")
Back to Journals » International Journal of Nanomedicine » Volume 11
Oxidative stress, inflammation, and DNA damage in multiple organs of mice acutely exposed to amorphous silica nanoparticles
Authors Nemmar A, Yuvaraju P, Beegam S, Pathan J , Kazzam EE, Ali BH
Received 12 July 2015
Accepted for publication 3 December 2015
Published 7 March 2016 Volume 2016:11 Pages 919—928
DOI https://doi.org/10.2147/IJN.S92278
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Abderrahim Nemmar,1 Priya Yuvaraju,1 Sumaya Beegam,1 Javed Yasin,2 Elsadig E Kazzam,2 Badreldin H Ali3
1Department of Physiology, 2Department of Internal Medicine, College of Medicine and Health Sciences, United Arab Emirates University, Al Ain, UAE; 3Department of Pharmacology, College of Medicine and Health Sciences, Sultan Qaboos University, Muscat, Al-Khoudh, Sultanate of Oman
Abstract: The use of amorphous silica (SiO2) in biopharmaceutical and industrial fields can lead to human exposure by injection, skin penetration, ingestion, or inhalation. However, the in vivo acute toxicity of amorphous SiO2 nanoparticles (SiNPs) on multiple organs and the mechanisms underlying these effects are not well understood. Presently, we investigated the acute (24 hours) effects of intraperitoneally administered 50 nm SiNPs (0.25 mg/kg) on systemic toxicity, oxidative stress, inflammation, and DNA damage in the lung, heart, liver, kidney, and brain of mice. Lipid peroxidation was significantly increased by SiNPs in the lung, liver, kidney, and brain, but was not changed in the heart. Similarly, superoxide dismutase and catalase activities were significantly affected by SiNPs in all organs studied. While the concentration of tumor necrosis factor α was insignificantly increased in the liver and brain, its increase was statistically significant in the lung, heart, and kidney. SiNPs induced a significant elevation in pulmonary and renal interleukin 6 and interleukin-1 beta in the lung, liver, and brain. Moreover, SiNPs caused a significant increase in DNA damage, assessed by comet assay, in all the organs studied. SiNPs caused leukocytosis and increased the plasma activities of lactate dehydrogenase, creatine kinase, alanine aminotranferase, and aspartate aminotransferase. These results indicate that acute systemic exposure to SiNPs causes oxidative stress, inflammation, and DNA damage in several major organs, and highlight the need for thorough evaluation of SiNPs before they can be safely used in human beings.
Keywords: amorphous silica nanoparticles, organ toxicity, oxidative stress, inflammation, DNA damage
Introduction
Engineered nanomaterials are commonly defined as materials designed and produced to have structural features with at least one dimension of ≤100 nm.1 Engineered nanoparticles display features such as small size, large surface area to mass ratio, shape, crystallinity, surface charge, reactive surface groups, dissolution rate, state of agglomeration, and dispersal that give them properties greatly different from larger particles of the same composition.1 In this context, nanoparticles have been demonstrated to cause greater biological responses and particle-mediated toxicity than larger particles per given mass.1
Amorphous silica (SiO2) nanoparticles (SiNPs) are nano-sized structures of SiO2 that are used in various areas, such as sunscreen lotions, drug delivery, cosmetics, food, and chemical industries entailing human exposure during production or use.2 These wide applications of SiNPs have prompted several investigators to perform in vitro studies on the effect of these nanoparticles in different cell types such as human endothelial cells, erythrocytes, platelets, epithelial cell line A549, monocyte-macrophages J774, or co-culture models such as alveolar–capillary barrier.3–8 It has been shown that SiNPs are internalized by the cells and induce their cytotoxicity through the release of reactive oxygen species and proinflammatory cytokines, leading to DNA and cell membrane damage.3–8
In vivo research examining the potential toxicity of SiNPs is essential for acquiring more relevant information about human exposure scenarios to nanoparticles. It has been previously reported that SiNPs could penetrate the skin and enter various tissues.9 Even after inhalation, nanoparticles have been reported to translocate rapidly to the systemic circulation and reach different organs.10–13 Moreover, with medical applications, injected nanoparticles can be distributed by the bloodstream and reach various parts of the body.2 Data related to the acute toxicity of systemically administered SiNPs are scarce. An in vivo study in mice using high intravenous (iv) doses (29.5–177.5 mg/kg) of SiNPs reported histopathological injury in the lung, liver, and spleen 14 days following the exposure.14 We have recently demonstrated that intraperitoneal (ip) administration of SiNPs to mice caused systemic inflammation and coagulation events and altered vascular reactivity 24 hours post administration.15 We have now extended these studies to investigate further the effects of acute (24 hours) administration of SiNPs on oxidative stress, inflammation, and DNA damage in major organs, viz, lung, heart, liver, kidney, and brain of mice. Such a study, as far as we are aware, has not been reported before.
Materials and methods
Ethics statement
This project was reviewed and approved by the Institutional Review Board of the College of Medicine and Health Sciences [CMHS], United Arab Emirates University [UAEU], and experiments were performed in accordance with protocols approved by the Institutional Animal Care and Research Advisory Committee. All surgeries were performed under sodium pentobarbital, and all efforts were made to minimize suffering.
Amorphous SiO2 nanoparticles
Amorphous siNPs (50 nm) were purchased from Polysciences, Inc. (Warrington, PA, USA). The structure of the particles and their shape, size, and charge have recently been reported.6,16
To confirm their size, we performed transmission electron microscopic analysis. Thus, droplets (10 μL) of a suspension of 0.1 mg of SiNPs in 500 μL were placed on matured formvar/carbon film for 30 seconds. The samples were then drained and inverted onto droplets of ultrapure water for 1 hour before being drained, dried, and examined in a Philips CM10 transmission electron microscope (Philips, Eindhoven, the Netherlands).
Animals and ip administration of amorphous SiNPs
SiNPs were suspended in normal saline (NaCl 0.9%) containing Tween 80 (0.01%). To minimize aggregation, particle suspensions were always sonicated (Clifton Ultrasonic Bath, Clifton, NJ, USA) for 15 minutes and vortexed before their dilution and prior to ip administration.
Male Tuck-Ordinary mice (obtained from our CMHS animal house, UAEU, UAE), weighing 28±4 g, were housed in light (12-hour light:12-hour dark cycle) and temperature-controlled (22°C±1°C) rooms. The animals had free access to normal commercial laboratory chow and tap water ad libitum.
Blood collection and analysis
Twenty-four hours after the ip administration of either saline or SiNPs (0.25 mg/kg), in a volume of 150 μL, the animals were anesthetized ip with sodium pentobarbital (45 mg/kg), and blood was drawn from the inferior vena cava in ethylenediaminetetraacetic acid (EDTA; 4%). A sample was used for leukocytes counting using an ABX Vet ABC hematology analyzer with a mouse card (ABX Diagnostics, Montpellier, France). The remaining blood was centrifuged at 4°C for 15 minutes at 3,000 rpm, and the plasma samples were stored at -80°C until further analysis of the activities of lactate dehydrogenase (LDH), creatine kinase (CK), alanine aminotranferease (ALT), and aspartate aminotransferase (AST); urea and creatinine were measured by standard laboratory methods using commercially available kits (Hoffman-La Roche Ltd., Basel, Switzerland) and a LX20 multiple automated analyzer (Beckman Coulter, Brea, CA, USA).
Measurement of interleukin 6, interleukin-1 beta, and tumor necrosis factor α in tissues
Following blood collection, animals were sacrificed by an overdose of sodium pentobarbital and their lung, heart, liver, kidney, and whole brain were quickly collected and rinsed with ice-cold phosphate-buffered saline (pH 7.4) before homogenization in 50 mM Tris buffer containing 400 mM NaCl and 0.5% Triton X-100 at 4°C.17 The homogenates were centrifuged at 14,000 rpm for 15 minutes at 4°C to remove cellular debris, and the supernatants were used for further analysis. Protein content in each organ was measured by Bradford’s method, as described earlier.18,19 The concentrations of interleukin 6 (IL-6), interleukin-1 beta (IL-1β), and tumor necrosis factor α (TNFα) in the tissues were determined using ELISA kits (Duo Set; R&D Systems, Inc., Minneapolis, MN, USA).
Measurement of tissue lipid peroxidation, superoxide dismutase, and catalase
In separate animals, organs were collected as described earlier before homogenization in 0.1 M phosphate buffer, pH 7.4, containing 0.15 M KCl, 0.1 mM EDTA, 1 mM dithiothreitol, and 0.1 mM phenylmethylsulfonyl fluoride at 4°C. The homogenates were centrifuged at 14,000 rpm for 20 minutes at 4°C, and protein was measured as reported earlier.18–20
Reduced nicotinamide adenine dinucleotide phosphate-dependent membrane lipid peroxidation (LPO) was measured as thiobarbituric acid reactive substance using malondialdehyde as standard (Sigma-Aldrich Co., St Louis, MO, USA).18,19 Superoxide dismutase (SOD) activity was measured as the conversion of nitroblue tetrazolium (NBT) to NBT-diformazan according to the vendor’s protocol (R&D Systems, Inc.). The extent of reduction in the appearance of NBT-formazan was used as a measure of SOD activity present in each organ.18–20 Catalase (CAT) activity was measured using a commercially available kit (Cayman Chemical, Ann Arbor, MI, USA).
DNA damage assessment by comet assay
Immediately after sacrifice, the lung, heart, liver, kidney, and brain were removed from each animal. Single-cell suspensions of the different lungs, hearts, livers, kidneys, and brains were obtained according to the method described by de Souza et al.21 Each collected organ was washed in a chilled medium (Roswell Park Memorial Institute medium 1640, 15% dimethyl sulfoxide [DMSO], 1.8% (w/v) NaCl). The lung, heart, liver, kidney, and brain tissues were placed in 1.5 mL medium and chopped finely into pieces in a Petri dish using scissors. The pieces were allowed to settle, and the supernatant was collected in a 15 mL tube. The obtained cell suspension was centrifuged at 1,000 rpm for 5 minutes at 4°C. The supernatant was discarded, and the pellets were resuspended in 0.5 mL of the medium. The cell suspensions were mixed with low melting point agarose solution (0.65%) and spread onto agarose (1.5%)-precoated microscope slides. For each treatment, five slides were prepared, which were incubated in ice-cold lysis buffer (2.5 M NaCl, 10 mM Tris, 100 mM EDTA, 1% Triton X-100, and 10% DMSO) at 4°C for at least 1 hour to remove the cell membranes. After the incubation, the slides were placed in a horizontal electrophoresis unit and incubated in electrophoresis buffer (0.2 M EDTA, 5 M NaCl, pH 10) for 20 minutes for DNA unwinding and the expression of alkali labile sites. Then, electrophoresis was conducted for 20 minutes at 25 V and 300 mA. After that, the slides were neutralized with Tris buffer (0.4 M Trizma base, pH 7.5) for 5 minutes and washed with methanol. Then the slides were stained with propidium iodide, as previously described.22 All these steps were performed in darkness to prevent additional DNA damage. The slides were mounted on a fluorescent microscope, and cell scoring was performed. Fifty cells from each treatment were scored and analyzed for DNA migration, and the average of the five slides from each group was calculated. The measurement of length of the DNA migration (ie, diameter of the nucleus plus migrated DNA) was calculated using image analysis AxioVision 3.1 software (Carl Zeiss Meditec AG, Jena, Germany).23
Statistics
All statistical analyses were performed with GraphPad Prism software Version 5 (GraphPad, San Diego, CA, USA). To determine whether parameters were normally distributed, the D’Agostino and Pearson normality test was applied. Normally distributed data were analyzed using the unpaired t-test for differences between groups. Nonnormally distributed data (ALT, AST, DNA migration [lung heart, liver, and kidney], LPO [liver, kidney, and brain], SOD [lung, liver, and brain], CAT [liver and brain], and TNFα [heart and kidney] and IL-6 [lung]) were analyzed using the Mann–Whitney test for differences between groups. All the data in figures are reported as mean ± standard error of the mean (SEM). P-values <0.05 are considered significant.
Results
Transmission electron microscopy analysis of SiNPs
Transmission electron microscopy of the SiNPs revealed a particle size of ~50 nm (Figure 1). This confirms the size provided by the manufacturer.
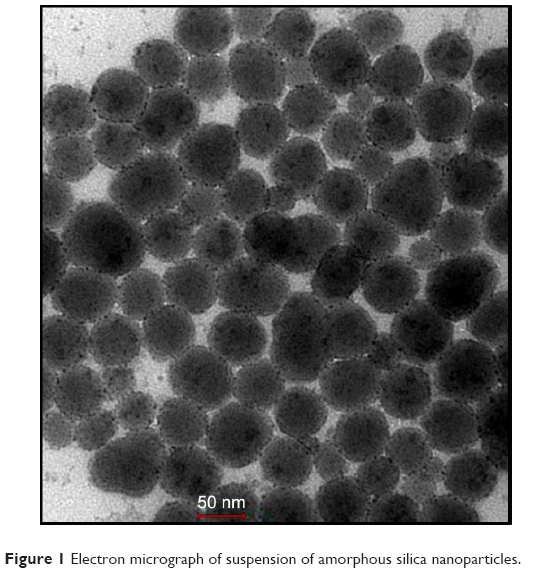 | Figure 1 Electron micrograph of suspension of amorphous silica nanoparticles. |
Effect of SiNPs on circulating leukocyte numbers and plasma LDH and CK
Figure 2A illustrates that acute exposure to SiNPs caused a significant increase (P<0.05) in circulating leukocytes compared with the control group.
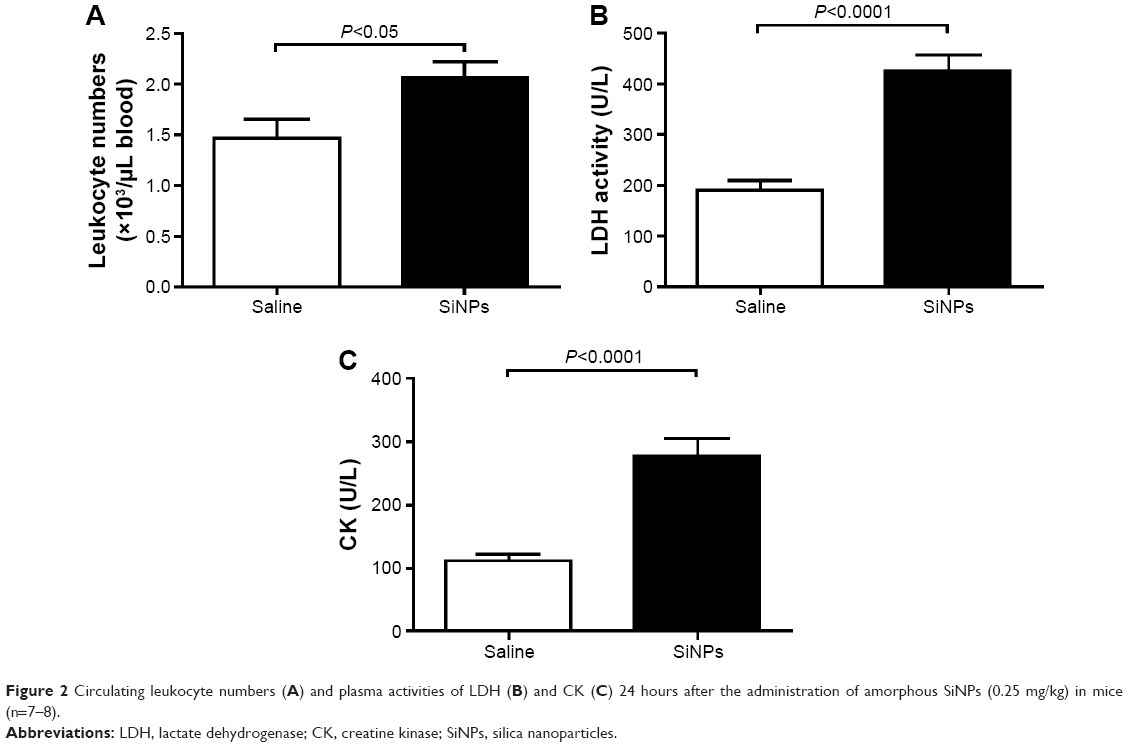 | Figure 2 Circulating leukocyte numbers (A) and plasma activities of LDH (B) and CK (C) 24 hours after the administration of amorphous SiNPs (0.25 mg/kg) in mice (n=7–8). |
The plasma concentration of LDH, suggestive of cytolysis, was significantly increased (P<0.0001) following administration of SiNPs compared with the control group (Figure 2B).
Compared with the control group, the administration of SiNPs caused a significant increase (P<0.0001) in the plasma CK activity (Figure 2C).
Effect of SiNPs on plasma levels of ALT, AST, urea, and creatinine
Figure 3 illustrates the effect of SiNPs on the activities of ALT and AST. Compared with the control group, acute ip administration of SiNP induced a significant increase (P<0.01) in ALT activity (Figure 3A). Likewise, the AST was significantly augmented (P<0.005) in SiNPs-treated mice compared with the saline-treated group (Figure 3B).
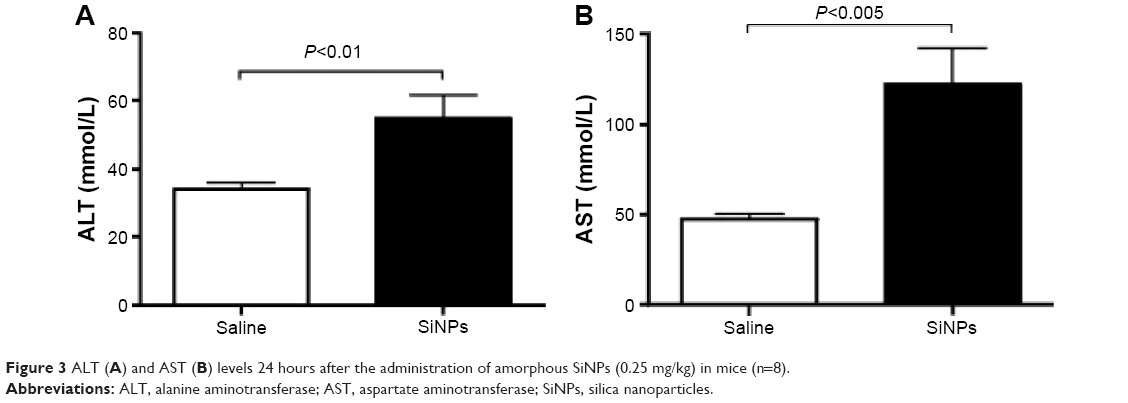 | Figure 3 ALT (A) and AST (B) levels 24 hours after the administration of amorphous SiNPs (0.25 mg/kg) in mice (n=8). |
The concentration of urea in the control group (7.02±1.0 mmol/L; n=8) was not significantly different from that observed in the SiNPs-treated ones (7.3±1.6 mmol/L; n=8; P>0.05). Likewise, the creatinine level in the control group (14.9±2.2 μmol/L; n=8) was not significantly different from that observed in the SiNPs group (16.1±2.9 μmol/L; n=8; P>0.05).
Effect of SiNPs on LPO concentration, and SOD and CAT activities in the lung, heart, liver, kidney, and brain
SiNPs induced a significant increase in LPO levels in the lung (P<0.0001), liver (P<0.005), kidney (P<0.005), and brain (P<0.01) compared with the control group. However, the concentration of LPO in the heart was not affected by SiNPs (Table 1).
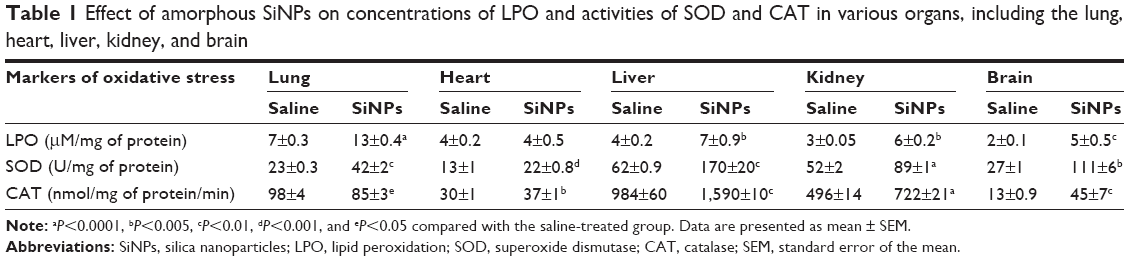 | Table 1 Effect of amorphous SiNPs on concentrations of LPO and activities of SOD and CAT in various organs, including the lung, heart, liver, kidney, and brain |
Compared with saline-treated mice, administration of SiNPs caused a significant increase in SOD activity in the lung (P<0.01), heart (P<0.001), liver (P<0.01), kidney (P<0.0001), and brain (P<0.01; Table 1).
Administration of SiNPs induced a significant decrease in CAT activity in the lung (P<0.05). However, a significant increase in CAT activity was observed in the heart (P<0.005), liver (P<0.005), kidney (P<0.0001), and brain (P<0.01) following the exposure to SiNPs (Table 1).
Effect of SiNPs on TNFα, IL-6, and IL-1β concentrations in the lung, heart, liver, kidney, and brain
Table 2 shows the concentrations of proinflammatory cytokines in the lung, heart, liver, kidney, and brain.
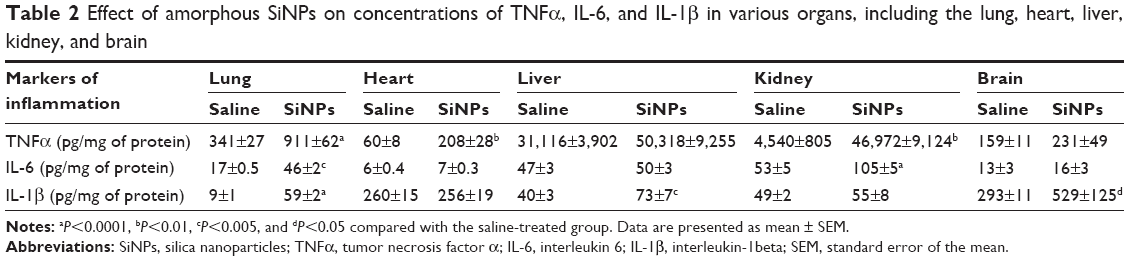 | Table 2 Effect of amorphous SiNPs on concentrations of TNFα, IL-6, and IL-1β in various organs, including the lung, heart, liver, kidney, and brain |
Compared with saline-treated mice, ip administration of SiNPs induced a significant increase in TNFα concentration in the lung (P<0.0001), heart (P<0.01), and kidney (P<0.01). However, the increase in TNFα concentration in the liver and brain did not reach statistical significance (Table 2).
Administration of SiNPs induced a significant increase in IL-6 in the lung (P<0.005) and kidney (P<0.0001) compared with the control group. IL-6 concentration insignificantly increased in the heart, liver, and brain (Table 2).
The concentration of IL-1β was significantly increased in the lung (P<0.0001), liver (P<0.005), and brain (P<0.05) of SiNPs-treated mice compared with the saline-treated ones. However, no increase was observed in the heart and kidney by the administration of SiNPs (Table 2).
Effect of SiNPs on DNA damage in the lung, heart, liver, kidney, and brain
Figure 4 shows that compared with the control group, the ip injection of SiNPs caused a significant increase in DNA migration in the lung (P<0.05), heart (P<0.01), liver (P<0.05), kidney (P<0.05), and brain (P=0.0005).
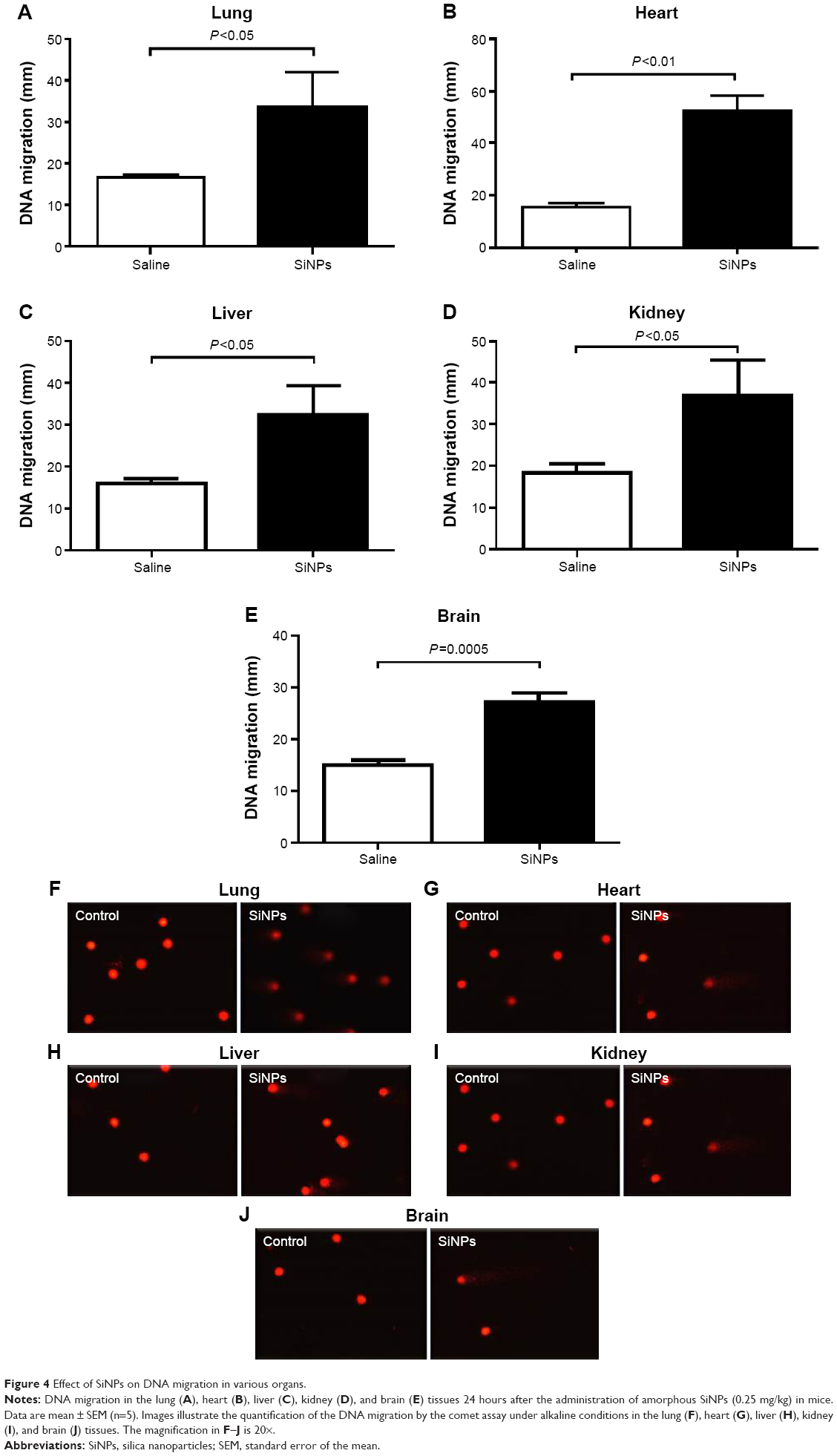 | Figure 4 Effect of SiNPs on DNA migration in various organs. |
Discussion
In this study, we have shown that acute (24 hours) ip administration of SiNPs caused an increase in circulating leukocytes, LDH, CK, and liver enzymes AST and ALT. Moreover, we have reported the occurrence of inflammation, oxidative stress, and DNA damage in several major organs, including lung, heart, liver, kidney, and brain, following the acute systemic administration of SiNPs.
We investigated the acute toxicity of systemically administered amorphous SiNPs. Acute toxicity studies provide specific information on the initial changes in various organs following exposure to SiNPs, which may constitute the underlying causal chain of reactions leading to the ultimate chronic effects of these nanoparticles. In order to imitate the effect of injected nanoparticles as it may occur in medical applications, SiNPs were administered ip.2,15 Moreover, even after inhalation, ingestion, or skin application, nanoparticles can penetrate into the systemic circulation and reach various organs.2,10,12,13 Previous studies assessed the toxicity of SiNPs given intravenously using very high doses of nanoparticles (29.5–177.5 mg/kg14 and 10–30 mg/kg).24 Here, we used a lower dose of SiNPs, ie, 0.25 mg/kg, which would correspond to a dose of 7 μg SiNPs for a mouse of 28 g. The approximate blood volume of a mouse is 80 μL/g, and for a 28 g mouse, this is equivalent to 2.2 mL. Assuming that 100% of the injected dose would reach the blood, a concentration of 3.2 μg SiNPs/mL would reach the plasma. Regarding a human exposure scenario (eg, nanomedicine), pharmacokinetic data related to the liposome-encapsulated doxorubicin nanoparticles, used in cancer therapy, established plasma concentrations of >10 μg/mL.25
Our data show that acute exposure to SiNPs caused leukocytosis and increased plasma activities of LDH and CK. Several studies have reported an increase in leukocyte numbers following exposure to particulate air pollution and engineered nanoparticles.26–28 Along with the leukocytosis, we found an increase in plasma activities of LDH and CK. Increase in LDH activity is suggestive of cytolysis. CK was also increased in SiNPs-treated mice. CK is an intracellular enzyme present in striated and smooth muscles and in the brain; it is an important enzyme regulator of high-energy phosphate production and utilization in contractile tissues, and is considered a marker of muscle damage.29 Both LDH and CK were reported to increase following exposure to nanoparticles.7,8,15,30 Moreover, the increase in ALT and AST shows that SiNPs cause hepatotoxicity. This finding indicates that the liver is a potential target organ for SiNPs. It has been previously reported that plain SiNPs induce hepatotoxicity in mice and that surface amino or carboxyl modification mitigated the liver toxicity of plain-surface SiNPs.31
A recent study demonstrated that iv injection of fluorophore-conjugated and radiolabeled amorphous SiNPs (20–25 nm) in mice caused accumulation of nanoparticles in various organs, including liver, kidney, lung, heart, stomach, and spleen.32 Moreover, SiO2-coated magnetic nanoparticles containing rhodamine B isothiocyanate within an SiO2 shell (50 nm) were found in the brain following ip injection in mice.33 Inspired by the observations that leukocyte numbers, LDH, CK, AST, and ALT were increased by administration of SiNPs, and because systemically administered nanoparticles can reach various organs, we wanted to verify whether and to what extent these nanoparticles can induce inflammation, oxidative stress, and DNA damage in major organs, including the lung, heart, liver, kidney, and brain. Such an approach would provide insight into the mechanisms underlying SiNPs-induced toxicity.
Oxidative stress results from an imbalance between radical-generating and radical-scavenging systems, leading to cell membrane impairment or DNA damage.34 To further evaluate the mechanism underlying the toxicity of SiNPs, we measured the degree of oxidative stress by determining LPO levels in lung, heart, liver, kidney, and brain. Our data show that exposure to SiNPs causes a significant increase in LPO in the lung, liver, kidney, and brain, but not the heart. Moreover, we have also measured the activities of two antioxidant enzymes (SOD and CAT). Our data show that SOD activity was significantly increased in all the studied organs. CAT activity was significantly decreased in the lung, but was significantly augmented in the heart, liver, kidney, and brain. An increase in antioxidants suggests that the rise in oxidative stress induced by SiNPs was accompanied by an increase in the activity of SOD and CAT, indicating that the development of oxidative stress is accompanied by an adaptive response that counterbalances the potentially damaging activity of oxygen free radicals by antioxidant defense mechanisms.35,36 However, the decrease in CAT observed in the lung suggests its consumption during the breakdown of free radicals.20 While several studies reported the occurrence of oxidative stress following exposure to SiNPs in vitro using erythrocytes, platelets, human endothelial cells, epithelial cell line A549, monocyte-macrophages J774, or co-culture models such as alveolar–capillary barrier,3–8 little is known about the occurrence of oxidative stress in various organs following systemic administration. The majority of the studies focused on histological, cytological, and serum markers of organ injury.2 Therefore, our findings are novel and provide insight into the mechanisms related to SiNPs-induced organ toxicity.
Our data also show that administration of SiNPs caused a significant increase in the concentrations of TNFα in the lung, heart, and kidney; IL-6 in the lung and kidney; and IL-1β in the lung, liver, and brain. Although the release of proinflammatory cytokines differed among the studied organs, at least one or more proinflammatory cytokines increased in each studied organ, indicating the occurrence of inflammation following administration of SiNPs. While the quantification of proinflammatory cytokines in various organs after systemic administration of SiNPs has not been reported, it has been shown that ip administration of SiNPs (50 nm) causes an increase in IL-1β and TNFα in the plasma.15,37 Also, an increase in IL-1β and TNFα has been reported in RAW 264.7 cells derived from mouse peritoneal macrophages that were exposed to SiNPs (average primary size 12 nm).37 In the same study, it was also shown that mice treated with a single dose of SiNPs (ip; 50 mg/kg) induced the activation of peritoneal macrophages, increased blood level of IL-1β and TNFα, and increased the level of nitric oxide released from peritoneal macrophages.37 Furthermore, a significant increase in TNFα and IL-6 has been reported in the bronchoalveolar lavage of rats exposed to SiNPs (5 mg/kg) by intratracheal instillation.38
For evaluating genetic damage, gel electrophoresis of a single cell (comet assay) was used to assess DNA damage in the lung, heart, liver, kidney, and brain following administration of SiNPs. Our data show the presence of DNA damage in all the studied organs 24 hours after the ip administration of a relatively low dose of SiNPs, ie, 0.25 mg/kg, in mice. These observations indicate that SiNPs induced DNA damage by promoting a milieu of oxidative stress and inflammation. It has been proposed that particulate-mediated DNA oxidation damage could originate from oxidative stress and/or inflammation.34 The literature regarding the genotoxic effects of amorphous SiNPs is conflicting. Using a much higher dose of SiNPs (50 mg/kg; 15 nm) injected intravenously in rats, Downs et al reported the presence of DNA damage in liver, lung tissue, and white blood cells at 48, 24, and 4 hours time points.39 However, a recent study performed in rats reported no DNA damage after iv administration of cumulative doses of 15, 30, and 60 mg/kg of SiNPs (24 nm) 48, 24, and 4 hours before sacrifice.40 Additional studies are needed to address these discrepancies.
Conclusion
We conclude that acute systemic exposure to SiNPs causes oxidative stress, inflammation, and DNA damage in multiple major organs, including the lung, heart, liver, and brain, and highlights the need for exhaustive appraisal of SiNPs before they can be used in human beings.
Acknowledgments
This work was supported by funds of the CMHS grant (31M215), and UAEU Program for Advanced Research (31M189) and center-based interdisciplinary (31R052) grants.
The authors wish to thank Saeed Tariq, Department of Anatomy, CMHS, UAEU, for his help in the electromagnetic analysis.
The authors wish to thank Professor Gerald Blunden (University of Portsmouth, UK) for critically reading the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Nemmar A, Holme JA, Rosas I, Schwarze PE, Alfaro-Moreno E. Recent advances in particulate matter and nanoparticle toxicology: a review of the in vivo and in vitro studies. Biomed Res Int. 2013;2013:279371. | ||
Napierska D, Thomassen LC, Lison D, Martens JA, Hoet PH. The nanosilica hazard: another variable entity. Part Fibre Toxicol. 2010;7:39. | ||
Napierska D, Thomassen LC, Rabolli V, et al. Size-dependent cytotoxicity of monodisperse silica nanoparticles in human endothelial cells. Small. 2009;5:846–853. | ||
Lison D, Thomassen LC, Rabolli V, et al. Nominal and effective dosimetry of silica nanoparticles in cytotoxicity assays. Toxicol Sci. 2008;104:155–162. | ||
Kasper J, Hermanns MI, Bantz C, et al. Inflammatory and cytotoxic responses of an alveolar-capillary coculture model to silica nanoparticles: comparison with conventional monocultures. Part Fibre Toxicol. 2011;8:6. | ||
Corbalan JJ, Medina C, Jacoby A, Malinski T, Radomski MW. Amorphous silica nanoparticles trigger nitric oxide/peroxynitrite imbalance in human endothelial cells: inflammatory and cytotoxic effects. Int J Nanomedicine. 2011;6:2821–2835. | ||
Nemmar A, Yuvaraju P, Beegam S, et al. In vitro platelet aggregation and oxidative stress caused by amorphous silica nanoparticles. Int J Physiol Pathophysiol Pharmacol. 2015;7:27–33. | ||
Nemmar A, Beegam S, Yuvaraju P, Yasin J, Shahin A, Ali BH. Interaction of amorphous silica nanoparticles with erythrocytes in vitro: role of oxidative stress. Cell Physiol Biochem. 2014;34:255–265. | ||
Nabeshi H, Yoshikawa T, Matsuyama K, et al. Systemic distribution, nuclear entry and cytotoxicity of amorphous nanosilica following topical application. Biomaterials. 2011;32:2713–2724. | ||
Nemmar A, Hoet PH, Vanquickenborne B, et al. Passage of inhaled particles into the blood circulation in humans. Circulation. 2002;105:411–414. | ||
Nemmar A, Vanbilloen H, Hoylaerts MF, Hoet PH, Verbruggen A, Nemery B. Passage of intratracheally instilled ultrafine particles from the lung into the systemic circulation in hamster. Am J Respir Crit Care Med. 2001;164:1665–1668. | ||
Pery AR, Brochot C, Hoet PH, Nemmar A, Bois FY. Development of a physiologically based kinetic model for 99m-Technetium-labelled carbon nanoparticles inhaled by humans. Inhal Toxicol. 2009;21:1099–1107. | ||
Oberdorster G, Sharp Z, Atudorei V, et al. Extrapulmonary translocation of ultrafine carbon particle following whole-body inhalation exposure of rats. J Toxicol Environ Health A. 2002;65:1531–1543. | ||
Yu Y, Li Y, Wang W, et al. Acute toxicity of amorphous silica nanoparticles in intravenously exposed ICR mice. PLoS One. 2013;8:e61346. | ||
Nemmar A, Albarwani S, Beegam S, et al. Amorphous silica nanoparticles impair vascular homeostasis and induce systemic inflammation. Int J Nanomedicine. 2014;9:2779–2789. | ||
Corbalan JJ, Medina C, Jacoby A, Malinski T, Radomski MW. Amorphous silica nanoparticles aggregate human platelets: potential implications for vascular homeostasis. Int J Nanomedicine. 2012;7:631–639. | ||
Blalock TD, Varela JC, Gowda S, et al. Ischemic skin wound healing models in rats. Wounds. 2001;13:35–44. | ||
Nemmar A, Raza H, Subramaniyan D, et al. Evaluation of the pulmonary effects of short-term nose-only cigarette smoke exposure in mice. Exp Biol Med (Maywood). 2012;237:1449–1456. | ||
Nemmar A, Raza H, Subramaniyan D, et al. Short-term systemic effects of nose-only cigarette smoke exposure in mice: role of oxidative stress. Cell Physiol Biochem. 2013;31:15–24. | ||
Nemmar A, Al HA, Al HN, et al. Early pulmonary events of nose-only water pipe (shisha) smoking exposure in mice. Physiol Rep. 2015;3:e12258. | ||
de Souza MF, Goncales TA, Steinmetz A, et al. Cocaine induces DNA damage in distinct brain areas of female rats under different hormonal conditions. Clin Exp Pharmacol Physiol. 2014;41:265–269. | ||
Olive PL, Banath JP, Fjell CD. DNA strand breakage and DNA structure influence staining with propidium iodide using the alkaline comet assay. Cytometry. 1994;16:305–312. | ||
Hartmann A, Speit G. The contribution of cytotoxicity to DNA-effects in the single cell gel test (comet assay). Toxicol Lett. 1997;90:183–188. | ||
Nishimori H, Kondoh M, Isoda K, Tsunoda S, Tsutsumi Y, Yagi K. Histological analysis of 70-nm silica particles-induced chronic toxicity in mice. Eur J Pharm Biopharm. 2009;72:626–629. | ||
Gabizon A, Isacson R, Libson E, et al. Clinical studies of liposome-encapsulated doxorubicin. Acta Oncol. 1994;33:779–786. | ||
Riediker M, Cascio WE, Griggs TR, et al. Particulate matter exposure in cars is associated with cardiovascular effects in healthy young men. Am J Respir Crit Care Med. 2004;169:934–940. | ||
Nemmar A, Melghit K, Al-Salam S, et al. Acute respiratory and systemic toxicity of pulmonary exposure to rutile Fe-doped TiO(2) nanorods. Toxicology. 2011;279:167–175. | ||
Nemmar A, Al-Salam S, Subramaniyan D, et al. Influence of experimental type 1 diabetes on the pulmonary effects of diesel exhaust particles in mice. Toxicol Lett. 2013;217:170–176. | ||
Baird MF, Graham SM, Baker JS, Bickerstaff GF. Creatine-kinase- and exercise-related muscle damage implications for muscle performance and recovery. J Nutr Metab. 2012;2012:960363. | ||
Iavicoli I, Leso V, Fontana L, Bergamaschi A. Toxicological effects of titanium dioxide nanoparticles: a review of in vitro mammalian studies. Eur Rev Med Pharmacol Sci. 2011;15:481–508. | ||
Lu X, Ji C, Jin T, Fan X. The effects of size and surface modification of amorphous silica particles on biodistribution and liver metabolism in mice. Nanotechnology. 2015;26:175101. | ||
Kumar R, Roy I, Ohulchanskky TY, et al. In vivo biodistribution and clearance studies using multimodal organically modified silica nanoparticles. ACS Nano. 2010;4:699–708. | ||
Kim JS, Yoon TJ, Yu KN, et al. Toxicity and tissue distribution of magnetic nanoparticles in mice. Toxicol Sci. 2006;89:338–347. | ||
Moller P, Danielsen PH, Karottki DG, et al. Oxidative stress and inflammation generated DNA damage by exposure to air pollution particles. Mutat Res Rev Mutat Res. 2014;762:133–166. | ||
Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5:9–19. | ||
Valenca SS, Silva BF, Lopes AA, et al. Oxidative stress in mouse plasma and lungs induced by cigarette smoke and lipopolysaccharide. Environ Res. 2008;108:199–204. | ||
Park EJ, Park K. Oxidative stress and pro-inflammatory responses induced by silica nanoparticles in vivo and in vitro. Toxicol Lett. 2009;184:18–25. | ||
Sayes CM, Reed KL, Warheit DB. Assessing toxicity of fine and nanoparticles: comparing in vitro measurements to in vivo pulmonary toxicity profiles. Toxicol Sci. 2007;97:163–180. | ||
Downs TR, Crosby ME, Hu T, et al. Silica nanoparticles administered at the maximum tolerated dose induce genotoxic effects through an inflammatory reaction while gold nanoparticles do not. Mutat Res. 2012;745:38–50. | ||
Guichard Y, Fontana C, Chavinier E, et al. Cytotoxic and genotoxic evaluation of different synthetic amorphous silica nanomaterials in the V79 cell line. Toxicol Ind Health. Epub 2015 Mar 10. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.