")
Back to Journals » Infection and Drug Resistance » Volume 11
Overview of meropenem-vaborbactam and newer antimicrobial agents for the treatment of carbapenem-resistant Enterobacteriaceae
Authors Petty LA, Henig O , Patel TS, Pogue JM, Kaye KS
Received 16 April 2018
Accepted for publication 3 July 2018
Published 12 September 2018 Volume 2018:11 Pages 1461—1472
DOI https://doi.org/10.2147/IDR.S150447
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Lindsay A Petty,1 Oryan Henig,1 Twisha S Patel,2 Jason M Pogue,3 Keith S Kaye1
1Department of Internal Medicine, Division of Infectious Diseases, University of Michigan Medical School, Ann Arbor, MI, USA; 2Department of Pharmacy Services, University of Michigan Hospitals and Health Centers, Ann Arbor, MI, USA; 3Department of Pharmacy Services, Sinai-Grace Hospital, Detroit Medical Center, Detroit, MI, USA
Abstract: There has been a worldwide increase in infections caused by drug-resistant Gram-negative pathogens, including carbapenem-resistant Enterobacteriaceae (CRE). Meropenem-vaborbactam, a carbapenem antibiotic and novel boronic acid-based beta-lactamase inhibitor, is a fixed-dose combination product with potent in vitro activity against Enterobacteriaceae that are Klebsiella pneumoniae carbapenemase producers. Meropenem-vaborbactam has been studied in two Phase III trials, Targeting Antibiotic Non-susceptible Gram-negative Organisms (TANGO)-I and TANGO-II. TANGO-I was a multicenter, international Phase III, randomized, double-blind, double-dummy, active-control trial to evaluate the efficacy and safety of meropenem-vaborbactam for the treatment of complicated urinary tract infection, including acute pyelonephritis. Among patients with complicated urinary tract infection and growth of a baseline pathogen, meropenem-vaborbactam was determined to be superior to piperacillin-tazobactam based on the composite outcome of symptom improvement or resolution and microbial eradication at the end of intravenous therapy. TANGO-II was a multicenter, international, Phase III, randomized, prospective, open-label, comparative trial to evaluate the efficacy and safety of meropenem-vaborbactam vs best available therapy for CRE infections. Treatment with meropenem-vaborbactam resulted in higher rates of clinical cure at the end of therapy (64.3%vs 33.3%, P=0.04). Additionally, 28-day all-cause mortality was 17.9% in the meropenem-vaborbactam group compared to 33.3% in the best available therapy group, a relative risk reduction of 46.5% (P=0.03). In addition to meropenem-vaborbactam, three other agents with activity against CRE are in late-stage development: imipenem-relebactam, plazomicin, and cefiderocol. The data from Phase II and III studies will help to further define the role of these agents. Overall, the recent approval of meropenem-vaborbactam and the active pipeline for other agents with broad Gram-negative activity are encouraging developments on the CRE therapeutic front.
Keywords: meropenem, vaborbactam, carbapenemase, CRE, UTI, KPC
Introduction
Worldwide, there has been a continued increase in drug-resistant Gram-negative infections, including carbapenemase-producing Enterobacteriaceae. Carbapenemases include a variety of beta-lactamases that can hydrolyze carbapenems, as well as penicillins, with variable hydrolysis of cephalosporins, and aztreonam. Typically, the activity of these enzymes is not inhibited by the traditional beta-lactamase inhibitors clavulanic acid, tazobactam, and sulbactam.1 In many locations globally, including the United States, the most common carbapenemase produced in carbapenem-resistant Enterobacteriaceae (CRE) is Klebsiella pneumoniae carbapenemase (KPC), an Ambler class A enzyme.2 However, Ambler class B (NDM, IMP, and VIM) and Ambler class D (OXA-48 like) enzymes have been reported from various parts of the world, as well as less frequently in the United States.3
CRE most often cause infection in health care settings, primarily in the hospital and long-term care facilities. In the majority of published literature, CRE are most commonly isolated from the urine, which is not surprising given the fact that Enterobacteriaceae are the most common urinary tract infection (UTI) pathogens.4 Across multiple types of infections, drug-resistant Enterobacteriaceae have been associated with poor outcomes, including increased mortality,4,5 hospital readmissions,6 and cost.7,8 Mortality rates for invasive CRE infections range from 26% to 44%.5 Treatment options for CRE are limited, as resistance to multiple classes of antimicrobials is mediated by a variety of resistance mechanisms, including efflux pumps, enzymatic degradation, porin mutations, and target-site alterations, that are usually present in addition to carbapenemases. As a result, in some cases, no effective first-line agent is active against CRE pathogens, and only older toxic agents, such as polymyxins and aminoglycosides, are therapeutic options.9
The growing incidence of resistant Gram-negative infections in general, and CRE in particular, has resulted in a critical need for new antibiotics.10,11 Ceftazidime-avibactam, a combination of a broad-spectrum antipseudomonal cephalosporin and a novel beta-lactamase inhibitor, was approved in 2015 for the treatment of complicated urinary tract infection (cUTI) and intra-abdominal infection and has in vitro activity against Ambler class A (eg, KPC) and some class D (ie, OXA-48 like) serine carbapenemases.12 It was also recently approved for the treatment of hospital-acquired/ventilator-associated pneumonia. Retrospective, observational data comparing treatment of CRE with ceftazidime-avibactam to that with colistin have demonstrated decreased mortality with this agent.13,14 Shields et al demonstrated an increased survival rate of 92% for ceftazidime-avibactam (12/13) compared to 55% (53/96) with any other regimen, which largely consisted of a combination or monotherapy with colistin, aminoglycosides, and carbapenems (P=0.01).13 Similarly, data from the Consortium of Resistance Against Carbapenems in Klebsiella and other Enterobacteriaceae (CRACKLE) database in the US demonstrated that therapy for CRE infection with ceftazidime-avibactam (N=38) was associated with significantly lower mortality rates when compared to initial therapy with colistin (N=99) (adjusted mortality rate 9% vs 32%, respectively; P=0.001).14 While these data are encouraging, the emergence of resistance to ceftazidime-avibactam in patients with CRE infections has been reported by several groups and is concerning.13,15–17 This highlights the need for good antimicrobial stewardship practices for appropriate use of these niche drugs with anti-CRE activity, such as ceftazidime-avibactam, as well as the continuing need for development of novel agents with anti-CRE activity such as meropenem-vaborbactam.13,15
This paper will provide a detailed review of meropenem-vaborbactam, an antibiotic recently approved for treatment of cUTI and acute pyelonephritis. In addition, three agents that are currently being evaluated in Phase III studies that are active against CRE also will be discussed.
Structure and mechanism of action
Meropenem-vaborbactam was designed as a fixed-dose combination product. Meropenem is a carbapenem antibiotic that inhibits the cell wall synthesis of most Gram-positive and Gram-negative bacteria by binding to penicillin-binding proteins and, therefore, inhibiting the cross-linking of peptidoglycan chains leading to cell lysis and ultimately cell death. Meropenem is stable in the presence of many hydrolyzing beta-lactamases, including penicillinases and cephalosporinases, largely due to its 6-trans-hydroxyethyl group (Figure 1). Vaborbactam (formerly RPX7009; Figure 1) is a novel cyclic boronic acid-based beta-lactamase inhibitor that potentiates the activity of meropenem. The cyclic boronate ester was designed to configure the compound in a preferred conformation to selectively improve substrate activity against beta-lactamases as compared to mammalian serine proteases. Experiments were conducted to explore the structure–activity relationships of added substituents and resultant potentiation of carbapenem activity. The addition of a 2-thienyl acetyl group found in the structure of vaborbactam increased the potency of the beta-lactamase inhibitor.18 Vaborbactam works by inhibiting serine beta-lactamases, including many Ambler class A and C enzymes, most notably the KPC enzyme.18 For cell entry of K. pneumoniae, vaborbactam crosses the outer membrane using porins OmpK35 and OmpK36.19 In regard to the stability of the medication, the intravenous (IV) infusion of the diluted solution must be completed within 4 hours if stored at room temperature or 22 hours if stored refrigerated at 2°C–8°C (36°F–46°F).20
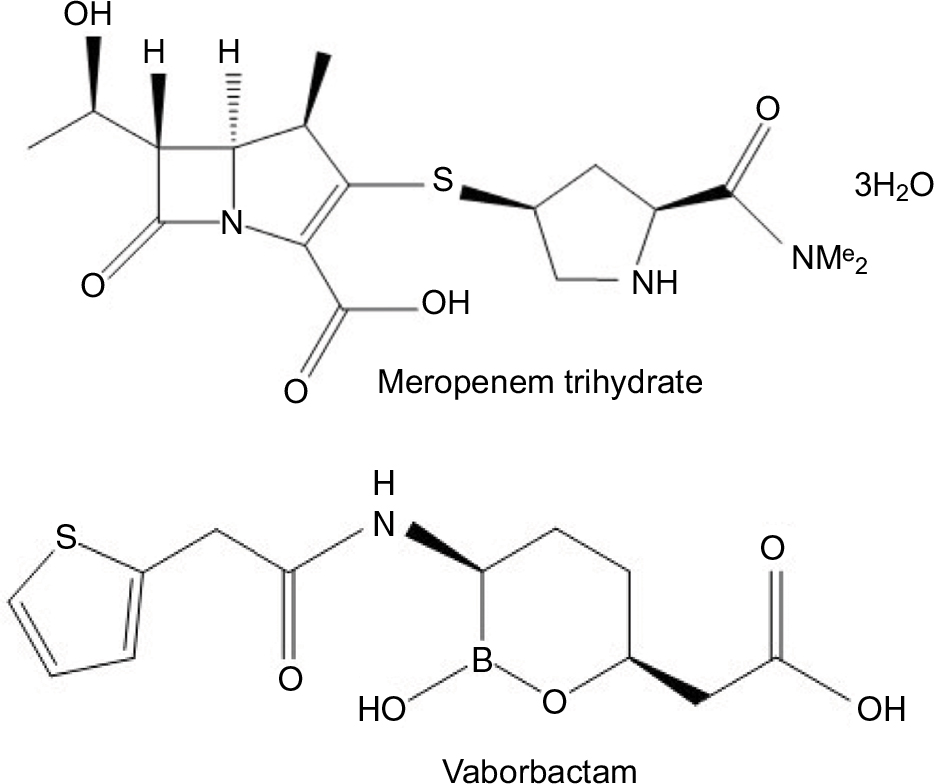 | Figure 1 Chemical structures of meropenem and vaborbactam. Note: Data from MEROPENEM package insert. Schaumburg, IL: SAGENT Pharmaceuticals; 2016.20 |
Spectrum of activity, in vitro potency, and resistance
Meropenem alone has broad-spectrum Gram-negative activity, including against multi-drug-resistant organisms, and remains stable in the presence of various beta-lactamases such as extended-spectrum beta-lactamases (ESBLs) and AmpC beta-lactamases. Vaborbactam, which alone has no antimicrobial activity, is a potent inhibitor of class A carbapenemases, including KPC.19 It is also an inhibitor of other class A (eg, CTX-M, SHV, and TEM) and class C (eg, P99, MIR, FOX, AmpC) beta-lactamases.19 However, the activity of meropenem-vaborbactam against organisms that produce these enzymes is similar to that of meropenem alone given the relative stability of meropenem in the presence of these beta-lactamases. For example, for a strain of CTX-M-producing Escherichia coli, both the meropenem and meropenem-vaborbactam minimum inhibitory concentrations (MICs) were ≤0.03 mg/L. Likewise, for an AmpC-producing strain of E. coli, the meropenem MIC (≤0.03 mg/L) was unchanged when vaborbactam was present.19
Meropenem-vaborbactam has potent in vitro activity against KPC-producing strains of CRE. Of 991 clinical isolates of KPC-producing Enterobacteriaceae collected from a global clinical isolate surveillance study, 99% (n=981) were susceptible to meropenem-vaborbactam using a clinical breakpoint of ≤4/8 mg/L (MIC50 0.06 mg/L and MIC90 1 mg/L). K. pneumoniae was the most common type of Enterobacteriaceae included in this study, and meropenem-vaborbactam displayed good activity against these organisms (MIC50 0.12 mg/L and MIC90 1 mg/L). Additionally, there were no differences between the activity of meropenem-vaborbactam against organisms producing KPC-2 (n=610, MIC50 0.06 mg/L and MIC90 1 mg/L) or KPC-3 (n=373, MIC50 0.12 mg/L and MIC90 1 mg/L).21 In a study by Castanheira et al, 10,426 Enterobacteriaceae isolates were tested against meropenem-vaborbactam. Vaborbactam combined with meropenem had improved activity compared to meropenem alone across all isolates. Against 265 CRE isolates, meropenem alone had little activity (MIC50 16 mg/L and MIC90 >32 mg/L), whereas meropenem-vaborbactam displayed improved in vitro activity (MIC50 0.5 mg/L and MIC90 32 mg/L). Of note, meropenem-vaborbactam activity was better against KPC-producing CRE (MIC50 0.12 mg/L and MIC90 0.5 mg/L) as compared to non-KPC-producing CRE (MIC50 4 mg/L and MIC90 >32 mg/L).22 The difference in MIC50 and MIC90 values is, in part, due to the fact that vaborbactam does not inhibit class B (metallo-beta-lactamases) or class D enzymes (oxacillinases), and in the presence of these enzymes, vaborbactam does not restore meropenem activity. For example, the MICs for meropenem-vaborbactam were the same as the MICs for meropenem in an engineered strain of NDM-1 (class B)-producing E. coli (MIC 16 mg/L) and in a strain of OXA-48 (class D)-producing E. coli (MIC 0.125 mg/L).19 Furthermore, in an analysis of 41 metallo-beta-lactamase producers (MIC50 32 mg/L and MIC90 >32 mg/L) and 25 OXA-48 producers (MIC50 16 mg/L and MIC90 >32 mg/L), the MIC50 and MIC90 for both meropenem and meropenem-vaborbactam were the same.22 This study also found that the addition of vaborbactam did not improve the in vitro activity of meropenem against Acinetobacter spp. (n=708, MIC50 32 mg/L and MIC90 >32 mg/L) or Pseudomonas aeruginosa (n=2,604, MIC50 0.5 mg/L and MIC90 8 mg/L).22 Carbapenem resistance in P. aeruginosa and Acinetobacter spp. is multifactorial and can be caused by a number of mechanisms including reduced outer membrane permeability, overexpression of efflux pumps, and production of Ambler class B and D enzymes.23 Therefore, the addition of vaborbactam would not be expected to improve the activity of meropenem against these pathogens.24
The activity of meropenem-vaborbactam was studied in Gram-negative bacteria that have porin mutations combined with overexpression of efflux pumps and has showed varying results. Lomovskaya et al studied engineered strains of KPC-producing K. pneumoniae with different downregulation or knockout mutations involving efflux pumps and porins and found that efflux by the multidrug resistance efflux pump AcrAB-TolC had minimal impact on the activity of meropenem-vaborbactam, but that mutations to the outer membrane porins, OmpK35 and OmpK36, reduced the activity of this agent. It is important to note that in spite of these resistance mechanisms, MICs remained within the susceptible range (at 2 mg/L), and thus, the clinical significance is unclear.19 However, in a separate analysis by Sun et al, KPC-producing isolates with various mutations to OmpK35 and OmpK36 had meropenem/vaborbactam MIC values ranging from ≤0.06/8 to 32/8 mg/L, demonstrating that this combination of resistance mechanisms can lead to frank resistance.25
Pharmacokinetics
In a Phase I sequential single- and multiple-dose-escalating trial, 80 healthy adult subjects were randomly assigned to receive doses ranging from 250 to 2000 mg of vaborbactam vs placebo as a 3-hour infusion (Table 1). At various time points following the infusion, plasma and urine samples were analyzed using high-performance liquid chromatography-tandem mass spectrometry. Exposure to vaborbactam (Cmax and area under the concentration–time curve [AUC]) increased proportionally by dose, and there was no evidence of accumulation with multiple doses.26 The terminal half-life was ~2 hours. The average serum protein binding level for vaborbactam was 33%, and volume of distribution was independent of the dose.26 Additionally, vaborbactam was measured at high concentrations in urine, with 80%–90% of the administered dose present in the urine unchanged.26
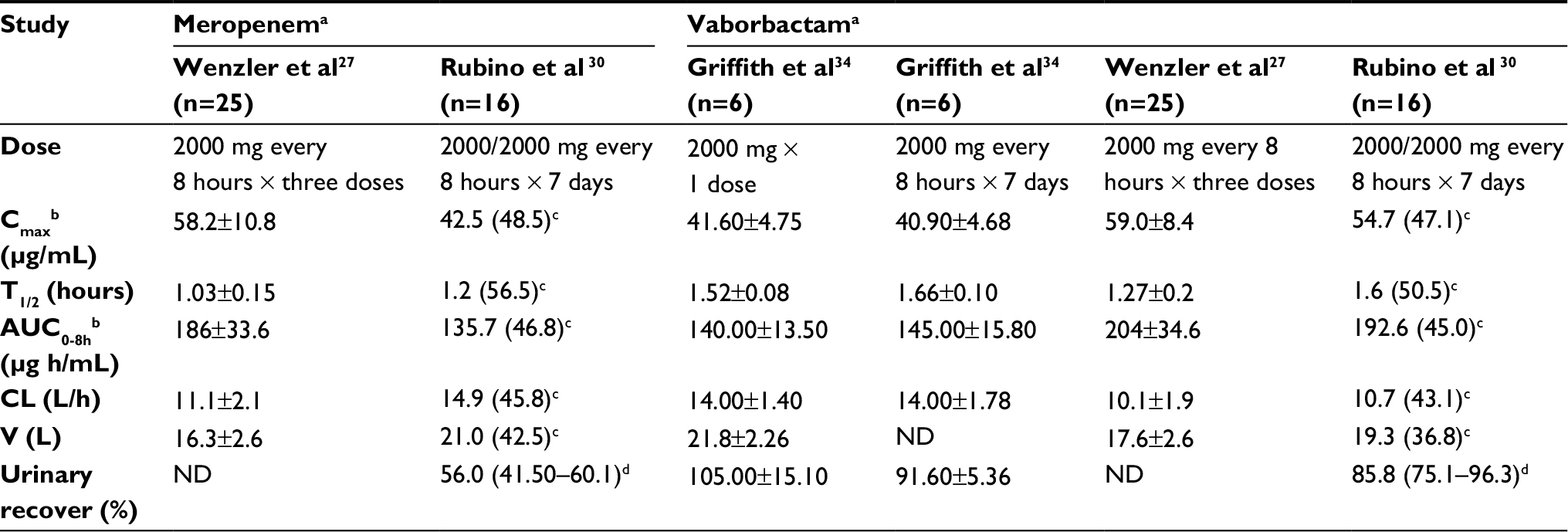 | Table 1 Pharmacokinetic parameters of meropenem and vaborbactam determined in healthy adults Notes: aAdministered as a 3-hour intravenous infusion; bdata represented are total plasma concentrations; cmean (geometric coefficient of variation); dmedian (range). Abbreviations: AUC, area under the concentration–time curve; CL, clearance; ND, not done. |
In a separate multiple-dose Phase I study by Wenzler et al, the pharmacokinetics of both agents were determined following the IV administration of a fixed-dose combination product of meropenem (2 g) and vaborbactam (2 g) (Table 1).27 An average total plasma Cmax of 58.2 and 59.0 µg/mL, total plasma AUC0-8h of 186 and 204 µg h/mL, volume of distribution of 16.3 and 17.6 L, and half-lives of 1 and 1.3 h for meropenem and vaborbactam at steady state, respectively, were determined following the administration of meropenem-vaborbactam 2 g/2 g given as a 3-hour IV infusion.27 Using these data and a protein binding of 2% for meropenem and 33% for vaborbactam, the estimated free-drug AUC0-24h was 547 and 410 µg h/mL, respectively.27 Data from both Phase I studies suggest that meropenem and vaborbactam display similar pharmacokinetics in plasma. Pharmacokinetic (PK) data from Phase III studies have not yet been published, although preliminary reports suggest a higher estimated free-drug AUC0-24h of 560 µg h/mL for vaborbactam.28,29
The study by Wenzler et al also assessed the pulmonary penetration of meropenem-vaborbactam. Following administration of meropenem-vaborbactam (2 g/2 g 3-hour infusion every 8 hours) for three doses in 25 healthy adults, multiple respiratory samples were obtained by bronchoscopy and bronchoalveolar lavage at various time points. The results demonstrated epithelial lining fluid (ELF) concentrations ranging from one-half to two times the simultaneous plasma concentrations, with ratios of ELF-to-plasma concentrations of meropenem and vaborbactam 65% and 79%, respectively.27
Both meropenem and vaborbactam are primarily excreted by the kidneys. Decreasing renal function results in increased plasma exposures and elimination half-life of both meropenem and vaborbactam.30 In a Phase I study, 41 subjects were assigned to one of five groups based on degree of renal impairment. The elimination half-life was longer, and exposures (Cmax and AUC) were greater for both meropenem and vaborbactam in patients with higher degrees of renal impairment. Both meropenem and vaborbactam are removed by hemodialysis, and thus, an increased total plasma clearance of 2.21-fold and 5.11-fold, respectively, is seen in patients undergoing hemodialysis.30 For meropenem and vaborbactam, 38.3% and 52.9% of the dose, respectively, were recovered in dialysate during hemodialysis sessions.30 For these reasons the dose of meropenem-vaborbactam should be adjusted based on renal function, and it should be dosed after hemodialysis in end-stage renal disease (Table 2). These labeled dosing recommendations are supported by a PK study that simulated exposures with labeled dosing in patients with various degrees of renal insufficiency. These data demonstrated high probabilities of attaining PK–PD (pharmacodynamic) targets with these doses at or above the current meropenem-vaborbactam MIC breakpoint.31
 | Table 2 Dosing of meropenem-vaborbactam Notes: aeGFR is calculated using the Modification of Diet in Renal Disease formula as follows: eGFR (mL/min/1.73 m2)=175 × (serum creatinine)–1.154 × (age) – 0.203 × (0.742 if female) × (1.212 if African American); bAdministered as a 3-hour infusion; cFor patients on hemodialysis, give the dose after a dialysis session. Abbreviation: eGFR, estimated glomerular filtration rate. |
Vaborbactam is not metabolized by the liver, so systemic clearance of meropenem-vaborbactam is not expected to be affected by hepatic impairment. Therefore, dose adjustment is not needed in the setting of hepatic dysfunction.
Pharmacodynamics
The pharmacodynamics of meropenem has been well described, and meropenem is known to display time-dependent bactericidal activity.32 Therefore, the best predictor of effective antimicrobial killing is the percentage of time that free-drug concentrations are above the MIC (%ƒT>MIC) during the dosing interval. For meropenem, animal models suggest that a %ƒT >MIC of 40 or greater is needed to achieve maximal bactericidal activity.32,33
Less published data are available regarding the pharmacodynamics of vaborbactam. In a study using KPC-producing Enterobacteriaceae with meropenem-vaborbactam MICs ranging from ≤0.06/8 to 64/8 mg/L, adjustments were made to an in vitro hollow-fiber model to mimic exposures seen in humans following the administration of meropenem and vaborbactam 2 g/2 g administered every 8 hours by 3-hour infusion. Using PK data from Phase I studies, the mean vaborbactam ƒAUC(0–24) in the model was 316.7 µg h/mL, resulting in a 6-log kill and suppression of resistance among isolates with a meropenem-vaborbactam MIC of up to 8 mg/L. Interestingly, when the vaborbactam exposures in the model were increased to those demonstrated in the Phase III studies, a 6-log kill and suppression of resistance was demonstrated in isolates with a meropenem-vaborbactam MIC of up to 16 mg/L.
In unpublished data, both an in vitro hollow-fiber model and a neutropenic murine thigh model were utilized to assess the pharmacodynamics of vaborbactam in the backdrop of meropenem 2 g every 8 hours as a 3-hour infusion (the labeled dose) against KPC producers. The ratio of free-drug AUC to meropenem-vaborbactam (vaborbactam fixed concentration of 8 mg/L) MIC (ƒAUC:MIC) was the vaborbactam target that best predicted restoration of meropenem antimicrobial activity. In the in vitro hollow-fiber model, ƒAUC:MIC ratios of 12 and 18 were associated with bacteriostasis and 1-log10 kill, respectively. In the neutropenic murine thigh model, ƒAUC:MIC ratios of 9 and 38 were associated with bacteriostasis and 1-log10 kill, respectively.34 Additionally, suppression of resistance was observed with ƒAUC:MIC ratios >24 in the hollow-fiber model. Taking into account a meropenem-vaborbactam clinical breakpoint of 4/8 mg/L and a PD target of ƒAUC:MIC ratio of 38 (based on the murine thigh model), the ƒAUC(0-24) of vaborbactam needed to ensure bactericidal activity is ≥152 µg h/mL. The Phase I PK data presented in Table 1 suggest that mean vaborbactam ƒAUC(0-24) values ranging from 290 to 410 µg h/mL were attained following administration of meropenem-vaborbactam 2 g/2 g every 8 hours, and the unpublished Phase III PK data suggest even higher mean exposures (~560 µg h/mL).28,31 Based on these data, it is likely that most patients will be able to achieve or exceed the vaborbactam target of ƒAUC(0-24) ≥152 µg h/mL. PD data from lung models have not yet been assessed and are warranted.
Efficacy and safety in clinical trials
Targeting Antibiotic Non-susceptible Gram-negative Organisms (TANGO)-I
TANGO-I was a multicenter, international, Phase III, randomized, double-blind, double-dummy, active-control trial to evaluate the efficacy and safety of meropenem-vaborbactam in cUTI, including acute pyelonephritis.35 It was conducted across 60 sites in 17 countries; 550 patients were randomized 1:1 to receive either meropenem-vaborbactam (2 g/2 g through 3-hour infusion) or piperacillin-tazobactam (4 g/0.5 g through 30-minute infusion) every 8 hours. Patients received a total duration of therapy of 10 days (IV ± transition to oral levofloxacin 500 mg after ≥15 doses of IV therapy). Randomization was stratified by geographic region and infection type. According to the protocol, when possible, foci of infection (ie, catheters) were to be removed within 12 hours of enrollment.
Patients were analyzed in three different populations. First, 544 patients were analyzed in the modified intent-to-treat (ITT) population and were required to have received ≥1 dose of study drug. Of those enrolled, 31% did not have a baseline pathogen present at ≥105 CFU/mL. The remaining 374 (69%) enrolled patients had a bacterial pathogen in baseline urine culture or the same bacterial pathogen in concurrent blood and urine cultures and were included in the microbiologic modified ITT population. The microbiologic evaluable population was composed of subjects from the microbiologic modified ITT who met more detailed and thorough follow-up criteria. The primary Food and Drug Administration (FDA) endpoint was overall success, a composite outcome including clinical cure or improvement and microbial eradication at the end of IV treatment in the microbiologic modified ITT population, using a noninferiority margin of 15%. The primary efficacy endpoint for the European Medicines Agency (EMA) was microbiologic cure, defined as reduction in bacteria to <103 CFU/mL at test of cure for the microbiologic modified ITT and the microbiologic evaluable populations.
Baseline characteristics for the study populations were similar between the two treatment groups (including type of infection, mean number of symptoms, and baseline urinary pathogen). E. coli (65.1% and 64.3%) and K. pneumoniae (15.6% and 15.4%) were the most commonly isolated pathogens. Of note, only one patient in the meropenem-vaborbactam arm had an isolate resistant to meropenem, due to OXA-48 production (also resistant to meropenem-vaborbactam; MIC >32 mg/L). Overall, there was about a 12% resistance rate to piperacillin-tazobactam, with the majority of resistance occurring in K. pneumoniae (15/30, 50% meropenem-vaborbactam arm vs 9/27, 33.3% piperacillin-tazobactam arm).
Most patients in both groups of the modified ITT population completed study treatment, and reasons for not completing included premature discontinuation of IV therapy, adverse events, and physician discretion. Treatment duration was similar across both groups, with mean duration of IV therapy 8 days, and of total therapy approximately 10 days. Step-down therapy with levofloxacin occurred in >90% of patients in both groups, and approximately 9% of patients in both groups were switched despite having a levofloxacin-resistant pathogen at baseline.
Overall success was 98.4% (189/192) in the meropenem-vaborbactam group compared to 94.0% (171/182) in the piperacillin-tazobactam group at the end of IV therapy (difference, 95% CI: 4.5%, 0.7, 9.1; P for noninferiority <0.001, P=0.01 for superiority). In the microbiologic modified ITT population, clinical cure occurred in 98.4% (189/192) of the meropenem-vaborbactam group vs 95.6% (174/182) of the piperacillin-tazobactam group at the end of IV treatment, and at test of cure was 90.6% (174/192) vs 86.3% (157/182), respectively. Of note, all patients with an isolate that was resistant to piperacillin-tazobactam (MIC≥64 mg/L) achieved clinical cure in both treatment groups. For the EMA primary endpoint, rate of microbiologic eradication at test of cure was similar between the two treatment arms, 66.7% (128/192) in the meropenem-vaborbactam group and 57.7% (105/182) in the piperacillin-tazobactam group (difference of 9.0% [95% CI: −0.9%, 18.7%]; P<0.001 for noninferiority).
In summary, among patients with cUTI including acute pyelonephritis and growth of a baseline pathogen, superiority was demonstrated for the primary FDA endpoint of symptom improvement or resolution and microbial eradication at the end of IV therapy for meropenem-vaborbactam compared to piperacillin-tazobactam.
TANGO II
TANGO II was a multicenter, international, Phase III, randomized, prospective, open-label, comparative trial to evaluate the efficacy and safety of meropenem-vaborbactam vs best available therapy (BAT) for CRE infections.36 The 72 adult patients enrolled with suspected CRE infection were randomized 2:1 to receive meropenem-vaborbactam (2 g/2 g over 3 hours, every 8 hours, 7–14 days) or BAT. BAT included mono- or combination therapy with polymyxin B or colistin, carbapenems, aminoglycosides, tigecycline, or ceftazidime-avibactam. Only one patient received treatment with ceftazidime-avibactam, administered as monotherapy; therefore, no conclusions can be drawn regarding the comparative efficacy of meropenem-vaborbactam and ceftazidime-avibactam. Eligible infections included cUTI or acute pyelonephritis, health care-associated pneumonia, ventilator-associated pneumonia, bloodstream infection (BSI), and complicated intra-abdominal infection. Enrollment was stratified by infection type and geographic region. Exclusion criteria included effective antibiotic therapy for >1 day (unless there was documented clinical failure to that regimen) or presence of an immediate life-threatening disease and known infection due to a pathogen producing NDM-, VIM-, IMI-, or OXA-encoded carbapenemase.
All patients who received ≥1 dose of study drug were included in the safety analysis. The primary study population was the microbiologic-CRE-modified ITT (mCRE-MITT) population, defined as patients who received ≥1 dose of study drug and had a baseline qualifying CRE isolate. Of those patients randomized, 43 (28 meropenem-vaborbactam; 15 BAT) had a baseline CRE infection. The primary efficacy endpoint was clinical success at test-of-cure visit.
In the mCRE-MITT population, patients were similar between treatment groups, except a higher proportion of patients with prior antibiotic failure were randomized to receive meropenem-vaborbactam (meropenem-vaborbactam n=9, BAT n=0). The most common infections were bacteremia (N=20, 46%) and cUTI or pyelonephritis (N=15, 35%); K. pneumoniae was the most frequent pathogen isolated (N=37, 86%).
In the mCRE-MITT population, meropenem-vaborbactam compared to BAT resulted in a higher rate of clinical cure at the end of therapy (N=18, 64.3% vs N=5, 33.3%, respectively; absolute difference 31.0%; 95% CI: 1.2%, 60.7%; P=0.04) and at test of cure (N=16, 57.1% vs N=4, 26.7%; absolute difference 30.5%; 95% CI: 1.5%, 59.4%; P=0.04). 28-day all-cause mortality was 17.9% (N=5) in the meropenem-vaborbactam group compared to 33.3% (N=5) in the BAT group, a relative risk reduction of 46.5%, P=0.03. In the mCRE-MITT population in which prior antibiotic failures were removed, meropenem-vaborbactam compared to BAT resulted in decreased 28-day all-cause mortality (meropenem-vaborbactam 1/19, 5.3% vs BAT 5/15, 33.3%), relative risk reduction of 84.1%. Overall, meropenem-vaborbactam was associated with fewer adverse events than BAT, including nephrotoxicity. Given the improved efficacy and decreased adverse events seen in the meropenem-vaborbactam group, the study was discontinued on July 21, 2017, on the recommendation of the Data Safety Monitoring Board. These data have been presented in preliminary fashion at a conference, but at the time of this publication have not yet been published.
Safety
In general, meropenem-vaborbactam has been shown in Phase I and Phase III trials to be well tolerated and associated with minimal adverse reactions. Table 3 outlines the most common adverse events experienced by subjects who received meropenem-vaborbactam. Headache (8.8%), phlebitis/infusion site reactions (4.4%), and diarrhea (3.3%) were the most common adverse events demonstrated in TANGO I and are consistent with the adverse events reported for meropenem. Although Clostridium difficile-associated diarrhea was not reported in the clinical trials, this adverse effect has been reported with meropenem, and thus, appropriate monitoring is warranted.20
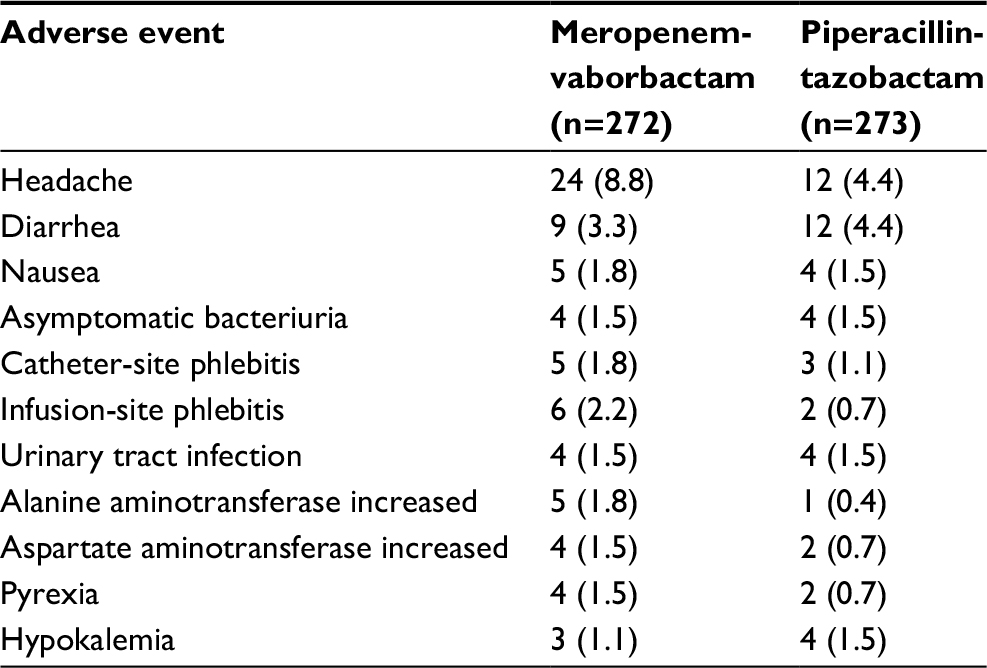 | Table 3 Common adverse events experienced by ≥1% of patients receiving meropenem-vaborbactam in TANGO I Note: Data are presented as n (%). Abbreviation: TANGO, Targeting Antibiotic Non-susceptible Gram-negative Organisms. |
Conclusions and indications for meropenem-vaborbactam use
TANGO I demonstrated the superiority of meropenem-vaborbactam compared to piperacillin-tazobactam in adults with cUTI including acute pyelonephritis. Based on these results, meropenem-vaborbactam was FDA-approved on August 29th, 2017, for treatment of cUTI including pyelonephritis. Additionally, meropenem-vaborbactam has potent in vitro activity against KPC-producing CRE. Thus, meropenem-vaborbactam is a good treatment option for cUTI caused by KPC-producing CRE. TANGO II demonstrated improved efficacy with decreased adverse events in patients treated with meropenem-vaborbactam compared to BAT for CRE infections. Given these findings, which more closely mirror anticipated real-world use of the agent, meropenem-vaborbactam should be considered a first-line option for patients with serious CRE infections caused by KPC-producing pathogens, and antimicrobial stewardship programs should restrict use of this agent to this indication to preserve future use. Additional real-world experience with meropenem-vaborbactam will help to define the role of this agent in treating CRE, particularly as the armamentarium of agents active against CRE grows.
Selected pipeline agents for CRE
Imipenem-relebactam
Relebactam (MK-7655) is a non-beta-lactam (diazabicyclooctane) beta-lactamase inhibitor that is active against class A beta-lactamases (including KPCs) and class C beta-lactamases. The structure of relebactam is similar to that of avibactam. Compared to avibactam, relebactam has less inhibitory activity against OXA-48, and neither beta-lactamase inhibitor is active against metallo-beta-lactamases.37
In vitro studies have demonstrated that relebactam, when added to imipenem/cilastatin, restores activity against imipenem-nonsusceptible strains that produce KPC. In the SMART study (Study for Monitoring Antimicrobial Resistance Trends) that included 1,451 GNB from the US during 2015, addition of 4 mg/L of relebactam restored susceptibility for 66.7% of 72 imipenem-nonsusceptible Enterobacteriaceae and decreased the MIC90 from 16 to 2 mg/L. Among the 24 Enterobacteriaceae that were nonsusceptible to imipenem/relebactam, four carried beta-lactamases (two carried a KPC-3, one carried an OXA-48, and one carried a GES-20 class A beta-lactamase). No acquired beta-lactamase was detected for the other 20 CRE that were nonsusceptible to imipenem/relebactam.38 In another study, among 111 K. pneumoniae with blakpc, the imipenem MIC50 (16 mg/L) and MIC90 (>16 mg/L) decreased to 0.25 and 1 mg/L, respectively.39 These data demonstrate the high level of in vitro activity for imipenem/relebactam for KPC-producing CRE.
Two randomized-controlled Phase II trials have evaluated the efficacy and safety of imipenem/relebactam. Both of the trials enrolled patients into three treatment groups (1:1:1): imipenem/cilastatin alone, imipenem/cilastatin with relebactam 125 mg q6 hours, and imipenem/cilastatin with relebactam 250 mg q6 hours. The first study enrolled adults with complicated intra-abdominal infections (IAI) (appendicitis, cholecystitis, and perforated viscus.) The overall microbiologic as well as clinical response were noninferior in all combination arms compared to imipenem/cilastatin alone (among the ME population, favorable clinical response at day of study drug discontinuation, 96.3% [78/81], 98.8% [85/86], and 95.2% [79/83] for relebactam 250 mg + imipenem/cilastatin, relebactam 125 mg + imipenem/cilastatin, and imipenem/cilastatin alone, respectively, P<0.001 for noninferiority for both relebactam arms compared to imipenem/cilastatin alone).40
The second Phase II trial enrolled adults who had cUTI or acute pyelonephritis. Similar to the IAI study, more than 95% of subjects achieved favorable microbiologic and clinical response at the time of IV treatment discontinuation with no difference in outcomes across treatment groups.41 Neither study was specific for multidrug-resistant pathogens and enrolled few patients with imipenem-nonsusceptible pathogens. Furthermore, imipenem-resistant isolates were commonly resistant to imipenem/relebactam.
In both trials, imipenem/relebactam was well tolerated. The most common adverse events were diarrhea, nausea, and headache, with no differences across treatment groups.41 These data supported moving forward in Phase III trials with the dose of imipenem/relebactam of 500 mg/250 mg every 6 hours.
One Phase III trial has been completed, and results were reported at a conference in April 2018. It is a multicenter, randomized, double-blind, comparator-controlled trial comparing the efficacy and safety of imipenem/relebactam and colistin-imipenem/cilastatin in patients with imipenem-resistant GNB infection (RESTORE-IMI 1). Enrolled patients had health care-associated pneumonia/ventilator-associated pneumonia, complicated intra-abdominal infection, or cUTI caused by an imipenem-nonsusceptible bacteria. Patients were randomized 2:1 to receive imipenem/relebactam or colistin + imipenem/cilastatin. In the microbiologic modified intent-to-treat (mMITT) population, the primary endpoint was favorable overall response, with 31 of 47 patients meeting mMITT criteria. Favorable overall response was comparable for the imipenem/relebactam arm (15/21; 71.4%) and the colistin + imipenem/cilistatin cohort (7/10; 70.0%).42 Of note, 77% of pathogens were P. aeruginosa, and only 16% of the bacteria were KPC producers, making this a different cohort than the prior studies discussed focusing on CRE. The RESTORE-IMI 2, a second Phase III randomized-controlled trial, which compares imipenem/relebactam to piperacillin/tazobactam for hospital-acquired and/or ventilator-associated pneumonia, is ongoing and is anticipated to be completed in 2019.43 Based on in vitro and limited clinical data, it is expected that imipenem/relebactam should provide an alternative option for KPC-producing CRE, but will not add any significant benefit for other mechanisms of carbapenem resistance in Enterobacteriaceae.
Plazomicin
The aminoglycosides are among the historic drug classes that are used for treatment of resistant GNB. Their use has been limited because of toxicity concerns as well as the emergence of resistance. Among Enterobacteriaceae, aminoglycoside resistance is most commonly mediated by aminoglycoside-modifying enzymes (AMEs) that deactivate aminoglycosides by N-acetylation (AAC), O-adenylylation, or O-phosphorylation.44
Plazomicin is a novel aminoglycoside, designed as a modification of sisomicin, to block interactions and inactivation by most of the AMEs among CRE, with the exception of AAC(2′). As an aminoglycoside, it inhibits bacterial protein synthesis and exhibits bactericidal activity in a dose-dependent manner.45 In in vitro studies, resistance to plazomicin has been reported to be mediated by ribosomal 16S methylation (ribosomal methyl transferase), which causes resistance to all commercially available aminoglycosides.46
Several in vitro studies reported broad-spectrum activity of plazomicin against Gram-positive and Gram-negative organisms, including CRE, ESBL producers, and gentamicin-nonsusceptible E. coli.47 Studies that investigated in vitro activity of plazomicin for CRE have demonstrated an MIC50 of plazomicin against CRE of <1 mg/L and an MIC90 of ≤4 mg/L.46
There is a suggestion that plazomicin activity might vary as a function of CRE mechanism. Among KPC producers, MICs between 0.25 and 2 mg/L were reported in most studies.45,48–50 Similarly, in a surveillance study from 16 hospitals in New York city, most of the Enterobacteriaceae that harbored blaKPC (36 E. coli and 25 K. pneumoniae) had a plazomicin MIC of 0.25–8 mg/L, whereas three isolates had an MIC >8 mg/L.51 Among K. pneumoniae and E coli producing a VIM enzyme, MICs were reported between 0.25 and 4 mg/L across various studies.48,50,52
Importantly, in two studies plazomicin demonstrated high MICs of 64–≥256 mg/L in isolates co-producing NDM-1. In both of these studies, the presence of 16S rRNA methylases (Rmtc, ArmA in one study, and ArmA in the second study) was reported.48,50 The association between NDM-1-producing isolates and co-production of 16S methylases such as Rmtc or ArmA should be further investigated.
Importantly, plazomicin has been shown to be more potent than other aminoglycosides against KPC-producing Enterobacteriaceae. For example, among 25 KPC-producing K. pneumoniae, the plazomicin MIC50 was 0.5 mg/L, whereas the MIC50 of gentamicin, amikacin, and tobramycin was 8, 32, and 32 mg/L, respectively. Similarly, the plazomicin MIC90 was 1 mg/L, whereas the MIC90 of gentamicin, amikacin, and tobramycin was 16, 32, and ≥64 mg/L, respectively.49
Plazomicin demonstrated encouraging efficacy and safety results in a Phase II trial for UTI, which supported the continuation to Phase III clinical trials, using a plazomicin dose of 15 mg/kg. Two Phase III clinical trials have evaluated the safety and efficacy of plazomicin, although neither are currently published and have only been presented at scientific conferences. In the EPIC (Evaluating Plazomicin In cUTI) trial,53 388 hospitalized adult patients who had cUTI or acute pyelonephritis caused by Enterobacteriaceae were randomized to receive IV plazomicin (15 mg/kg once daily) or IV meropenem (1 g three times a day) for 4–7 days, followed by oral step-down to levofloxacin to a total 7–10 days of treatment. The primary outcome was composite cure (both clinical and microbiologic cure) at the test-of-cure visit. Among patients with cUTI, composite cure occurred in 78.5% (84/107) and 68.9% (82/119) in the plazomicin and meropenem treatment groups, respectively. Among patients with acute pyelonephritis, composite cure occurred in 85.7% (72/84) and 71.8% (56/78) of subjects in the plazomicin and meropenem treatment groups, respectively (difference of 13.9%, 95% CI: 0.4, 27.1).
In the Combating Antibiotic-Resistant Enterobacteriaceae (CARE) trial,54 two cohorts of adults who had invasive CRE infections (BSI, ventilator-associated bacterial pneumonia, hospital-acquired bacterial pneumonia, and cUTI) were enrolled. In the first cohort, patients were randomized to receive either IV plazomicin (15 mg/kg every 24 hours) or colistin (300 mg colistin base activity [CBA] loading dose, followed by 5 mg/kg CBA divided to two or three times a day) each combined with meropenem or tigecycline. The mMITT population included 37 patients (17 in the plazomicin arm). In this population, 28-day all-cause mortality was lower in the plazomicin-based treatment arm than the colistin-based treatment arm (2/17 [11.8%] vs 8/20 [40%], an absolute difference of −28.2, 90% CI −0.7, –52.5). Similar findings were seen in a subset of patients with proven BSI due to CRE (n=29 patients, n=14 in the plazomicin arm). In this population, 28-day all-cause mortality was lower in the plazomicin-based therapy treatment arm than in the colistin-based treatment arm (1/14 [7.1%] vs 6/15 [40%]), an absolute difference of −32.9 (90% CI −60, –4). The survival benefit was maintained through day 60 (HR for death through day 60, 0.37, 90% CI 0.15, 0.91). Clearance of CRE bacteremia at day 5 was higher in the plazomicin-based therapy (12/14, 85.7%) compared to colistin-based therapy (7/15, 46.7%) (estimated difference of 39.0, 90% CI 9.4, 65.5). The second cohort was an observational cohort of patients who did not meet criteria for inclusion in the randomized study (n=27, 14 patients with BSI). Subjects were treated with IV plazomicin and a second active agent. Twenty-eight-day mortality occurred in 3 (11.1%) patients in this cohort.
In both the EPIC and CARE studies, plazomicin was well tolerated. The most common adverse events were gastrointestinal related, headache, and hypertension, with no difference between treatment groups. Mild reversible ototoxicity occurred in one patient in each of the treatment groups in the EPIC study, and in none of the patients in the CARE study.
Increase in creatinine by at least 0.5 mg/dL while on study drugs occurred in 11 (3.7%) patients in the plazomicin treatment arm, compared to 9 (3%) in the meropenem treatment arm in the EPIC study. Creatinine levels returned to baseline in all but two patients in the plazomicin treatment arm. In the CARE study, lower incidence of creatinine elevation occurred in the plazomicin-based therapy compared to colistin-based therapy (2/12 [16.7%] vs 8/16 [50%] subjects had an increased creatinine level of ≥0.5 mg/dL at any time during the study).
Although published data with plazomicin are lacking, the enhanced in vitro activity and potency compared to traditional aminoglycosides and the superiority data over meropenem for cUTI from the EPIC trial and over colistin-based therapy from the CARE trial are encouraging and suggest that plazomicin will have an important role in the management of CRE infections, particularly in cUTI.
Cefiderocol
Cefiderocol is a novel siderophore cephalosporin with a unique mechanism of action that confers activity against a broad spectrum of resistant GNB, including Enterobacteriaceae, P. aeruginosa, and A. baumannii. It possesses a catechol moiety on the 3-position side chain. This component facilitates cefiderocol binding to ferric iron, and by creating a complex with the iron ion, it then uses an iron-transport system to penetrate the outer cell membrane of GNB. Subsequently, when cefiderocol is across the outer membrane, the iron dissociates and the cephalosporin component of cefiderocol inhibit cell wall synthesis by binding to penicillin-binding proteins.55 Within the periplasmic space, the cephalosporin/catechol structure increases its stability against hydrolysis by clinically relevant beta-lactamases, including serine (including KPCs and OXA-48) and metallo-beta lactamases.56 Moreover, given the use of the iron-transport pathway, cefiderocol does not require classic beta-lactam porin channels to penetrate the cells and therefore retaining activity in the presence of porin channel mutations. Cefiderocol also appears unaffected by common efflux pumps present in Enterobacteriaceae.55
Although limited data are currently published, in vitro studies have demonstrated excellent activity of cefiderocol against class A, B, and D carbapanemase-producing Enterobacteriaceae.57,58 MICs ≤4 mg/L were reported in 90.7% of the meropenem-nonsusceptible Enterobacteriaceae (100% [47/47] of class A producers [KPC]; 89.9% [62/69] of class B beta-lactamase producers [NDM, VIM, IMP]; and 100% [25/25] of class D beta-lactamase producers).58
Currently, one Phase III trial has been completed for cefiderocol compared to imipenem/cilistatin for the treatment of cUTI (for 7–14 days) in 452 adults. While these data have not yet been published, high-level results were presented at a scientific conference. The primary outcome was a composite of clinical and microbiologic response at the time of test of cure, which occurred 7 days after the end of therapy. With regard to the primary outcome, cefiderocol demonstrated superiority over imipenem/cilistatin (183/252, 72.6% vs 65/119, 54.6%, P=0.0002).59 Additionally, cefiderocol was well tolerated. Only five patients (1.7%) discontinued cefiderocol because of either Clostridium difficile, hypersensitivity (itching), increased liver enzymes, or diarrhea.59
Two additional clinical trials are ongoing and are expected to be completed by October 2018 and February 2019. The first (CREDIBLE-CR) is a randomized controlled trial comparing cefiderocol to BAT among patients with a variety of severe infections caused by carbapenem-resistant GNB60; the second trial is a randomized controlled trial comparing cefiderocol to meropenem for the treatment of nosocomial pneumonia caused by GNB (APEKS-NP).61 The results from these trials will further delineate the role of cefiderocol for the management of CRE. Importantly, cefiderocol appears to be stable against Class A, B, and D carbapenemases, which should give it a broader spectrum of activity than the other agents discussed in this review.
Summary and conclusion
Although the threat of Gram-negative antimicrobial resistance remains high, there has been notable recent progress in antibiotic development. Meropenem-vaborbactam is a welcomed therapeutic addition for CRE, and several agents in the pipeline are promising with regard to CRE treatment. Data for the use of these agents for the treatment of resistant Gram-negative pathogens in real-world settings are needed. Additionally, as these new agents become available, it will be crucial to pair their use with antimicrobial stewardship strategies for optimal use and maximum impact.
Disclosures
KS Kaye, LA Petty, and JM Pogue are funded by the National Institute of Allergy and Infectious Diseases (DMID Protocol Number: 10-0065). KS Kaye and JM Pogue are funded by the National Institute of Allergy and Infectious Diseases (R01-AI119446-01). JM Pogue is a paid consultant, has received research support, and/or has served on the Speaker’s Bureau for The Medicines Company, Melinta, Merck, Allergand, Achaogen, Tetraphase, Shionogi, and Zavante. KS Kaye is a paid consultant and advisor for Melinta Therapeutics, The Medicines Company and Allergan. The authors have no other conflicts of interest in this work.
References
Queenan AM, Bush K. Carbapenemases: the versatile beta-lactamases. Clin Microbiol Rev. 2007;20(3):440–458. | ||
Munoz-Price LS, Poirel L, Bonomo RA, et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis. 2013;13(9):785–796. | ||
Perez F, El Chakhtoura NG, Papp-Wallace KM, Wilson BM, Bonomo RA. Treatment options for infections caused by carbapenem-resistant Enterobacteriaceae: can we apply “precision medicine” to antimicrobial chemotherapy? Expert Opin Pharmacother. 2016;17(6):761–781. | ||
van Duin D, Kaye KS, Neuner EA, Bonomo RA. Carbapenem-resistant Enterobacteriaceae: a review of treatment and outcomes. Diagn Microbiol Infect Dis. 2013;75(2):115–120. | ||
Falagas ME, Tansarli GS, Karageorgopoulos DE, Vardakas KZ. Deaths attributable to carbapenem-resistant Enterobacteriaceae infections. Emerg Infect Dis. 2014;20(7):1170–1175. | ||
Messina JA, Cober E, Richter SS, et al. Hospital readmissions in patients with carbapenem-resistant Klebsiella pneumoniae. Infect Control Hosp Epidemiol. 2016;37(3):281–288. | ||
Maragakis LL, Perencevich EN, Cosgrove SE. Clinical and economic burden of antimicrobial resistance. Expert Rev Anti Infect Ther. 2008;6(5):751–763. | ||
Mauldin PD, Salgado CD, Hansen IS, Durup DT, Bosso JA. Attributable hospital cost and length of stay associated with health care-associated infections caused by antibiotic-resistant gram-negative bacteria. Antimicrob Agents Chemother. 2010;54(1):109–115. | ||
Kaye KS, Pogue JM, Tran TB, Nation RL, Li J. Agents of last resort: polymyxin resistance. Infect Dis Clin North Am. 2016;30(2):391–414. | ||
Boucher HW, Talbot GH, Bradley JS, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48(1):1–12. | ||
Prevention CfDCa. Antibiotic resistance threats in the United States, 2013; 2013:1–114. Available from: http://www.cdc.gov/drugresistance/threat-report-2013/. Accessed March 30 2018. | ||
Zhanel GG, Lawson CD, Adam H, et al. Ceftazidime-avibactam: a novel cephalosporin/β-lactamase inhibitor combination. Drugs. 2013;73(2):159–177. | ||
Shields RK, Nguyen MH, Chen L, et al. Ceftazidime-avibactam is superior to other treatment regimens against carbapenem-resistant Klebsiella pneumoniae bacteremia. Antimicrob Agents Chemother. 2017;61(8):1–7. | ||
van Duin D, Lok JJ, Earley M, et al. Colistin versus ceftazidime-avibactam in the treatment of infections due to carbapenem-resistant enterobacteriaceae. Clin Infect Dis. 2018;66(2):163–171. | ||
Shields RK, Potoski BA, Haidar G, et al. Clinical outcomes, drug toxicity, and emergence of ceftazidime-avibactam resistance among patients treated for carbapenem-resistant Enterobacteriaceae infections. Clin Infect Dis. 2016;63(12):1615–1618. | ||
Haidar G, Clancy CJ, Shields RK, Hao B, Cheng S, Nguyen MH. Mutations in bla KPC-3 that confer ceftazidime-avibactam resistance encode novel KPC-3 variants that function as extended-spectrum β-lactamases. Antimicrob Agents Chemother. 2017;61(5):e02534–16. | ||
Giddins MJ, Macesic N, Annavajhala MK, et al. Successive emergence of ceftazidime-avibactam resistance through distinct genomic adaptations in blaKPC-2-harboring Klebsiella pneumoniae sequence type 307 isolates. Antimicrob Agents Chemother. 2018;62(3):1–8. | ||
Hecker SJ, Reddy KR, Totrov M, et al. Discovery of a cyclic boronic acid β-lactamase inhibitor (RPX7009) with utility vs class A serine carbapenemases. J Med Chem. 2015;58(9):3682–3692. | ||
Lomovskaya O, Sun D, Rubio-Aparicio D, et al. Vaborbactam: spectrum of beta-lactamase inhibition and impact of resistance mechanisms on activity in Enterobacteriaceae. Antimicrob Agents Chemother. 2017;61(11):1–15. | ||
MEROPENEM package insert. Schaumburg, IL: SAGENT Pharmaceuticals; 2016. | ||
Hackel MA, Lomovskaya O, Dudley MN, Karlowsky JA, Sahm DF. In vitro activity of meropenem-vaborbactam against clinical isolates of KPC-positive Enterobacteriaceae. Antimicrob Agents Chemother. 2018;62(1):1–10. | ||
Castanheira M, Huband MD, Mendes RE, Flamm RK. Meropenem-vaborbactam tested against contemporary gram-negative isolates collected worldwide during 2014, including carbapenem-resistant, KPC-producing, multidrug-resistant, and extensively drug-resistant Enterobacteriaceae. Antimicrob Agents Chemother. 2017;61(9): 5454–5458. | ||
Pai H, Kim J, Kim J, Lee JH, Choe KW, Gotoh N. Carbapenem resistance mechanisms in Pseudomonas aeruginosa clinical isolates. Antimicrob Agents Chemother. 2001;45(2):480–484. | ||
Viehman JA, Nguyen MH, Doi Y. Treatment options for carbapenem-resistant and extensively drug-resistant Acinetobacter baumannii infections. Drugs. 2014;74(12):1315–1333. | ||
Sun D, Rubio-Aparicio D, Nelson K, Dudley MN, Lomovskaya O, Selection M-VR. Resistance prevention, and molecular mechanisms in mutants of KPC-producing Klebsiella pneumoniae. Antimicrob Agents Chemother. 2017;61(12):1–14. | ||
Griffith DC, Loutit JS, Morgan EE, Durso S, Dudley MN. Phase 1 study of the safety, tolerability, and pharmacokinetics of the β-lactamase inhibitor vaborbactam (RPX7009) in healthy adult subjects. Antimicrob Agents Chemother. 2016;60(10):6326–6332. | ||
Wenzler E, Gotfried MH, Loutit JS, et al. Meropenem-RPX7009 concentrations in plasma, epithelial lining fluid, and alveolar macrophages of healthy adult subjects. Antimicrob Agents Chemother. 2015;59(12):7232–7239. | ||
Trang M, Griffith DC, Bhavnani SM, et al. Population pharmacokinetics of meropenem and vaborbactam in healthy volunteers and infected patients. In: 2nd ASM Microbe; June 1–5, 2017; New Orleans, LA, USA. | ||
Bhavnani SM, Hammel JP, Rubino CM, et al. Meropenem–vaborbactam pharmacokinetic pharmacodynamic analyses for efficacy based on data from patients enrolled in phase 3 studies. In: 2nd ASM Microbe; June 1–5, 2017; New Orleans, LA, USA. | ||
Rubino CM, Bhavnani SM, Loutit JS, Lohse B, Dudley MN, Griffith DC. Meropenem-vaborbactam: single-dose pharmacokinetics and safety in subjects with chronic renal impairment. Antimicrob Agents Chemother. 2018:e02103–17. | ||
Bhavnani SM, Trang M, Griffith DC. Meropenem–vaborbactam pharmacokinetic-pharmacodynamic (PK-PD) target attainment analyses as support for dose selection in patients with normal renal function and varying degrees of renal impairment. Open Forum Infect Dis. 2017;4(suppl_1):S530–S531. | ||
Nicolau DP. Pharmacokinetic and pharmacodynamic properties of meropenem. Clin Infect Dis. 2008;47 (Suppl 1):S32–40. | ||
Ong CT, Tessier PR, Li C, Nightingale CH, Nicolau DP. Comparative in vivo efficacy of meropenem, imipenem, and cefepime against Pseudomonas aeruginosa expressing MexA-MexB-OprM efflux pumps. Diagn Microbiol Infect Dis. 2007;57(2):153–161. | ||
Griffith D, Sabet M, Tarazi Z, Lomovskaya O, Dudley MN. Pharmacodynamics of vaborbactam when administered in combination with meropenem. In: 2nd ASM Microbe; 1-5 June, 2017; New Orleans, LA, USA. | ||
Kaye KS, Bhowmick T, Metallidis S, et al. Effect of meropenem-vaborbactam vs piperacillin-tazobactam on clinical cure or improvement and microbial eradication in complicated urinary tract infection: the TANGO I randomized clinical trial. JAMA. 2018;319(8):788–799. | ||
Wunderink R, Giamarellos-Bourboulis E, Rahav G, et al. Meropenem-vaborbactam vs. best available therapy for carbapenem-resistant Enterobacteriaceae infections in TANGO II: primary outcomes by site of infection. Open Forum Infect Dis. 2017;4(Issue suppl 10):S536–S537. | ||
Livermore DM, Warner M, Mushtaq S. Activity of MK-7655 combined with imipenem against Enterobacteriaceae and Pseudomonas aeruginosa. J Antimicrob Chemother. 2013;68(10):2286–2290. | ||
Lob SH, Hackel MA, Kazmierczak KM, et al. In vitro activity of imipenem-relebactam against gram-negative ESKAPE pathogens isolated by clinical laboratories in the United States in 2015 (results from the SMART Global Surveillance Program). Antimicrob Agents Chemother. 2017;61(6):e02209–16. | ||
Lapuebla A, Abdallah M, Olafisoye O, et al. Activity of imipenem with relebactam against gram-negative pathogens from New York City. Antimicrob Agents Chemother. 2015;59(8):5029–5031. | ||
Lucasti C, Vasile L, Sandesc D, et al. Phase 2, dose-ranging study of relebactam with imipenem-cilastatin in subjects with complicated intra-abdominal infection. Antimicrob Agents Chemother. 2016;60(10):6234–6243. | ||
Sims M, Mariyanovski V, Mcleroth P, et al. Prospective, randomized, double-blind, Phase 2 dose-ranging study comparing efficacy and safety of imipenem/cilastatin plus relebactam with imipenem/cilastatin alone in patients with complicated urinary tract infections. J Antimicrob Chemother. 2017;72(9):2616–2626. | ||
Motsch J, Stus V, Koksal I. RESTORE-IMI 1: a multicenter, randomized, double-blind, comparator-controlled trial comparing the efficacy and safety of imipenem/relebactam versus colistin plus imipenem in patients with imipenem-non-susceptible bacterial infections. In: European Congress of Clinical Microbiology and Infectious Diseases, 21-24 April, 2018; Madrid. | ||
ClinicalTrials.gov. Imipenem/relebactam/cilastatin versus piperacillin/tazobactam for treatment of participants with bacterial pneumonia (MK-7655A-014) (RESTORE-IMI 2). Available from: https://clinicaltrials.gov/ct2/show/NCT02493764. Accessed April 13, 2018. | ||
Aggen JB, Armstrong ES, Goldblum AA, et al. Synthesis and spectrum of the neoglycoside ACHN-490. Antimicrob Agents Chemother. 2010;54(11):4636–4642. | ||
Almaghrabi R, Clancy CJ, Doi Y, et al. Carbapenem-resistant Klebsiella pneumoniae strains exhibit diversity in aminoglycoside-modifying enzymes, which exert differing effects on plazomicin and other agents. Antimicrob Agents Chemother. 2014;58(8):4443–4451. | ||
Karaiskos I, Souli M, Giamarellou H. Plazomicin: an investigational therapy for the treatment of urinary tract infections. Expert Opin Investig Drugs. 2015;24(11):1501–1511. | ||
Walkty A, Adam H, Baxter M, et al. In vitro activity of plazomicin against 5,015 gram-negative and gram-positive clinical isolates obtained from patients in canadian hospitals as part of the CANWARD study, 2011-2012. Antimicrob Agents Chemother. 2014;58(5):2554–2563. | ||
Livermore DM, Mushtaq S, Warner M, et al. Activity of aminoglycosides, including ACHN-490, against carbapenem-resistant Enterobacteriaceae isolates. J Antimicrob Chemother. 2011;66(1):48–53. | ||
Endimiani A, Hujer KM, Hujer AM, et al. ACHN-490, a neoglycoside with potent in vitro activity against multidrug-resistant Klebsiella pneumoniae isolates. Antimicrob Agents Chemother. 2009;53(10):4504–4507. | ||
Zhang Y, Kashikar A, Bush K. In vitro activity of plazomicin against β-lactamase-producing carbapenem-resistant Enterobacteriaceae (CRE). J Antimicrob Chemother. 2017;72(10):2792–2795. | ||
Landman D, Babu E, Shah N, et al. Activity of a novel aminoglycoside, ACHN-490, against clinical isolates of Escherichia coli and Klebsiella pneumoniae from New York City. J Antimicrob Chemother. 2010;65(10):2123–2127. | ||
Galani I, Souli M, Daikos GL, et al. Activity of plazomicin (ACHN-490) against MDR clinical isolates of Klebsiella pneumoniae, Escherichia coli, and Enterobacter spp. from Athens, Greece. J Chemother. 2012;24(4):191–194. | ||
Cloutier DJ, Komirenko AS, Cebrik DS, et al. Plazomicin versus meropenem for complicated urinary tract infection and acute pyelonephritis: diagnosis-specific results from the Phase 3 EPIC study. Open Forum Infect Dis. 2017;4(Issue suppl 1):S532. | ||
McKinnell JA, Connolly LE, Pushkin R, et al. Improved outcomes with plazomicin compared with colistin in patients with bloodstream infections caused by carbapenem-resistant Enterobacteriaceae (CRE): results from the CARE study. Open Forum Infect Dis. 2017;4(Issue suppl 1):S531. | ||
Dobias J, Dénervaud-Tendon V, Poirel L, Nordmann P. Activity of the novel siderophore cephalosporin cefiderocol against multidrug-resistant Gram-negative pathogens. Eur J Clin Microbiol Infect Dis. 2017;36(12):2319–2327. | ||
Ito-Horiyama T, Ishii Y, Ito A, et al. Stability of novel siderophore cephalosporin S-649266 against clinically relevant carbapenemases. Antimicrob Agents Chemother. 2016;60(7):4384–4386. | ||
Hackel MA, Tsuji M, Yamano Y, Echols R, Karlowsky JA, Sahm DF. In vitro activity of the siderophore cephalosporin, cefiderocol, against a recent collection of clinically relevant gram-negative bacilli from North America and Europe, including carbapenem-nonsusceptible isolates (SIDERO-WT-2014 Study. Antimicrob Agents Chemother. 2017;61(9):e00093–17. | ||
Kohira N, West J, Ito A, et al. In vitro antimicrobial activity of a siderophore cephalosporin, S-649266, against Enterobacteriaceae clinical isolates, including carbapenem-resistant strains. Antimicrob Agents Chemother. 2016;60(2):729–734. | ||
Portsmith S, Echols R, Machida M, Juan Camillo, Arjona Ferreira, Mari Ariyasu, Tsutae Den Nagata. Cefiderocol compared with imipenem/cilastatin in the treatment of adults with complicated urinary tract infections with or without pyelonephritis or acute uncomplicated pyelonephritis: results from a multicenter, double-blind, randomized study (APEKS-cUTI). In: 27th European Congress of Clinical Microbiology and Infectious Diseases, April 2017; Vienna, Austria. | ||
ClinicalTrials.gov. Study of S-649266 or best available therapy for the treatment of severe infections caused by carbapenem-resistant Gram negative pathogens (CREDIBLE e CR). Available from: https://clinicaltrials.gov/ct2/show/NCT02714595?term=S-649266&rank=1. Accessed February 8, 2018. | ||
ClinicalTrials.gov. Clinical study of S-649266 for the treatment of nosocomial pneumonia caused by gram negative pathogens (APEKS-NP). Available from: https://clinicaltrials.gov/ct2/show/NCT03032380?term=S-649266&rank=3. Accessed February 9, 2018. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.