")
Back to Journals » Journal of Pain Research » Volume 11
Ozone induces autophagy in rat chondrocytes stimulated with IL-1β through the AMPK/mTOR signaling pathway
Authors Zhao X , Li Y, Lin X, Wang J , Zhao X, Xie J, Sun T, Fu Z
Received 11 August 2018
Accepted for publication 28 September 2018
Published 27 November 2018 Volume 2018:11 Pages 3003—3017
DOI https://doi.org/10.2147/JPR.S183594
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Michael Schatman
Xu Zhao, Yun Li, Xiaowen Lin, Junnan Wang, Xuejun Zhao, Juntian Xie, Tao Sun, Zhijian Fu
Department of Pain Management, Shandong Provincial Hospital Affiliated to Shandong University, Shandong University, Jinan, Shandong Province 250021, P.R. China
Background: Ozone injection is generally used for the management of pain in diseases such as osteoarthritis (OA). Recent studies have shown that reduced autophagy in chondrocytes plays an important role in the development of OA. The purpose of this study was to determine whether ozone treats OA by inducing autophagy in OA chondrocytes.
Materials and methods: In this study, primary chondrocytes were stimulated with IL-1β for 24 hours to simulate an OA chondrocyte model, followed by treatment with ozone (30 µg/mL) or pretreatment with 3-methyladenine or compound C before ozone treatment. Then, cell viability was detected by a CCK-8 kit, and the AMPK/mTOR signaling pathway and autophagy were detected by Western blotting and immunofluorescence. The mRNA expression levels of IL-6, TNF-α, MMP-13 and TIMP-1 were measured by quantitative real-time PCR. Finally, autophagosomes in chondrocytes were observed by transmission electron microscopy.
Results: Ozone improved cell viability in chondrocytes stimulated by IL-1β. The decreased level of autophagy in IL-1β-stimulated chondrocytes improved with ozone treatment through activation of the AMPK/mTOR signaling pathway. In addition, the mRNA expression levels of IL-6 and TNF-α were suppressed by ozone treatment in chondrocytes stimulated with IL-1β. Ozone increased the mRNA level of TIMP-1 and decreased the mRNA level of MMP-13 in chondrocytes stimulated with IL-1β.
Conclusion: These results suggested that ozone improved the decreased level of autophagy in chondrocytes stimulated with IL-1β through activation of the AMPK/mTOR signaling pathway. Moreover, ozone treatment suppressed inflammation and helped maintain metabolic balance in chondrocytes stimulated with IL-1β.
Keywords: ozone, osteoarthritis, chondrocyte, AMPK, mTOR signaling pathway, autophagy
Introduction
Osteoarthritis (OA), with clinical manifestations of joint swelling and chronic pain, seriously affects quality of life. Moreover, OA is characterized by articular cartilage degeneration and chronic inflammation. Chronic inflammation includes IL-1, IL-6, TNF-α and other inflammatory factors related to decomposition.1,2 However, the central link in the pathogenesis of OA is articular cartilage degeneration.3 Articular cartilage degeneration is caused by chondrocyte degeneration, extracellular matrix degradation and subchondral bone sclerosis.4 Therefore, articular cartilage degeneration is also affected by the balance between matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs).5,6
Decreased autophagy in chondrocytes is one of the main mechanisms of articular cartilage degeneration in OA.7 The promotion of autophagy in chondrocytes provides a novel therapeutic target in the treatment of OA.8 Autophagy is widely present in eukaryotic cells at a low level under normal physiological conditions. Autophagy is induced when cells are stimulated by starvation, nutritional deficiencies and oxygen stress. Intracellular long-lived proteins, soluble proteins, damaged organelles and other intracellular substances are transported to lysosomes for degradation and then reused to meet the cell’s needs, and this process is essential for cell survival.9 Therefore, decreasing autophagy may induce various human diseases. Autophagy seems to play an important role in maintaining cell survival and biosynthesis due to the low rate of proliferation of chondrocytes.8 It has been reported that the occurrence of OA is associated with a decrease in the autophagy level in chondrocytes.7
The AMP-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) signaling pathway and the AKT/mTOR signaling pathway are involved in autophagy in OA chondrocytes.10,11 The activation of AMPK or the inhibition of AKT promotes autophagy in chondrocytes and suppresses the inflammatory reaction in OA.10,11 It has been reported that rapamycin acts on the mTOR signaling pathway in mouse knee joints by suppressing a target of mTOR and by promoting LC3 II, which is a main marker of autophagy, and the degree of cartilage degeneration with rapamycin treatment was significantly reduced compared with nontreatment.12,13 More importantly, metformin (an AMPK activator) inhibits inflammatory cytokine production, such as IL-6 and TNF-α, in inflammatory arthritis by regulating autophagy in chondrocytes.14 Therefore, the AMPK/mTOR signaling pathway is a potential treatment target in OA.
Ozone therapy was first reported by Wolff in 1974.15 In recent decades, ozone therapy has been widely used in the clinical treatment of acute and chronic inflammatory pain, including spinal pain, OA and soft-tissue injury.16 It has been clinically observed that ozone has a distinct positive effect on patients with disk herniation and OA.17,18 Some researchers have revealed that ozone activates antioxidant protection systems to induce the activation of intracellular signaling pathways, such as the AMPK signaling pathway, to attenuate chronic constriction injury (CCI)-induced neuropathic pain.19,20 Only some technical tips for performing ozone therapy are currently available, but the specific mechanism of ozone treatment remains unclear, especially for OA. Therefore, this study aimed to investigate whether ozone activates autophagy in chondrocytes pretreated with IL-1β in vitro through the AMPK/mTOR signaling pathway.
Materials and methods
Isolation and culture of rat chondrocytes
Chondrocytes were isolated from newborns Wistar rats within 72 hours. The newborn Wistar rats were purchased from the Experimental Animal Center of Shandong University. All procedures involving the use of animals were conducted in compliance with the guidelines of the National Institute of Health and were approved by the Animal Care and Use Committee of the Shandong Provincial Hospital affiliated with Shandong University. All possible efforts were made to minimize animal suffering and the number of animals used.
The rats were sacrificed by cervical dislocation under general anesthesia and then placed in povidone iodine and 75% ethanol for sterilization for 5 minutes. Rat knee cartilage was isolated and cut to approximately 1 mm3. The cartilage samples were first digested with 0.25% trypsin containing ethylenediaminetetraacetic acid (EDTA) for 30 minutes and then with 0.2% type II collagenase for 2 hours. A total of 5 mL of DMEM/F12 medium containing 10% fetal bovine serum (FBS) was added, and the supernatant was discarded by centrifugation. The cells were resuspended in DMEM/F12 medium containing 10% FBS and antibiotics (100 U/mL penicillin and 100 U/mL streptomycin). The cells were counted using a hemocytometer to adjust the cell concentration to 1×105/ mL and were then placed in culture flasks at 37°C under humidified conditions and 5% CO2 for culture. The medium was replaced 3 days later. Approximately 5 days later, the cells had spread and grown to confluence, and they were then digested with 0.25% trypsin containing EDTA and passaged at a 1:2 ratio. In this study, we used third-generation cells. Chondrocytes were identified by immunofluorescence staining for type II collagen.
Osteoarthritis chondrocyte model
Cells were stimulated with IL-1β (10 ng/mL) (PeproTech, USA) for 24 hours before O3 treatment similar to OA chondrocytes. 3-methyladenine (3MA) and compound C (com C) were added 12 hours before ozone treatment.
Ozone exposure
Chondrocytes were treated with different doses of ozone at referential levels in a computer-controlled in vitro exposure chamber for 30 minutes.21,22 The ozone concentration was analyzed by an ozone analyzer (Model MD-050-12-f-4, Perma Pure Inc., Toms River, NJ) and was managed by a computerized system. All experiments with ozone treatment for 30 minutes or 1 hour were analyzed after 30 minutes.
Cell viability assay
Cell viability was detected by using a CCK-8 kit (Dojindo, Kumamoto, Japan). The procedure was performed as follows. Chondrocytes (5×104/ mL) were seeded in each well of a 96-well plate. The cells were maintained in an incubator at 5% CO2 and at 37°C for 24 hours.
Briefly, 10 µL of solution from the CCK-8 kit was added to each well of the 96-well plates, and the plates were incubated in the dark for 4 hours. The absorbance was measured at 450 nm with a microplate reader. Cell viability was expressed as a percentage of live cells compared with the control group, which was set to 1.
Quantitative real-time PCR assay
Total RNA was extracted from chondrocytes using Trizol reagent (Takara, Shiga, Japan) in accordance with the manufacturer’s instructions. Total RNA (1 µg) was reverse-transcribed into complementary DNA (cDNA) using a PrimeScript RT reagent kit (RR047A; Takara, Shiga, Japan). mRNA expression was measured by quantitative real-time PCR (qRT-PCR) using SYBR® GreenERTM SuperMix (Takara, Shiga, Japan) with specific primers for β-actin: forward 5′-GGAGATTACTGCCCTGGCTCCTA-3′, reverse 5′-GACTCATCGTACTCCTGCTTGCTG-3′, IL-6: forward 5′-ATTGTATGAACAGCGATGATGCAC-3′, reverse 5′-CCAGGTAGAAACGGAACTCCAGA-3′, TNF-α: forward 5′-TCAGTTCCATGGCCCAGAC-3′, reverse 5′-GTTGTCTTTGAGATCCATGCCATT –3′, MMP-13: forward 5′-TGATGATGAAACCTGGACAAGCA-3′, reverse 5′-GAACGTCATCATCTGGGAGCA-3′, and TIMP-1: forward 5′-CATCTCTGGCCTCTGGCATC-3′, reverse 5′-CATAACGCTGGTATAAGGTGGTCTC-3′. Fold changes in gene expression were calculated by the following formula: 2−Δct, where ΔCt is the difference in the threshold cycle of the sample.
Western blotting
Chondrocytes were lysed by radio immunoprecipitation assay (RIPA) lysis buffer with phenylmethylsulfonyl fluoride (PMSF) and a phosphatase inhibitor. The concentration of proteins was measured using a bicinchoninic acid (BCA) protein assay kit (Beyotime, Beyotime Biotechnology, China). The proteins (30 µg) were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and were transferred to polyvinylidenedifluoride (PVDF) membranes. The membranes were blocked in a nonfat dry milk solution for 1 hour. Then, the membranes were incubated overnight at 4°C with anti-AMPK (CST, #2532, 1:1000 dilution), anti-p-AMPK (CST, #2535, 1:1000 dilution), anti-mTOR (CST, #2983, 1:1000 dilution), anti-p-mTOR (CST, #5536, 1:1000 dilution), anti-Beclin-1 (Abcam, ab207612, 1:1000 dilution), anti-ULK1 (Abcam, ab133747, 1:1000 dilution), anti-LC3 II (Abcam, ab48394, 1:1000 dilution), anti-P62 (Abcam, ab56416, 1:2000 dilution), anti-Bcl2 (Abcam, ab194583, 1:2000 dilution) and anti-Bax (Abcam, ab32503, 1:1000 dilution) primary antibodies in dilution buffer. Next, the membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse IgG (Zhongshan Golden Bridge Biotechnology, 1:5000 dilution) at room temperature for 1 hour. Then, the membranes were developed using the enhanced chemiluminescence substrate LumiGLO (Millipore, Bedford, MA, USA).
Immunofluorescence assay
Cells were seeded in 14 mm glass-bottomed microwell dishes. Chondrocytes were treated with ozone when they reached approximately 40% confluence. They were then washed with PBS three times and fixed with 4% paraformaldehyde for 15 minutes. Next, 0.3% TritonX-100 was used for cell permeabilization for 5 minutes. The cells were blocked with goat serum at room temperature for 1 hour and were incubated with anti-LC3 II (Abcam, ab48394, 1:100) and P62 (Abcam, ab56416, 1:100) overnight at 4°C. The next day, the cells were incubated with the secondary antibody (Abcam, ab150080, ab150117, 1:400) at room temperature for 1 hour. The 4′,6-diamidino-2-phenylindole (DAPI) counterstain was used to show the chondrocyte nuclei. The stained chondrocytes were observed by immunofluorescence microscopy.
Transmission electron microscopy (TEM)
Chondrocytes were treated with ozone after 30 minutes as described above. The cells were washed with PBS three times and were collected by a cell scraper. Then, the cells were centrifuged at 1000 g for 10 minutes. The cell clumps were fixed with ice-cold 3% glutaraldehyde in 0.1 M cacodylate buffer and were post-fixed in osmium tetroxide. The cell clumps were cut into ultrathin sections after they were embedded in Epon epoxy resin. The ultrathin sections were stained with 0.1% lead citrate and 10% uranyl acetate and were observed with a transmission electron microscope.
Statistical analysis
The data are presented as the mean ± SD and were analyzed using variance SPSS 22.0 (IBM software, Chicago, IL, USA). The differences in the means were determined by a one-way analysis of variance (ANOVA) for multiple comparisons followed by the least significant difference (LSD) test for two-group comparisons among the multiple comparisons. A P-value of less than 0.05 was considered statistically significant. All measurements were carried out at least three times.
Results
Morphological observations and identification of cultured chondrocytes
Under an inverted phase contrast microscope, cells were observed to be attached to the wall of the culture flask 24 hours after inoculation, and approximately 3 days later, the cells exhibited a cobblestone-like appearance (Figure 1A, B). The cells grew rapidly at the second and third generation, but slowly after the fourth generation, with the cell morphology exhibiting long spindles. Therefore, the third generation of cells was chosen for use in this study.
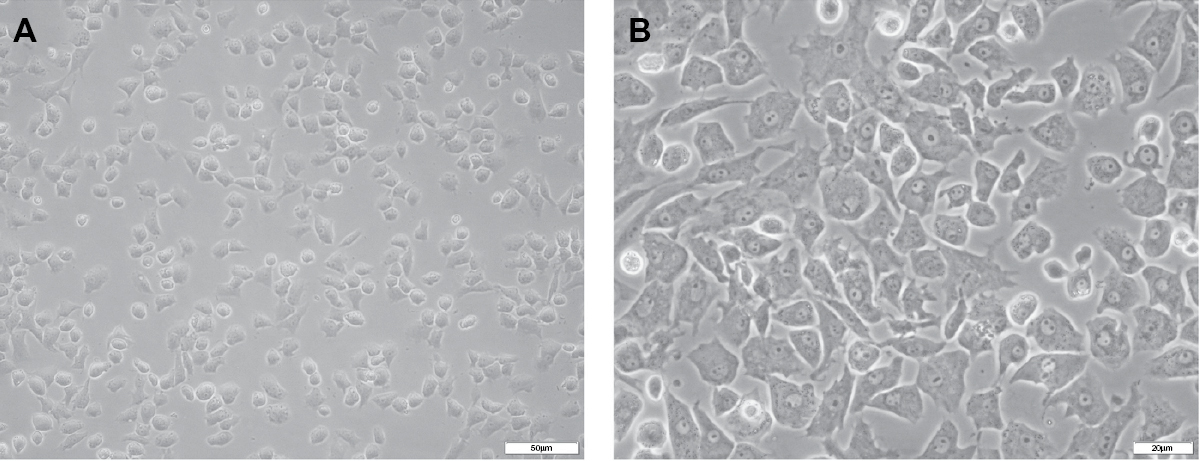 | Figure 1 Rat chondrocyte culture and morphological observations. Notes: On day 3 of culture in vitro, chondrocytes were growing well and were observed under an inverted phase contrast microscope. Under the inverted phase contrast microscope at different magnifications of 20× (A) and 40× (B), chondrocytes were observed to be attached to the wall of the culture flask and had spread. A cobblestone-like appearance was clearly observed (B). |
To ensure cell purity, immunofluorescence staining with an anti-collagen II polyclonal antibody was performed. The results revealed that more than 95% of the cultured cells showed positive expression for collagen II (Figure 2A, B).
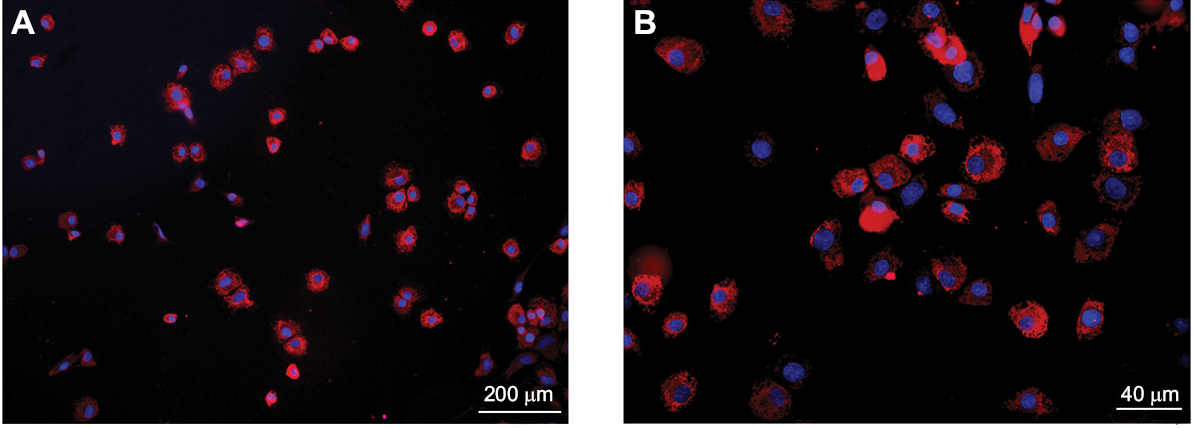 | Figure 2 Chondrocytes were stained with an anti-type II collagen (col II) polyclonal antibody. Notes: More than 95% of the cultured cells (the third generation) showed positive expression of col II in vitro. The number of col II-positive cells was counted per area and is presented as a percentage of the total number of cells. At least 15 photomicrographs were evaluated randomly per treatment, and each treatment was performed in triplicate. |
Ozone (30 µg/mL) improves the cell viability of chondrocytes stimulated with IL-1β
The viability of chondrocytes stimulated by IL-1β (10 ng/mL) for 24 hours was significantly reduced after ozone treatment in the range of 40 μg/ml to 60 μg/ml. There was a significant difference between chondrocytes stimulated with only IL-1β and IL-1β-stimulated chondrocytes treated with 60 µg/mL ozone (Figure 3A). Treatment with 30 µg/mL ozone dramatically and significantly improved cell viability compared with stimulation with only IL-1β (Figure 3A). The currently recommended concentration of ozone for intra-articular injection, as a clinical treatment for OA, is 30 µg/mL.23 Therefore, an ozone concentration of 30 µg/mL for 30 minutes was selected for the subsequent studies.
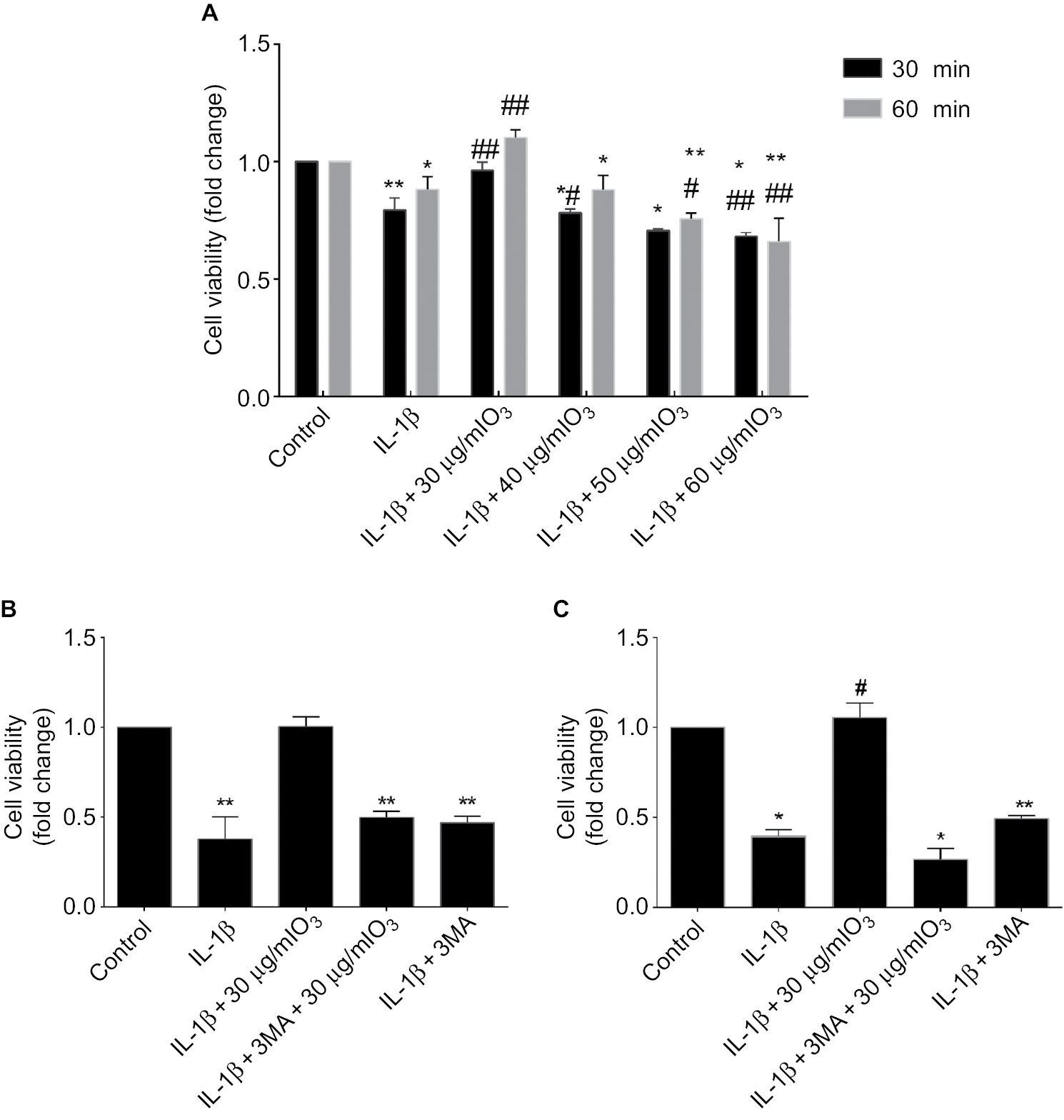 | Figure 3 The increased cell viability in chondrocytes stimulated with IL-1β and treated with ozone was reduced by 3MA or com C. Notes: (A) Chondrocytes stimulated with IL-1β (10 ng/mL) for 24 hours were treated with different doses of ozone for different time periods, and treatment with 30 µg/mL ozone for 30 minutes promoted cell viability in chondrocytes stimulated with IL-1β, as determined by the CCK-8 assay. (B) Chondrocytes stimulated with IL-1β (10 ng/mL) for 24 hours were treated with ozone (30 µg/mL) in the absence or presence of 3MA (100 nM), and the improvement in cell viability induced by ozone was abated by 3MA. (C) Chondrocytes stimulated with IL-1β (10 ng/mL) for 24 hours were treated with ozone (30 µg/mL) in the absence or presence of com C (5 µM), and the improvement in cell viability induced by ozone was abated by com C. The data are presented as the mean ± SD for three independent experiments. *P<0.05, **P<0.01, compared with the control; #P<0.05, ##P<0.01, compared with IL-1β. |
To study the signaling pathway or mechanism involved in the ozone-induced autophagy of IL-1β-stimulated chondrocytes, cells were pretreated with the autophagy inhibitor 3MA (100 nM) before treatment with 30 µg/mL ozone. 3MA suppressed the increased cell viability that was induced by 30 µg/mL ozone compared with the control group (Figure 3B). These data suggested that a dose of 30 µg/mL ozone promoted cell viability in chondrocytes stimulated with IL-1β by inducing autophagy. As shown in Figure 3C, an AMPK inhibitor, com C (5 µM), inhibited the improvement in cell viability of IL-1β-stimulated chondrocytes that was induced by the ozone treatment.
Treatment with 30 µg/mL ozone induces autophagy of IL-1β-stimulated chondrocytes
To better determine whether the increased cell viability due to ozone treatment was related to autophagy, the effect of 3MA, an autophagy inhibitor, on chondrocytes stimulated by IL-1β treatment with ozone was examined. The autophagy-related protein Beclin-1 was measured by Western blotting, and LC3 II and P62 were measured by Western blotting and immunofluorescence.
As shown in Figure 4, LC3 II decreased significantly, and P62 obviously increased in chondrocytes stimulated with IL-1β. These data revealed that the level of autophagy declined in IL-1β-stimulated chondrocytes. Furthermore, LC3 II and Beclin-1 distinctly increased, while P62 slightly decreased with ozone treatment. Moreover, more autophagosomes were observed in chondrocytes stimulated with IL-1β treated with ozone (Figure 5). However, the increased autophagy process induced by ozone was restrained by 3MA pretreatment (Figure 5). This finding suggested that ozone treatment increased the decreased level of autophagy in chondrocytes stimulated with IL-1β but did not change the low level of autophagy in IL-1β-stimulated chondrocytes pretreated with 3MA (Figure 4). Overall, the combined treatment experiments indicated that 30 µg/mL ozone induced autophagy in IL-1β-stimulated chondrocytes.
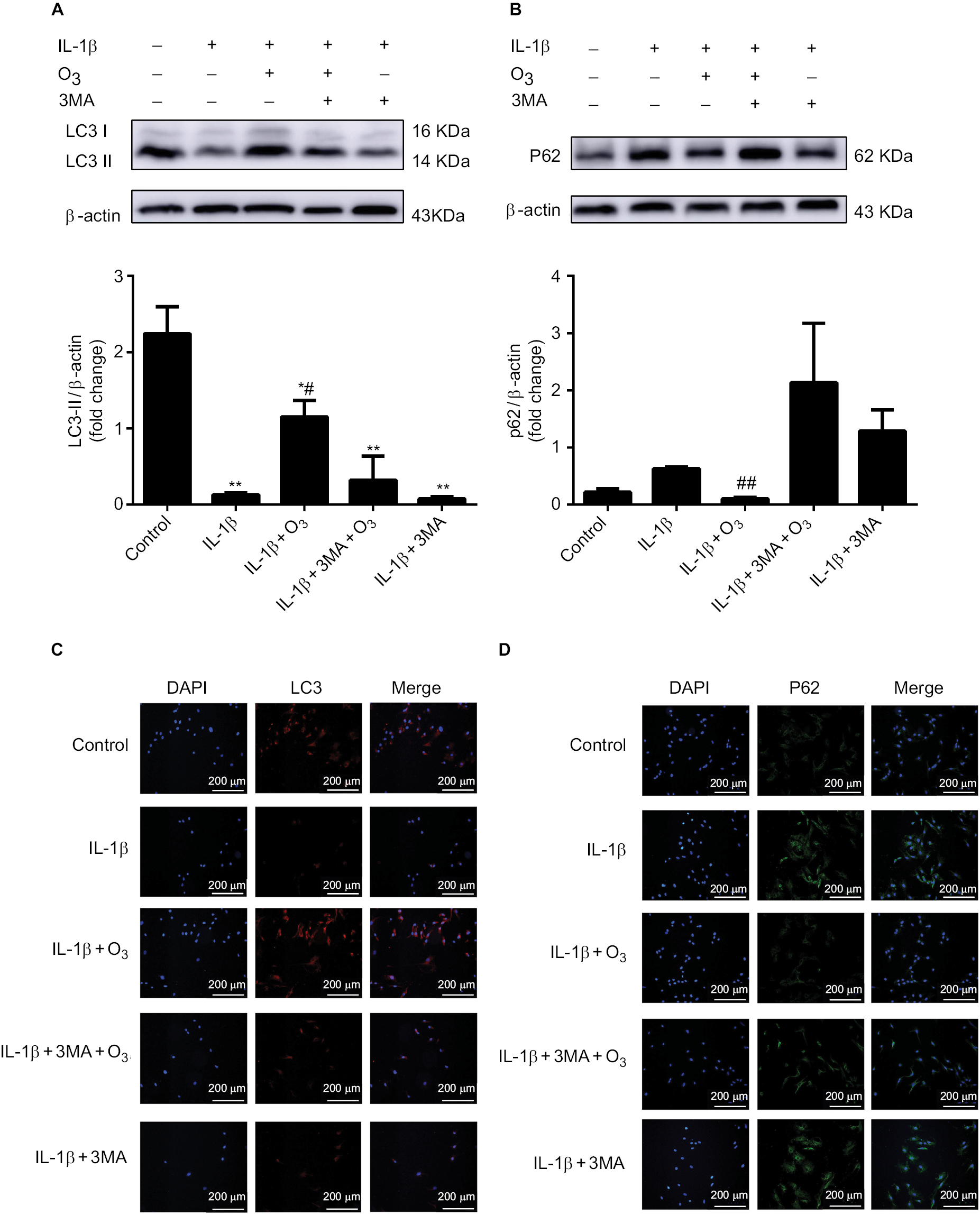 | Figure 4 Role of ozone-induced autophagy in chondrocytes stimulated with IL-1β (10 ng/mL). Notes: Cells were treated with IL-1β for 24 hours, with or without pretreatment with the autophagy inhibitor 3MA (100 nM) for 12 hours. The levels of LC3 II (A, C) and P62 (B, D) were measured by Western blotting and immunofluorescence. The data are presented as the mean ± SD of three independent experiments. |
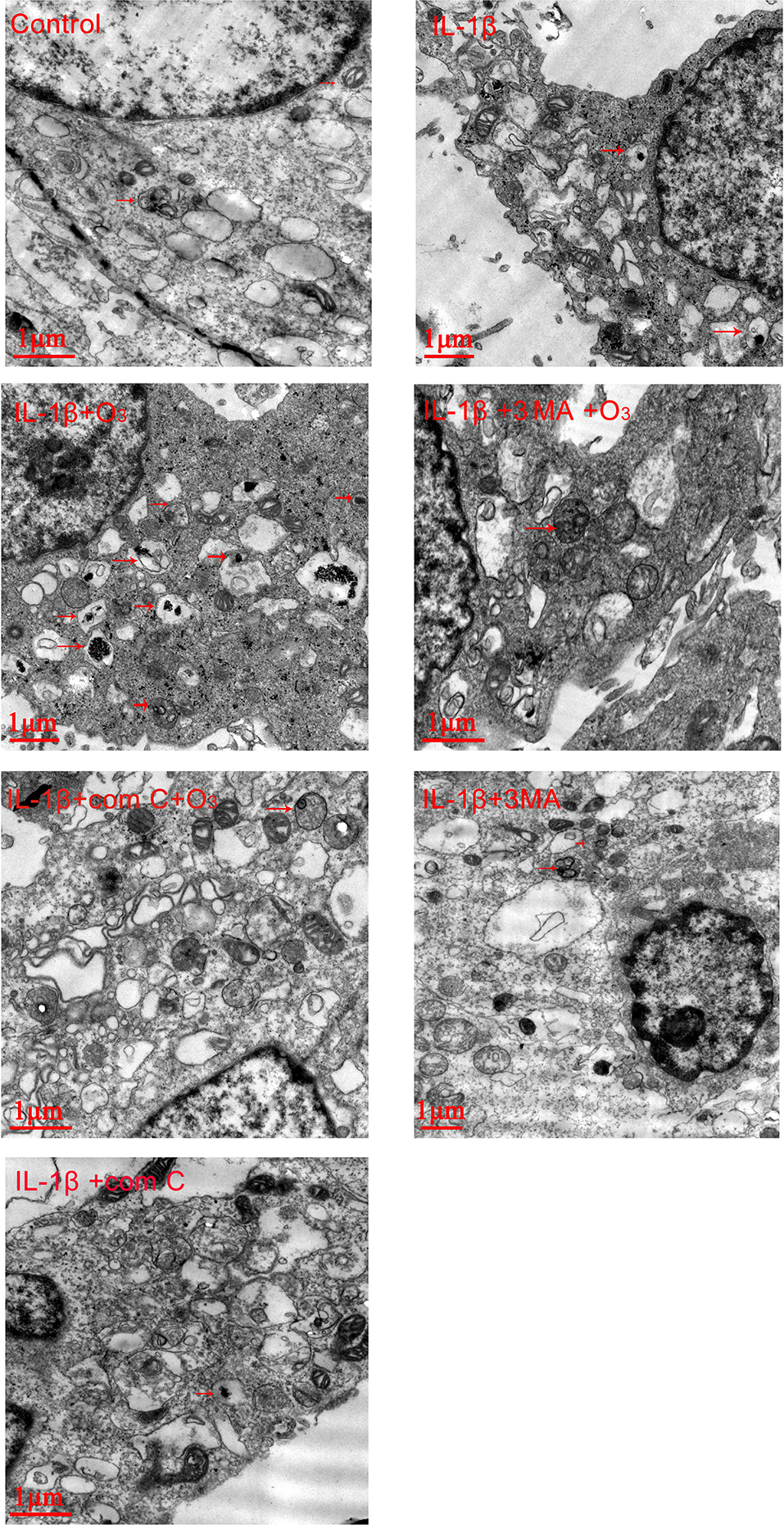 | Figure 5 Ozone increased the autophagosomes in chondrocytes stimulated with IL-1β (10 ng/mL), which was blocked by 3MA and com C. Notes: The arrows indicate autophagosomes. The data are from three independent experiments. |
Ozone-induced autophagy is involved in activation of the AMPK/mTOR signaling pathway in chondrocytes stimulated with IL-1β
It has been reported that mTOR participates in cell growth regulation and is closely related to autophagy.24 Moreover, the AMPK/mTOR signaling pathway participates in autophagy, and AMPK plays a role as a pivotal activator of autophagy.25 To investigate whether the ozone-induced autophagy in IL-1β-stimulated chondrocytes occurred through the AMPK/mTOR pathway, p-AMPK and p-mTOR proteins were detected by Western blotting. As shown in Figure 6, the protein expression of p-AMPK decreased in IL-1β-stimulated chondrocytes compared with control chondrocytes. Therefore, the protein expression of p-mTOR in IL-1β-stimulated chondrocytes was significantly increased compared with control chondrocytes (Figure 6A, B). More importantly, ozone treatment promoted the level of p-AMPK in chondrocytes stimulated with IL-1β. However, pretreatment with 3MA restrained the high expression of p-AMPK in chondrocytes stimulated by IL-1β and treated with ozone (Figure 5A). Additionally, the p-mTOR protein level in IL-1β-stimulated chondrocytes that were pretreated with 3MA increased slightly, which was attributed to the decreased p-AMPK level (Figure 5B).
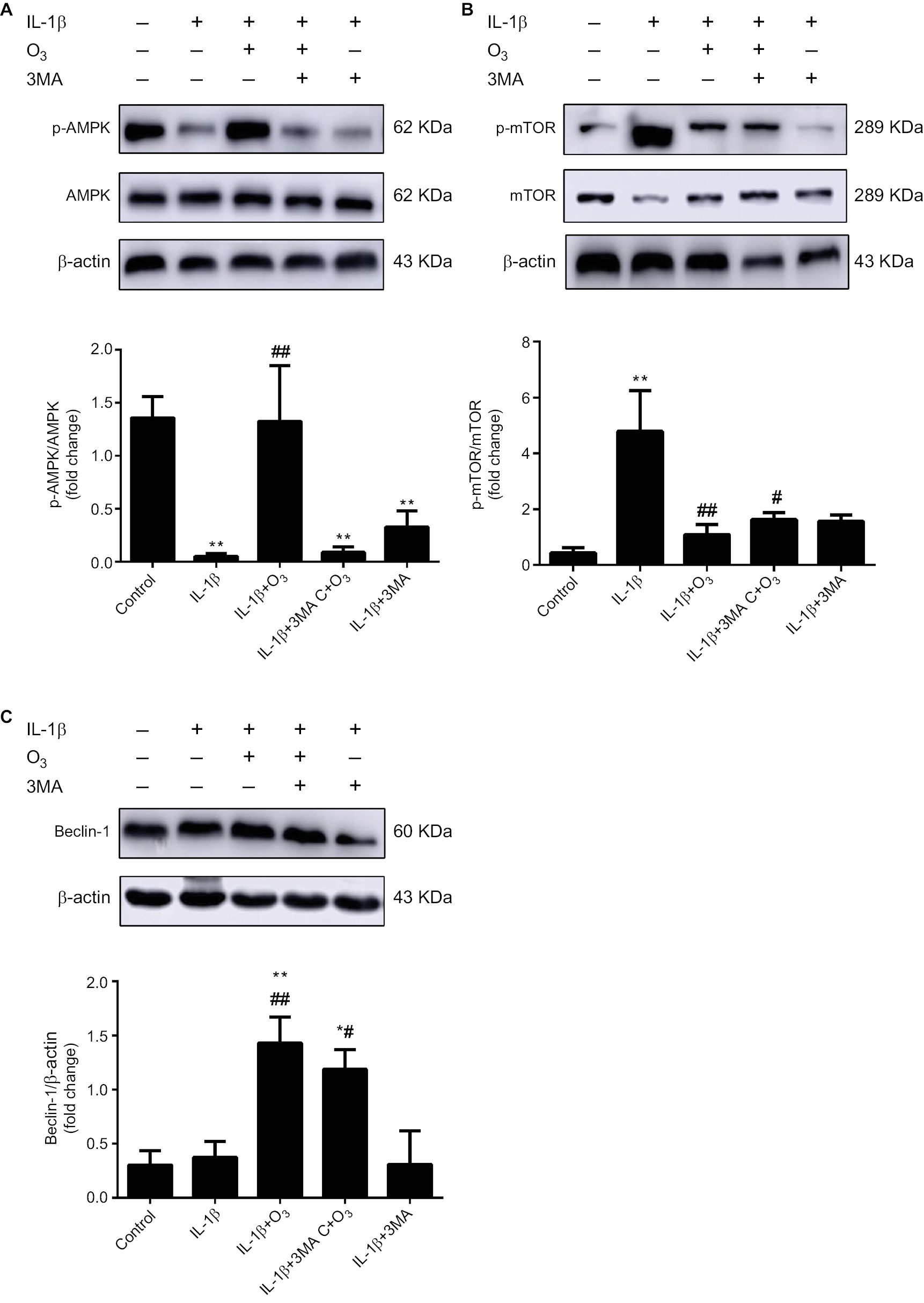 | Figure 6 Ozone activated the AMPK/mTOR signaling pathway in chondrocytes stimulated with IL-1β (10 ng/mL), and this effect was inhibited by 3MA. Notes: Cells were treated with IL-1β for 24 hours, with or without pretreatment with the autophagy inhibitor 3MA (100 nM) for 12 hours. The levels of p-AMPK (A), p-mTOR (B) and Beclin-1 (C) in chondrocytes stimulated with IL-1β were measured by Western blotting. The data are presented as the mean ± SD of three independent experiments. *P<0.05, **P<0.01, compared with the control; #P<0.05, ##P<0.01, compared with IL-1β. |
Ozone inhibits the mRNA expression levels of IL-6 and TNF-α in chondrocytes stimulated with IL-1β
It was previously reported that inflammation, such as the level of NLRP3 inflammasomes, is inhibited through the activation of autophagy.26 To determine whether inflammation is related to ozone-induced autophagy in IL-1β-stimulated chondrocytes, the mRNA expression of IL-6 and TNF-α was detected by qRT-PCR. The mRNA expression levels of IL-6 and TNF-α in chondrocytes stimulated by IL-1β with ozone treatment were dramatically decreased, whereas high expression levels were maintained with 3MA pretreatment (Figure 7A, B). Thus, it is speculated that the inhibited mRNA expression of IL-6 and TNF-α by ozone treatment may be related to ozone-induced autophagy in chondrocytes stimulated with IL-1β. In summary, ozone negatively affects IL-1β-induced expression of chondrocyte IL-6 and TNF-α mRNA, which may be related to the improvement in autophagy.
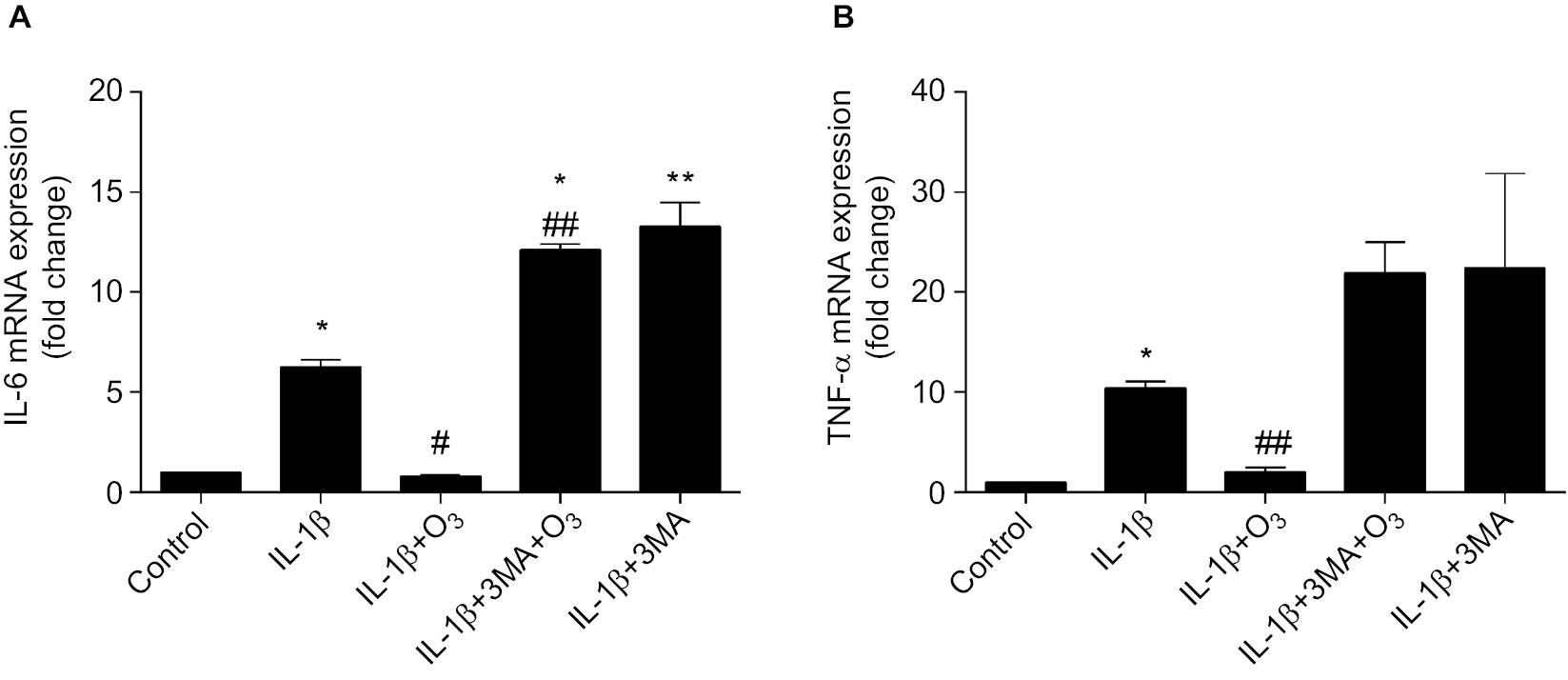 | Figure 7 Ozone suppresses inflammatory cytokines in chondrocytes stimulated with IL-1β (10 ng/mL). Notes: Cells were treated with IL-1β (10 ng/mL) for 24 hours, with or without pretreatment with the autophagy inhibitor 3MA (100 nM) for 12 hours. The mRNA expression of IL-6 (A) and TNFα (B) in chondrocytes stimulated with IL-1β was measured by qRT-PCR. The data are presented as the mean ± SD of three independent experiments. *P<0.05, **P<0.01, compared with the control; #P<0.05, ##P<0.01, compared with IL-1β. |
The AMPK inhibitor compound C blocks ozone-induced autophagy in chondrocytes stimulated with IL-1β
To further determine whether ozone-induced autophagy resulted from activation of the AMPK/mTOR signaling pathway, the effect of the AMPK inhibitor com C on ozone-induced autophagy in chondrocytes stimulated with IL-1β was detected. Figure 8 shows that p-AMPK protein expression was inhibited by pretreatment with com C, and the protein expression of p-mTOR was obviously increased in IL-1β-stimulated chondrocytes treated with ozone (Figure 8A, B). However, the protein expression of ULK1 was significantly improved with ozone treatment, and this result did not visibly decline with com C pretreatment (Figure 8C).
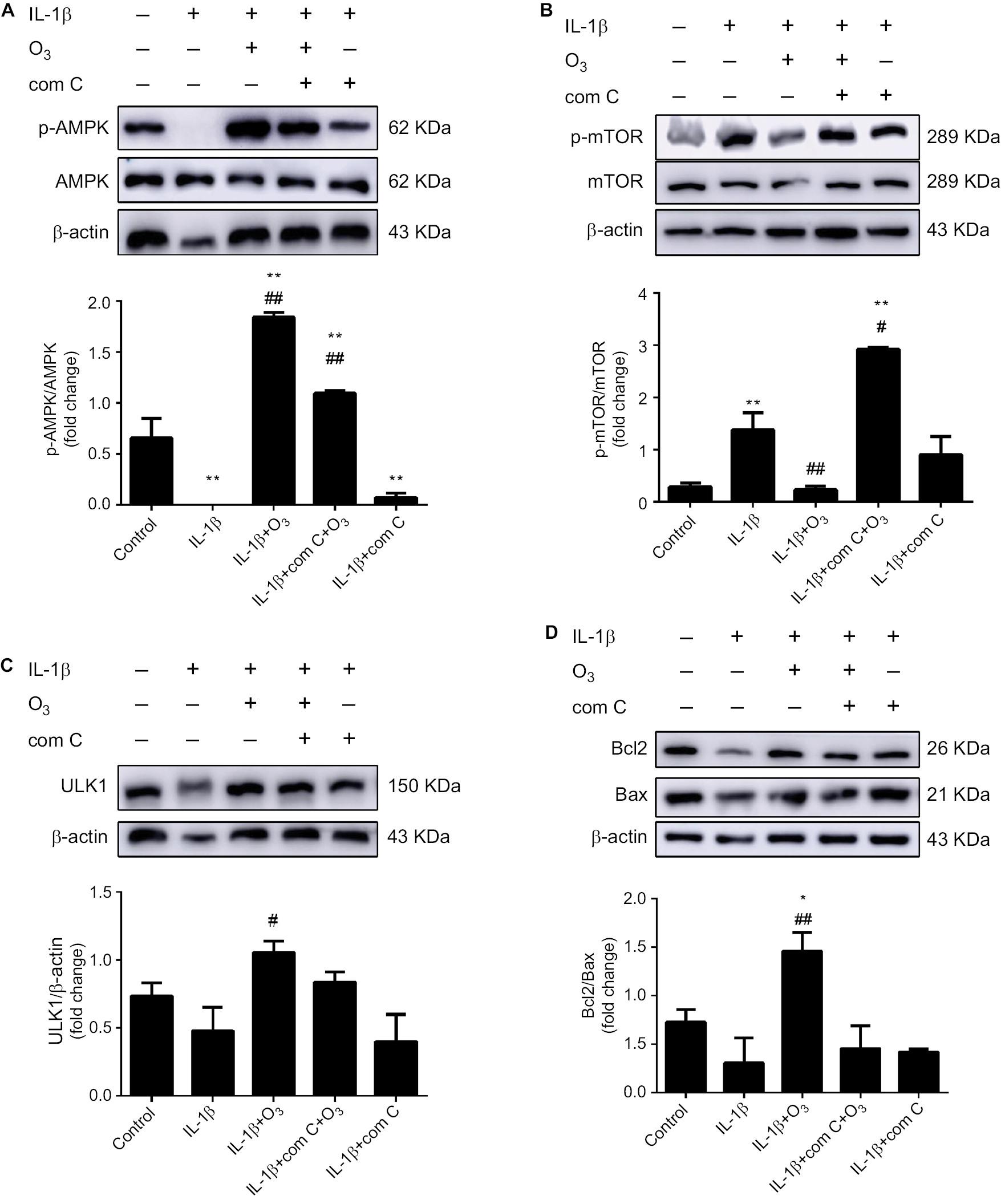 | Figure 8 Ozone activated the AMPK/mTOR signaling pathway in chondrocytes stimulated with IL-1β, and this effect was inhibited by com C. Notes: Cells were treated with IL-1β for 24 hours, with or without pretreatment with the AMPK inhibitor com C (5 µM) for 12 hours. The levels of p-AMPK (A), p-mTOR (B) and ULK1 (C) and the Bcl2/Bax ratio (D) in chondrocytes stimulated with IL-1β were measured by Western blotting. The data are presented as the mean ± SD of three independent experiments. *P<0.05, **P<0.01, compared with the control; #P<0.05, ##P<0.01, compared with IL-1β. |
Furthermore, the autophagy-related protein LC3 II decreased and P62 sharply increased in chondrocytes stimulated with IL-1β and pretreated with com C (Figure 9). In addition, the formation process of autophagosomes was inhibited by com C (Figure 5). Moreover, the apoptosis-associated proteins Bcl2 and Bax play important roles in regulating apoptosis. An increased Bcl2/Bax ratio prevents chondrocyte apoptosis via the AMPK signaling pathway.27 The Bcl2/Bax ratio was significantly higher in chondrocytes stimulated with IL-1β treated with ozone compared with those pretreated with com C (Figure 8D). Com C alone did not markedly affect the Bcl2/Bax ratio in IL-1β-stimulated chondrocytes (Figure 8D). In conclusion, ozone-induced autophagy inhibited apoptosis by activating the AMPK/mTOR pathway, but this result was blocked by com C.
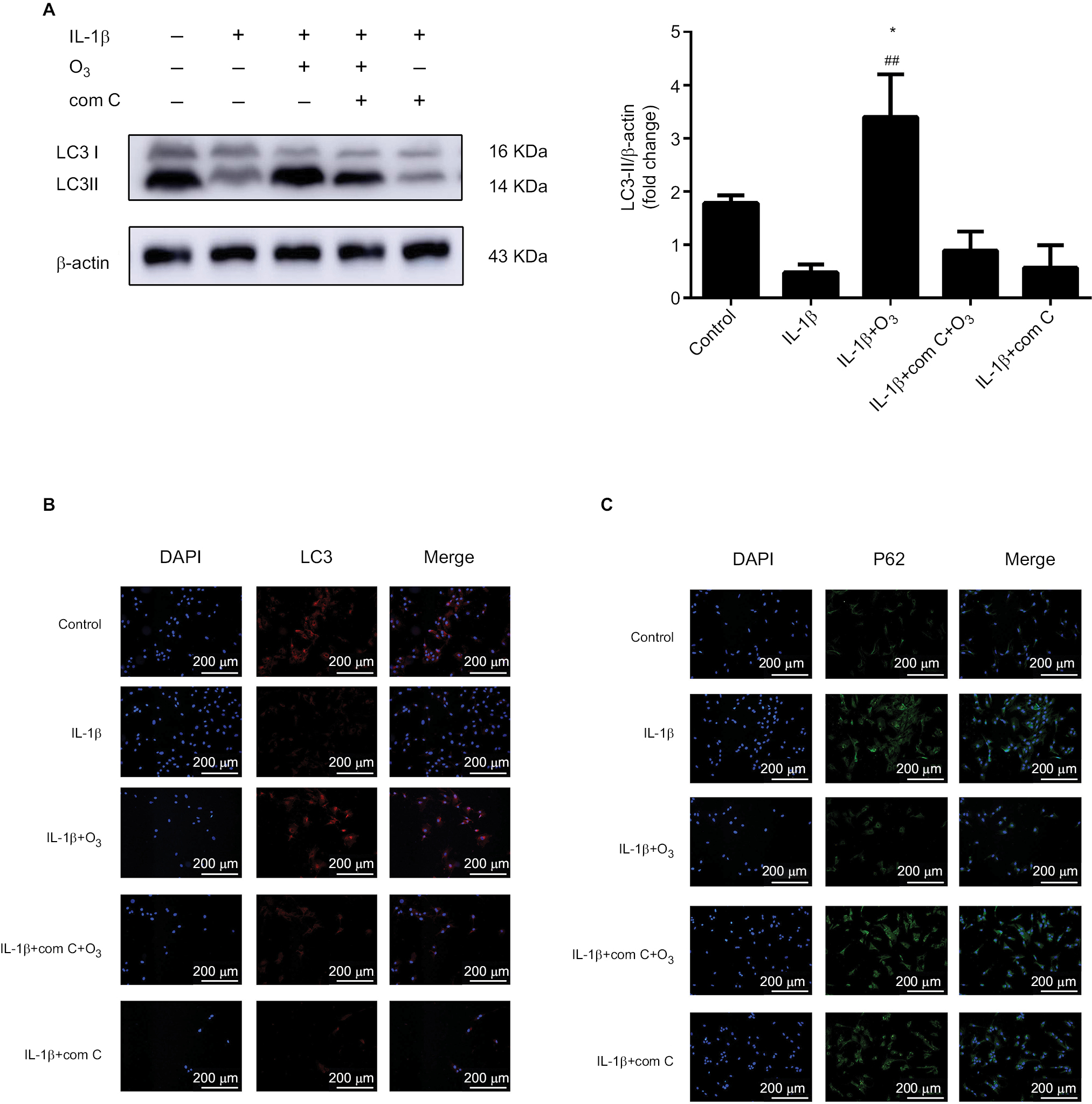 | Figure 9 Ozone induced autophagy in chondrocytes stimulated with IL-1β (10 ng/mL), and this effect was blocked by com C. Notes: Cells were treated with IL-1β for 24 hours, with or without pretreatment with the AMPK inhibitor com C (5 µM) for12 h. The levels of LC3 (A, B) and P62 (C) were measured by Western blotting and immunofluorescence. The data are presented as the mean ± SD of three independent experiments. *P<0.05, **P<0.01, compared with the control; #P<0.05, ##P<0.01, compared with IL-1β. |
The AMPK inhibitor compound C blocks the inhibition of IL-6 and TNFα mRNA expression in chondrocytes stimulated with IL-1β
Research shows that inflammatory cytokines, such as IL-6 and TNF-α, and multiple chemokines promote OA progression by altering chondrocyte function and viability.28 To determine whether the inhibition of mRNA expression levels of IL-6 and TNF-α by ozone resulted from activation of the AMPK/mTOR pathway in chondrocytes stimulated with IL-1β, the mRNA expression of IL-6 and TNF-α after pretreatment with com C was measured by qRT-PCR. The results showed that com C led to high mRNA levels of IL-6 and TNF-α, and this partly inhibited the suppression of TNF-α mRNA expression caused by ozone (Figure 10A, B). These data indicated that the AMPK/mTOR pathway might be involved in the inhibition of IL-6 and TNF-α mRNA expression in chondrocytes stimulated with IL-1β and treated with ozone.
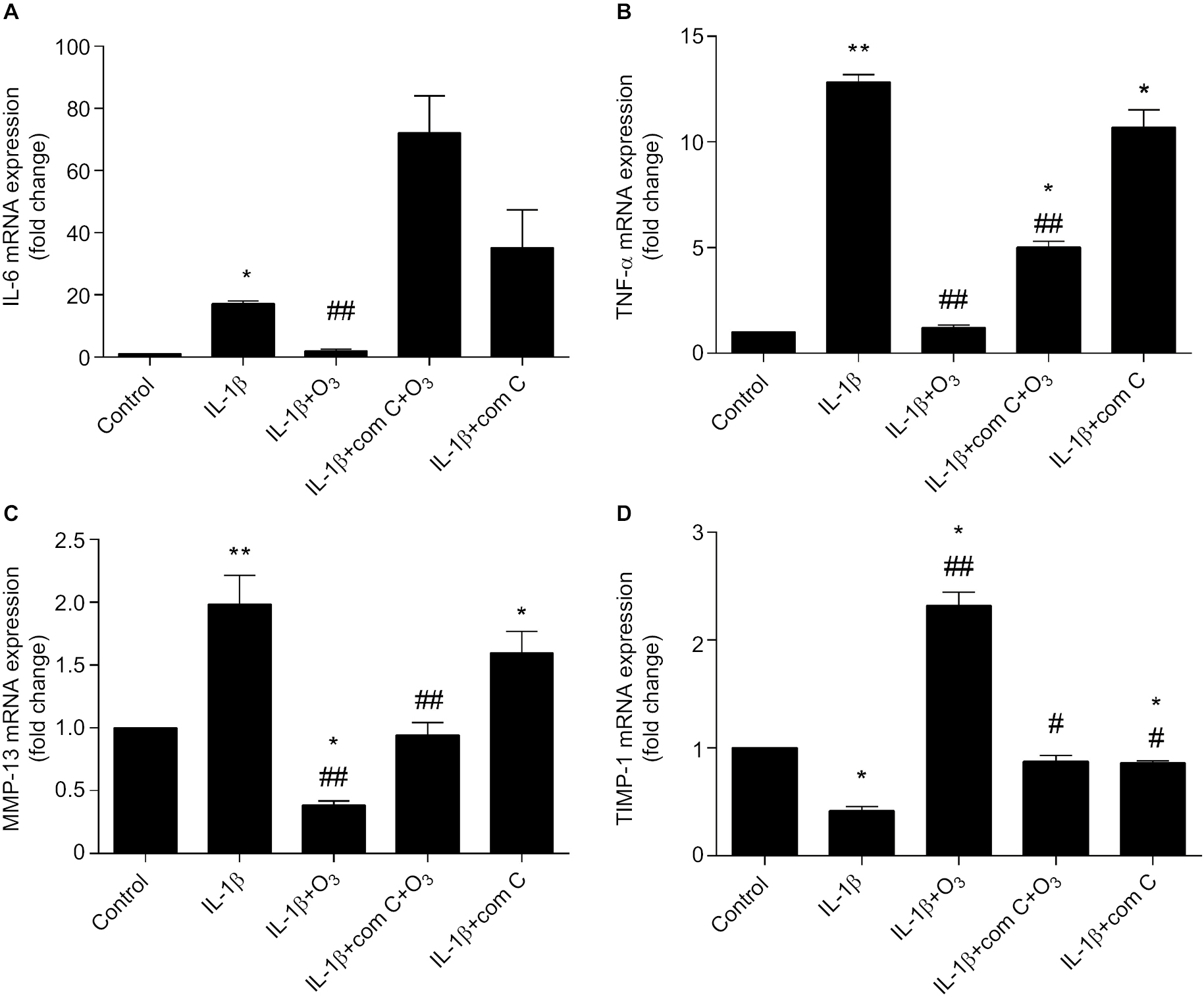 | Figure 10 Ozone suppressed inflammatory cytokines and regulated chondrocyte metabolism, and this effect was blocked by com C. Notes: The cells were treated with IL-1β for 24 hours, with or without pretreatment with the AMPK inhibitor com C (5 µM) for 12 hours. The mRNA expression of IL-6 (A), TNF-α (B), MMP-13 (C) and TIMP-1 (D) in chondrocytes stimulated with IL-1β was measured by qRT-PCR. The data are presented as the mean ± SD of three independent experiments. *P<0.05, **P<0.01, compared with the control; #P<0.05, ##P<0.01, compared with IL-1β. |
Ozone maintains the balance between extracellular matrix synthesis and degradation
Chondrocytes are indispensable for maintaining a healthy cartilage extracellular matrix.29 As shown in Figure 10C, D, the mRNA expression of MMP-13 obviously increased and TIMP-1 decreased in IL-1β-stimulated chondrocytes. However, ozone reversed these effects, inhibiting MMP-13 and promoting TIMP-1 mRNA levels. Moreover, com C partially inhibited the effect of ozone on chondrocytes stimulated with IL-1β. The results verified that ozone contributed to improving the balance between extracellular matrix synthesis and degradation.
Discussion
OA is a general joint disease that affects approximately 3.3% to 3.6% of the population globally, this representing a global burden.30,31 The main symptoms of OA are joint pain, arthroncus, stiffness and limited range of motion. Autophagy is constitutively activated in normal chondrocytes, which is a key cell survival mechanism under various forms of stress.11 Nevertheless, there are some side effects of ozone, such as neurotoxicity and respiratory injury. Ozone therapy has been widely used in the treatment of OA and has yielded positive results.32 However, the clinical application of ozone seems to be empirical, and there are limited studies showing its molecular biological mechanisms.
Our study demonstrated that ozone dramatically improved autophagy in chondrocytes stimulated with IL-1β, suppressed inflammation and promoted the balance between extracellular matrix synthesis and degradation in OA. Ozone treatment significantly increased LC3 II, Beclin-1 and ULK1 and decreased mTOR and P62 in IL-1β-stimulated chondrocytes; these proteins are autophagy-related proteins that are involved in key molecular signaling pathways. Furthermore, ozone treatment profoundly upregulated the levels of p-AMPK, and the improved autophagy in chondrocytes stimulated with IL-1β was suppressed by com C. In addition, ozone treatment significantly suppressed inflammation and regulated metabolism in IL-1β-stimulated chondrocytes. Above all, AMPK activation plays an essential role in promoting autophagy in chondrocytes stimulated with IL-1β and treated with ozone, allowing a new perspective regarding the mechanism of ozone treatment for OA to be proposed.
In this study, we initially sought to establish the effect of ozone treatment on IL-1β-stimulated chondrocytes as well as its impact on the autophagy of chondrocytes with IL-1β. We explored the suitable concentration of ozone for chondrocytes stimulated with IL-1β using a CCK-8 kit. Our results showed that ozone at 40, 50 and 60 µg/mL reduced cell viability in a concentration-dependent manner. However, 30 µg/mL ozone was the most suitable for chondrocytes simulated with IL-1β, and it promoted cell viability (Figure 3A). These results indicated that an ozone concentration of 30 µg/mL could be suitable for the treatment of chondrocytes stimulated with IL-1β.
Autophagy is considered an important cell viability mechanism under diversified situations of stress.33 Moreover, normal autophagy in articular cartilage plays a crucial role in cellular homeostasis.10 Our results demonstrated that the level of autophagy was decreased in IL-1β-stimulated chondrocytes in vitro; the autophagy-related marker LC3 II was decreased, whereas P62 was increased. Consistent with our results, the protein expression of LC3 II was significantly downregulated and P62 was upregulated in chondrocytes stimulated with IL-1β in vitro.34 More importantly, our data showed that ozone promoted a low level of autophagy in chondrocytes stimulated with IL-1β. Furthermore, the ozone-induced autophagy of chondrocytes stimulated by IL-1β was inhibited by 3MA (Figure 4). However, Beclin-1, a key autophagy-related marker, was not affected by 3MA and was still expressed at a high level with ozone treatment (Figure 6C). In addition, autophagosomes and degradative autophagosomes were observed by TEM. The ozone-induced autophagy was inhibited by 3MA (Figure 5), suggesting that ozone-induced autophagy might not be affected by Beclin-1.
It has been reported that the level of autophagy is related to ROS and that autophagy is induced by ROS through the AMPK/mTOR signaling pathway.35,36 In order of preference, ozone reacts with polyunsaturated fatty acids, antioxidants such as ascorbic and uric acids, thiol compounds with -SH groups such as cysteine, reduced glutathione (GSH) and albumin. All of these compounds act as electron donors and undergo oxidation. The main reaction shows the simultaneous formation of one mole of hydrogen peroxide (included among reactive oxygen species, ROS) and two moles of lipid oxidation products.37In other words, the effect of ozone treatment activates the antioxidation system mediated by ROS. Therefore, we further studied whether the AMPK/mTOR signaling pathway was involved in ozone-induced autophagy in chondrocytes stimulated with IL-1β. Our data demonstrated that ozone induced AMPK phosphorylation and suppressed mTOR phosphorylation in chondrocytes stimulated with IL-1β. In addition, 3MA was a barrier for ozone activation of the AMPK/mTOR signaling pathway (Figure 5A, B). Furthermore, com C inhibited ozone-induced autophagy in IL-1β-stimulated chondrocytes (Figures 5 and 7). Therefore, AMPK is a key regulator of autophagy in chondrocytes stimulated with IL-1β. Additionally, metformin, which is mainly used for clinical treatment of diabetes, is an agonist of AMPK. However, a previous clinical study reported that metformin does not reduce the risk of OA.38
ULK1 is another key regulator of autophagy that is also regulated by AMPK and mTOR. AMPK can promote autophagy by directly activating ULK1 when the organism lacks energy. Otherwise, the increased mTOR activity inhibits ULK1 to suppress autophagy and interrupts the interconnection between ULK1 and AMPK.39 Our results showed that com C did not completely inhibit the protein level of ULK1, but autophagy was decreased with com C pretreatment in chondrocytes stimulated with IL-1β (Figures 8C and 10). Therefore, ULK1 is not the crucial factor for ozone-induced autophagy in chondrocytes. Activating the AMPK/mTOR signaling pathway might be the key mechanism of ozone-induced autophagy in chondrocytes stimulated with IL-1β.
Moreover, inflammation suppresses autophagy in OA, which may aggravate the degree of OA. During the course of inflammation, proinflammatory cytokines, such as TNF-α, IL-1β and IL-6, are secreted by activated chondrocytes and stimulate the production of other inflammatory cytokines and matrix degrading enzymes.40 Karouzakis et al and Vaillant et al reported that ozone therapy reduces TNF-α concentrations, which is a proinflammatory cytokine and can activate the nuclear factor-κB (NF-κB) pathway, leading to a downstream cascade of other inflammatory cytokines and giving rise to a vicious cycle that perpetuates the chronic inflammatory process.41,42 Our data suggested that ozone-induced autophagy might contribute to inhibiting the expression of proinflammatory cytokines in IL-1β-stimulated chondrocytes. However, the downregulation of TNF-α mRNA was not completely inhibited by com C in chondrocytes with IL-1β (Figure 9A, B). These results indicated that activation of the AMPK/mTOR signaling pathway by ozone participates in suppressing the expression of inflammatory cytokines and that ozone inhibits the expression of TNF-α by other signaling pathways besides AMPK/mTOR. These results were consistent with the previous results showing that the activation of AMPK inhibits inflammation by aggravating autophagy.43
Furthermore, these proinflammatory cytokines play important roles in disrupting the cartilaginous metabolic balance between MMPs and TIMPs, which mediate cartilage degradation.44 These proinflammatory cytokines increase the release of MMPs and aggrecanase, which are enzymes that degrade the matrices, causing cartilage damage, disturbance of the metabolic balance of the cartilage matrix and eventually apoptosis.45 Therefore, we evaluated whether ozone regulated the cartilaginous metabolic balance interrupted by inflammatory cytokines, and the mRNA expression levels of MMP-13 and TIMP-1 were measured. Our data demonstrated that ozone-induced autophagy might regulate the cartilaginous metabolism interrupted by inflammatory cytokines in chondrocytes stimulated with IL-1β. As Jiang et al reported, adipose-derived stem cells induced autophagy and inhibited the catabolic response of chondrocytes treated with IL-1β.46 Ozone reduced the amount of MMP-13 to lower levels than those detected in control chondrocytes and chondrocytes stimulated with only IL-1β, but com C inhibited the reduction caused by ozone to a certain degree. Likewise, com C slightly suppressed the enhancement of the TIMP-1 level caused by ozone. Our data suggested that, in addition to the AMPK/mTOR signaling pathway, ozone-induced autophagy regulating cartilaginous metabolism also acts upon other signaling pathways, such as NF-κB.41,42
Conclusion
We demonstrated that ozone induced autophagy by activating the AMPK/mTOR signaling pathway in chondrocytes stimulated with IL-1β. We propose a potential mechanism for ozone treatment of OA, and we provide powerful evidence for the application of ozone for the treatment of OA.
Acknowledgment
This research was funded by grants from the National Natural Science Foundation of China (81771199, 81271346).
Disclosure
The authors report no conflicts of interest in this work.
References
Felson DT, Zhang Y. An update on the epidemiology of knee and hip osteoarthritis with a view to prevention. Arthritis Rheum. 1998;41(8):1343–1355. | ||
Greene MA, Loeser RF. Aging-related inflammation in osteoarthritis. Osteoarthritis Cartilage. 2015;23(11):1966–1971. | ||
Yu D, Xu J, Liu F, Wang X, Mao Y, Zhu Z. Subchondral bone changes and the impacts on joint pain and articular cartilage degeneration in osteoarthritis. Clin Exp Rheumatol. 2016;34(5):929–934. | ||
Liu-Bryan R. Inflammation and intracellular metabolism: new targets in OA. Osteoarthritis Cartilage. 2015;23(11):1835–1842. | ||
Cawston T. Matrix metalloproteinases and TIMPs: properties and implications for the rheumatic diseases. Mol Med Today. 1998;4(3):130–137. | ||
Tetlow LC, Adlam DJ, Woolley DE. Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum. 2001;44(3):585–594. | ||
Li YS, Zhang FJ, Zeng C, et al. Autophagy in osteoarthritis. Joint Bone Spine. 2016;83(2):143–148. | ||
Caramés B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62(3):791–801. | ||
Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. | ||
Almonte-Becerril M, Navarro-Garcia F, Gonzalez-Robles A, Vega-Lopez MA, Lavalle C, Kouri JB. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of osteoarthritis within an experimental model. Apoptosis. 2010;15(5):631–638. | ||
Mizushima N. Autophagy in Infection and Immunity[M]. Berlin, Heidelberg: Springer; 2009. | ||
Caramés B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis. 2012;71(4):575–581. | ||
Takayama K, Kawakami Y, Kobayashi M, et al. Local intra-articular injection of rapamycin delays articular cartilage degeneration in a murine model of osteoarthritis. Arthritis Res Ther. 2014;16(6):482. | ||
Yan H, Zhou HF, Hu Y, Pham CT. Suppression of experimental arthritis through AMP-activated protein kinase activation and autophagy modulation. J Rheum Dis Treat. 2015;1(1):5. | ||
Wolff HH. Die Behandlung perripherer Durchblutungsstorungen mit Ozon [The treatment of peripheral circulatory disturbances with ozone]. Erfahr Hk. 1974;23:181–184. German. | ||
Bocci V, Borrelli E, Zanardi I, Travagli V. The usefulness of ozone treatment in spinal pain. Drug Des Devel Ther. 2015;9:2677–2685. | ||
Hashemi M, Poorfarokh M, Mohajerani SA, et al. Injection of intradiscal o2-o3 to reduce pain and disability of patients with low back pain due to prolapsed lumbar disk. Anesth Pain Med. 2014;4(5):e19206. | ||
Al-Jaziri AA, Mahmoodi SM. Painkilling effect of ozone-oxygen injection on spine and joint osteoarthritis. Saudi Med J. 2008;29(4):553–557. | ||
Sagai M, Bocci V. Mechanisms of action involved in ozone therapy: is healing induced via a mild oxidative stress? Med Gas Res. 2011;1:29. | ||
Lu L, Pan C, Chen L, et al. AMPK activation by peri-sciatic nerve administration of ozone attenuates CCI-induced neuropathic pain in rats. J Mol Cell Biol. 2017;9(2):132–143. | ||
Li Y, Lin X, Zhao X, et al. Ozone (O3) elicits neurotoxicity in spinal cord neurons (SCNs) by inducing ER Ca(2+) release and activating the CaMKII/MAPK signaling pathway. Toxicol Appl Pharmacol. 2014;280(3):493–501. | ||
Manzer R, Wang J, Nishina K, Mcconville G, Mason RJ. Alveolar epithelial cells secrete chemokines in response to IL-1beta and lipopolysaccharide but not to ozone. Am J Respir Cell Mol Biol. 2006;34(2):158–166. | ||
Raeissadat SA, Rayegani SM, Forogh B, Hassan Abadi P, Moridnia M, Rahimi Dehgolan S. Intra-articular ozone or hyaluronic acid injection: which one is superior in patients with knee osteoarthritis? A 6-month randomized clinical trial. J Pain Res. 2018;11:111–117. | ||
Tee A. The target of rapamycin and mechanisms of cell growth. Int J Mol Sci. 2018;19(3):880. | ||
Huang X, Wu H, Jiang R, et al. The antidepressant effects of a-tocopherol are related to activation of autophagy via the AMPK/mTOR pathway. Eur J Pharmacol. 2018;833:1–7. | ||
Chang YP, Ka SM, Hsu WH, et al. Resveratrol inhibits NLRP3 inflammasome activation by preserving mitochondrial integrity and augmenting autophagy. J Cell Physiol. 2015;230(7):1567–1579. | ||
Zhou Y, Liu SQ, Yu L, et al. Berberine prevents nitric oxide-induced rat chondrocyte apoptosis and cartilage degeneration in a rat osteoarthritis model via AMPK and p38 MAPK signaling. Apoptosis. 2015;20(9):1187–1199. | ||
Chevalier X, Eymard F, Richette P. Biologic agents in osteoarthritis: hopes and disappointments. Nat Rev Rheumatol. 2013;9(7):400–410. | ||
Blaney Davidson EN, van Caam AP, van der Kraan PM. Osteoarthritis year in review 2016: biology. Osteoarthritis Cartilage. 2017;25(2):175–180. | ||
Bijlsma JW, Berenbaum F, Lafeber FP. Osteoarthritis: an update with relevance for clinical practice. Lancet. 2011;377(9783):2115–2126. | ||
Sen R, Hurley JA. Osteoarthritis. Treasure Island (FL): StatPearls Publishing; 2018. | ||
Manoto SL, Maepa MJ, Motaung SK. Medical ozone therapy as a potential treatment modality for regeneration of damaged articular cartilage in osteoarthritis. Saudi J Biol Sci. 2018;25(4):672–679. | ||
Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol. 2009;335:71–84. | ||
Xue JF, Shi ZM, Zou J, Li XL. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of articular chondrocytes and attenuates inflammatory response in rats with osteoarthritis. Biomed Pharmacother. 2017;89:1252–1261. | ||
Ma KG, Shao ZW, Yang SH, et al. Autophagy is activated in compression-induced cell degeneration and is mediated by reactive oxygen species in nucleus pulposus cells exposed to compression. Osteoarthritis Cartilage. 2013;21(12):2030–2038. | ||
Chen JW, Ni BB, Zheng XF, Li B, Jiang SD, Jiang LS. Hypoxia facilitates the survival of nucleus pulposus cells in serum deprivation by down-regulating excessive autophagy through restricting ROS generation. Int J Biochem Cell Biol. 2015;59:1–10. | ||
Pryor WA, Squadrito GL, Friedman M. The cascade mechanism to explain ozone toxicity: the role of lipid ozonation products. Free Radic Biol Med. 1995;19(6):935–941. | ||
Barnett LA, Jordan KP, Edwards JJ, van der Windt DA. Does metformin protect against osteoarthritis? An electronic health record cohort study. Prim Health Care Res Dev. 2017;18(6):623–628. | ||
Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. | ||
de Luna-Preitschopf A, Zwickl H, Nehrer S, Hengstschläger M, Mikula M. Rapamycin Maintains the Chondrocytic Phenotype and Interferes with Inflammatory Cytokine Induced Processes. Int J Mol Sci. 2017;18(7):1494. | ||
Karouzakis E, Neidhart M, Gay RE, Gay S. Molecular and cellular basis of rheumatoid joint destruction. Immunol Lett. 2006;106(1):8–13. | ||
Vaillant JD, Fraga A, Díaz MT, et al. Ozone oxidative postconditioning ameliorates joint damage and decreases pro-inflammatory cytokine levels and oxidative stress in PG/PS-induced arthritis in rats. Eur J Pharmacol. 2013;714(1-3):318–324. | ||
Xu C, Wang W, Zhong J, et al. Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem Pharmacol. 2018;152:45–59. | ||
Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7(1):33–42. | ||
Kondo M, Yamaoka K, Tanaka Y. Acquiring chondrocyte phenotype from human mesenchymal stem cells under inflammatory conditions. Int J Mol Sci. 2014;15(11):21270–21285. | ||
Jiang LB, Lee S, Wang Y, Xu QT, Meng DH, Zhang J. Adipose-derived stem cells induce autophagic activation and inhibit catabolic response to pro-inflammatory cytokines in rat chondrocytes. Osteoarthritis Cartilage. 2016;24(6):1071–1081. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.