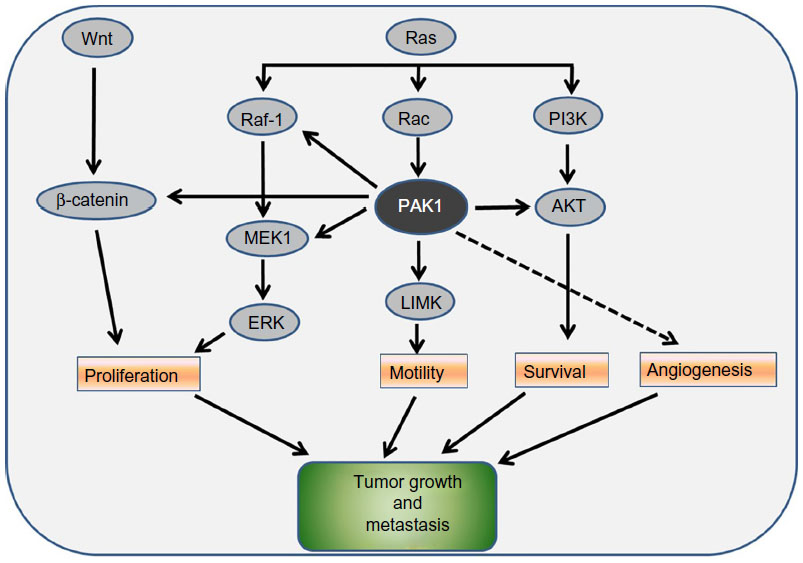 | Figure 2 PAK1 functions as a node for multiple signaling pathways. |
Accumulating evidence indicates that micro (mi)RNAs play a key role in a wide range of biological functions, including cellular proliferation, differentiation, and apoptosis in cancer. Emerging evidence shows that PAKs are regulated by a number of miRNAs that are also recognized to promote hyperactivation of oncogenic Kras signaling. MiRNA-433 inhibits proliferation of hepatocellular carcinoma cells by downregulation of PAK4.57 miRNA-145 inhibits P-ERK expression by targeting PAK4 and leads to inhibition of colon cancer growth.57 Similarly, miRNA-133 inhibits gastric cancer growth by downregulation of CDC42-PAK1 pathway.58 Further investigation of the mechanism(s) of regulation by aberrantly expressed miRNAs of PAKs in cancer and its implications for oncogenic signaling would advance the development of successful therapies targeting PAKs.
PAKs can stimulate cell survival through interaction with the proteins of the Bcl-2 family. PAK1,25,59 PAK4,60 and PAK561,62 can phosphorylate Bad on S112 and/or S136, disabling it from interacting with and inhibiting the prosurvival Bcl-2 and Bcl-xL.62 PAK1 and PAK5 can phosphorylate and translocate Raf-1 to the mitochondria, where it can in turn phosphorylate Bad on S112 to inhibit its proapoptotic activity.24,25,63 In addition, PAK1 can phosphorylate BimL, another proapoptotic protein, dissociating it from Bcl-2, thereby enhancing cell survival.64 Another mechanism employed by PAKs to protect cells from apoptosis involves stimulating the transcription factor nuclear factor kappa B, which regulates genes important for cell survival and proliferation.3 Also, PAK1 can directly phosphorylate and inhibit the proapoptotic transcription factor FKHR by sequestering it in the cytosol, preventing it from activating transcription of proapoptotic genes.65 Unlike PAK1, PAK4, or PAK5, PAK2 is unique in containing a caspase site that is cleaved by caspase 3 upon apoptotic signaling, generating a constitutively active fragment known as PAK-2p34,66 which can regulate morphological changes during late apoptosis67 and stimulate cell death in Jurkat cells.68 The full-length PAK2 localizes in the cytoplasm and promotes cell survival in a manner similar to PAK1 phosphorylation of Bad. PAK2 is reported to phosphorylate and inhibit caspase-7 in breast cancer cells, hence reducing cell apoptosis.69 The dual role of PAK2 in promoting cell survival or apoptosis is due to differential regulation of subcellular localization of PAK2 and PAK-2p34, where the latter is localized to the nucleus to activate a proapoptotic substrate.66
Cell migration requires formation of filopodia and lamellipodia at the leading edge, establishing new adhesions at the forefront, detaching adhesions at the trailing edge, and contracting to propel the cell forward. PAKs coordinate the formation of new adhesions at the leading edge with contraction and detachment at the trailing edge of human microvascular endothelial cells.70 PAK1 was observed to localize to focal adhesions in fibroblasts, and when stimulated by platelet-derived growth factor, PAK1 redistributed into the dorsal and membrane ruffles and into the edges of lamellipodia, where it colocalized with polymerized actin.71,72 Constitutively active PAK1 induced rapid formation of filopodia and membrane ruffles in mammalian cells73 and enhanced cell motility, invasiveness, and anchorage-independent growth.74,75 PAKs appear to control actin filaments turnover and assembly via its downstream target LIM kinase. PAK121 phosphorylates LIM kinase on T508, which in turn phosphorylates cofilin to prevent actin depolymerization.76,77 LIM kinase is also reported to be activated in a PAK2-dependent manner.78 In addition, PAK1 phosphorylates the p41-ARC subunit of the Apr2/3 complex, which is important for branching of the network of actin filaments in the cell cortex, contributing to extension of the leading edge.22 PAKs can enhance cell motility by controlling microtubule stability, mediated by the microtubule-destabilizing protein Op18/Stathmin. PAK1 mediates the phosphorylation of Stathmin by Rac1 and Cdc42, blocking its ability to destabilize microtubule, resulting in net growth at the cell’s leading edge.79,80 PAK1 can directly phosphorylate the tubulin cofactor B, and this is essential for polymerization of new microtubules.81 PAK-mediated phosphorylation of myosin light chain kinase inhibits phosphorylation of myosin light chain, leading to reduced stress fiber formation.23,70,74 For a tumor cell to invade and metastasize, elevated cell motility must be parallel to destruction and reorganization of the extracellular matrix.26 Matrix metalloproteinases (MMPs) control the restructure of the extracellular matrix and mediate human tumor metastasis.82 Increased activity of PAK1 in breast cancer cells induces expression of MMP1 and MMP3 and increased cell invasion.83,84 PAK5 regulates breast cancer and glioma cell migration and invasion, possibly through Egr1-MMP2 signaling pathway.85,86 PAK4 associated with MMP2, and inhibition of PAK4 by small interfering RNA decreased tumor growth by inhibiting MMP-2, β3-integrin, and phosphorylated epidermal growth factor levels in tumors.87 In addition, PAKs are speculated to further facilitate this process by downregulation of adhesion junctions to increase cell permeability and by stimulation of angiogenesis.27,88–91
PAKs as therapeutic targets
The unique central position of PAKs in many intersecting signaling pathways controlling cell proliferation, survival, transformation, and mobility that are frequently deregulated in tumorigenesis and the observation of highly upregulated expression of PAKs in various cancers have validated the rationale for targeting PAKs in treatment of disease, including cancers.
Functional inhibition of PAK1 has been achieved experimentally using a few forms of dominant-negative mutants, aiming to exploit the protein–protein interaction properties of PAKs, including kinase-dead mutant (K299A) and expressing the AID alone. Either approach had been successful to some degree in suppressing kinase-dependent functions of PAKs both in vitro and in vivo.55,92 For example, PAK1 transgenic mice expressing a kinase-dead form of PAK1 ameliorate some symptoms in a mouse model of fragile X syndrome.93 However, PAKs do have kinase-independent activities, most of which are implicated in the scaffolding and cytoskeletal reorganization. Attempts to block their functions in this respect include a PAK1-specific cell-permeable small peptide called WR-PAK18, which contains a PIX-binding site. This peptide was demonstrated to block Ras-induced malignant transformation in fibroblasts.94 Another small peptide containing a Nck-binding motif effectively disrupted PAK1-Nck interaction and consequentially, blocked cell migration, contractility, and tube formation in endothelial cells.91 Despite this, the use of dominant-negative forms or peptides to inhibit PAK1 still faces the question of specificity and functional redundancy as it is difficult to delineate the functions of PAK1 from PAK2 and PAK3.
The next approach is using RNA interference. The advantage is that it overcomes the issue of non-specificity presented by the dominant-negative forms, providing improved discrimination between the different PAK isoforms. However, the difficulties associated with RNA interference are unoptimized delivery method, stability, and off-target effects.95
Early generations of PAK inhibitors have focused on ATP-competitive compounds, a typical approach to inhibit protein kinases. Extensive structural studies have revealed the ATP binding and substrate catalysis pocket deeply tucked in the cleft between the N-terminal and C-terminal lobes of PAK1.43 This catalytic pocket is unusually large and highly flexible, and in addition to the highly mobile N lobe, presents a challenge in developing a PAK inhibitor.43 Several broad-range kinase inhibitors demonstrate potent PAK inhibition; however, with poor selection due to the strong similarity between the ATP binding pocket of kinases.43 Such non-selective compounds have limited use clinically because of undesired side effects. Attempts to achieve higher selectivity identified the bulky ATP antagonist, CEP-1347. Although CEP-1347 inhibited both PAK1 and MLKs and suppressed PAK-dependent growth of Ras-transformed cells,96 disappointingly, it was later demonstrated to have more selectivity for MLK3 and poor potency for PAK1, with an IC50 (half maximal inhibitory concentration) value of more than 1 μM, which is too low to be of clinical importance.97 Further attempts to exploit the capacious ATP binding pocket of PAKs identified an organoruthenium compound F172, which showed improved selectivity and potency for PAK1, with an IC50 value of around 100 nM.98 Nevertheless, compounds based on organometal conjugates like this usually display poor solubility and high toxicity, so their use in clinical settings is questionable.27,43
An ATP-competitive pyrrolopyrazole pan-PAK inhibitor, PF-3758309, was the first PAK inhibitor to enter clinical trials and was developed by Pfizer.99 Although designed as a PAK4 inhibitor, it effectively inhibits all PAK members in addition to other off-target protein kinases. PF-3758309 has highest affinity for PAK4 (Kd 2.7 nM), followed by PAK1, PAK5, and PAK6 (Kd 14–18 nM), and lowest affinity for PAK2 and PAK3 (Kd 100–190 nM). Preclinical evaluation demonstrated great potency of PF-3758309 in suppressing proliferation of a panel of tumor cell lines, with an impressive IC50 of less than 10 nM and high antitumor effects in human xenograft tumor models.100 However, a clinical trial in patients with advanced solid tumors was prematurely terminated in Phase I due to low bioavailability, lack of responses in some instances, and in particular adverse effects43 (http://clinicaltrials.gov/show/NCT00932126).
Recently, Licciulli et al discovered a group I-specific ATP-competitive PAK inhibitor, FRAX597,101 and illustrated selectivity and potency of FRAX597 against group I PAKs, with a biochemical IC50 value of 7–20 nM, while group II PAKs were spared from inhibition even at a concentration higher than 10 μM. Despite this, FRAX597 also exhibits potency against other kinases, especially the receptor tyrosine kinases. FRAX597 effectively blocked in vitro growth of NF2-deficient Schwann cells as well as tumor formation involving these cells in a xenograft model. In a Kras-driven skin cancer model, treatment with FRAX597 inhibited tumorigenesis associated with near-abolished PAK activation, the reduction of total PAK1 and PAK2 levels and the reduced activity of both ERK and AKT to a level comparable with that of PAK1 knockout mice.53 These data highlight the importance of PAK1 signaling cross-talk with the two Ras effector pathways. Interestingly, the decreased PAK1 and PAK2 expression but not their kinase activity is abolished in cells treated with the proteasome inhibitor, MG132, suggesting that FRAX597 has a dual inhibitory effect on group I PAKs, ie, acting as an ATP-competitive inhibitor and a destabilizing agent.27,53 It is worth noting that the inhibitory effects observed in the two models here are consistent with the pan-PAK inhibitor, PF-3758309, yielding similar results. Although both FRAX597 and PF-3758309 have off-target effects on other kinases, these targets are largely non-overlapping. This suggests that the similar antitumor effects exerted by both compounds can be, at least in part, attributed to inhibition of PAKs.
In addition, Zhang et al identified LCH-7749944 as a potent inhibitor of PAK4.102 LCH-7749944 inhibited PAK4 kinase activity with an IC50 of 14.93 μM, and had less of an inhibitory effect against PAK1, PAK5, and PAK6. They predicted a binding model of LCH-7749944 in the PAK4 ATP binding pocket, suggesting that LCH-7749944 acts as an ATP-competitive inhibitor. They further reported that LCH-7749944 inhibited gastric cancer growth and migration/invasion via suppression of PAK4-mediated signaling pathways.
Considering the wide breadth of off-target effects the ATP-competitive compounds have in addition to their intended target due to the highly conserved catalytic pocket present in all kinases, the successful ATP-competitor imatinib (Gleevec®)3 achieved high kinase selectivity by binding to a less conserved region near the ATP binding pocket, which accentuated the feasibility of developing inhibitors interacting with less conserved regions of the kinase. This alternative approach seems especially appealing in the tightly regulated group I PAKs by autoinhibition, which presents an opportunity for allosteric inhibition during the multistep conformational changes accompanying kinase activation. Indeed, Deacon et al reported the discovery of a small molecule allosteric inhibitor, IPA-3,103 which selectively inhibited group I PAKs, with greatest potency for PAK1, displaying isoform selectivity within group I PAKs. Mechanistically, IPA-3 was found to bind covalently to the AID, preventing binding of Cdc42 to PAK, thus effectively blocked Cdc42-triggered conformational changes and subsequent activation of PAKs.104 In support of this finding, IPA-3 was essentially ineffective in inhibiting preactivated PAKs. As expected, group II PAKs are insensitive to IPA-3 because their activation is not regulated by AID and is therefore Cdc42-independent. In addition, IPA-3 showed a very limited inhibitory effect on a panel of diverse kinases, demonstrating high kinase specificity. Regrettably, the disulfide covalent bond that is critical for the IPA-3 inhibitory effect can be reduced in the cytoplasm by reducing agents, thereby reversing the binding of IPA-3 to and inhibition of PAKs. These limitations render IPA-3 unsuitable for clinical use. Nonetheless, allosteric inhibition provided an approach from a different angle, ie, targeting the PAK activation mechanism rather than its kinase activity.
Future prospects
A substantial amount of research has been conducted to decipher the roles of PAKs in a range of diseases, with a focus on cancer. Increasing experimental evidence affirm PAKs as potential drug targets due to these observations: PAKs are frequently upregulated in various forms of human cancers, with their overexpression being correlated with disease progression. PAKs mediate the effects of major signaling pathways, which are often deregulated during cell transformation, such as the Ras/Raf/MEK/ERK, PI3K/AKT/mammalian target of rapamycin and Wnt/β-catenin pathways. PAKs occupy a unique central position where many oncogenic signaling pathways intersect, enabling cross-talk between them. PAKs also have an established role in many cellular processes that are the hallmarks of cancer initiation, growth, and metastasis. Thus, inhibition of PAKs is attracting increasing interest. The search for PAK inhibitors started in the late 1990s and has intensified over the years. Despite this, only a handful of inhibitors have been developed to date due to the challenges mentioned in the previous section. The currently available inhibitors still face the issue of kinase selectivity and isoform specificity. Such limitations preclude the advancement of these compounds to clinical trials. In cancers, the ideal choice of target should have a critical role in disease progression and persistence while having a more dispensable role for the survival of the whole organism as to enable a more cancer-targeted effect that is tolerable by the subject undergoing treatment. In the case of PAK1, PAK1 knockout mice develop an overall normal phenotype with normal life expectancy and health,105 suggesting that PAK1-targeted therapy may be well tolerated by the patient. However, given the highly conserved features among the PAKs, especially group I PAKs, isoform specificity in targeting PAKs may trigger compensatory effects by other isoforms due to functional redundancy. This has a more serious implication in the treatment of cancer, ie, acquired resistance to therapy. As PAKs also play important roles physiologically, targeting a single isoform may cause less side effects than targeting all of them. After all, combined knockout of all PAK isoforms has not been achieved and therefore the consequences are still unknown. On the other hand, multiple isoform inhibition of PAK may be necessary to circumvent therapeutic resistance caused by functional redundancy, thereby increasing the risk of side effects. The current understanding of the functions of PAKs has advanced and continues to improve. Nonetheless, there are still gaps in our knowledge that need to be filled, such as the precise underlying mechanism for PAK-targeted therapeutic resistance and identification of substrates unique to specific isoforms. Such knowledge will help to advance the development and improvement of PAK inhibitors, which can conquer the current obstacles of isoform specificity in parallel with functional redundancy, off-target effects, and acquired resistance to anti-PAK therapies.
Disclosure
The authors report no conflicts of interest in this work.
References
Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. | |
Molli PR, Li DQ, Murray BW, Rayala SK, Kumar R. PAK signaling in oncogenesis. Oncogene. 2009;28:2545–2555. | |
Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 2009;28:51–63. | |
Whale A, Hashim FN, Fram S, Jones GE, Wells CM. Signalling to cancer cell invasion through PAK family kinases. Front Biosci (Landmark Ed). 2011;16:849–864. | |
Rane CK, Minden A. P21 activated kinases: structure, regulation, and functions. Small GTPases. March 21, 2014. [Epub ahead of print.] | |
King H, Nicholas NS, Wells CM. Role of p-21-activated kinases in cancer progression. Int Rev Cell Mol Biol. 2014;309:347–387. | |
Wells CM, Jones GE. The emerging importance of group II PAKs. Biochem J. 2010;425:465–473. | |
Szczepanowska J. Involvement of Rac/Cdc42/PAK pathway in cytoskeletal rearrangements. Acta Biochim Pol. 2009;56:225–234. | |
Ching YP, Leong VY, Wong CM, Kung HF. Identification of an autoinhibitory domain of p21-activated protein kinase 5. J Biol Chem. 2003;278:33621–33624. | |
Baskaran Y, Ng YW, Selamat W, Ling FT, Manser E. Group I and II mammalian PAKs have different modes of activation by Cdc42. EMBO Rep. 2012;13:653–659. | |
Ha BH, Davis MJ, Chen C, et al. Type II p21-activated kinases (PAKs) are regulated by an autoinhibitory pseudosubstrate. Proc Natl Acad Sci U S A. 2012;109:16107–16112. | |
Menges CW, Sementino E, Talarchek J, et al. Group I p21-activated kinases (PAKs) promote tumor cell proliferation and survival through the AKT1 and Raf-MAPK pathways. Mol Cancer Res. 2012;10:1178–1188. | |
Huynh N, Liu KH, Baldwin GS, He H. P21-activated kinase 1 stimulates colon cancer cell growth and migration/invasion via ERK- and AKT-dependent pathways. Biochim Biophys Acta. 2010;1803:1106–1113. | |
Beeser A, Jaffer ZM, Hofmann C, Chernoff J. Role of group A p21-activated kinases in activation of extracellular-regulated kinase by growth factors. J Biol Chem. 2005;280:36609–36615. | |
King AJ, Sun H, Diaz B, et al. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature. 1998;396:180–183. | |
Tran NH, Frost JA. Phosphorylation of Raf-1 by p21-activated kinase 1 and Src regulates Raf-1 autoinhibition. J Biol Chem. 2003;278:11221–11226. | |
He H, Huynh N, Liu KH, et al. P-21 activated kinase 1 knockdown inhibits beta-catenin signalling and blocks colorectal cancer growth. Cancer Lett. 2012;317:65–71. | |
Park MH, Kim DJ, You ST, et al. Phosphorylation of beta-catenin at serine 663 regulates its transcriptional activity. Biochem Biophys Res Commun. 2012;419:543–549. | |
Balasenthil S, Sahin AA, Barnes CJ, et al. p21-activated kinase-1 signaling mediates cyclin D1 expression in mammary epithelial and cancer cells. J Biol Chem. 2004;279:1422–1428. | |
Zhao ZS, Lim JP, Ng YW, Lim L, Manser E. The GIT-associated kinase PAK targets to the centrosome and regulates Aurora-A. Mol Cell. 2005;20:237–249. | |
Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. | |
Vadlamudi RK, Li F, Barnes CJ, Bagheri-Yarmand R, Kumar R. p41-Arc subunit of human Arp2/3 complex is a p21-activated kinase-1-interacting substrate. EMBO Rep. 2004;5:154–160. | |
Sanders LC, Matsumura F, Bokoch GM, de Lanerolle P. Inhibition of myosin light chain kinase by p21-activated kinase. Science. 1999;283:2083–2085. | |
Jin S, Zhuo Y, Guo W, Field J. p21-activated kinase 1 (Pak1)-dependent phosphorylation of Raf-1 regulates its mitochondrial localization, phosphorylation of BAD, and Bcl-2 association. J Biol Chem. 2005;280:24698–24705. | |
Ye DZ, Jin S, Zhuo Y, Field J. p21-Activated kinase 1 (PAK1) phosphorylates BAD directly at serine 111 in vitro and indirectly through Raf-1 at serine 112. PLoS One. 2011;6:e27637. | |
Kichina JV, Goc A, Al-Husein B, Somanath PR, Kandel ES. PAK1 as a therapeutic target. Expert Opin Ther Targets. 2010;14:703–725. | |
Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer. 2014;14:13–25. | |
Baker NM, Yee Chow H, Chernoff J, Der CJ. Molecular pathways: targeting RAC-p21-activated serine-threonine kinase signaling in RAS-driven cancers. Clin Cancer Res. 2014;20:4740–4746. | |
Ye DZ, Field J. PAK signaling in cancer. Cell Logist. 2012;2:105–116. | |
Ong CC, Jubb AM, Haverty PM, et al. Targeting p21-activated kinase 1 (PAK1) to induce apoptosis of tumor cells. Proc Natl Acad Sci U S A. 2011;108:7177–7182. | |
Bostner J, Skoog L, Fornander T, Nordenskjold B, Stal O. Estrogen receptor-alpha phosphorylation at serine 305, nuclear p21-activated kinase 1 expression, and response to tamoxifen in postmenopausal breast cancer. Clin Cancer Res. 2010;16:1624–1633. | |
Bostner J, Ahnstrom Waltersson M, Fornander T, Skoog L, Nordenskjold B, Stal O. Amplification of CCND1 and PAK1 as predictors of recurrence and tamoxifen resistance in postmenopausal breast cancer. Oncogene. 2007;26:6997–7005. | |
Brown LA, Kalloger SE, Miller MA, et al. Amplification of 11q13 in ovarian carcinoma. Genes Chromosomes Cancer. 2008;47:481–489. | |
Zhu G, Wang Y, Huang B, et al. A Rac1/PAK1 cascade controls beta-catenin activation in colon cancer cells. Oncogene. 2012;31:1001–1012. | |
Carter JH, Douglass LE, Deddens JA, et al. PAK-1 expression increases with progression of colorectal carcinomas to metastasis. Clin Cancer Res. 2004;10:3448–3456. | |
Gong W, An Z, Wang Y, et al. P21-activated kinase 5 is overexpressed during colorectal cancer progression and regulates colorectal carcinoma cell adhesion and migration. Int J Cancer. 2009;125:548–555. | |
Chen S, Auletta T, Dovirak O, et al. Copy number alterations in pancreatic cancer identify recurrent PAK4 amplification. Cancer Biol Ther. 2008;7:1793–1802. | |
Kimmelman AC, Hezel AF, Aguirre AJ, et al. Genomic alterations link Rho family of GTPases to the highly invasive phenotype of pancreas cancer. Proc Natl Acad Sci U S A. 2008;105:19372–11937. | |
Begum A, Imoto I, Kozaki K, et al. Identification of PAK4 as a putative target gene for amplification within 19q13.12–q13.2 in oral squamous-cell carcinoma. Cancer Sci. 2009;100:1908–1916. | |
Kissil JL, Wilker EW, Johnson KC, Eckman MS, Yaffe MB, Jacks T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Mol Cell. 2003;12:841–849. | |
Hirokawa Y, Tikoo A, Huynh J, et al. A clue to the therapy of neurofibromatosis type 2: NF2/merlin is a PAK1 inhibitor. Cancer J. 2004;10(1):20–26. | |
Fawdar S, Trotter EW, Li Y, et al. Targeted genetic dependency screen facilitates identification of actionable mutations in FGFR4, MAP3K9, and PAK5 in lung cancer. Proc Natl Acad Sci U S A. 2013;110:12426–12431. | |
Rudolph J, Crawford JJ, Hoeflich KP, Wang W. Inhibitors of p21-activated kinases (PAKs). J Med Chem. 2015;58:111–129. | |
Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. | |
Andreyev HJ, Norman AR, Cunningham D, Oates JR, Clarke PA. Kirsten ras mutations in patients with colorectal cancer: the multicenter “RASCAL” study. J Natl Cancer Inst. 1998;90:675–684. | |
Arrington AK, Heinrich EL, Lee W, et al. Prognostic and predictive roles of KRAS mutation in colorectal cancer. Int J Mol Sci. 2012;13:12153–12168. | |
Bamford S, Dawson E, Forbes S, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004; 91:355–358. | |
Benhattar J, Losi L, Chaubert P, Givel JC, Costa J. Prognostic significance of K-ras mutations in colorectal carcinoma. Gastroenterology. 1993;104:1044–1048. | |
Chaudhary A, King WG, Mattaliano MD, et al. Phosphatidylinositol 3-kinase regulates Raf1 through PAK phosphorylation of serine 338. Curr Biol. 2000;10:551–554. | |
Frost JA, Steen H, Shapiro P, et al. Cross-cascade activation of ERKs and ternary complex factors by Rho family proteins. EMBO J. 1997;16:6426–6438. | |
Higuchi M, Onishi K, Kikuchi C, Gotoh Y. Scaffolding function of PAK in the PDK1-Akt pathway. Nat Cell Biol. 2008;10:1356–1364. | |
Li LH, Zheng MH, Luo Q, et al. P21-activated protein kinase 1 induces colorectal cancer metastasis involving ERK activation and phosphorylation of FAK at Ser-910. Int J Oncol. 2010;37:951–962. | |
Chow HY, Jubb AM, Koch JN, et al. p21-Activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 2012;72:5966–5975. | |
Tabusa H, Brooks T, Massey AJ. Knockdown of PAK4 or PAK1 inhibits the proliferation of mutant KRAS colon cancer cells independently of RAF/MEK/ERK and PI3K/AKT signaling. Mol Cancer Res. 2013;11:109–121. | |
He H, Shulkes A, Baldwin GS. PAK1 interacts with beta-catenin and is required for the regulation of the beta-catenin signalling pathway by gastrins. Biochim Biophys Acta. 2008;1783:1943–1954. | |
Arias-Romero LE, Villamar-Cruz O, Huang M, Hoeflich KP, Chernoff J. Pak1 kinase links ErbB2 to beta-catenin in transformation of breast epithelial cells. Cancer Res. 2013;73:3671–3682. | |
Xue J, Chen LZ, Li ZZ, Hu YY, Yan SP, Liu LY. MicroRNA-433 inhibits cell proliferation in hepatocellular carcinoma by targeting p21 activated kinase (PAK4). Mol Cell Biochem. 2015;399:77–86. | |
Cheng Z, Liu F, Wang G, Li Y, Zhang H, Li F. miR-133 is a key negative regulator of CDC42-PAK pathway in gastric cancer. Cell Signal. 2014;26:2667–2673. | |
Schurmann A, Mooney AF, Sanders LC, et al. p21-activated kinase 1 phosphorylates the death agonist BAD and protects cells from apoptosis. Mol Cell Biol. 2000;20:453–461. | |
Gnesutta N, Qu J, Minden A. The serine/threonine kinase PAK4 prevents caspase activation and protects cells from apoptosis. J Biol Chem. 2001;276:14414–14419. | |
Wang X, Gong W, Qing H, et al. p21-activated kinase 5 inhibits camptothecin-induced apoptosis in colorectal carcinoma cells. Tumour Biol. 2010;31:575–582. | |
Cotteret S, Jaffer ZM, Beeser A, Chernoff J. p21-Activated kinase 5 (Pak5) localizes to mitochondria and inhibits apoptosis by phosphorylating BAD. Mol Cell Biol. 2003;23(16):5526–5539. | |
Wu X, Carr HS, Dan I, Ruvolo PP, Frost JA. p21 activated kinase 5 activates Raf-1 and targets it to mitochondria. J Cell Biochem. 2008;105:167–175. | |
Vadlamudi RK, Bagheri-Yarmand R, Yang Z, et al. Dynein light chain 1, a p21-activated kinase 1-interacting substrate, promotes cancerous phenotypes. Cancer Cell. 2004;5:575–585. | |
Mazumdar A, Kumar R. Estrogen regulation of Pak1 and FKHR pathways in breast cancer cells. FEBS Lett. 2003;535:6–10. | |
Jakobi R, McCarthy CC, Koeppel MA, Stringer DK. Caspase-activated PAK-2 is regulated by subcellular targeting and proteasomal degradation. J Biol Chem. 2003;278:38675–38685. | |
Rudel T, Bokoch GM. Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science. 1997;276:1571–1574. | |
Rudel T, Zenke FT, Chuang TH, Bokoch GM. p21-activated kinase (PAK) is required for Fas-induced JNK activation in Jurkat cells. J Immunol. 1998;160:7–11. | |
Li X, Wen W, Liu K, et al. Phosphorylation of caspase-7 by p21-activated protein kinase (PAK) 2 inhibits chemotherapeutic drug-induced apoptosis of breast cancer cell lines. J Biol Chem. 2011;286:22291–22299. | |
Kiosses WB, Daniels RH, Otey C, Bokoch GM, Schwartz MA. A role for p21-activated kinase in endothelial cell migration. J Cell Biol. 1999;147:831–844. | |
Dharmawardhane S, Sanders LC, Martin SS, Daniels RH, Bokoch GM. Localization of p21-activated kinase 1 (PAK1) to pinocytic vesicles and cortical actin structures in stimulated cells. J Cell Biol. 1997;138:1265–1278. | |
Sells MA, Pfaff A, Chernoff J. Temporal and spatial distribution of activated Pak1 in fibroblasts. J Cell Biol. 2000;151:1449–1458. | |
Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210. | |
Sells MA, Boyd JT, Chernoff J. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J Cell Biol. 1999;145:837–849. | |
Vadlamudi RK, Adam L, Wang RA, et al. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J Biol Chem. 2000;275:36238–36244. | |
Bagheri-Yarmand R, Mazumdar A, Sahin AA, Kumar R. LIM kinase 1 increases tumor metastasis of human breast cancer cells via regulation of the urokinase-type plasminogen activator system. Int J Cancer. 2006;118:2703–2710. | |
Delorme V, Machacek M, DerMardirossian C, et al. Cofilin activity downstream of Pak1 regulates cell protrusion efficiency by organizing lamellipodium and lamella actin networks. Dev Cell. 2007;13:646–662. | |
Misra UK, Deedwania R, Pizzo SV. Binding of activated alpha2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK. J Biol Chem. 2005;280:26278–26286. | |
Daub H, Gevaert K, Vandekerckhove J, Sobel A, Hall A. Rac/Cdc42 and p65PAK regulate the microtubule-destabilizing protein stathmin through phosphorylation at serine 16. J Biol Chem. 2001;276:1677–1680. | |
Wittmann T, Bokoch GM, Waterman-Storer CM. Regulation of microtubule destabilizing activity of Op18/stathmin downstream of Rac1. J Biol Chem. 2004;279:6196–6203. | |
Vadlamudi RK, Barnes CJ, Rayala S, et al. p21-activated kinase 1 regulates microtubule dynamics by phosphorylating tubulin cofactor B. Mol Cell Biol. 2005;25:3726–3736. | |
Murphy G, Nagase H. Progress in matrix metalloproteinase research. Mol Aspects Med. 2008;29:290–308. | |
Li Q, Mullins SR, Sloane BF, Mattingly RR. p21-Activated kinase 1 coordinates aberrant cell survival and pericellular proteolysis in a three-dimensional culture model for premalignant progression of human breast cancer. Neoplasia. 2008;10:314–329. | |
Rider L, Oladimeji P, Diakonova M. PAK1 regulates breast cancer cell invasion through secretion of matrix metalloproteinases in response to prolactin and three-dimensional collagen IV. Mol Endocrinol. 2013;27:1048–1064. | |
Wang XX, Cheng Q, Zhang SN, et al. PAK5-Egr1-MMP2 signaling controls the migration and invasion in breast cancer cell. Tumour Biol. 2013;34:2721–2729. | |
Han ZX, Wang XX, Zhang SN, et al. Downregulation of PAK5 inhibits glioma cell migration and invasion potentially through the PAK5-Egr1-MMP2 signaling pathway. Brain Tumor Pathol. 2014;31:234–241. | |
Kesanakurti D, Chetty C, Rajasekhar Maddirela D, Gujrati M, Rao JS. Functional cooperativity by direct interaction between PAK4 and MMP-2 in the regulation of anoikis resistance, migration and invasion in glioma. Cell Death Dis. 2012;3:e445. | |
Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–1234. | |
Rettig M, Trinidad K, Pezeshkpour G, et al. PAK1 kinase promotes cell motility and invasiveness through CRK-II serine phosphorylation in non-small cell lung cancer cells. PLoS One. 2012;7:e42012. | |
Guilluy C, Zhang Z, Bhende PM, et al. Latent KSHV infection increases the vascular permeability of human endothelial cells. Blood. 2011;118:5344–5354. | |
Kiosses WB, Hood J, Yang S, et al. A dominant-negative p65 PAK peptide inhibits angiogenesis. Circ Res. 2002;90:697–702. | |
Frost JA, Khokhlatchev A, Stippec S, White MA, Cobb MH. Differential effects of PAK1-activating mutations reveal activity-dependent and -independent effects on cytoskeletal regulation. J Biol Chem. 1998;273:28191–28198. | |
Hayashi ML, Rao BS, Seo JS, et al. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci U S A. 2007;104:11489–11494. | |
He H, Hirokawa Y, Manser E, Lim L, Levitzki A, Maruta H. Signal therapy for RAS-induced cancers in combination of AG 879 and PP1, specific inhibitors for ErbB2 and Src family kinases, that block PAK activation. Cancer J. 2001;7:191–202. | |
Uprichard SL. The therapeutic potential of RNA interference. FEBS Lett. 2005;579:5996–6007. | |
Nheu TV, He H, Hirokawa Y, et al. The K252a derivatives, inhibitors for the PAK/MLK kinase family selectively block the growth of RAS transformants. Cancer J. 2002;8:328–336. | |
Maroney AC, Finn JP, Connors TJ, et al. Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem. 2001;276:25302–25308. | |
Maksimoska J, Feng L, Harms K, et al. Targeting large kinase active site with rigid, bulky octahedral ruthenium complexes. J Am Chem Soc. 2008;130:15764–15765. | |
Guo C, McAlpine I, Zhang J, et al. Discovery of pyrroloaminopyrazoles as novel PAK inhibitors. J Med Chem. 2012;55:4728–4739. | |
Murray BW, Guo C, Piraino J, et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc Natl Acad Sci U S A. 2010;107:9446–9451. | |
Licciulli S, Maksimoska J, Zhou C, et al. FRAX597, a small molecule inhibitor of the p21-activated kinases, inhibits tumorigenesis of neurofibromatosis type 2 (NF2)-associated schwannomas. J Biol Chem. 2013;288:29105–29114. | |
Zhang J, Wang J, Guo Q, et al. LCH-7749944, a novel and potent p21-activated kinase 4 inhibitor, suppresses proliferation and invasion in human gastric cancer cells. Cancer Lett. 2012;317:24–32. | |
Deacon SW, Beeser A, Fukui JA, et al. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem Biol. 2008;15:322–331. | |
Viaud J, Peterson JR. An allosteric kinase inhibitor binds the p21-activated kinase autoregulatory domain covalently. Mol Cancer Ther. 2009;8:2559–2565. | |
Hofmann C, Shepelev M, Chernoff J. The genetics of Pak. J Cell Sci. 2004;117 Pt 19:4343–4354. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.