")
Back to Journals » Journal of Pain Research » Volume 14
P-Selectin Blockade in the Treatment of Painful Vaso-Occlusive Crises in Sickle Cell Disease: A Spotlight on Crizanlizumab
Received 19 January 2021
Accepted for publication 13 March 2021
Published 30 March 2021 Volume 2021:14 Pages 849—856
DOI https://doi.org/10.2147/JPR.S278285
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Robert B. Raffa
Nabin Raj Karki, Abdullah Kutlar
Division of Hematology/Oncology, Augusta University, Augusta, GA, USA
Correspondence: Nabin Raj Karki
Division of Hematology Oncology, Georgia Cancer Center at Augusta University, 1410, Laney Walker Blvd, CN 5347, Augusta, GA, 30912, USA
Tel +1-706-721-2505
Fax +1-706-721-5566
Email [email protected]
Abstract: Microvascular vaso-occlusion driven pain crisis is the hallmark of sickle cell disease with profound morbidity and increased mortality. Selectins, most notably P-selectins have an integral role in this phenomenon. P-selection was first identified in 1989. In 2019, after 3 decades of basic, translational, and clinical work with this pathway, the US Food and Drug Administration approved a P-selectin antibody, crizanlizumab to reduce frequency of pain crisis in patients more than 16 years with sickle cell disease. We review the fundamentals of P-selectin pathobiology, P-selectin blocking agents, clinical data with the use of crizanlizumab and prospects of this novel class of drugs in the context of other treatments for painful vaso-occlusive episodes.
Keywords: P-selectin, sickle cell disease, pain crisis, crizanlizumab
Introduction
Sickle cell disease (SCD) is an inherited red blood cell (RBC) disorder resulting from a GAG→GTG substitution (glutamic acid→valine) in the sixth codon of the β-globin subunit of human adult hemoglobin. The resultant variant hemoglobin, Hb S, polymerizes upon deoxygenation, leading to distortion of the RBC shape and rheologic impairment. The downstream effects of this phenomenon include chronic hemolytic anemia, microvascular occlusion, and activation of microvascular endothelium and inflammation, which culminate in chronic organ damage, impaired quality of life, and early mortality. Painful vaso-occlusive episodes (VOEs) or sickle cell-related pain crises are characteristic of SCD and are responsible for 73.7% of emergency department visits and ≈95% of hospital admissions.1,2 VOEs are manifested as acute and recurrent attacks of painful extremities and of the back, joint, abdomen and chest lasting for ≈10 days.3 VOEs result in a reduced quality of life, low self-esteem, anxiety, depression, body image dissatisfaction, poor school performance, social isolation, decreased participation in normal daily activities, poor peer and family relationships, and an increased mortality risk.4
Polymerization of deoxygenated hemoglobin causes changes in the morphology of erythrocytes, apparent as sickled RBCs. While sickled RBCs cause occlusion of smaller vessels, activated and adherent leukocytes are drivers of vaso-occlusion in postcapillary venules.5 The vaso-occlusion leads to tissue ischemia, and VOEs are often an antecedent to acute chest syndrome. Hepatic sequestration, splenic sequestration, priapism, and bacterial infections are more serious but less common acute complications seen in patients with SCD.6 Long-term manifestations of SCD include chronic pain, stroke, pulmonary hypertension, leg ulcers, avascular necrosis of hips or shoulders, retinopathy, and renal failure.6
SCD encompasses multiple autosomal recessive genotypes in which hemoglobin S may exist in a homozygous state (HbSS) or in a heterozygous state where it co-exists with other globin mutations, such as hemoglobin C (HbSC), hemoglobin D (HbSD), hemoglobin E (HbSE), or β-thalassemia (HbSβ0 or HbSβ+). There is large phenotypic variability within each SCD genotype, both in terms of VOEs and other sickle cell-related complications. In general, the sickle cell anemia (SCA) genotypes HbSS and HbSβ0 are typically more severe than other milder forms, such as HbSC and HbSβ+. HbSD runs a course similar to that of SCA but can uniquely be associated with cerebrovascular complications.7 Patients with HbSE have mild anemia, without typical SCD manifestations.
P-Selectin and Vaso-Occlusion
There are 3 members of the selectin family of cytoadhesion molecules: E-selectin, P-selectin, and L-selectin. P-selectin is found in storage granules of resting endothelial cells (Weibel-Palade bodies) and platelets (alpha granules) and is expressed on cell membranes upon activation.8 P-selectin expression in platelets is a marker of platelet activation. Some potent activator signals are thrombin, complement components, reactive oxygen species, hypoxia, hypoxia-reoxygenation, and heme. P-selectin glycoprotein ligand-1 (PSGL-1) is present on leukocytes; a PSGL-1-like ligand is also expressed on RBCs.9 The P-selectin and PSGL-1 pairing leads to interactions between endothelial cells and sickled RBCs, endothelial cells and platelets, platelets and leukocytes, and platelets and sickled RBCs that underlie SCD complications including VOEs. P-selectin mediates platelet activation, coagulation, and inflammation and has been found to be the most important of all selectins for initiation and maintenance of the cascade of events triggered by leukocyte adhesion to vascular endothelium during VOEs.10 Vaso-occlusion is a complex process heralded by adhesion of sickled RBCs and leukocytes to the endothelium, leading to vascular obstruction and tissue ischemia.11 Activated platelet P-selectin also promotes lung vaso-occlusion in SCD by forming arteriolar neutrophil-platelet microemboli.12 The combination of hypoxia/reperfusion injury, ischemic tissue damage, inflammation, and altered nociception culminates in a full-blown VOE.3
P-Selectin and Vascular Hemodynamics
Abnormal blood flow in the microvasculature is the hallmark of SCD. While viscosity is a more important determinant of blood flow in large vessels, individual RBC deformability is of greater consequence to microcirculatory flow.13 The tight squeeze of flowing RBCs through the smallest vessels exposes the entire circumference to endothelial adhesion molecules.14 In SCD, endothelial cells chronically express P-selectin.15,16 Contact between sickled RBCs and endothelial P-selectin is central to initiation of the vaso-occlusive cascade by downstream engagement of multiple adhesion receptor/ligand pairs.17 The adhesion of sickled RBCs to endothelial cells results in increased levels of reactive oxygen species within endothelial cells, which upregulates the expression of P-selectin on the cell surface.18,19 This positive feedback cycle accounts for the slower microcirculatory flow of sickled RBCs.13 While P-selectin-mediated adhesion is a key event in the initiation of VOEs, multiple mechanisms are established subsequently.20
P-Selectin Inhibition in Mice Models with SCD
Adhesion by selectin requires hemodynamic shear stress.21 Inhibition of P-selectin-mediated adhesion pathways significantly reduces the adherence of sickled RBCs and leucocytes to endothelium and improves microvascular flow velocities in transgenic mice expressing HbSS.22 Chimeric mice with HbSS but deficient in P-selectin and E-selectin created by transplanting SCD mice bone marrow in nonsickle P- and E-selectin knockout mice are protected from vaso-occlusion.23 Not surprisingly, transgenic P-selectin-deficient SCD mice are protected from lung vaso-occlusion, which proves the crucial role of P-selectin in acute chest syndrome as well.24
P-Selectin Inhibition in Humans with SCD
Thus, P-selectin inhibition has long been postulated to be of more value in preventing a VOE or, at the most, aborting a VOE in its early stage than in treating an established crisis. Low-dose unfractionated heparin (12- to 50-fold lower concentration than required for anticoagulation) can block P-selectin, and pentosan polysulfate sodium (PPS) has even greater P-selectin-blocking activity than heparin.25,26 In open-label PPS Phase 1/2 studies, it was shown that blocking P-selectin via an oral agent in patients with SCD improved microvascular flow and led to a significant reduction in soluble vascular cell adhesion molecule 1 levels—a marker of endothelial injury that is frequently responsive to chronic transfusions and hydroxyurea.27 PPS was not developed further. Rivipansel, a pan-selectin inhibitor, failed to meet its endpoints in a Phase 3 study, although it had showed a reduction in the time to VOE resolution and intravenous opioid use in a Phase 2 trial.28,29 Sevuparin is a modified form of heparin and a P-selectin and L-selectin inhibitor. It failed to show benefit in treating VOEs, although it was shown to prevent VOEs and to normalize blood flow in animal studies.30 Both drugs were ineffective at treating VOEs and accelerating recovery, in agreement with the concept that P-selectin inhibition is not an effective strategy after a VOE has set in. Recently, crizanlizumab—a monoclonal antibody that inhibits P-selectin function—was studied for its prophylactic use and yielded positive results that led to regulatory approval.31
Crizanlizumab
Crizanlizumab is a humanized immunoglobulin G2κ monoclonal antibody that binds to and inhibits P-selectin, thereby blocking its interaction with its ligand, PSGL-1 (Figure 1). The drug-binding P-selection on the surface of activated endothelium and platelets blocks the interaction between endothelial cells, platelets, sickled RBCs, and leukocytes—leading to improved microvascular flow dynamics.32 Crizanlizumab may also mitigate a VOE because of its ability to reverse some, but not all, established aberrant adhesive interactions.10 The half-life (t1/2) of crizanlizumab is 7.6 days, and it is degraded into small peptides via catabolism.32 The effect of renal or hepatic impairment on crizanlizumab is not known. Moreover, hydroxyurea does not seem to have a meaningful impact on its pharmacokinetics.32 In vitro studies have confirmed the role of crizanlizumab in preventing a VOE by blocking leukocyte adhesions; however, this also poses the theoretical risk of increased infections due to reduced leukocyte adhesions.10 Crizanlizumab did not have any effect on hemolysis.
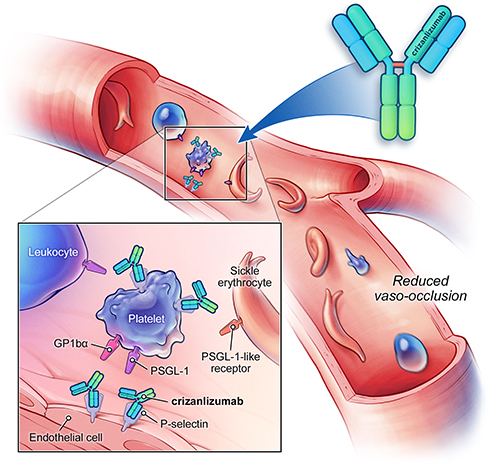 |
Figure 1 Mechanism of action of crizanlizumab. Crizanlizumab is a monoclonal antibody that binds to P-selectin. Blockade of P-selectin may prevent cell–cell interactions between blood cells and endothelial cells, and thus eliminate or reduce vaso-occlusion. Figure reproduced with permission from© 2020 Novartis AG. Abbreviations: GP1bα, glycoprotein 1bα; PSGL-1, P-selectin glycoprotein ligand-1. |
SUSTAIN Trial
SUSTAIN was a phase 2 multicenter, multinational, randomized, placebo-controlled, double-blind, 12-month study that assessed the safety and efficacy of crizanlizumab with or without hydroxyurea therapy in 198 patients with SCD with painful VOEs.33 Eligible patients were 16 to 65 years of age, encompassed all SCD genotypes, and had 2 to 10 VOEs in the 12 months before enrollment. Although patients were not allowed to initiate hydroxyurea, those already receiving hydroxyurea for ≥6 months and on a stable dose were allowed to continue receiving the same dose. Subjects were randomized to placebo, low-dose crizanlizumab (2.5 mg/kg), or high-dose crizanlizumab (5 mg/kg) in a 1:1:1 ratio. High-dose crizanlizumab (5 mg/kg, 2 loading doses 2 weeks apart and then every 4 weeks (maintenance) for a total of 14 doses) resulted in a 45.3% lower annual rate of sickle cell pain crises compared with placebo (median, 1.63 vs 2.98). The median time to first crisis was significantly longer with high-dose crizanlizumab than with placebo (4.07 vs 1.38 months), as was the median time to the second crisis (10.32 vs 5.09 months), representing crisis-free intervals that were almost twice as long. This positive effect was seen regardless of concomitant hydroxyurea use. All genotypes of SCD (HbSS, HbSC, HbSβ0, HbSβ+, and others) derived benefit. A reduction in the annual crisis rate was observed in subjects who had experienced 2 to 4 crises and 5 to 10 crises in the previous year (Figure 2).34 The most common adverse events (arthralgia, diarrhea, pruritus, vomiting, and chest pain) occurred in ≥10% of patients in the active treatment group and at a frequency that was higher than in the placebo group. The most frequent serious adverse events were pyrexia, influenza, and pneumonia. However, crizanlizumab did not show an increased risk of infectious complications compared with placebo. Acute chest syndrome, hepatic/splenic sequestration, and priapism were rare (median rate, 0.00 in all groups). No patients developed a detectable antibody response against crizanlizumab.
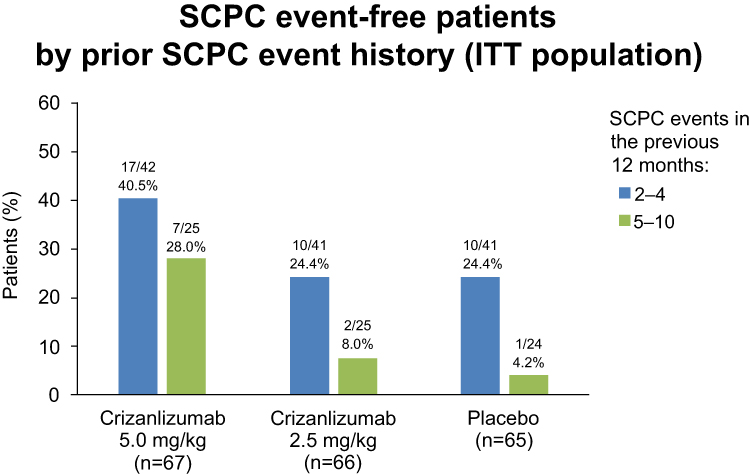 |
Figure 2 SCPC event-free patients in the high-dose crizanlizumab, low-dose crizanlizumab, and placebo subgroups in an ITT population with respect to the annualized crisis rate in the preceding year.Note: Data from Kutlar et al.34 Abbreviations: SCPC, sickle cell pain crisis; ITT, intention to treat. |
In the SUSTAIN study, 35.8% of subjects in the high-dose crizanlizumab had no crisis events during the 52-week study period—a much higher percentage than in the low-dose crizanlizumab (18.2%) and placebo (16.9%) groups (Figure 2).34,35 A post hoc subgroup analysis and a retrospective trial (SUCCESSOR trial) assessed the efficacy of crizanlizumab, using both the data set from the same original SUSTAIN trial and data corroborating the activity of high-dose crizanlizumab at preventing VOEs.35–37 These findings led to the US Food and Drug Administration (FDA) approval of crizanlizumab for the prevention of VOEs in patients with SCD of all genotypes who are >16 years of age.31 In the subgroup analysis, patients who received only high-dose crizanlizumab had a lower annual crisis rate than those who received crizanlizumab with concomitant hydroxyurea (difference from placebo: −50.0% vs −32.1%).38 Similarly, among the patients receiving hydroxyurea, low-dose crizanlizumab was associated with a greater reduction in the annual rate of VOEs (44.1% lower median rate compared with placebo) than high-dose crizanlizumab (32.1% lower median rate compared with placebo).33 The concentration-related interaction with hydroxyurea leading to a reduced effect at higher doses of crizanlizumab was erroneously thought to account for this phenomenon by some authors.38,39 The trial was not powered to detect differences between subgroups, and the sample sizes were small in each subgroup. Patients with more severe disease are more likely to be put on hydroxyurea, potentially influencing the results.38 We believe that high-dose crizanlizumab provides additional benefit when used in combination with hydroxyurea.
Infusion-Related Reactions with Crizanlizumab
Several cases of severe pain during or within 24 hours of the infusion (infusion-related reactions [IRRs] resembling complement activation-mediated pseudo-allergy)—similar to those seen with other monoclonal antibodies—have been noted in postmarketing adverse event reporting.40,41 More than two-thirds (16/22) of patients reporting these IRRs required hospitalization. In the SUSTAIN trial, mild to moderate pain was observed in 5% of patients after infusions, and no severe hypersensitivity was reported. Seven cases of complications—such as acute chest syndrome, fat embolization syndrome, hemolytic crisis, pneumonia, and multiorgan failure—have been reported following the infusion, but all cases resolved.41 Slowing or discontinuation of infusions and appropriate treatment (eg, analgesics, antihistamines) are suggested for mild to moderate IRRs. Steroids are recommended only for anaphylaxis.
Crizanlizumab in Patients on Hydroxyurea
Hydroxyurea is an inhibitor of ribonucleotide reductase and may cause mild bone marrow suppression. Hydroxyurea was approved in 1998 for SCA, and, for almost 2 decades, was the only drug available that modifies the natural history of the disease. Hydroxyurea results in a 44% reduction in the median annual rate of painful crises.42 The majority of patients (74%) report low adherence despite its clinical benefits.43 Although hydroxyurea is still the recommended first-line agent for preventing VOEs in SCA (although not for all patients with SCD), the inclusion of patients on hydroxyurea in the SUSTAIN trial paved the way for its approval and subsequently for the use of crizanlizumab as an adjunct to hydroxyurea.35,44
High-dose crizanlizumab provided additional benefit in combination with hydroxyurea, which is only additive and not synergistic.35,38 The positive effects of hydroxyurea are likely due to increased fetal hemoglobin, improved RBC rheology, and higher circulating nitric oxide levels.45 Hydroxyurea also exerts cytotoxic effects that cause a decreased production of neutrophils, reticulocytes, and platelets.46 In contrast, crizanlizumab targets intercellular adhesion involving RBCs, platelets, neutrophils, and endothelial cells. While hydroxyurea targets RBCs, crizanlizumab targets the vasculature.35 This forms the basis of additional benefit seen with crizanlizumab for patients already on hydroxyurea. Thus far, there is no biological basis for a synergistic or antagonistic interaction between crizanlizumab and hydroxyurea.
Crizanlizumab Vis-à-Vis Other SCD Treatments
In the past, multiple pathophysiological targets have been explored to abrogate VOEs: anticoagulation (aspirin, ticlopidine), antioxidation (ω-3 fatty acid, N-acetylcysteine, l-glutamine, vitamins C and E), and antisickling (sodium bicarbonate, promazine, cromolyn sodium, piracetam, and dietary supplements such as folic acid or zinc).47 Apart from hydroxyurea, an antioxidant and an antiadhesion antibody have been approved by the FDA in last 3 years to prevent/treat VOEs. L-Glutamine was evaluated in a phase 3 trial and is now approved to prevent pain crises in SCD.48
Role of P-Selectin Beyond Vaso-Occlusive Pain Crisis
Platelet P-selectin enhances platelet aggregation and thrombus formation through binding to PSGL-1.53 The increased risk of thromboses in SCD is well known—in tandem with vaso-occlusion, in which selectins are intricately involved.54,55 One patient in the placebo arm and no patients in the crizanlizumab arm had a thrombotic event in the SUSTAIN study.33 In Philadelphia chromosome-negative, JAK2V617F-positive myeloproliferative neoplasms, constitutively active JAK2 results in downstream activation of JAK2 kinase that initiates intracellular signaling of various type 1 cytokine receptors such as erythropoietin and thrombopoietin receptors. In addition to an overproduction of myeloid cells via this mechanism, as many as one-third of patients experience arterial and venous thromboses complications during their lifetime.56 JAK2 is also present in endothelial cells. In a recent investigation using human and mice endothelial cells overexpressing JAK2V617F, the authors showed that JAK2V617F-expressing endothelial cells promote thromboses through the induction of endothelial P-selectin expression and release, also a consequence of STAT3 phosphorylation, that can be reversed by hydroxyurea or a P-selectin-blocking antibody.57 This study showed that hydroxyurea reduced levels of STAT3 phosphorylation and, in effect, decreased endothelial membrane P-selectin expression. The antithrombotic effect of hydroxyurea may not be due only to a lowering of blood cell counts. GATA1low mouse models show that P-selectin plays a role in the progression to myelofibrosis in mice that also had a high risk of thromboses.58 Ongoing trials to evaluate the role of crizanlizumab in sickle cell nephropathy and prevention of priapism in SCD are underway.59,60 P-selectin blockade has been shown to block the development of acute chest syndrome in a mouse model with SCA.61 More studies are required to elucidate the impact of P-selectin inhibition in non-VOE sickle cell complications.
Conclusion
Crizanlizumab, a monoclonal antibody that inhibits P-selectin, is effective at preventing vaso-occlusive pain crises in SCD. It may not be useful as a treatment, but it remains to be studied. While this class of drug may reduce the risk of thromboses and prevent the development of acute chest syndrome, these effects were not meaningfully tested in the landmark SUSTAIN trial. It remains to be seen whether P-selectin inhibition modifies the natural history of SCD and what effect, if any, it has on other vaso-occlusion-related, end-organ complications. More agents that target P-selectin (and other selectins) inhibition are expected to result in a new class of armamentarium against SCD.
Acknowledgment
English review was performed by an independent agency “Clinical Thinking”, paid for by Novartis©. Both Clinical Thinking and Novartis had no influence over content of the review.
Disclosure
Dr Abdullah Kutlar reports grants, personal fees, non-financial support from Novartis Pharmaceuticals, and is a Data Safety Monitoring Board member for Bluebird Bio, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Lanzkron S, Carroll CP, Haywood CJ. The burden of emergency department use for sickle-cell disease: an analysis of the national emergency department sample database. Am J Hematol. 2010;85(10):797–799. doi:10.1002/ajh.21807
2. Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol. 2005;79(1):17–25. doi:10.1002/ajh.20336
3. Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood. 2012;120(18):3647–3656. doi:10.1182/blood-2012-04-383430
4. van Tuijn CFJ, van Beers EJ, Schnog -J-JB, Biemond BJ. Pain rate and social circumstances rather than cumulative organ damage determine the quality of life in adults with sickle cell disease. Am J Hematol. 2010;85(7):532–535. doi:10.1002/ajh.21731
5. Manwani D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood. 2013;122(24):3892–3898. doi:10.1182/blood-2013-05-498311
6. Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Prim. 2018;4(1):18010. doi:10.1038/nrdp.2018.10
7. Schmugge M, Frischknecht H, Yonekawa Y, Baumgartner RW, Boltshauser E, Humbert J. Stroke in hemoglobin (SD) sickle cell disease with moyamoya: successful hydroxyurea treatment after cerebrovascular bypass surgery. Blood. 2001;97(7):2165–2167. doi:10.1182/blood.V97.7.2165
8. McEver RP. Selectins. Curr Opin Immunol. 1994;6(1):75–84. doi:10.1016/0952-7915(94)90037-X
9. Matsui NM, Borsig L, Rosen SD, Yaghmai M, Varki A, Embury SH. P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood. 2001;98(6):1955–1962. doi:10.1182/blood.V98.6.1955
10. Man Y, Goreke U, Kucukal E, et al. Leukocyte adhesion to P-selectin and the inhibitory role of Crizanlizumab in sickle cell disease: a standardized microfluidic assessment. Blood Cells Mol Dis. 2020;83:102424. doi:10.1016/j.bcmd.2020.102424
11. Francis RJ, Johnson CS. Vascular occlusion in sickle cell disease: current concepts and unanswered questions. Blood. 1991;77(7):1405–1414. doi:10.1182/blood.V77.7.1405.1405.
12. Bennewitz MF, Jimenez MA, Vats R, et al. Lung vaso-occlusion in sickle cell disease mediated by arteriolar neutrophil-platelet microemboli. JCI Insight. 2017;2(1):e89761. doi:10.1172/jci.insight.89761
13. Kutlar A, Embury SH. Cellular adhesion and the endothelium: p-selectin. Hematol Oncol Clin North Am. 2014;28(2):323–339. doi:10.1016/j.hoc.2013.11.007
14. Hebbel RP. Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest. 1997;100(11 Suppl):S83–6.
15. Wood K, Russell J, Hebbel RP, Granger DN. Differential expression of E- and P-selectin in the microvasculature of sickle cell transgenic mice. Microcirculation. 2004;11(4):377–385. doi:10.1080/10739680490437559
16. Solovey A, Lin Y, Browne P, Choong S, Wayner E, Hebbel RP. Circulating activated endothelial cells in sickle cell anemia. N Engl J Med. 1997;337(22):1584–1590. doi:10.1056/NEJM199711273372203
17. Lawrence MB, Springer TA. Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell. 1991;65(5):859–873. doi:10.1016/0092-8674(91)90393-D
18. Sultana C, Shen Y, Rattan V, Johnson C, Kalra VK. Interaction of sickle erythrocytes with endothelial cells in the presence of endothelial cell conditioned medium induces oxidant stress leading to transendothelial migration of monocytes. Blood. 1998;92(10):3924–3935. doi:10.1182/blood.V92.10.3924
19. Takano M, Meneshian A, Sheikh E, et al. Rapid upregulation of endothelial P-selectin expression via reactive oxygen species generation. Am J Physiol Heart Circ Physiol. 2002;283(5):H2054–61. doi:10.1152/ajpheart.01001.2001
20. Conran N, Franco-Penteado CF, Costa FF. Newer aspects of the pathophysiology of sickle cell disease vaso-occlusion. Hemoglobin. 2009;33(1):1–16. doi:10.1080/03630260802625709
21. Lawrence MB, Kansas GS, Kunkel EJ, Ley K. Threshold levels of fluid shear promote leukocyte adhesion through selectins (CD62L,P,E). J Cell Biol. 1997;136(3):717–727. doi:10.1083/jcb.136.3.717
22. Gutsaeva DR, Parkerson JB, Yerigenahally SD, et al. Inhibition of cell adhesion by anti-P-selectin aptamer: a new potential therapeutic agent for sickle cell disease. Blood. 2011;117(2):727–735. doi:10.1182/blood-2010-05-285718
23. Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A. 2002;99(5):3047–3051. doi:10.1073/pnas.052522799
24. Bennewitz MF, Tutuncuoglu E, Gudapati S, et al. P-selectin–deficient mice to study pathophysiology of sickle cell disease. Blood Adv. 2020;4(2):266–273. doi:10.1182/bloodadvances.2019000603
25. Koenig A, Norgard-Sumnicht K, Linhardt R, Varki A. Differential interactions of heparin and heparan sulfate glycosaminoglycans with the selectins. Implications for the use of unfractionated and low molecular weight heparins as therapeutic agents. J Clin Invest. 1998;101(4):877–889. doi:10.1172/JCI1509
26. Höpfner M, Alban S, Schumacher G, Rothe U, Bendas G. Selectin-blocking semisynthetic sulfated polysaccharides as promising anti-inflammatory agents. J Pharm Pharmacol. 2003;55(5):697–706. doi:10.1211/002235703765344621
27. Kutlar A, Ataga KI, McMahon L, et al. A potent oral P-selectin blocking agent improves microcirculatory blood flow and a marker of endothelial cell injury in patients with sickle cell disease. Am J Hematol. 2012;87(5):536–539. doi:10.1002/ajh.23147
28. Telen MJ, Wun T, McCavit TL, et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood. 2015;125(17):2656–2664. doi:10.1182/blood-2014-06-583351
29. Pfizer. Pfizer announces phase 3 top-line results for rivipansel in patients with sickle cell disease experiencing a vaso-occlusive crisis [Internet]; [cited December 6, 2020]. Available from: https://www.pfizer.com/news/press-release/press-release-detail/pfizer_announces_phase_3_top_line_results_for_rivipansel_in_patients_with_sickle_cell_disease_experiencing_a_vaso_occlusive_crisis.
30. Biemond BJ, Tombak A, Kilinc Y, et al. Efficacy and safety of sevuparin, a novel non-anti-coagulant heparinoid, in patients with acute painful vaso-occlusive crisis; a global, multicenter double-blind, randomized, placebo-controlled phase 2 trial (TVOC01). Blood. 2019;134(Supplement_1):614. doi:10.1182/blood-2019-124653
31. US FDA. FDA approves first targeted therapy to treat patients with painful complication of sickle cell disease. FDA news release [Internet]; 2019 [cited January 15, 2021]; Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-targeted-therapy-treat-patients-painful-complication-sickle-cell-disease.
32. Novartis. ADAKVEO (crizanlizumab-tmca) for IV infusion: full prescribing information [Internet]; [cited December 6, 2020]. Available from: https://www.novartis.us/sites/www.novartis.us/files/adakveo.pdf.
33. Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376(5):429–439. doi:10.1056/NEJMoa1611770
34. Kutlar A, Kanter J, Liles D, et al. Crizanlizumab, a P-selectin inhibitor, increases the likelihood of experiencing a sickle cell-related pain crisis while on treatment: results from Phase II SUSTAIN study. In:
35. Kutlar A, Kanter J, Liles DK, et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: a SUSTAIN study analysis. Am J Hematol. 2019;94(1):55–61. doi:10.1002/ajh.25308
36. Liles D, Shah N, Scullin B, et al. SUCCESSOR: a multicenter retrospective non-interventional follow-up study in patients with sickle cell pain crisis who previously participated in the SUSTAIN trial in the United States. In:
37. Han J, Saraf SL, Gordeuk VR. Systematic review of crizanlizumab: a new parenteral option to reduce vaso-occlusive pain crises in patients with sickle cell disease. Pharmacotherapy. 2020;40(6):535–543. doi:10.1002/phar.2409
38. Yu Z, Blankenship L, Jaiyesimi I. Crizanlizumab in sickle cell disease. N Engl J Med. 2017;376:1795–1796.
39. Riley TR, Riley TT. Profile of crizanlizumab and its potential in the prevention of pain crises in sickle cell disease: evidence to date. J Blood Med. 2019;10:307–311. doi:10.2147/JBM.S191423
40. Karkoska K, Quinn CT, Clapp K, McGann PT. Severe infusion-related reaction to crizanlizumab in an adolescent with sickle cell disease. Am J Hematol. 2020;95(12):E338–9. doi:10.1002/ajh.26002
41. Novartis. Novartis provides update on safety information about crizanlizumab. [Internet]; [cited February 17, 2021]. Available from: https://www.crizanlizumab.info/.
42. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332(20):1317–1322. doi:10.1056/NEJM199505183322001
43. Badawy SM, Thompson AA, Lai J-S, Penedo FJ, Rychlik K, Liem RI. Adherence to hydroxyurea, health-related quality of life domains, and patients’ perceptions of sickle cell disease and hydroxyurea: a cross-sectional study in adolescents and young adults. Health Qual Life Outcomes. 2017;15(1):136. doi:10.1186/s12955-017-0713-x
44. Darbari DS, Sheehan VA, Ballas SK. The vaso-occlusive pain crisis in sickle cell disease: definition, pathophysiology, and management. Eur J Haematol. 2020;105(3):237–246. doi:10.1111/ejh.13430
45. Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood. 2010;115(26):5300–5311. doi:10.1182/blood-2009-04-146852
46. Platt OS. Hydroxyurea for the treatment of sickle cell anemia. N Engl J Med. 2008;358(13):1362–1369. doi:10.1056/NEJMct0708272
47. Sins JWR, Mager DJ, Davis SCAT, Biemond BJ, Fijnvandraat K. Pharmacotherapeutical strategies in the prevention of acute, vaso-occlusive pain in sickle cell disease: a systematic review. Blood Adv. 2017;1(19):1598–1616. doi:10.1182/bloodadvances.2017007211
48. Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of l-Glutamine in sickle cell disease. N Engl J Med. 2018;379(3):226–235. doi:10.1056/NEJMoa1715971
49. Niihara Y, Zerez CR, Akiyama DS, Tanaka KR. Oral L-glutamine therapy for sickle cell anemia: i. Subjective clinical improvement and favorable change in red cell NAD redox potential. Am J Hematol. 1998;58(2):117–121. doi:10.1002/(SICI)1096-8652(199806)58:2<117::AID-AJH5>3.0.CO;2-V
50. Daak AA, Ghebremeskel K, Hassan Z, et al. Effect of omega-3 (n-3) fatty acid supplementation in patients with sickle cell anemia: randomized, double-blind, placebo-controlled trial. Am J Clin Nutr. 2013;97(1):37–44. doi:10.3945/ajcn.112.036319
51. Salinas Cisneros G, Thein SL. Recent advances in the treatment of sickle cell disease. Front Physiol. 2020;11:435. doi:10.3389/fphys.2020.00435
52. Vichinsky E, Hoppe CC, Ataga KI, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 2019;381(6):509–519. doi:10.1056/NEJMoa1903212
53. Théorêt JF, Yacoub D, Hachem A, Gillis M-A, Merhi Y. P-selectin ligation induces platelet activation and enhances microaggregate and thrombus formation. Thromb Res. 2011;128(3):243–250. doi:10.1016/j.thromres.2011.04.018
54. Naik RP, Streiff MB, Haywood CJ, Nelson JA, Lanzkron S. Venous thromboembolism in adults with sickle cell disease: a serious and under-recognized complication. Am J Med. 2013;126(5):443–449. doi:10.1016/j.amjmed.2012.12.016
55. Shet AS, Lizarralde-Iragorri MA, Naik RP. The molecular basis for the prothrombotic state in sickle cell disease. Haematologica. 2020;105(10):2368–2379. doi:10.3324/haematol.2019.239350
56. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood. 2013;122(13):2176–2184. doi:10.1182/blood-2013-03-460154
57. Guy A, Gourdou-Latyszenok V, Le Lay N, et al. Vascular endothelial cell expression of JAK2(V617F) is sufficient to promote a pro-thrombotic state due to increased P-selectin expression. Haematologica. 2019;104(1):70–81. doi:10.3324/haematol.2018.195321
58. Zetterberg E, Verrucci M, Martelli F, et al. Abnormal P-selectin localization during megakaryocyte development determines thrombosis in the gata1low model of myelofibrosis. Platelets. 2014;25(7):539–547. doi:10.3109/09537104.2013.840720
59. Novartis. NCT04053764: study exploring the effect of crizanlizumab on kidney function in patients with chronic kidney disease caused by sickle cell disease (STEADFAST) [Internet]; [cited December 6, 2020]. Available from: https://clinicaltrials.gov/ct2/show/NCT04053764.
60. Novartis. NCT03938454: a study to evaluate the safety and efficacy of crizanlizumab in sickle cell disease related priapism (SPARTAN) [Internet]; [cited December 6, 2020]. Available from: https://clinicaltrials.gov/ct2/show/NCT03938454.
61. Ghosh S, Flage B, Weidert F, Ofori-Acquah SF. P-selectin plays a role in haem-induced acute lung injury in sickle mice. Br J Haematol. 2019;186(2):329–333. doi:10.1111/bjh.15807
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.