")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Ox-LDL Causes Endothelial Cell Injury Through ASK1/NLRP3-Mediated Inflammasome Activation via Endoplasmic Reticulum Stress
Authors Hang L, Peng Y, Xiang R, Li X , Li Z
Received 20 September 2019
Accepted for publication 29 January 2020
Published 24 February 2020 Volume 2020:14 Pages 731—744
DOI https://doi.org/10.2147/DDDT.S231916
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Liwei Hang, 1–5,* Yan Peng, 6,* Rui Xiang, 7 Xiangdong Li, 8 Zhiliang Li 1–4
1Department of Cardiology, Heart Center, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong 510280, People’s Republic of China; 2Laboratory of Heart Center and Department of Cardiology, Heart Center, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong 510280, People’s Republic of China; 3Guangdong Provincial Biomedical Engineering Technology Research Center for Cardiovascular Disease, Guangdong, Guangdong 510280, People’s Republic of China; 4Sino-Japanese Cooperation Platform for Translational Research in Heart Failure, Guangzhou, Guangdong 510280, People’s Republic of China; 5Department of Cardiology, Dongsheng People’s Hospital, Erdos City, Inner Mongolia 017000, People’s Republic of China; 6Department of Critical Care Medicine, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, People’s Republic of China; 7Department of Cardiology, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, People’s Republic of China; 8Fuwai Hospital, National Center of Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhiliang Li
Department of Cardiology, Heart Center, Zhujiang Hospital, Southern Medical University, No. 253 Industrial Avenue, Haizhu District, Guangzhou, Guangdong 510280, People’s Republic of China
Tel +86-020-62782250
Email [email protected]
Xiangdong Li
Fuwai Hospital, National Center of Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, No. 167 Beilishi Road, Beijing 100037, People’s Republic of China
Tel +86-010-88392557
Email [email protected]
Objective: This study was to investigate the mechanism of inflammatory pathology modification induced by ox-LDL in endothelial cells.
Methodology: In this study, we firstly investigated the efflux of cholesterol of endothelial cells under the treatment of ox-LDL, and cell proliferation, ROS production, cell apoptosis was measured. Further, proteins of ASK1, NLRP3 inflammasomes and endoplasmic reticulum stress response were detected. Afterwards, ASK1 inhibitor (GS-4997) or endoplasmic reticulum stress (ERS) inhibitor (4-PBA) was used to measure the performance of endothelial cells.
Results: In this study, endothelial cells were treated with ox-LDLs alone or in combination with a GS-4997 or 4-PBA. Results showed that ox-LDLs attenuated the efflux of cholesterol from endothelial cells in a dose-dependent manner. Ox-LDLs inhibited the proliferation of endothelial cells, and induced their apoptosis and production of reactive oxygen species (ROS). Additionally, ox-LDLs upregulated the levels of phosphorylated ASK1, ERS-related proteins (chop, p-PERK, GRP78, and p-IRE-1), and inflammation-associated proteins (NLRP3, IL-1β, and caspase 1) in endothelial cells. Moreover, we proved that GS-4997 could partly reverse ox-LDL-mediated cell proliferation, apoptosis, ROS production, and inflammation in endothelial cells, and increase cholesterol efflux. We also found that 4-PBA could attenuate the effects of ox-LDLs on endothelial cell cholesterol efflux, proliferation, apoptosis, ROS production, and inflammation.
Conclusion: Our results suggest that cholesterol efflux from endothelial cells is reduced by ox-LDLs, and these reductions in cholesterol efflux are accompanied by increased NLRP3 inflammasome signaling, ASK1 and higher levels of endoplasmic reticulum stress. Our results suggest this axis as potential targets for treating atherosclerosis.
Keywords: ox-LDL, endoplasmic reticulum stress, cholesterol efflux, inflammatory corpuscles, endothelial cell, apoptosis signal-regulating kinase 1
Introduction
Atherosclerotic cardiovascular disease (ACVD) is the leading cause of death among humans, and its prevention and treatment are all important areas of medical research.1 The pathological features of ACVD are extremely complex, and include endothelial cell injury, lipid infiltration, and the secretion of inflammatory mediators; the sum of which eventually leads to the formation of large numbers of plaques on the walls of large and middle-sized arteries.2,3 Lipid uptake, macrophage foam cell formation, and inflammatory responses are key events needed for the initiation of atherosclerosis.4 Therefore, it is highly important to study biological mechanisms that might inhibit the formation of foam cells and reduce inflammatory reactions that lead to the development and progression of atherosclerosis.
Oxidized low-density lipoproteins (ox-LDLs) are vital pathogenic factors of atherosclerotic lesions. Ox-LDLs are mediated by scavenger receptors, consumed by monocytes/macrophages and smooth muscle cells, and inhibit cholesterol efflux, resulting in large accumulations of cholesterol in foam cells.5 Previous studies have shown that apoptosis of macrophage-derived foam cells occurs in the plaques of most patients with acute coronary syndrome. In addition, foam cell death and lipid infiltration can lead to extracellular lipid expansion and enhance plaque instability.5,6 Therefore, the outcome of foam cells has a significant effect on the development of atherosclerotic plaque and occurrence of complications.7 Moreover, cholesterol efflux is the process by which intracellular-free cholesterol flows out of cells through specific areas of the cell membrane.8 Therefore, promoting cholesterol efflux might be a strategy for delaying the development of atherosclerotic plaque. However, the regulatory mechanism and biological effects of ox-LDL-mediated cholesterol efflux on atherosclerosis have not been fully elucidated.
Recent studies have suggested that endoplasmic reticulum stress (ERS) plays an essential role in inflammatory responses, and is involved in the pathological process of inflammatory diseases.9,10 The ERS downstream pathway inositol-requiring kinase 1 (IRE1) regulates apoptosis via the ASK1-c-terminal kinase (JNK) pathway.11 Therefore, ASK1 is correlated with ERS. NLRP3 inflammatory corpuscles are intracellular protein complexes formed by the nod-like receptor (NLR) family (NLRP3). Those protein complexes can induce the maturation and secretion of pro-inflammatory factors such as IL-1β and IL-18, and thereby promote the occurrence of inflammatory responses.12 NLRP3 inflammatory corpuscles can be activated by a variety of pathogenic microorganisms and risk signals, and participate in the occurrence of numerous human diseases.13,14 There is also evidence that apoptosis signal-regulating kinase 1 (ASK1) is required for NLRP3 inflammasome activation.15 However, it is not clear whether ox-LDL-mediated cholesterol efflux is associated with ERS, ASK1, and NLRP3 inflammatory corpuscles.
Nowadays, the certain mechanism of cholesterol efflux remains unclear. Of the mechanisms that have been described, pathway included ABCA1/ABCG1 was widely studied.16–19 As far as we know, cholesterol efflux transport relies on some specific proteins, such as ABCA1 and ABCG1, which has been regarded as gatekeeper for mediating tissue cholesterol.20 ABCA1 and ABCG1 present additional activities during enhancing process of reverse cholesterol transport in vivo.21 Mice being knock-out of ABCA1 and ABCG1 have been demonstrated to lack of ability to scavenge macrophage foam cells as results.22 Frambach et al indicate that small molecule pharmacological strategies targeting ABCA1 and ABCG1 can be recommended as a promising option for cardiovascular disease.16 Therefore, we also wondered that whether ABCA1 and ABCG1 can be mediated by endoplasmic reticulum stress response to facilitate cholesterol efflux in endothelial cell.
In this study, we investigated the effects of ox-LDLs on cholesterol efflux, cellular apoptosis, ERS, inflammatory responses, reactive oxygen species (ROS) production, and the proliferation of endothelial cells. In addition, we explored the underlying mechanisms by which ox-LDLs participate in the pathogenesis of atherosclerosis, such as participation by influencing the apoptosis signal-regulating kinase 1 (ASK1) signaling pathway and endoplasmic reticulum (ER) stress. Our study revealed the mechanism by which ox-LDLs promote atherosclerosis development, and suggests potential therapeutic targets for preventing the development and progression of atherosclerosis.
Materials and Methods
Cell Culture and Treatments
Human aortic endothelial cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM medium (Hyclone, Logan, UT, USA) containing 10% fetal bovine serum (FBS, Gibco, Waltham, MA, USA), 100 μL penicillin G, 100 mg/L streptomycin, and 100 μg/mL endothelial cell growth factor (ECGF, Sigma, St. Louis, MO, USA) in a 37°C incubator with 5% CO2. Human ox-LDL was purchased from Yiyuan Biotechnologies (Cat. No. YB-002, Guangzhou, China), and it was produced by oxidized LDL with Cu2SO4 (oxidant) in PBS at 37°C. Endothelial cells were incubated with ox-LDLs (0–200 mg/L) for 16 hrs prior to receiving any other treatment. Afterwards, the cells were treated for 24 hrs with an ASK1 inhibitor (GS-4997) or an ER inhibitor (4-PBA) at concentrations of 10 µM and 5 mM, respectively.
Detection of Cholesterol Efflux
Cholesterol efflux is closely related to the development of atherosclerosis.23 In this study, Cholesterol efflux assays were performed using a commercial kit (BioVision Inc., Milpitas, CA, USA, Cat. K582-100), according to instructions provided by the manufacturer. Briefly, 1 × 105 endothelial cells were seeded into each well of a 96-well plate and incubated for 2 hrs. Next fluorescent-labeled cholesterol was added to each well and incubated for 15 hrs, followed by the addition of ox-LDLs. Next, the supernatants were collected and measured for their fluorescence (Ex/Em = 482/515 nm). Endothelial cells were also collected and lysed. The supernatants of the lysed cells were transferred into a 96-well plate for measurements of their fluorescence (Ex/Em = 482/515 nm). Finally, cholesterol efflux was calculated using the formula below:

Western Blot Assays
The total proteins were extracted from endothelial cells by using RIPA Lysis Buffer (Vazyme, Nanjing, China, FD008) containing PMSF (Solarbio, China, P0100-1.5). The protein concentration in each extract was determined using a BCA Protein Quantification Kit (Vazyme, Nanjing, China, E112-01). Samples of total protein were separated by 10% SDS-PAGE, and the protein bands were transferred onto PVDF membranes (Millipore, Burlington, MA, USA, cat. no. 10083), which were subsequently blocked with 5% skim milk for 2 hrs. Next, the membranes were incubated with primary antibodies at 4°C overnight, followed by incubation with a Goat Anti-Mouse HRP secondary antibody (Vazyme, Nanjing, China, Ab201-01) or Goat Anti-Rabbit HRP secondary antibody (Vazyme, Nanjing, China, Ab203-01) for 1 h at room temperature. The membranes were then treated with reagents in an Enhanced ECL Luminescence Detection Kit (Vazyme, Nanjing, China, E411-04), and the immunostained protein bands were scanned with a FluorChem™ M System. The primary antibodies used in this study included ASK1 (Abcam, Cambridge, UK, ab131506), p-AKS1 (Abcam, ab81283), ABCA1 (Abcam, ab66217), ABCG1 (Abcam, ab201776), NLRP3 (Abcam, ab214185), ASC (Abcam, ab175449), caspase 1 (Abcam, ab1872), IL-1β (Abcam, ab2105), Chop (Abcam, ab10444), p-PERK (Abcam, ab192591), PERK (Abcam, ab65142), GRP78 (Abcam, ab21685), p-IRE-1 (Abcam, ab48187), IRE-1 (Abcam, ab37073), and GAPDH (Abcam, ab125247). GAPDH served as an internal control.
Edu Staining
Cell proliferation was determined using an Edu staining assay. Briefly, 50 μM EdU solution was prepared by diluting EdU solution (Solarbio Life Sciences, Beijing, China, cat. no. CA1170) with cell culture medium (1:1000). The treated endothelial cells (1 × 105 cells/well) were placed into 24-well plates and incubated with 100 μL of EdU solution (50 μM) for 2 hrs. After washing, the cells were fixed with methyl alcohol for 15 mins and then decolored with 50 μL of glycine (2 mg/mL) for 5 mins. Finally, images were obtained using a fluorescence microscope.
Apoptosis Assay
Cell apoptosis was assessed using an Annexin V-FITC/PI Apoptosis Kit (Solarbio Life Sciences, cat. no. CA1020). The treated endothelial cells were collected by centrifugation (1000g, 5 mins). After washing, the cells were suspended in 1× Binding buffer, and 100 μL of cell suspension was mixed with 5 μL of FITC and 5 μL of PI. After incubation for 15 mins in the dark, cell apoptosis was measured using a FACSCalibur flow cytometer (BD, FACSCalibur).
Detection of ROS
ROS levels were examined using a Fluorometric Intracellular Ros Kit (CA1410, Solarbio Life Sciences, cat. No CA1410). The treated endothelial cells were labeled with DCFH-DA fluorescent probes (Dilution: 1/1000) for 20 min at 37°C, and then washed three times with serum-free medium. Fluorescence intensity was determined by flow cytometry (BD, FACSCalibur, Becton Dickinson, Franklin Lakes, NJ, USA).
ELISA Assays
The levels of IL-1β and IL-18 were detected using ELISA kits (Affymetrix, Santa Clara, CA, USA, cat. no. 88-7261-86 and Sigma, cat. no. RAB0543, respectively) according to instructions provided by the manufacturers.
Determination of Lactic Dehydrogenase (LDH) Levels
LDH levels were analyzed using a LDH-Cytotoxicity Assay Kit (Thermo Fisher Scientific, cat. no. 88953) according to the manufacturer’s instructions.
Statistical Analysis
All data were analyzed using GraphPad Prism Software (Ver. Prism 7), and results are expressed as the mean ± SD. The data were compared by one-way ANOVA, and a P-value < 0.05 was considered statistically to be significant. Each experiment was performed independently in triplicate (N=3).
Results
Effects of Ox-LDLs on Cholesterol Efflux and ABCA1 and ABCG1 Expression in Endothelial Cells
To investigate the influence of ox-LDLs on cholesterol efflux from endothelial cells, the levels of cholesterol efflux were examined in ox-LDL-treated endothelial cells. We found that treatment with a low dose of ox-LDLs induced a significant increase in cholesterol efflux relative to cholesterol efflux in control cells. However, cholesterol efflux became significantly reduced as the dose of ox-LDLs increased (Figure 1A, P < 0.05), and cells treated with a higher dose of ox-LDLs displayed lower levels of cholesterol efflux when compared to cells treated with a low dose of ox-LDLs (Figure 1A, P < 0.05). Moreover, the levels of ABCA1 and ABCG1 expression showed a similar tendency, as both proteins were more highly expressed in ox-LDL-treated cells, and their expression levels became downregulated as the ox-LDL concentration increased (Figure 1B).
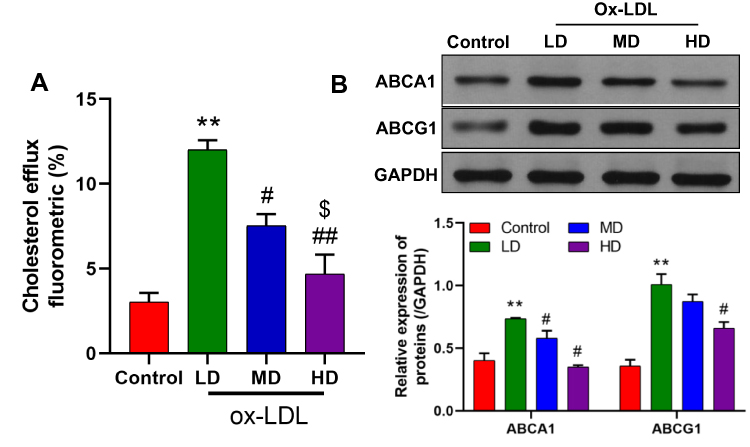 |
Figure 1 Effects of ox-LDLs on cholesterol efflux and ABCA1/ABCG1 expression in endothelial cells. (A) Cholesterol efflux was assessed in low dose (50 mg/L), middle dose (100 mg/L), and high dose (200 mg/L) ox-LDL-treated endothelial cells by using a Cholesterol Efflux Assay Kit. (B) Western blot analyses of ABCA1 and ABCG1 expression in ox-LDL-treated endothelial cells. **P < 0.01 vs control group; #P < 0.05, ##P < 0.01 vs LD; $P < 0.05 vs MD. |
Increasing Concentrations of Ox-LDLs Suppressed Endothelial Cell Proliferation, but Induced Apoptosis and ROS Production
To determine the effects of ox-LDLs on endothelial cell proliferation, apoptosis, and ROS production, groups of endothelial cells were treated with three different concentrations of ox-LDL. EdU staining revealed that ox-LDL-treated cells had lower rates of proliferation than control cells (Figure 2A). Annexin V FITC/PI double staining showed that ox-LDL significantly increased the apoptosis rate of endothelial cells in a dose-dependent manner (Figure 2B). In addition, ox-LDL treatment produced a gradual increase in ROS levels in endothelial cells as the ox-LDL concentration increased (Figure 2C). These data suggest that ox-LDLs inhibited proliferation and promoted apoptosis and ROS production in endothelial cells.
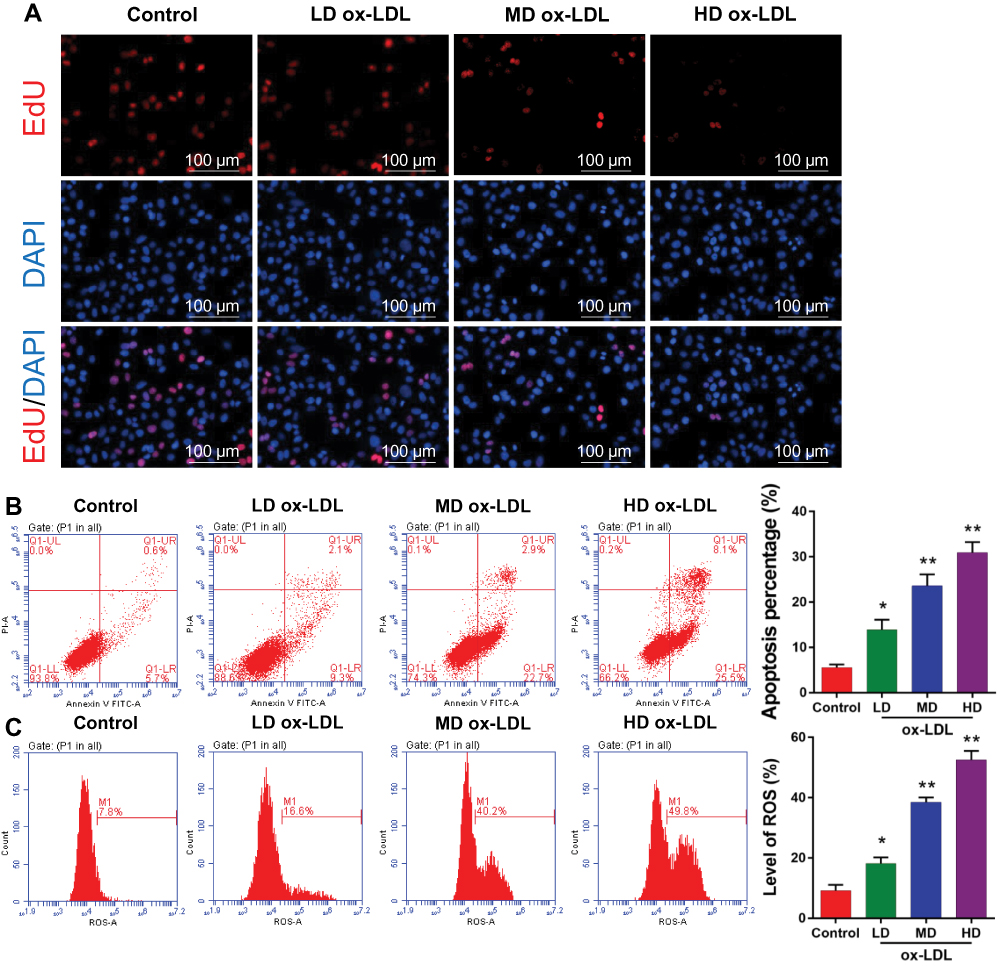 |
Figure 2 Ox-LDLs suppressed proliferation and induced apoptosis and ROS production in endothelial cells. Endothelial cells were treated with different concentrations of ox-LDL. (A) The effect of ox-LDLs on endothelial cell viability was determined by EdU staining; magnification, ×100. (B) Annexin V FITC/PI double staining was used to assess the apoptosis of ox-LDL-treated endothelial cells. (C) ROS levels were examined by flow cytometry. *P < 0.05, **P < 0.01 vs control group. |
Ox-LDLs Upregulated the Expression of ASK1, ERS- and Inflammasome-Related Proteins in a Dose-Dependent Manner in Endothelial Cells
To further confirm the possible regulatory mechanisms of ox-LDLs in endothelial cells, endothelial cells were treated with ox-LDLs, and their levels of apoptosis-related proteins (ASK1 and p-ASK1) were examined by Western blotting. We found that ox-LDLs markedly upregulated p-ASK1 expression in a dose–response manner (Figure 3A). To further understand the regulatory mechanisms by which ASK1 mediates endothelial cell injuries, we examined the effect of ox-LDLs on ERS and NLRP3 inflammasome signaling. Western blot analyses showed that the levels of chop, p-PERK, GRP78, and p-IRE-1 expression were dramatically upregulated in the ox-LDL treatment groups when compared with their levels in the control group, and there was an obvious dose–effect relationship (Figure 3B). We next examined the influence of ox-LDLs on inflammasome-associated proteins, and found that the levels of NLRP3, IL-1β, and caspase 1 expression became gradually increased as the ox-LDL concentration increased, while the ASC levels in endothelial cells remained unchanged after ox-LDL treatment (Figure 3C). Furthermore, ELISA assays revealed that the concentrations of IL-1β and IL-18 in endothelial cells increased as the ox-LDL concentration increased (P < 0.05, Figure 3D). Finally, we found that ox-LDLs markedly improved the LDH levels in endothelial cells (P < 0.05, Figure 3E). These results indicated that inflammasome and ERS signaling were enhanced in the ox-LDL-induced endothelial cells. Subsequent experiments were conducted using an ox-LDL concentration of 100 mg/L.
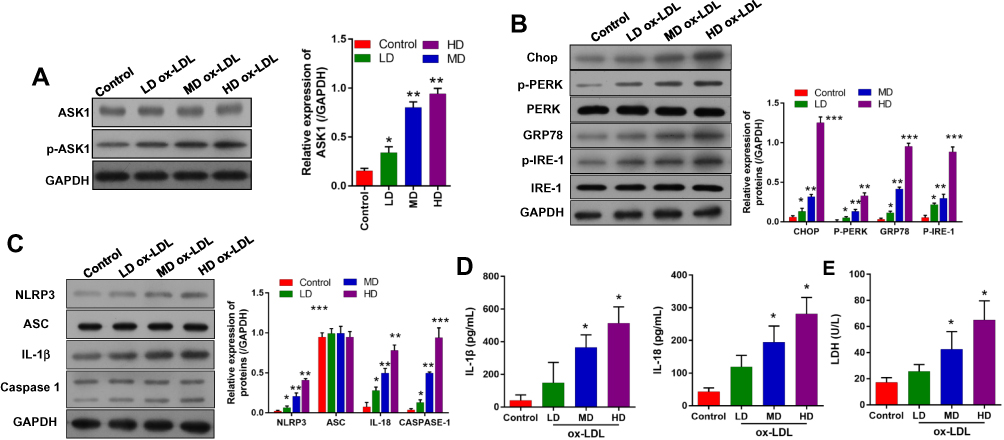 |
Figure 3 Ox-LDLs upregulated the expression of ASK1, ERS -and inflammasome-related proteins in a dose-dependent manner. Endothelial cells were treated with different concentrations of ox-LDL. (A) ASK1 and p-ASK1 expression were measured by Western blotting (B) The expression of ERS-related proteins was examined by Western blotting. (C) Western blotting was used to investigate the expression of inflammasome-associated proteins. (D) The concentrations of IL-1β and IL-18 were examined by ELISA. (E) LDH levels were evaluated using a LDH-Cytotoxicity Assay Kit. *P < 0.05, **P < 0.01, ***P < 0.001 vs control group. |
ASK1 Inhibition Augmented Cholesterol Efflux, Increased Cell Proliferation, and Reduced ROS Production and Cell Apoptosis
Because our previous experiments showed that inflammasome and ERS signaling were enhanced in ox-LDL-induced endothelial cells, we treated endothelial cells with an ASK1 inhibitor (GS-4997) to investigate the role played by ASK1 in ox-LDL-induced cholesterol efflux, cell proliferation, apoptosis, and ROS production. We found that GS-4997 further enhanced the cholesterol efflux that was induced by ox-LDLs in endothelial cells (P < 0.05, P < 0.01, Figure 4A). Moreover, Western blot assays showed that GS-4997 further promoted the upregulation of ABCA1 and ABCG1 expression mediated by ox-LDL treatment in endothelial cells (Figure 4B).
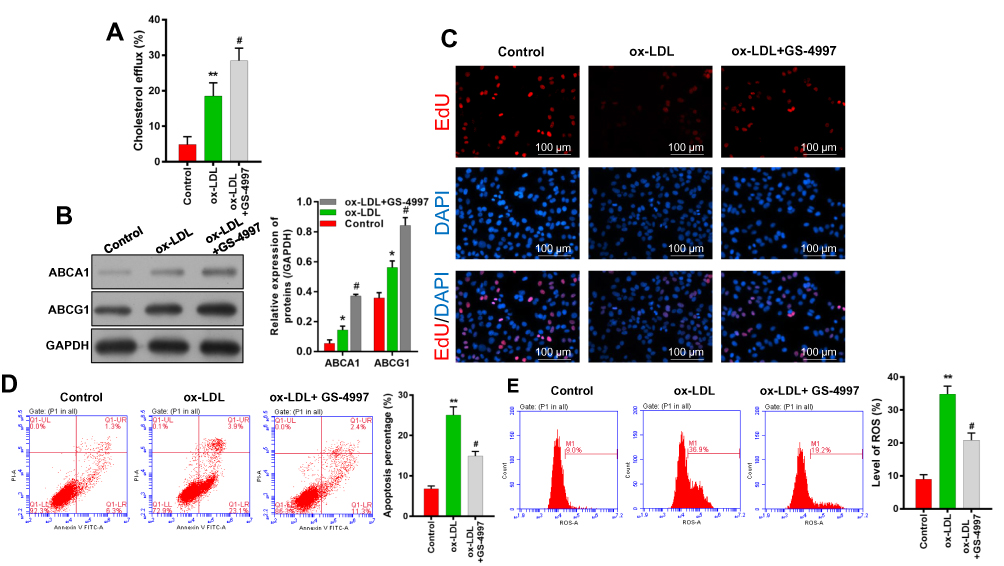 |
Figure 4 ASK1 inhibition increased cholesterol efflux and cell proliferation, and reduced ROS production and cell apoptosis. An ASK1 inhibitor (GS-4997) was used to treat ox-LDL-induced endothelial cells. (A) A Cholesterol Efflux Assay Kit was used to analyze the levels of cholesterol efflux. (B) ABCA1 and ABCG1 expression were evaluated by Western blotting. (C) EdU staining was used to determine the viability of endothelial cells; magnification, ×100. (D) Cell apoptosis was determined by Annexin V FITC/PI double staining. (E) ROS levels were examined by flow cytometry. *P < 0.05, **P < 0.01 vs control group; #P < 0.05 vs ox-LDL group. |
Results of EdU staining showed that the inhibitory effect of ox-LDLs on endothelial cell proliferation could be attenuated by treatment with GS-4997 (Figure 4C). Moreover, the inductive effect of ox-LDLs on endothelial cell apoptosis could be weakened by GS-4997 treatment (P < 0.01, P < 0.001, Figure 4D). Furthermore, the promotive effect of ox-LDLs on ROS levels could also be weakened by GS-4997 treatment (P < 0.05, P < 0.001, Figure 4E). These results indicated that inhibition of ASK1 could enhance cell survival and proliferation, and block ROS production in ox-LDL-induced endothelial cells by promoting cholesterol efflux via upregulation of ABCA1 and ABCG1.
ASK1 Inhibition Attenuated NLRP3 Inflammasome Protein Expression Rather Than ERS-Related Protein Expression in Ox-LDL-Treated Endothelial Cells
We next investigated whether ASK1 regulated the activation of ERS and inflammasomes in ox-LDL-treated endothelial cells. We found that subsequent treatment of ox-LDL-induced endothelial cells with an ASK1 inhibitor (GS-4997) significantly inhibited p-ASK1 expression in those cells (Figure 5A). An examination of ERS-related proteins showed that ox-LDLs could upregulate chop, p-PERK, GRP78, and p-IRE-1 expression (Figure 5B), while the expression of ERS-related proteins was not affected by GS-4997 (Figure 5B, P > 0.05). Furthermore, we also verified that the increases in NLRP3, IL-1β, and caspase 1 expression mediated by ox-LDLs in endothelial cells were obviously inhibited by GS-4997, while ASC expression was unaffected (Figure 5C). ELISA results also showed that GS-4997 treatment could partially reverse the elevation of IL-1β and IL-18 levels mediated by ox-LDLs in endothelial cells (Figure 5D). Moreover, the LDH levels in endothelial cells were enhanced by ox-LDLs and inhibited by GS-4997 (P < 0.05, Figure 5E). These results suggest that ASK1 regulates the activation of inflammasomes in ox-LDL-treated endothelial cells, but not the activation of ERS.
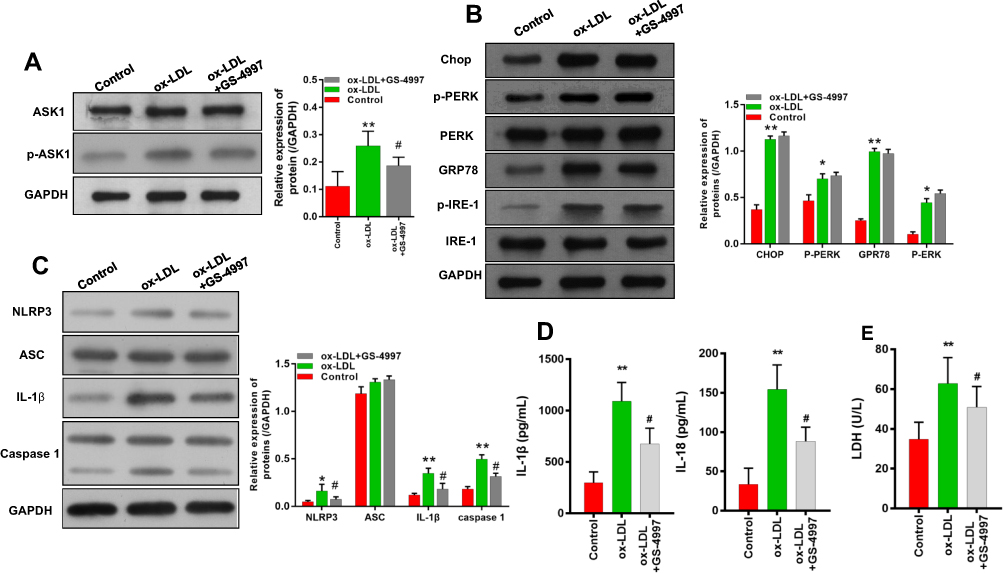 |
Figure 5 ASK1 inhibition attenuated the expression of NLRP3 inflammasome-related proteins rather than ERS-related proteins in ox-LDL-treated endothelial cells. Ox-LDL-induced endothelial cells were treated with an ASK1 inhibitor (GS-4997). (A) The levels of ASK1 and p-ASK1 expression were examined by Western blotting. (B) The expression of ERS-related proteins was investigated by Western blotting. (C) Western blot analyses of inflammasome-associated proteins. (D) IL-1β and IL-18 levels as detected by ELISA. (E) LDH levels as detected using a LDH-Cytotoxicity Assay Kit. *P < 0.05, **P < 0.01 vs control group; #P < 0.05 vs ox-LDL group. |
An ERS Inhibitor Enhanced Cholesterol Efflux, Increased Cell Proliferation, and Reduced Cell Apoptosis and ROS Production
A 4-PBA inhibitor was used to investigate the effect of ERS in ox-LDL-induced endothelial cells. As shown in Figure 6A and B, cholesterol efflux and ABCA1/ABCG1 expression were enhanced in ox-LDL-induced endothelial cells after the cells were subsequently treated with 4-PBA. Also, overall cell survival was improved by an increased rate of cell proliferation and a reduced rate of apoptosis (Figure 6C and D). EdU staining results showed that 4-PBA could partially abolish the inhibitory effect of ox-LDLs on endothelial cell proliferation (Figure 6C). Flow cytometry results showed that the promoting effects of ox-LDLs on endothelial cell apoptosis (P < 0.05, P < 0.01, Figure 6D) and ROS production (P < 0.05, P < 0.01, Figure 6E) could be attenuated by 4-PBA. In summary, these results indicated that ERS inhibition could block the effects of ox-LDLs on cell survival and ROS production.
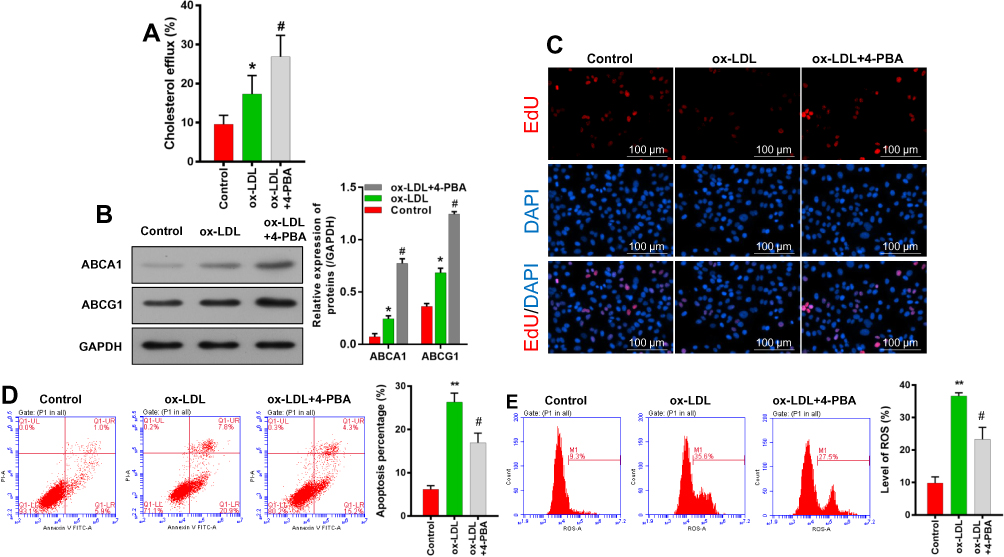 |
Figure 6 An ERS inhibitor enhanced cholesterol efflux, increased cell proliferation, and reduced cell apoptosis and ROS production. Endothelial cells were incubated with ox-LDL or 4-PBA. (A) Cholesterol efflux was detected using a Cholesterol Efflux Assay Kit. (B) Western blot analyses of ABCA1 and ABCG1 levels. (C) Cell viability was determined by EdU staining; magnification, ×100. Cell apoptosis (D) and (E) ROS levels were examined by flow cytometry. *P < 0.05, **P < 0.01 vs control group; #P < 0.05 vs ox-LDL group. |
An ERS Inhibitor Weakened the Downstream ASK1/NLRP3 Inflammatory Signaling Pathway in Endothelial Cells
In terms of mechanism, we used an ER inhibitor (4-PBA) to treat ox-LDL-induced endothelial cells, and found that the upregulation of p-ASK1 expression induced by ox-LDLs in those cells could be attenuated by 4-PBA (Figure 7A). In addition, 4-PBA also partially reversed the upregulation of chop, p-PERK, GRP78, and p-IRE-1 levels mediated by ox-LDLs in endothelial cells (Figure 7B), and attenuated the increases in NLRP3, IL-1β, and caspase 1 expression mediated by ox-LDLs (Figure 7C). Furthermore, our results suggested that the increases in IL-1β and IL-18 levels mediated by ox-LDLs in endothelial cells could be markedly prevented by 4-PBA (P < 0.05, P < 0.01, Figure 7D). Moreover, while treatment with ox-LDLs increased the LDH levels in endothelial cells, those increases were attenuated by 4-PBA (P < 0.05, Figure 7E). These results indicated that the ASK1/inflammasome pathway could be blocked by an ERS inhibitor.
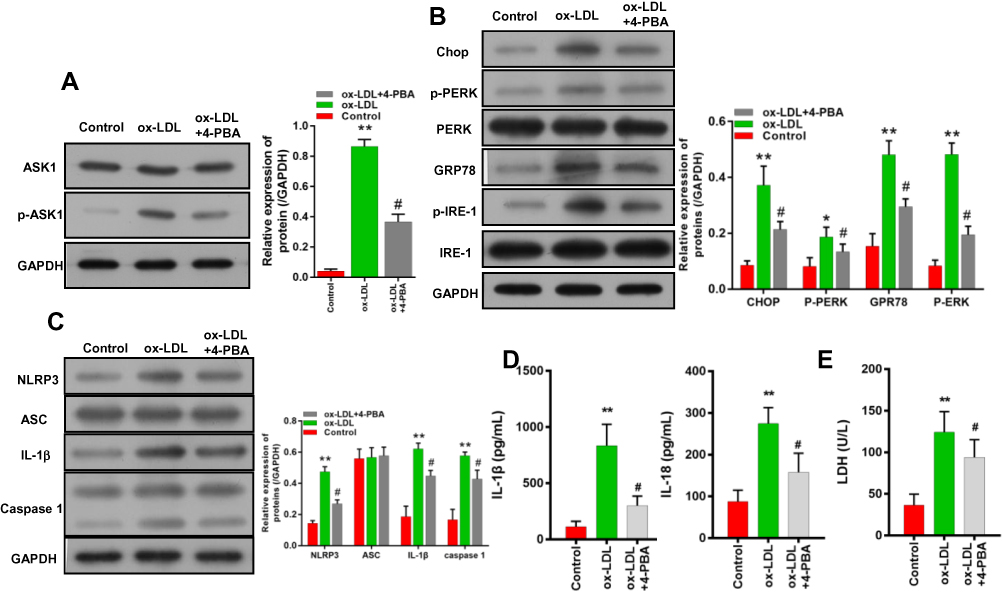 |
Figure 7 An ERS inhibitor weakened the downstream ASK1/NLRP3 inflammatory signaling pathway in endothelial cells. Endothelial cells were co-treated with or without an ER inhibitor (4-PBA). (A) ASK1 and p-ASK1 expression were measured by Western blotting. (B) The levels of ERS-related proteins were detected by Western blotting. (C) The levels of inflammasome-associated proteins were analyzed. (D) IL-1β and IL-18 levels were measured by ELISA. (E) LDH levels were evaluated using a LDH-Cytotoxicity Assay Kit. *P < 0.05, **P < 0.01 vs control group; #P < 0.05 vs ox-LDL group. |
Discussion
Atherosclerosis is a chronic inflammatory process that can be initiated by numerous factors, including hyperlipidemia, hypertension, hyperglycemia, hyperuricemia, and obesity, and result in vascular endothelial injury, vascular wall thickening, and changes in vascular elasticity.24,25 Previous studies suggested that several signaling pathways, including signals produced by inflammasomes and endoplasmic reticulum stress are involved in the occurrence and development of atherosclerosis.14,26,27 However, the regulatory mechanism of atherosclerosis is not fully understood. Our present study showed that in endothelial cells, ox-LDLs induced the activation of inflammasomes and endoplasmic reticulum stress via ASK1, resulting in shorter cell survival times and increased ROS production.
Under normal physiological conditions, endothelial cells prevent harmful substances in the blood from damaging vascular smooth muscle cells.28 However, when vascular endothelial cells are exposed to an external stimulus, their structure and function become impaired, leading to large accumulations of lipids in blood vessels and changes in vascular permeability.29 At the same time, these changes promote the proliferation of vascular smooth muscle cells, resulting in thrombus formation and platelet adhesion, and ultimately, the formation of atherosclerotic plaque.30,31 In our study, we used ox-LDL-treated endothelial cells as a model in which to investigate the pathogenesis of atherosclerotic disease, and found that ox-LDLs could inhibit cholesterol efflux, and downregulate ABCA1 and ABCG1 expression in a dose-dependent manner. We also proved that ox-LDLs could suppress endothelial cell proliferation, and induce apoptosis and ROS production. Previous studies have also suggested that ROS play an essential role in the formation of atherosclerotic plaque.32,33 In particular, high concentrations of hydrogen peroxide have been detected at the sites of atherosclerotic endothelial injury. Hydrogen peroxide can stimulate macrophages to express pro-inflammatory cytokines and chemokines that intensify the inflammatory activity of macrophages.34,35 In this study, endothelial cell proliferation was attenuated by ox-LDL at concentration of 50 mg/L. While cell apoptosis and ROS production were also enhanced by ox-LDL significantly. Yang et al found that proliferation of endothelial progenitor cells can be inhibited obviously by ox-LDL at concentration 30–100 mg/L after 12 h exposure.36–40 However, report also indicates that ox-LDL can enhance cell proliferation in human aortic smooth muscle cells or Endothelial cell at a relatively low concentration.41–44 This difference might be due to cell performance differs to cell type when expose to ox-LDL. Additionally, Zhu et al suggest that ox-LDL plays dual effect on inflammatory response by triggering LOX-1/PPARγ pathway.45 Our results in this study verified that ox-LDLs can inhibit cholesterol efflux retracement from endothelial cells with ox-LDL concentration increasing.
NLRP3 inflammasomes are protein complexes that contain intracellular receptors (primarily the NOD-like receptor), caspase precursors, and apoptosis-associated speck-like proteins.46 The activation of NLRP3 inflammasomes can induce caspase-1 activation and regulate the processes of its substrates, including interleukin-1β (IL-1β) and interleukin-18 (IL-18), leading to inflammatory responses. Therefore, that mechanism has been regarded as a major mediator of atherosclerosis.26 In recent years, numerous studies have confirmed that NLRP3 inflammasomes can be activated by two abundant components of atherosclerotic plaque (ox-LDLs and crystalline cholesterol).26,47–49 Hoseini et al reported that high numbers of inflammasomes were present in atherosclerotic plaque and ox-LDL-induced cells in vitro.26 However, the mechanism by which inflammasomes function in atherosclerosis is complex, and involves several signaling pathways, including the JNK-1 and ASK1 pathways.26 In our study, we showed that inflammasomes were activated in ox-LDL-induced endothelial cells, and that activation was characterized by elevated levels of IL-18, IL-1β, and LDH. In this study, we also found that low level of ox-LDL (50 mg/L) increased cholesterol efflux compared to control. While, the inflammasome was also promoted at the same time. This seems to be a conceptual disconnect to the afterwards experiments. The conceptual disconnect might be due to the baseline of content of total cholesterol was higher than control. While, with increasing of ox-LDL concentration, cell function disorder, such as evoked inflammation, endoplasmic reticulum stress response, impairs the normal process of cholesterol efflux. In this study, NLRP3 inflammasome was promoted by concentration of ox-LDL increased (As shown in Figure 3). Besides, as shown in Figure 1, cholesterol efflux process was obviously blocked by ox-LDL. Therefore, we suggested that promotion of NLRP3 inflammasome will harm process of cholesterol efflux. This is similar to conclusion by Chen et al.50 According to Chen et al, inhibition of the NLRP3 inflammasome decreases foam cell formation of THP-1 macrophages via suppression of ox-LDL uptake and enhancement of cholesterol efflux. However, report also suggests that Cholesterol accumulation in myeloid cells activates the NLRP3 inflammasome, which enhances neutrophil accumulation and neutrophil extracellular trap formation in atherosclerotic plaques, and promotion of cholesterol efflux suppress inflammasome activation.51 It means that there is a negative correlation between cholesterol efflux and inflammasome activation. In this study, the relationship between them was not included directly. We wish this content will be investigated in the future.
To the best of our knowledge, very few literature reports have mentioned the possible role of ASK1 in atherosclerosis. Liu et al52 found that TNF-α could induce the activation of ASK1 in endothelial cells, and that inhibition of ASK1 could attenuate cytokine signaling.52 Another study suggested ASK1 (apoptosis signal-regulating kinase 1) as an upstream regulator of inflammasome activation,53 and showed that knockdown of ASK1 abrogated IL-1α-driven inflammation in neutrophilic dermatosis. In this study, we showed that ox-LDL induction activated NLRP3 inflammasomes in endothelial cells, and that inhibition of ASK1 reversed the inflammation activation induced by ox-LDL by decreasing NLRP3 and IL-1β expression.
As a member of the mitogen-activated protein kinase (MAPK) family, ASK1 balances and integrates numerous endogenous and exogenous stimulations, making it possible for cells to appropriately respond to different stimuli.54 However, the continuous stimulation of pathogenic factors can cause the persistent abnormal activation of ASK1, leading to tissue and cell damage during the course of disease. Previous research has shown that the ox-LDLs is closely related to ASK1-induced apoptosis.55 Another study demonstrated that phosphorylated ASK1 plays a key role in atherosclerosis development,56 and that an ASK inhibitor (AGI-1067) exerts an anti-inflammatory effect by inhibiting the dissociation of thioredoxin in vascular endothelial cells.57 In addition, another study showed that thioredoxin-1 (Trx-1) could protect macrophages from ox-LDL-induced foam cell formation and apoptosis by interacting with ASK1.58 In our study, we further demonstrated that ox-LDLs decreased cell survival times and activated inflammasomes by upregulating ASK1. In summary, we found that ox-LDLs inhibited endothelial cell proliferation, and accelerated endothelial cell apoptosis and ROS production by regulating ASK1.
While ERS is an adaptive mechanism, sustained or excessive ERS induces apoptosis and tissue damage.59 Studies have shown that various risk factors may independently or in concert participate in the occurrence and development of atherosclerosis via ERS-induced apoptosis.60,61 While studies have confirmed that ox-LDLs can induce ERS,62,63 the mechanism by which ox-LDLs regulate ERS in atherosclerosis remains unclear. In our study, we found the ox-LDLs inhibited cholesterol efflux from endothelial cells, and also promoted apoptosis, ROS production, inflammation, and increased endothelial cell proliferation, which was closely associated with ERS. Studies have suggested that under conditions of ERS, activated IRE1α not only induces protein expression, but also combines with TNF-receptor associated factor 2 (TRAF2) to form a IRE1α-TRAF2 complex, which activates ASK1. Activated ASK1 finally promotes apoptosis via a series of cascade reactions.64,65 Recent studies have also proven that ERS is involved in the activation of NLRP3 inflammasomes66 After activating the unfolded protein response (UPR), ERS can promote the transcription of thioredoxin interaction protein (TXNIP) by binding IRE1α and PERK, and inhibit TXNIP by modulating miRNA-17, thereby regulating TXNIP to induce ROS production and activate NLRP3 inflammasomes.66–68 Above all, we verified that the ERS/ASK1/NLRP3 inflammasome pathway is involved in the ox-LDL-induced cholesterol efflux that occurs in atherosclerosis.
Conclusions
In this study, we proved that ox-LDLs dramatically injured endothelial cells and activated NLRP3 inflammasomes by upregulating ASK1 expression and inducing ERS in endothelial cells. In addition, our results showed that ox-LDLs inhibited proliferation, and induced apoptosis, ROS production, and inflammatory reactions in endothelial cells via the ERS/ASK1 axis (Figure 8). Our results suggest some new therapeutic targets for treating atherosclerosis.
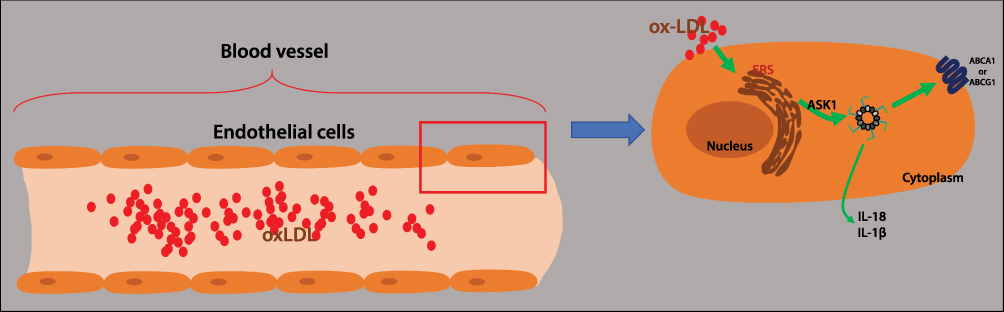 |
Figure 8 Graphic depiction of the mechanism in this study. Ox-LDLs induced endoplasmic reticulum stress in endothelial cells by activating ASK1 and NLRP3 inflammasomes. |
Abbreviations
ASK1, apoptotic signal-regulating kinase 1; ABCA1, ATP binding cassette subfamily A member 1; ABCG1, ATP binding cassette subfamily G member 1; CHOP, C/EBP-homologous protein; CVD, cardiovascular disease; ER, endoplasmic reticulum; ERS, endoplasmic reticulum stress; NLRP3, NLR family pyrin domain containing 3; FITC, fluorescein isothiocyanate; GRP78, glucose-regulated protein 78 kD; IL-18, interleukin 18; IL-1β, interleukin 1β; IRE-1, Inositol-requiring enzyme-1; LD, low dose; MD, middle dose; HD, high dose; PERK, protein kinase R (PKR)-like endoplasmic reticulum kinase; PI, propidium iodide; ROS, reactive oxygen species; TNF-α, tumor necrosis factor α; UPR, unfolded protein response.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article.
Author Contributions
LWH designed the experiments; LWH and YP performed the experiments and collected the data; LWH and RX analyzed and interpreted the data; XDL validated the data analysis; LWH and ZLL drafted the manuscript and ZLL and XDL revised and approved the manuscript. All authors approved the manuscript before submission. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This research was financially supported by the National Natural Science Foundation of China (81774292).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rahman MS, Woollard K. Atherosclerosis. Adv Exp Med Biol. 2017;1003:121–144.
2. Cancel LM, Ebong EE, Mensah S, Hirschberg C, Tarbell JM. Endothelial glycocalyx, apoptosis and inflammation in an atherosclerotic mouse model. Atherosclerosis. 2016;252:136–146. doi:10.1016/j.atherosclerosis.2016.07.930
3. Ou H, Huang Z, Mo Z, Xiao J. The characteristics and roles of advanced oxidation protein products in atherosclerosis. Cardiovasc Toxicol. 2017;17:1–12. doi:10.1007/s12012-016-9377-8
4. Chistiakov DA, Bobryshev YV, Orekhov AN. Macrophage-mediated cholesterol handling in atherosclerosis. J Cell Mol Med. 2016;20:17–28. doi:10.1111/jcmm.12689
5. Chistiakov DA, Melnichenko AA, Orekhov AN, Bobryshev YV. Paraoxonase and atherosclerosis-related cardiovascular diseases. Biochimie. 2017;132:19–27. doi:10.1016/j.biochi.2016.10.010
6. Fu Y, Zhao Y, Huang B. Tribbles homolog 1 enhances cholesterol efflux from oxidized low-density lipoprotein-loaded THP-1 macrophages. Exp Ther Med. 2017;14:862–866. doi:10.3892/etm.2017.4551
7. Luo Y, Duan H, Qian Y, et al. Macrophagic CD146 promotes foam cell formation and retention during atherosclerosis. Cell Res. 2017;27:352–372. doi:10.1038/cr.2017.8
8. Berryman CE, Fleming JA, Kris-Etherton PM. Inclusion of almonds in a cholesterol-lowering diet improves plasma HDL subspecies and cholesterol efflux to serum in normal-weight individuals with elevated LDL cholesterol. J Nutr. 2017;147:1517–1523. doi:10.3945/jn.116.245126
9. Cao SS, Luo KL, Shi L. Endoplasmic reticulum stress interacts with inflammation in human diseases. J Cell Physiol. 2016;231:288–294. doi:10.1002/jcp.v231.2
10. Sprenkle NT, Sims SG, Sanchez CL, Meares GP. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegener. 2017;12:42. doi:10.1186/s13024-017-0183-y
11. Bozi LHM, Takano APC, Campos JC, et al. Endoplasmic reticulum stress impairs cardiomyocyte contractility through JNK-dependent upregulation of BNIP3. Int J Cardiol. 2018;272:194–201. doi:10.1016/j.ijcard.2018.08.070
12. Baroja-Mazo A, Martin-Sanchez F, Gomez AI, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15:738–748.
13. Gaidt MM, Hornung V. The NLRP3 inflammasome renders cell death pro-inflammatory. J Mol Biol. 2018;430:133–141. doi:10.1016/j.jmb.2017.11.013
14. Baldrighi M, Mallat Z, Li X. NLRP3 inflammasome pathways in atherosclerosis. Atherosclerosis. 2017;267:127–138. doi:10.1016/j.atherosclerosis.2017.10.027
15. Place DE, Samir P, Karki R, Briard B, Vogel P, Kanneganti TD. ASK family kinases are required for optimal NLRP3 inflammasome priming. Am J Pathol. 2018;188:1021–1030. doi:10.1016/j.ajpath.2017.12.006
16. Frambach S, de Haas R, Smeitink JAM, Rongen GA, Russel FGM, Schirris TJJ. Brothers in arms: ABCA1- and ABCG1-mediated cholesterol efflux as promising targets in cardiovascular disease treatment. Pharmacol Rev. 2020;72:152–190.
17. Wang D, Hiebl V, Xu T, et al. Impact of natural products on the cholesterol transporter ABCA1. J Ethnopharmacol. 2019;249:112444.
18. Meng XD, Yao HH, Wang LM, et al. Knockdown of GAS5 inhibits atherosclerosis progression via reducing EZH2-mediated ABCA1 transcription in ApoE(-/-) mice. Mol therNucleic Acids. 2019;19:84–96. doi:10.1016/j.omtn.2019.10.034
19. Yvan-Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol. 2010;30:139–143. doi:10.1161/ATVBAHA.108.179283
20. Stefulj J, Panzenboeck U, Becker T, et al. Human endothelial cells of the placental barrier efficiently deliver cholesterol to the fetal circulation via ABCA1 and ABCG1. Circ Res. 2009;104:600–608. doi:10.1161/CIRCRESAHA.108.185066
21. Wang X, Collins HL, Ranalletta M, Fuki IV, Rader DJ. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest. 2007;117:2216–2224. doi:10.1172/JCI32057
22. Yvan-Charvet L, Ranalletta M, Wang N, et al. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117:3900–3908. doi:10.1172/JCI33372
23. Tan Y-L, H-x O, Zhang M, et al. Tanshinone IIA promotes macrophage cholesterol efflux and attenuates atherosclerosis of apoE-/- mice by omentin-1/ABCA1 pathway. Curr Pharm Biotechnol. 2019;20:422–432. doi:10.2174/1389201020666190404125213
24. Brown RA, Shantsila E, Varma C, Lip GY. Current understanding of atherogenesis. Am J Med. 2017;130:268–282. doi:10.1016/j.amjmed.2016.10.022
25. Gistera A, Hansson GK. The immunology of atherosclerosis. Nat Rev Nephrol. 2017;13:368–380. doi:10.1038/nrneph.2017.51
26. Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: its regulation and involvement in atherosclerosis. J Cell Physiol. 2018;233:2116–2132. doi:10.1002/jcp.v233.3
27. Zeeshan HM, Lee GH, Kim HR, Chae HJ. Endoplasmic reticulum stress and associated ROS. Int J Mol Sci. 2016;17:327. doi:10.3390/ijms17030327
28. Milliat F, François A, Isoir M, et al. Influence of endothelial cells on vascular smooth muscle cells phenotype after irradiation: implication in radiation-induced vascular damages. Am J Pathol. 2006;169:1484–1495. doi:10.2353/ajpath.2006.060116
29. Sturtzel C. Endothelial Cells. Adv Exp Med Biol. 2017;1003:71–91.
30. Cichon N, Lach D, Dziedzic A, Bijak M, Saluk J. The inflammatory processes in atherogenesis. Polski Merkuriusz Lekarski. 2017;42:125–128.
31. Quillard T, Franck G, Mawson T, Folco E, Libby P. Mechanisms of erosion of atherosclerotic plaques. Curr Opin Lipidol. 2017;28:434–441. doi:10.1097/MOL.0000000000000440
32. Geng C, Zhang Y, Hidru TH, et al. Sonodynamic therapy: a potential treatment for atherosclerosis. Life Sci. 2018;207:304–313. doi:10.1016/j.lfs.2018.06.018
33. Thent ZC, Chakraborty C, Mahakkanukrauh P, et al. The molecular concept of atheromatous plaques. Curr Drug Targets. 2017;18:1250–1258. doi:10.2174/1389450117666160502151600
34. Schurmann C, Rezende F, Kruse C, et al. The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur Heart J. 2015;36:3447–3456. doi:10.1093/eurheartj/ehv460
35. Wang Y, Li L, Zhao W, et al. Targeted therapy of atherosclerosis by a broad-spectrum reactive oxygen species scavenging nanoparticle with intrinsic anti-inflammatory activity. ACS Nano. 2018;12:8943–8960. doi:10.1021/acsnano.8b02037
36. Yang J, Yu J, Li D, et al. Store-operated calcium entry-activated autophagy protects EPC proliferation via the CAMKK2-MTOR pathway in ox-LDL exposure. Autophagy. 2017;13:82–98. doi:10.1080/15548627.2016.1245261
37. Gao W, Cui H, Li Q, et al. Upregulation of microRNA-218 reduces cardiac microvascular endothelial cells injury induced by coronary artery disease through the inhibition of HMGB1. J Cell Physiol. 2020;235:3079–3095. doi:10.1002/jcp.29214
38. Qian W, Cai X, Qian Q, et al. Astragaloside IV protects endothelial progenitor cells from the damage of ox-LDL via the LOX-1/NLRP3 inflammasome pathway. Drug Des Devel Ther. 2019;13:2579–2589. doi:10.2147/DDDT.S207774
39. Wang M, Liu Y, Li C, Zhang Y, Zhou X, Lu C. Long noncoding RNA OIP5-AS1 accelerates the ox-LDL mediated vascular endothelial cells apoptosis through targeting GSK-3beta via recruiting EZH2. Am J Transl Res. 2019;11:1827–1834.
40. Ji G, Song X, Wang L, Li Z, Dong H. Golgi apparatus fragmentation participates in oxidized low-density lipoprotein-induced endothelial cell injury. J Cell Biochem. 2019;120. doi:10.1002/jcb.29205
41. Cai T, Cui X, Zhang K, Zhang A, Liu B, Mu JJ. LncRNA TNK2-AS1 regulated ox-LDL-stimulated HASMC proliferation and migration via modulating VEGFA and FGF1 expression by sponging miR-150-5p. J Cell Mol Med. 2019;23:7289–7298. doi:10.1111/jcmm.14575
42. Bao MH, Li GY, Huang XS, Tang L, Dong LP, Li JM. Long noncoding RNA LINC00657 acting as a miR-590-3p sponge to facilitate low concentration oxidized low-density lipoprotein-induced angiogenesis. Mol Pharmacol. 2018;93:368–375. doi:10.1124/mol.117.110650
43. Qiu J, Wang G, Zheng Y, Hu J, Peng Q, Yin T. Coordination of Id1 and p53 activation by oxidized LDL regulates endothelial cell proliferation and migration. Ann Biomed Eng. 2011;39:2869–2878. doi:10.1007/s10439-011-0382-6
44. Zhang C, Adamos C, Oh MJ, et al. oxLDL induces endothelial cell proliferation via Rho/ROCK/Akt/p27(kip1) signaling: opposite effects of oxLDL and cholesterol loading. Am J Physiol Cell Physiol. 2017;313:C340–c351. doi:10.1152/ajpcell.00249.2016
45. Zhu H, Xia M, Hou M, et al. Ox-LDL plays dual effect in modulating expression of inflammatory molecules through LOX-1 pathway in human umbilical vein endothelial cells. Front Biosci. 2005;10:2585–2594. doi:10.2741/1722
46. Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin Exp Rheumatol. 2016;34:12–16.
47. Rhoads JP, Lukens JR, Wilhelm AJ, et al. Oxidized low-density lipoprotein immune complex priming of the Nlrp3 inflammasome involves TLR and FcgammaR cooperation and is dependent on CARD9. J Immunol. 2017;198:2105–2114. doi:10.4049/jimmunol.1601563
48. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi:10.1038/nature08938
49. Rajamaki K, Lappalainen J, Oorni K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765. doi:10.1371/journal.pone.0011765
50. Chen L, Yao Q, Xu S, Wang H, Qu P. Inhibition of the NLRP3 inflammasome attenuates foam cell formation of THP-1 macrophages by suppressing ox-LDL uptake and promoting cholesterol efflux. Biochem Biophys Res Commun. 2018;495:382–387. doi:10.1016/j.bbrc.2017.11.025
51. Westerterp M, Fotakis P, Ouimet M, et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation. 2018;138:898–912. doi:10.1161/CIRCULATIONAHA.117.032636
52. Liu Y, Yin G, Surapisitchat J, Berk BC, Min W. Laminar flow inhibits TNF-induced ASK1 activation by preventing dissociation of ASK1 from its inhibitor 14-3-3. J Clin Invest. 2001;107:917–923. doi:10.1172/JCI11947
53. Tartey S, Gurung P, Dasari TK, Burton A, Kanneganti TD. ASK1/2 signaling promotes inflammation in a mouse model of neutrophilic dermatosis. J Clin Invest. 2018;128:2042–2047.
54. Sakauchi C, Wakatsuki H, Ichijo H, Hattori K. Pleiotropic properties of ASK1. Biochim Biophys Acta Gen Subj. 2017;1861:3030–3038. doi:10.1016/j.bbagen.2016.09.028
55. Tang SY, Wan YP, Wu YM. Death domain associated protein (Daxx), a multi-functional protein. Cell Mol Biol Lett. 2015;20:788–797. doi:10.1515/cmble-2015-0048
56. Yamada S, Noguchi H, Tanimoto A. Critical and diverse in vivo roles of apoptosis signal-regulating kinase 1 in animal models of atherosclerosis and cholestatic liver injury. Histol Histopathol. 2017;32:11840.
57. Zheng S, Long L, Li Y, et al. A novel ASK inhibitor AGI-1067 inhibits TLR-4-mediated activation of ASK1 by preventing dissociation of thioredoxin from ASK1. Cardiovasc Pharm Open Access. 2015;4. doi:10.4172/2329-6607.1000132
58. Zhang H, Liu Q, Lin JL, et al. Recombinant human thioredoxin-1 protects macrophages from oxidized low-density lipoprotein-induced foam cell formation and cell apoptosis. Biomol Ther. 2018;26:121–129. doi:10.4062/biomolther.2016.275
59. Hu YB, Wu X, Qin XF, Wang L, Pan PH. Role of endoplasmic reticulum stress in silica-induced apoptosis in RAW264.7 cells. Biomed Environ Sci. 2017;30:591–600. doi:10.3967/bes2017.078
60. Dong Y, Fernandes C, Liu Y, et al. Role of endoplasmic reticulum stress signalling in diabetic endothelial dysfunction and atherosclerosis. Diabetes Vasc Dis Res. 2017;14:14–23. doi:10.1177/1479164116666762
61. Ivanova EA, Orekhov AN. The role of endoplasmic reticulum stress and unfolded protein response in atherosclerosis. Int J Mol Sci. 2016;17:193.
62. Guo C, Ma R, Liu X, et al. Silica nanoparticles promote oxLDL-induced macrophage lipid accumulation and apoptosis via endoplasmic reticulum stress signaling. Scie Total Environ. 2018;570:631–632.
63. Yao S, Tian H, Miao C, et al. D4F alleviates macrophage-derived foam cell apoptosis by inhibiting CD36 expression and ER stress-CHOP pathway. J Lipid Res. 2015;56:836–847. doi:10.1194/jlr.M055400
64. Starosyla SA, Volynets GP, Lukashov SS, et al. Identification of apoptosis signal-regulating kinase 1 (ASK1) inhibitors among the derivatives of benzothiazol-2-yl-3-hydroxy-5-phenyl-1,5-dihydro-pyrrol-2-one. Bioorg Med Chem. 2015;23:2489–2497. doi:10.1016/j.bmc.2015.03.056
65. Zeng T, Peng L, Chao H, et al. IRE1alpha-TRAF2-ASK1 complex-mediated endoplasmic reticulum stress and mitochondrial dysfunction contribute to CXC195-induced apoptosis in human bladder carcinoma T24 cells. Biochem Biophys Res Commun. 2015;460:530–536. doi:10.1016/j.bbrc.2015.03.064
66. Bronner DN, Abuaita BH, Chen X, et al. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and Caspase-2-driven mitochondrial damage. Immunity. 2015;43:451–462. doi:10.1016/j.immuni.2015.08.008
67. Li J, Wang Y, Wang Y, et al. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J Mol Cell Cardiol. 2015;86:62–74. doi:10.1016/j.yjmcc.2015.07.010
68. Yuan X, Zheng Y, Chen C, Wang C. Anisodamine inhibits endoplasmic reticulum stress-associated TXNIP/NLRP3 inflammasome activation in rhabdomyolysis-induced acute kidney injury. Apoptosis. 2017;22:1524–1531. doi:10.1007/s10495-017-1414-y
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.