")
Back to Journals » Cancer Management and Research » Volume 13
Overexpression of Taspase 1 Predicts Poor Prognosis in Patients with Hepatocellular Carcinoma
Authors Jiang J , Liu B, Liu R, Yang W
Received 10 December 2020
Accepted for publication 25 February 2021
Published 16 March 2021 Volume 2021:13 Pages 2517—2537
DOI https://doi.org/10.2147/CMAR.S296069
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eileen O'Reilly
Jie Jiang,1 Bin Liu,2 Ruilin Liu,3 Wenzhuo Yang1
1Department of Gastroenterology and Hepatology, Shanghai Tongji Hospital, Tongji University School of Medicine, Shanghai, People’s Republic of China; 2Department of Gastroenterology, The Fifth Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, People’s Republic of China; 3Department of Pulmonary, Shanghai Tongji Hospital, Tongji University School of Medicine, Shanghai, People’s Republic of China
Correspondence: Ruilin Liu
Department of Pulmonary, Shanghai Tongji Hospital, Tongji University School of Medicine, 389 Xincun Road, Shanghai, 200065, People’s Republic of China
Email [email protected]
Wenzhuo Yang
Department of Gastroenterology and Hepatology, Shanghai Tongji Hospital, Tongji University School of Medicine, 389 Xincun Road, Shanghai, 200065, People’s Republic of China
Email [email protected]
Background: Taspase 1 (TASP1) is a highly conserved protease involved in site-specific proteolysis. Existing researches have revealed a link between TASP1 expression and carcinogenesis. However, limited data are available regarding the prognosis and functions of TASP1 in hepatocellular carcinoma (HCC).
Methods: Western Blotting and qRT-PCR were employed to evaluate the level of TASP1 in HCC cell lines and clinical specimens. TASP1 expression was further calculated in clinical specimens by immunohistochemistry and the mRNA level of TASP1 in HCC was analyzed using Oncomine and UALCAN databases. The TASP1 promoter methylation modification was shown via MEXPRESS and UALCAN. The association between TASP1 expression and postoperative prognosis was evaluated using Kaplan–Meier and Cox regression analysis in clinical patients. The effect of TASP1 on HCC prognosis was analyzed via Kaplan-Meier plotter, GEPIA and UALCAN. Additionally, the regulators, kinases, miRNA and transcription factor targets of TASP1 were identified using LinkedOmics. Moreover, cBioPortal was used to detect the genetic alteration of TASP1. Finally, TIMER was utilized to assess the relation between TASP1 and the immune cell infiltration, whereas the correlation of TASP1 with three immune factors was detected through TISIDB.
Results: TASP1 expression was increased in HCC cell lines and HCC tissues. CNV and DNA methylation of TASP1 were changed. Survival analysis revealed that high TASP1 expression was correlated with overall survival (OS). Functional network analysis about TASP1 in HCC showed that the double-strand break repair, peptidyl-threonine modification, spindle organization, peptidyl-lysine modification and microtubule-based movement were modulated. Furthermore, TASP1 expression revealed puissant relation to the infiltration of immune cells and three immune factors in HCC.
Conclusion: These data indicate that TASP1 may act as a potential prognostic marker in HCC and regulate HCC via multiple mechanisms.
Keywords: hepatocellular carcinoma, TASP1, prognosis, biological functional networks
Introduction
Liver cancer is one of the most common malignant tumors worldwide, and its incidence is rising especially in developing countries.1,2 Hepatocellular carcinoma (HCC), accounting for almost 90% of the primary liver cancer, is difficult to be diagnosed at an early stage and many patients have a poor prognosis due to the high likelihood of metastasis and recurrence.3,4 Though many types of treatments, including surgery, liver transplantation and radiation, are used in clinical patients, the underlying mechanisms of HCC remain unknown, leading to the difficulty for effective treatments.5 Therefore, it is greatly important to identify novel markers to diagnose HCC, which may help find new targets for prognosis and treatment.
Site-specific proteolysis regulates critical aspects of biology by carrying out selective cleavage of specific substrates.6 Site-specific proteolysis is also essential for many pathological processes, including cancers.7 However, there is limited knowledge about the correlation between proteolysis and tumor biology. Therefore, it is remarkably important to understand the underlying mechanisms of proteolysis in cancers, so as to explore better therapeutic methods for cancers.
Taspase 1 (threonine aspartase 1; TASP1), encoding a 50 kDa proenzyme, is one of the highly conserved proteases, which cleaves the substrates to modulate diverse biological processes.8 TASP1 was originally recognized as the protease to regulate homeobox (HOX) expression through cleaving mixed lineage leukemia 1 (MLL1). Except MLL1, some other TASP1 substrates, such as MLL2, transcription factor IIA (TFIIA), TFIIA-Like Factor (ALF), and host cell factor 1 (HCF-1), are identified.9–11 These substrates are broad-acting nuclear factors which regulate gene transcription levels, indicating that TASP1 plays an important role in orchestrating numerous biological processes.12 TASP1 has been shown to affect the development of glioblastoma and melanoma through modulating apoptosis and proliferation.13 It has also been reported that the expression of TASP1 is abnormal in various cancers, such as leukaemia, breast cancer, gallbladder cancer and breast cancer.13–15 In addition, though one study shows that TASP1 has been involved in the networks of Hepatocyte Nuclear Factor 4 Alpha (HNF-4α) in HCC,16 the detailed functions and mechanisms of TASP1 in HCC are still unknown.
In this study, we identified that the TASP1 expression levels were increased in HCC tissues. Additionally, Kaplan–Meier and Cox regression multivariate analyses indicated that TASP1 expression is an independent prognostic factor of overall survival (OS) in HCC patients. Furthermore, the bioinformatics approaches were also utilized to explore the expression and mutation of TASP1 in HCC via various online databases. In addition, we evaluated genomic alterations, function networks, and prognostic value of TASP1 and explored the effect of TASP1 on tumor immunity in HCC by multi-dimensional methods. Therefore, we may provide new understandings about TASP1 in HCC, and reveal a potentially diagnostic, prognostic and therapeutic target for HCC.
Materials and Methods
Tissue Samples
A total of 100 patients, who were surgically treated for HCC between October 2005 and May 2011 at the Fifth Affiliated Hospital of Zhengzhou University, were enrolled in this study. Patients, who received anticancer therapy, such as chemotherapy, radiotherapy, and other adjuvant treatments, were excluded. Patient clinical data were obtained and this study was approved by the Ethics Committee of the Fifth Affiliated Hospital of Zhengzhou University, Zhengzhou, China. All the participants signed an informed consent form before this study. And this study was conducted in accordance with the Declaration of Helsinki.
Reverse-Transcription Quantitative PCR (qPCR)
Total RNA was extracted from the HCC cell lines, HCC patients and their paired adjacent normal tissues, using Trizol (Takara). Total RNA was reversely transcribed into cDNA with the PrimeScriptTM RT Reagent Kit (Takara). qPCR was carried out using SYBR Green (Takara), and the results were normalized to GAPDH expression. The primer sequences of TASP1 are: FORWARD (5′-3′) GTTGCCAACAGACTCTTATGTG, REVERSE (5′-3′) GTATTCCATGATCTACTGCCCA. The primer sequences of GAPDH are FORWARD (5′-3′) AATGGGCAGCCGTTAGGAAA, REVERSE (5′-3′) GCGCCCAATACGACCAAATC.
Western Blot Analysis
Proteins of the liver tissue of HCC patients, paired adjacent normal tissues and cultured cells were extracted with RIPA lysis buffer (Beyotime) supplemented with a protease inhibitor (Beyotime) and were quantified by means of the BCA Protein Assay Kit (Thermo Scientific). Western blotting was performed using antibodies against TASP1 (1: 1000; #16739‐1‐AP; Proteintech), GAPDH (1: 25000; #60004‐1‐lg; Proteintech) and then with horseradish peroxidase-conjugated secondary antibodies (Thermo Fisher, USA) for 1 hr at room temperature. ChemiDoc XRS systems (Bio-Rad Laboratories, CA, United States) and the AlphaView software were used to image and analyze the Western blot.
Immunohistochemistry
Briefly, the specimens were dewaxed routinely and boiled in citrate buffer. After the endogenous peroxidase activity in the specimen was inactivated with 3% H2O2, the specimen was incubated with polyclonal antibody against human TASP1 (1: 200; #16739‐1‐AP; Proteintech) for 1 h at 37°C. After washed with PBS, the specimen was incubated with secondary antibody for 0.5 h at 37°C. The specimens were stained with diaminobenzidine (DAB) and counterstained with hematoxylin. And the film was sealed. Two pathologists were assigned to evaluate the stained slides in a double-blinded manner. The IHC staining intensity scoring criteria were as follows: 0–3 (none = 0, weak = 1, medium = 2, and strong = 3). The scoring criteria for the proportion of positive tumor cells were as follows: 0, <5%; 1, 5–25%; 2, 26–50%; 3, 51–75%; and 4, >75%.
Oncomine Analysis
The Oncomine database (www.oncomine.org) was utilized for detecting the mRNA level and copy number variation of TASP1 in different cancers, including HCC.17 We paid attention to several datasets, such as Chen liver, Guichard Liver, Roessler liver. Differences associated with p < 0.05 were regarded statistically significant.
Kaplan–Meier Survival Curve Analysis
An online survival analysis tool that Kaplan–Meier Plotter (http://kmplot.com/analysis) was utilized to assess the prognostic significance of genes in 21 different cancers containing over 10,000 samples, including 371 LIHC.18 HCC patients were categorized into two groups according to the median expression level of TASP1 (high vs low) to analyze survival rate via Kaplan–Meier Plotter. A hazard ratio (HR) with a 95% confidence interval and log-rank P-value were calculated. P-value <0.05 was considered significant.
GEPIA Analysis
The Gene Expression Profiling Interactive Analysis (GEPIA) dataset (http://gepia.cancer-pku.cn/) is an interactive web server to visualize and analyze RNA sequencing expression data of 9736 tumors and 8587 normal samples from the TCGA and GTEx projects.19 GEPIA was utilized to examine the expression of TASP1 and generate the survival rate based on the TASP1 mRNA level in HCC.
UALCAN Analysis
UALCAN (http://ualcan.path.uab.edu) draws on TCGA level 3 RNA-seq and clinical data from 31 cancer types to analyze the gene expression data in depth.20 UALCAN was utilized to detect the relative expression and methylation of TASP1 across HCC tissues and normal tissues, as well as in various sub-groups defined according to cancer age, race, gender, weight, disease stage, tumor grade, nodal metastasis and TP53 mutation. P < 0.05 was regarded statistically significant.
MEXPRESS Analysis
MEXPRESS (https://mexpress.be/) is a web tool for integrating and visualizing the expression, DNA methylation and clinical data, as well as the relationships between them.21 In this study, MEXPRESS stool was utilized for detecting the relationship between the TASP1 expression and methylation status in HCC.
LinkedOmics Analysis
The LinkedOmics database (http://www.linkedomics.org/login.php) is a web-based platform for analyzing 32 TCGA cancer-associated multi-dimensional datasets.22 The LinkFinder module of LinkedOmics was used to study genes that were expressed differentially in correlation with LIHC via Pearson’s correlation coefficient. The LinkFinder was also utilized to create plots for individual genes. And the results were represented in the form of volcano plots, heatmaps, or scatter plots. The LinkInterpreter, another module of LinkedOmics, was drawn on to conduct pathway and network analyses. The comprehensive functional category database in the Web-based Gene Set AnaLysis Toolkit (WebGestalt) was applied. Gene set enrichment analysis (GSEA) was used to perform analysis of GO (CC, BP and MF), KEGG pathways, kinase-target enrichment, miRNA-target enrichment and transcription factor-target enrichment based on the data from the LinkFinder results. The rank criterion was an FDR <0.05, and 500 stimulations were conducted.
cBioPortal Analysis
The cBio Cancer Genomics Portal (cBioPortal) (http://cbioportal.org) is an open-access resource characterized by the multidimensional cancer genomics data sets, containing 225 cancer studies.23 The TASP1 alterations in TCGA LIHC, were analyzed using cBioPortal, and the search parameters consisted of mutation, copy number variation (CNV) and mRNA expression. In addition, the tab OncoPrint represents an overview of genetic alterations per sample in TASP1.
String Analysis
The Search Tool for the Retrieval of Interacting Genes (STRING) (https://string-db.org) was applied to perform the protein–protein interaction (PPI) network.24 As for the combined score >0.4 for PPI pairs in our study, we further used built Cytoscape to build a PPI network model and to identify hub genes.
TIMER Analysis
The TIMER dataset (https://cistrome.shinyapps.io/timer/), a comprehensive web server, is used to analyzing the tumor-infiltrating immune cells (TILCs) by gene expression profiling in 32 cancer types from over 10,000 samples.25 The TIMER database was employed to investigate the TASP1 expression and its relationship with the abundance of TILCs, including B cells, CD8+ T cells, CD4+ T cells, macrophages, neutrophils and dendritic cells, through the “gene” module.
TISIDB Analysis
The mRNA expression of TASP1 and its correlation with immunomodulators and chemokines were analyzed using TISIDB database (http://cis.hku.hk/TISIDB/index.php), which is a web portal for integrating genes of tumor and immune system interactions from seven public databases.26
Statistical Analysis
The Chi-square test was used to evaluate the correlations between TASP1 expression and clinicopathological features. The Kaplan–Meier method and Log rank test were used to analyze survival curves. The Cox repression analyses were utilized to assess the risk of death associated with clinically related factors. P < 0.05 was considered statistically significant. All Statistical analyses were performed using SPSS 20.0software package.
Results
Clinical Characteristics of HCC Patients
A total of 100 patients with HCC were enrolled in this study, including 54 males and 46 females; the median age of HCC patients was 63 (55–69) years. In respect of their disease features, 49 patients were AJCC stage I + II and 51 patients were AJCC stage III + IV; 72 patients were T stage I + II and 28 patients were T stage III + IV; 75 patients were pathologic differentiation moderate + well and 25 patients were pathologic differentiation poor; 68 patients had single tumor and 32 patients had multiple tumor; there were 64 patients with lymph node metastasis, and 36 patients without lymph node metastasis; there were 43 patients with vascular invasion, and 57 patients without vascular invasion; the largest tumor size of 56 patients was less than 5.0 cm, while that of 44 patients was equal to or greater than 5.0 cm. And the median levels of TBIL, Creatinine, AFP, INR and MELD were 21.38 (0.76–1.89) mg/dL, 0.97 (0.8–1.20) mg/dL, 52.5 (23.75–126.25) ng/mL, 1.21 (1.03–1.91) and 10 (8–14), respectively. The detailed clinicopathological characteristic data of HCC patients were shown in Table 1.
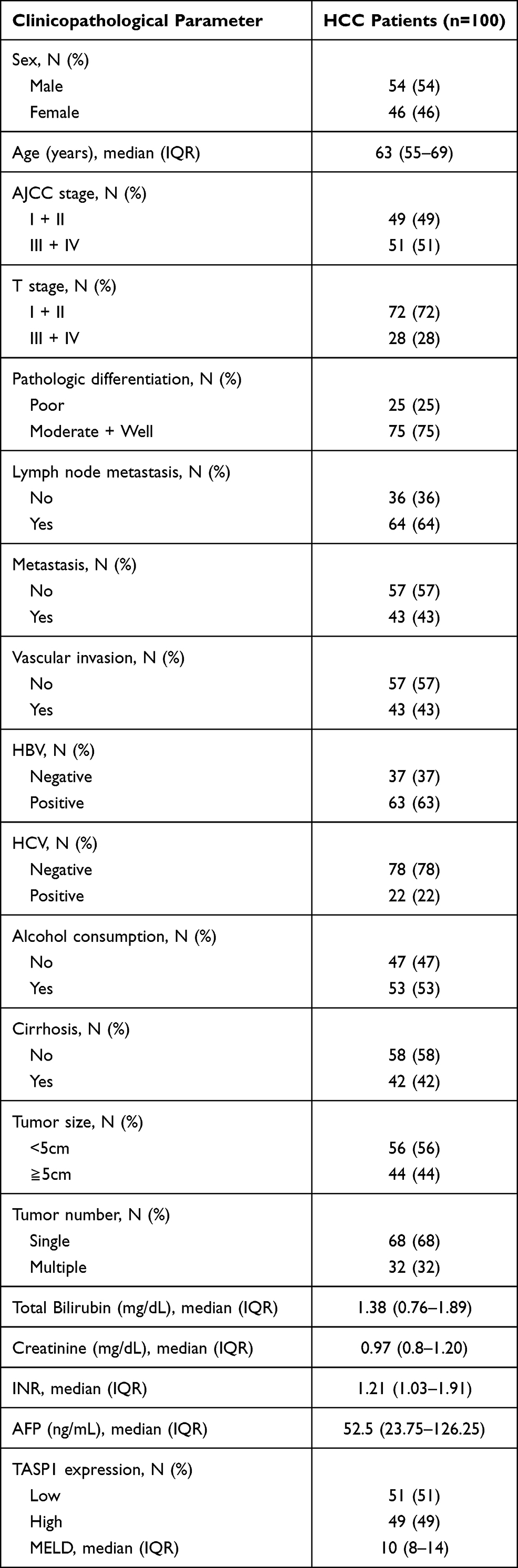 |
Table 1 Clinicopathological Characteristics of HCC Patients |
TASP1 Was Overexpressed in HCC Tissues and Correlated with Poor Prognosis of HCC Patients
Firstly, we used qRT-PCR and Western blotting to detect the mRNA and protein expression of TASP1 in HCC cell lines and normal liver cell line. The results showed that the mRNA and protein expression of TASP1 in HCC cell lines including SMMC7721, Huh7, HepG2, LM3 and Hep3B, was higher than that in the normal liver cell line LO2 (p < 0.05) (Figure 1A–C). Next, we examined TASP1 mRNA and protein expression in tumor and adjacent tissues, finding that TASP1 expression level was significantly higher in tumor tissues compared with adjacent tissues (p < 0.05) (Figure 1D–F).
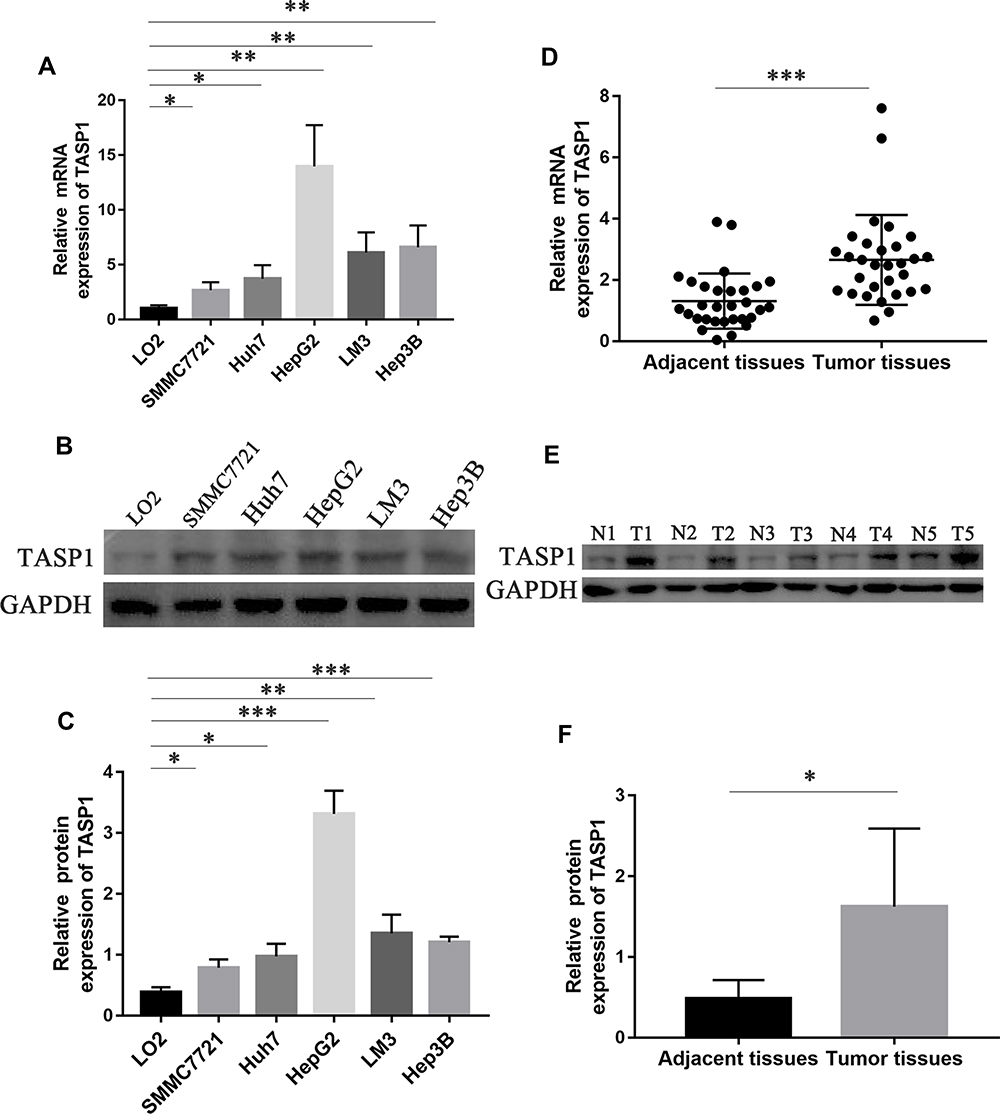 |
Figure 1 The mRNA and protein expression of TASP1 in HCC cell lines, HCC tissues and the adjacent tissues. Abbreviations: T, tumor tissue; N, adjacent tissue. Notes: (A) Relative mRNA expression of TASP1 in five HCC cell lines and the normal human liver cell line LO2. (B and C) TASP1 protein expression in five HCC cell lines and the normal human liver cell line LO2. (D) Relative mRNA expression of TASP1 in HCC tissues and the adjacent tissues. (E and F) TASP1 protein expression in HCC tissues and the adjacent tissues. *P<0.05, **P<0.01, ***P<0.01. |
Since the expression of TASP1 was different between HCC and normal tissues, we further performed immunohistochemistry to detect TASP1 protein expression in tumor and adjacent tissues. Results revealed that the expression of TASP1 in the tumor tissue was higher than that in the adjacent tissue (Figure 2A–D). In our clinical samples, we also found that upregulation of TASP1 expression was associated with pathologic differentiation (p= 0.028) and levels of AFP (p < 0.001) (Table 2). However, TASP1 expression was not correlated with sex, age, AJCC stage, T stage, lymph node metastasis, metastasis, vascular invasion, HBV, HCV, alcohol consumption, cirrhosis, tumor size, tumor number (Table 2). Then, we evaluated the prognostic value of TASP1 expression in HCC patients. Kaplan–Meier survival analysis showed that HCC patients with high TASP1 expression in our experiment had a poor prognosis (Figure 2E). These results collectively show that TASP1 was upregulated in HCC tumor tissues and was clinically significant.
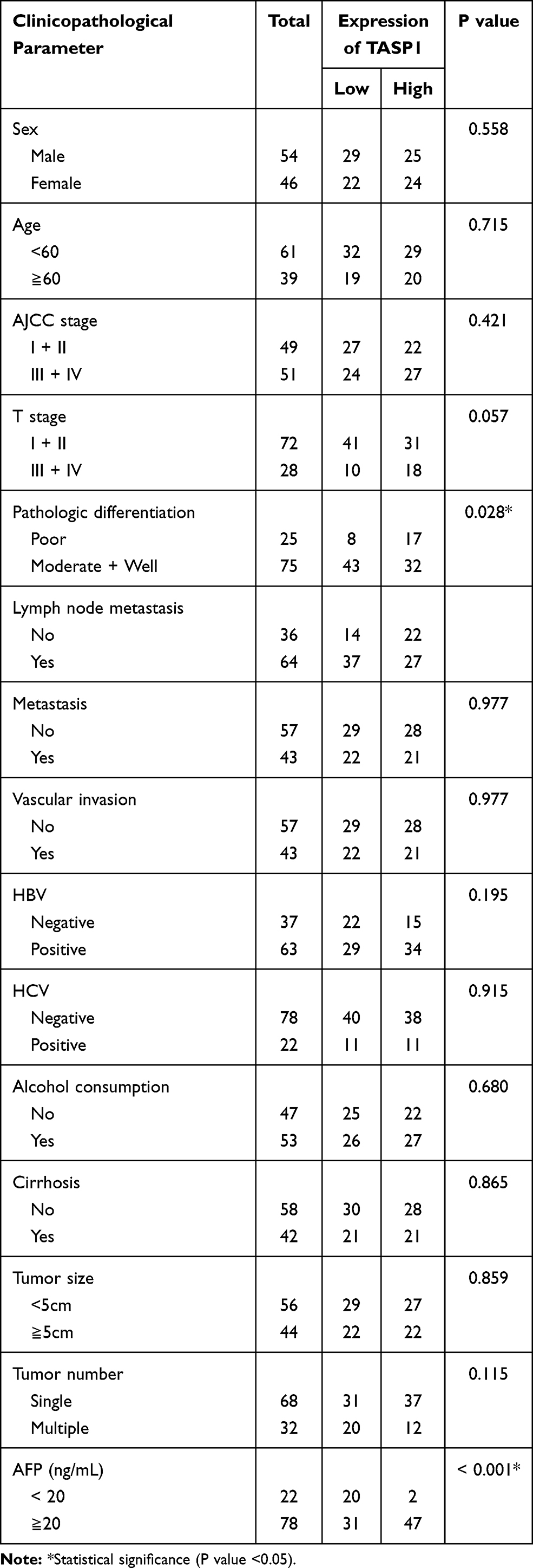 |
Table 2 Association Between TASP1 Expression and Clinicopathological Characteristics of Patients |
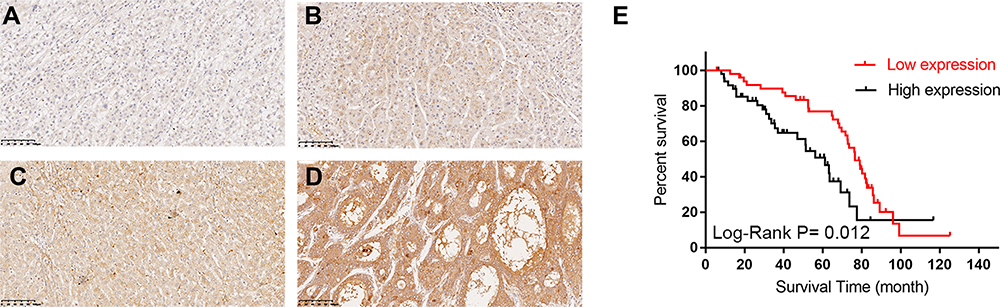 |
Figure 2 The Immunohistochemical staining of TASP1 expression in HCC tissues and survival curve. Notes: (A–D) TASP1 expression by immunohistochemical staining; (A) negative expression of TASP1 in tumor cells of HCC; (B) weak TASP1 positive staining in tumor cells of HCC; (C) moderate TASP1 positive staining in tumor cells of HCC; (D) strong TASP1 positive staining in tumor cells of HCC; (E) Kaplan–Meier analysis of overall survival (OS) with variable TASP1 expression in HCC patients. The red curve represents TASP1 low expression group, and the back curve represents TASP1 high expression group. |
TASP1 Was an Independent Risk Factor for Prognosis in Clinical HCC Patients
To test whether TASP1 performed a clinically independent prognostic value in HCC patients, we used univariate and multivariate analysis to analyzed independent prognostic factors, including TASP1, for survival in patients with HCC. The univariate analysis results (Table 3) showed that age (HR =1.936, 95% CI [1.146–3.271]; p = 0.014), pathologic differentiation (HR = 0.332, 95% CI [0.182–0.605]; p < 0.001), and TASP1 expression (HR = 2.025, 95% CI [1.169–3.508]; p = 0.012) were significantly associated with the OS of HCC patients. Multivariate survival analysis (Table 3) showed that TASP1 expression was statistically significant predictor of OS (HR = 1.832, 95% CI [1.028–3.267]; p = 0.040) and that age (HR = 2.032, 95% CI [1.187–3.477]; p = 0.010), and pathologic differentiation (HR = 0.387, 95% CI [0.209–0.717]; p = 0.003) were independent predictive factors for OS. In summary, TASP1 expression may be an independent prognostic factor in HCC patients.
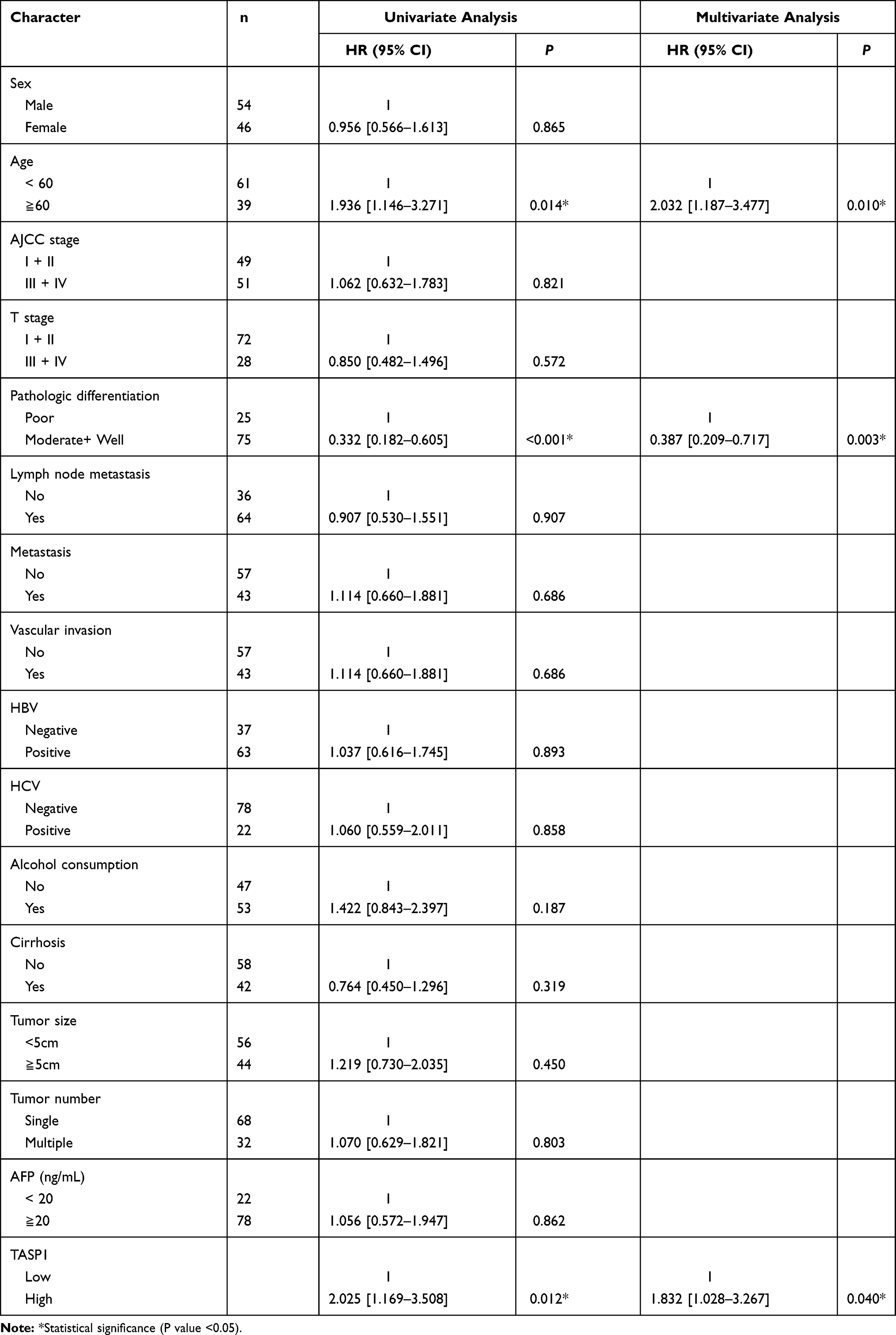 |
Table 3 Univariate and Multivariate Analysis of Various Prognostic Parameters in Patients with HCC |
The Expression of TASP1 in HCC and Other Cancers in Public Databases
To clarify the role of TASP1 in HCC, we performed bioinformatics analysis to detect the transcription level of TASP1 both in HCC tissues and normal tissues using Oncomine database. We found that the mRNA level of TASP1 was significantly higher in tumor tissues of HCC than that in normal tissues (Figure 3A–C). Aligned with the result, we also confirmed that the high expression of TASP1 in tumor tissues via UALCAN database (Figure 4A). Additionally, we partially verified the relationship between TASP1 and clinicopathological parameters by conducting a comprehensive analysis of the expression of TASP1 in HCC via UALCAN and the results showed that TASP1 mRNA expression in HCC samples was higher than that in normal samples based on age, race, gender, weight, disease stage, tumor grade, nodal metastasis and TP53 mutation (Figure 4B–I). In addition, we focused on the difference of DNA levels of TASP1, including copy number variation (CNV) and methylation. The results from Guichard livers in Oncomine database represented that TASP1 CNV in HCC tissues was higher compared with that in normal tissues (Figure 3D–F). Furthermore, we identified that TASP1 promoter methylation in tumor tissues was lower compared to that in normal tissues using UALCAN (Figure 4J). Similar results were found in MEXPRESS (Figure 5A). Interestingly, the expression of TASP1 was positively related with DNA methyltransferase (DNMT) expression (Figure 5B–E). Taken together, TASP1 may act as a potential diagnostic biomarker for HCC.
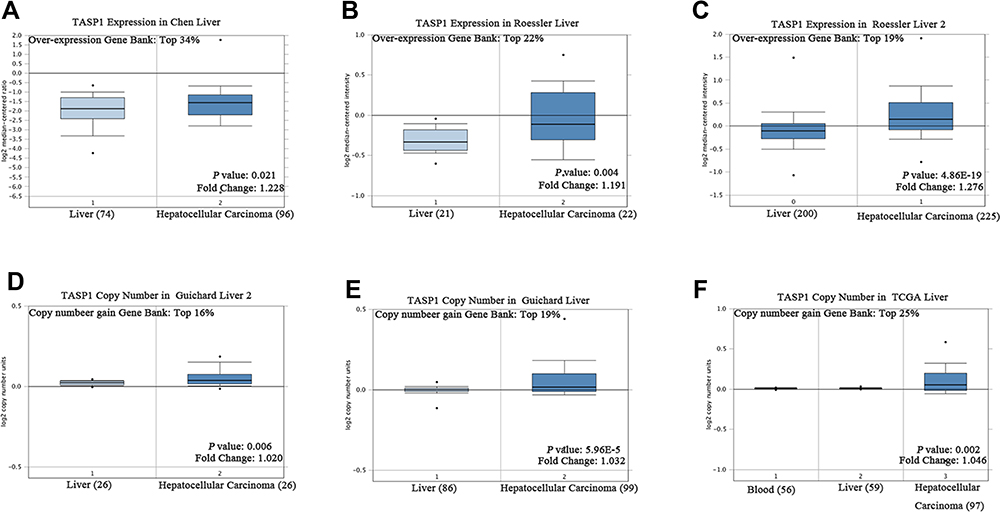 |
Figure 3 TASP1 transcription in hepatocellular carcinoma (Oncomine). Notes: (A–C) Box plot showing TASP1 mRNA levels in, respectively, the Chen liver, Guichard Liver, Roessler liver datasets. (D–F) Box plot showing TASP1 copy number in The Cancer Genome Atlas (TCGA) Liver and Guichard Liver and Guichard Liver 2 datasets, respectively. |
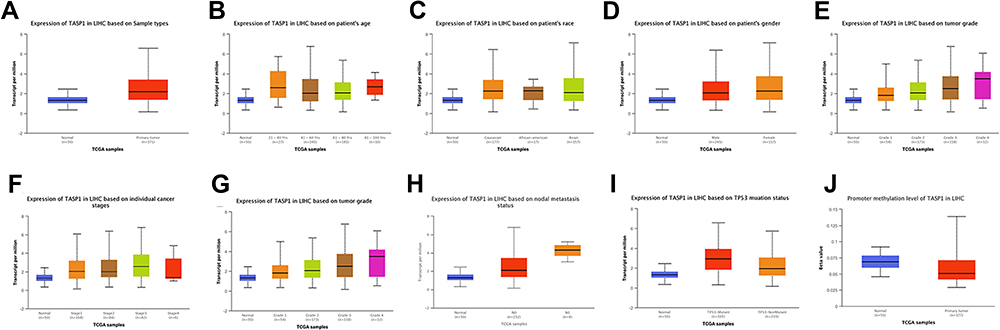 |
Figure 4 TASP1 expression in hepatocellular carcinoma (HCC) (UALCAN). Notes: (A) Boxplot showing the relative expression of TASP1 in normal and LIHC samples. (B) Boxplot showing relative expression of TASP1 in normal individuals of any ethnicity or in LIHC patients of Caucasian, African-American or Asian ethnicity. (C) Boxplot showing relative expression of TASP1 in normal individuals of either gender or male or female LIHC patients. (D) Boxplot showing relative expression of TASP1 in normal individuals of any age or in LIHC patients aged 21–40, 41–60, 61–80, or 81–100 yr. (E) Boxplot showing relative expression of TASP1 in normal individuals or in LIHC patients in stages 1, 2, 3 or 4. (F) Boxplot showing relative expression of TASP1 in normal individuals or LIHC patients with grade 1, 2, 3 or 4 tumors. (G) Boxplot showing relative expression of TASP1 in normal individuals or in LIHC patients in normal weight, extreme weight, obese, or extremely obese. (H) Boxplot showing relative expression of TASP1 in normal individuals or in LIHC patients with or without nodal metastasis. (I) Boxplot showing relative expression of TASP1 in normal individuals or in LIHC patients with or without TP53 mutation. (J) Boxplot showing the promoter methylation level of TASP1 in normal individuals or in LIHC patients. |
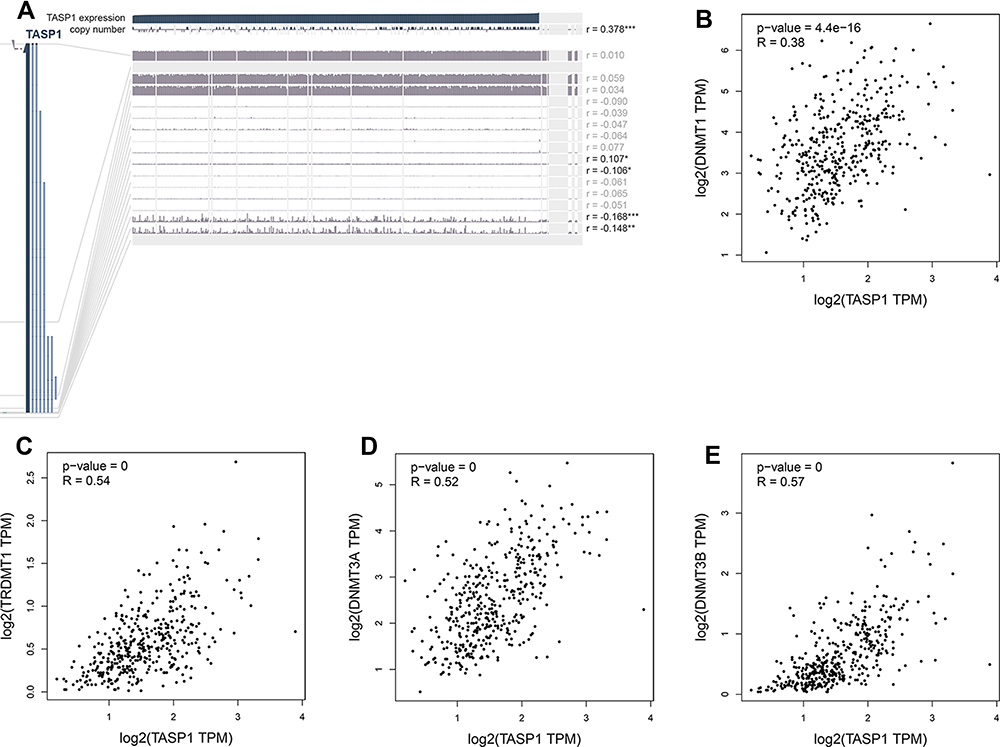 |
Figure 5 The methylation modification of TASP1 promoter in HCC. Notes: (A) TASP1 DNA methylation level in HCC via MEXPRESS. (B-E) The relationship between TASP1 transcription level and DNA methyltransferase (DNMT) expression via GEPIA.. |
Prognostic Significance of TASP1 Expression in HCC Patients in Public Databases
In cancers, the survival rate of patients is significantly correlated with the abnormal expression of genes. Then we used Kaplan–Meier plotter to examine the prognostic significance of TASP1 expression in HCC, finding that HCC patients with high TASP1 expression had a shorter overall survival, but not a disease-free survival, than those with low TASP1 expression (Figure 6A and B). Furthermore, we verified the results using GEPIA database (Figure 6C and D). Similarly, the result that patients with high TAPS1 expression had poor survival was also validated (Figure S1). These results indicated that TASP1 was tightly correlated with the prognosis of HCC.
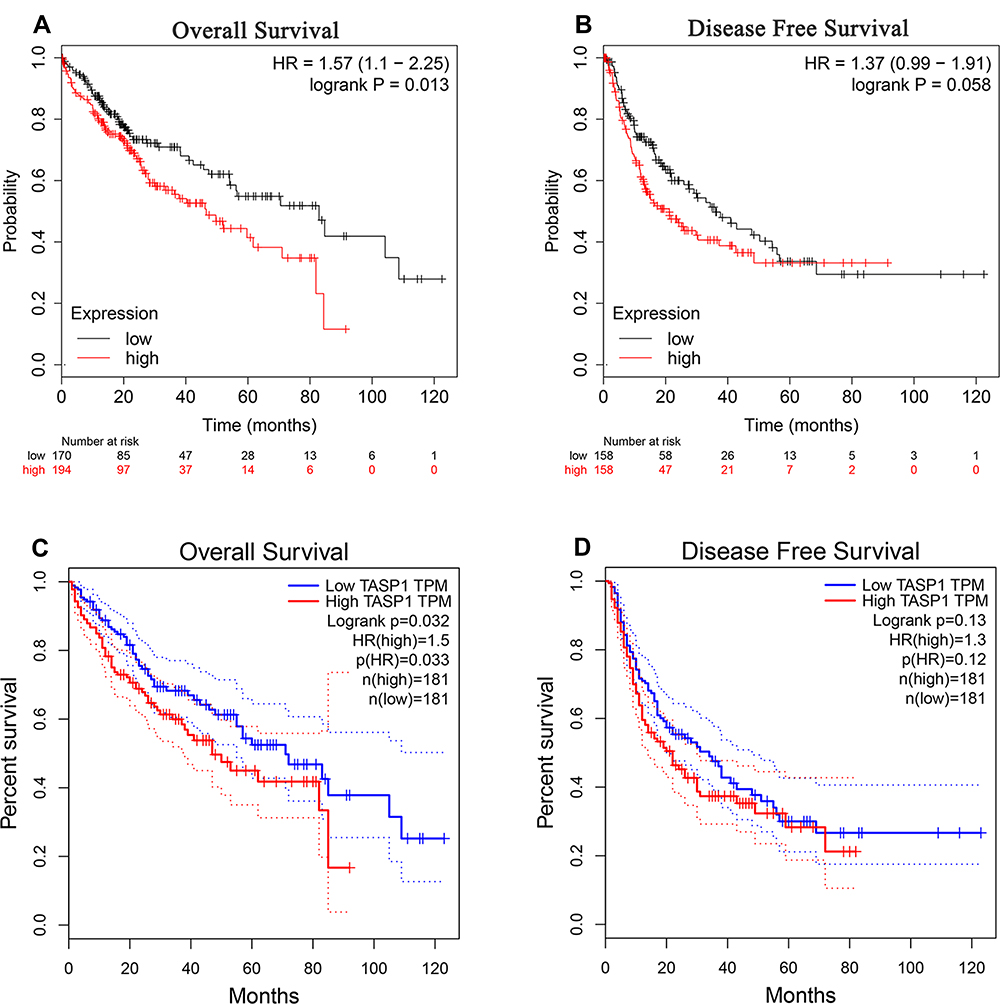 |
Figure 6 The survival outcome of TASP1 in HCC. Notes: (A) Overall survival (OS) (B) disease-free survival (DFS) of TASP1 in Kaplan–Meier Plotter database. (C) OS (D) DFS of TASP1 in GEPIA. |
Genomic Alterations of TASP1 in HCC
The alternation of TASP1 was analyzed using cBioPortal. TASP1 was altered in 22 of 367 (6%) HCC patients and the mutation, copy-number variations, and mRNA expression were the main parameters of TASP1 alternation (Figure 7A). Additionally, the expression of TASP1 in gain group was higher than that in the diploid group (Figure 7B). Moreover, we detected the frequency distribution of TASP1 CNV patients in different grade and stage groups (Figure 7C), and the results indicated that TASP1 CNV alteration had a strong correlation with the high occurrence and an early-event.
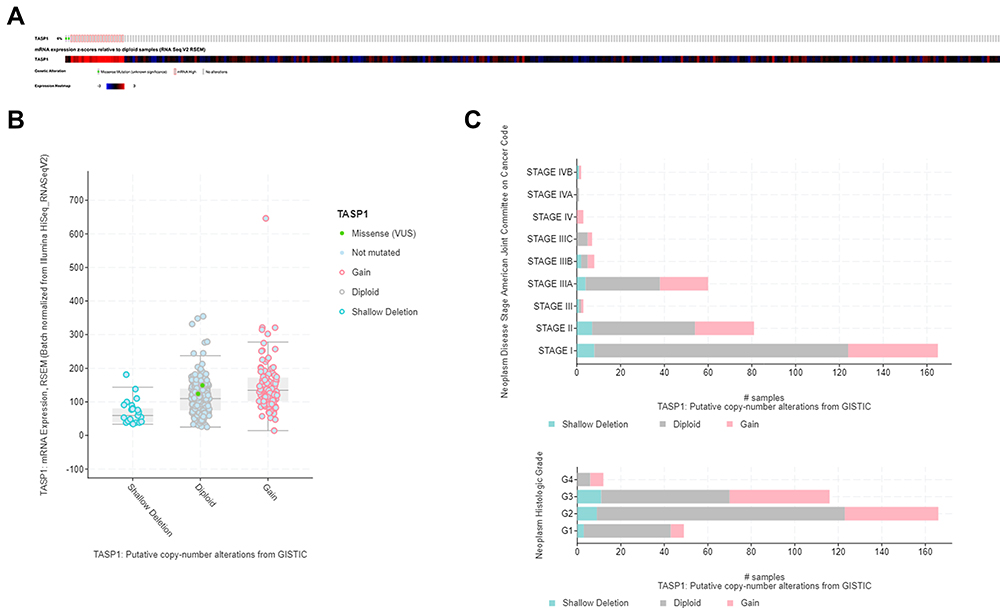 |
Figure 7 Visual summary of TASP1 alterations in HCC (cBioPortal). Notes: (A) OncoPrint of TASP1 alterations in LIHC cohort. (B) Boxplot showing the copy-number variation (CNV) of TASP1 in HCC by GISTIC. (C) The frequency of TASP1 CNV in subgroups such as stage and grade. |
The Biological Function of Co-Expression Genes Related with TASP1 in HCC
In order to explore the biological meaning of TASP1 in HCC and the correlation between TASP1 and TASP1-related genes, we used the function module of LinkedOmics to investigate the mRNA sequencing data of 371 LIHC patients in TCGA project. The volcano plot showed the correlated genes of TASP1 including 7549 positive genes and 2941 negative genes (Figure 8A). In addition, the results represented the top 50 significant genes positively and negatively correlated with TASP1 in the heatmap (Figure 8B and C). Furthermore, the all details of co-expressed genes were in Table S1. Thus, the result indicated that TASP1 played an important role in the transcriptome. TASP1 was positively correlated with top three genes, including ESF1 (r = 0.5959, p = 4.415E-36), CSNK2A1 (r = 0.5911, p = 2.2E-35), and XRN2 (r = 0. 5888, p = 4.676E-35) (Figure 8D–F), while TASP1 expression showed a negative association with expression of BLOC1S1 (r =-0.5188, p = 2.908E-26), RILP (r = -0.4794, p = 3.863E-22), and UQCRQ (r = -0.4773, p = 6.173E-22) (Figure 8G–I). These genes were correlated with TASP1 reflect changes in cell adhesion, proliferation, transcription and cell cycle. Additionally, the expression of top genes also affected the overall survival on Kaplan–Meier plotter (Figure S2). Furthermore, GO term analysis of GSEA showed that genes differentially expressed correlated with TASP1 were located mainly in double-strand break repair, peptidyl-threonine modification, spindle organization, peptidyl-lysine modification and microtubule-based movement (Figure 9A–C and Tables S2–4). And the KEGG pathway was enriched in Fanconi anemia pathway, TGF-β pathway, Thyroid hormone signaling pathway, Hippo signaling pathway, et al (Figure 9D and Table S5).
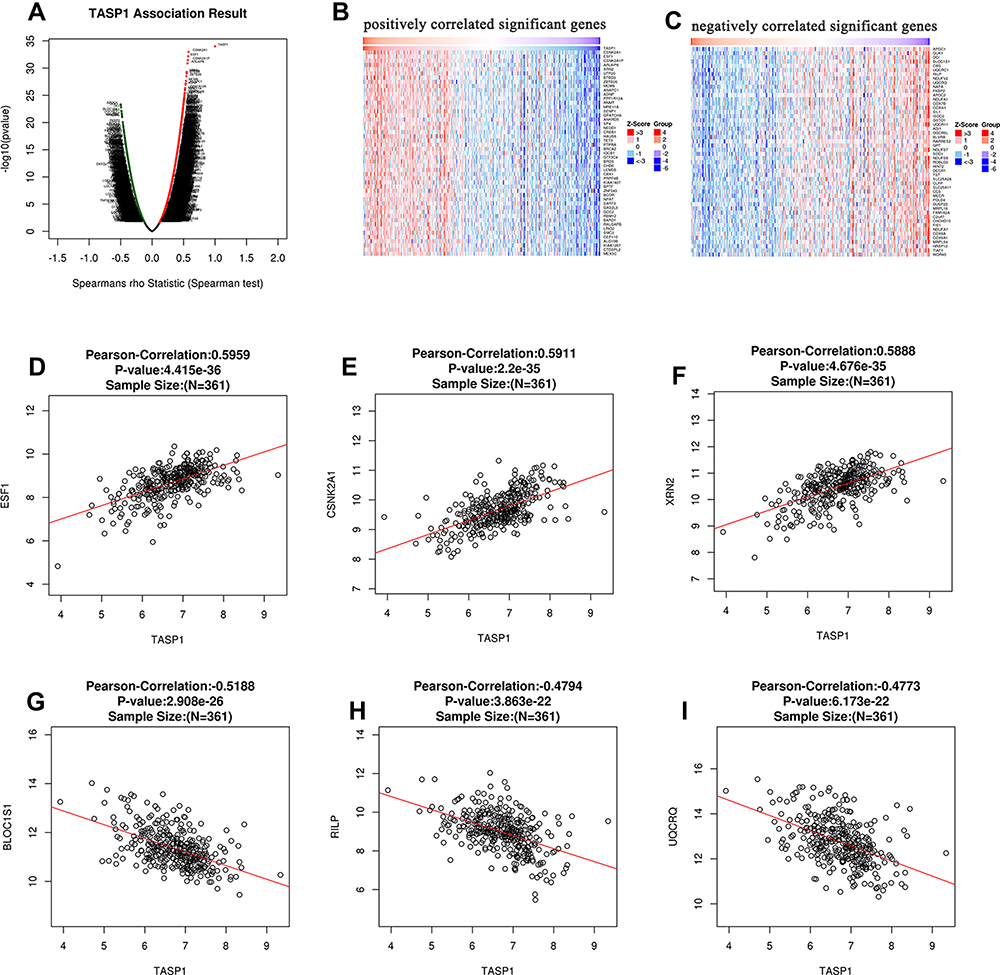 |
Figure 8 The co-expression genes of TASP1 in HCC. Notes: (A) A person test was used to explore correlations between TASP1 and genes differentially expressed in HCC. (B and C) The top 50 genes were positively or negatively correlated with TASP1 in HCC were shown in heat maps. (D–F) The positive correlation of TASP1 with the top 3 genes (SF1, CSNK2A1, XRN2). (G–I) The negative correlation of TASP1 with the top 3 genes (BLOC1S1, RILP, UQCRQ). |
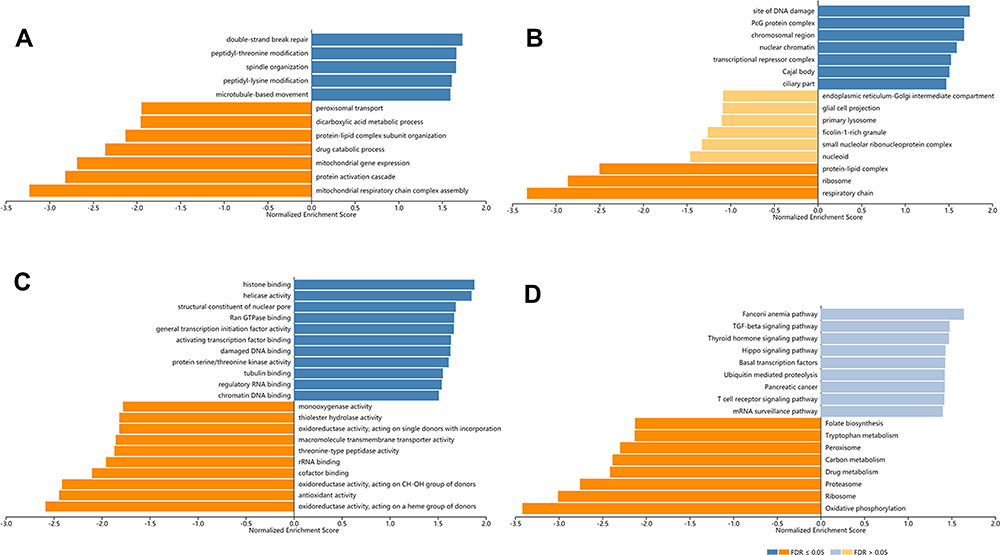 |
Figure 9 Biological function of TASP1 correlated genes in HCC (LinkedOmics). Notes: GO annotations and KEGG pathways by GSEA were performed to clarify the biological function of TASP1 correlated genes in HCC. (A) Cellular components. (B) Biological processes. (C) Molecular functions. (D) KEGG pathway analysis. |
TASP1 Networks of Kinases, miRNAs, Transcription Factor Targets in HCC
To further explore the regulators of TASP1 in HCC, we analyzed the kinases, miRNAs, and transcription factors using the GSEA module in LinkedOmics. The top 5 significant kinase targets of TASP1 were ATM serine/threonine kinase (ATM), ATR serine/threonine kinase (ATR), cyclin dependent kinase 1 (CDK1), polo like kinase 1 (PLK1) and checkpoint kinase 1 (CHEK1) (Table 4 and Table S6). Interestingly, the expression levels of these kinases, were higher in HCC tissues than those in normal tissues via UALCAN (Figure S3), and except ATM, they were also related with the OS of HCC via Kaplan-Meier plotter (Figure S4). The top 5 correlated miRNA-target networks were (ATAGGAA) MIR-202, (ATGTACA) MIR-493, (ATAAGCT) MIR-21, (ATAACCT) MIR-154, and (TTGGAGA) MIR-515-5P, MIR-519E (Table 4 and Table S7). And the top 5 enrichments of transcript factors were TAANNYSGCG_UNKNOWN, V$E2F1_Q6, V$E2F1DP1_01, V$E2F1DP2_01 and V$E2F4DP2_01 (Table 4 and Table S8). In addition, Gustavo et al study found that E2F-driven transcriptional program played a critical role in the development and progression of HCC. Therefore, TASP1 may affect the development and progression of HCC by modulating these kinases, microRNAs and transcript factors.
 |
Table 4 The Kinase, miRNA and Transcription Factor-Target Networks of TASP1 in Hepatocellular Carcinoma (LinkedOmics) |
Networks Analysis of TASP1 with Interacted Genes
We performed PPI network to identify the interactions between TASP1 and co-expressed genes in HCC using STRING database. The top 20 genes evaluated via connectivity degrees from the PPI network through Cytoscape were selected as hub genes, including KMT2A, WDR5, MEN1, PSIP1, KMT2B, MLLT3, RBBP5, MLLT1 and AFF1. The results indicated that these top genes with TASP1 were mainly enriched in transcriptional misregulation in cancers (Figure 10).
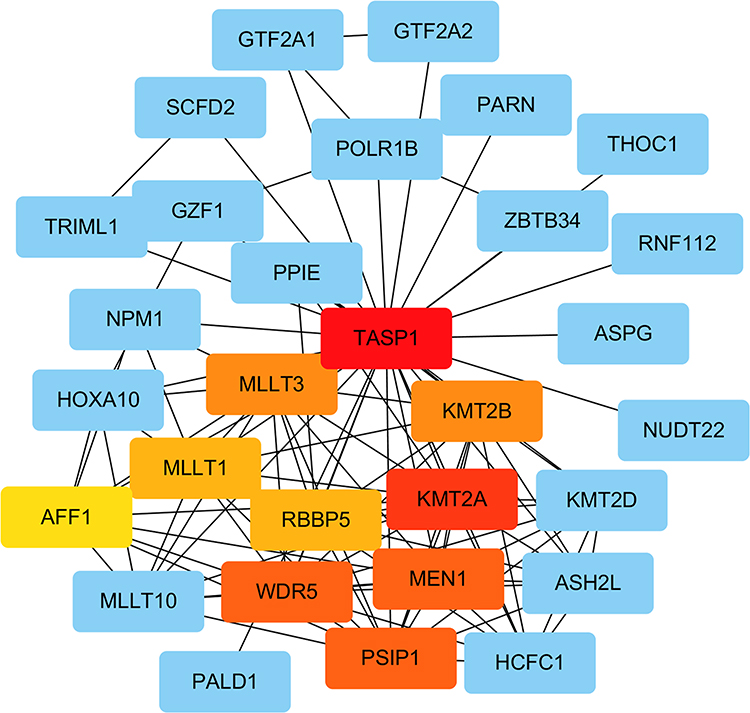 |
Figure 10 Analysis of TASP1 related proteins based on PPI network (STRING). Note: On line tool of STRING database analysis of the PPI network for the differentially expressed genes. |
Correlations Between TASP1 Expression and Immune Infiltrations in HCC
Growing studies showed that lymphocyte infiltration plays a key role in cancers, acting as an independent predictive factor for overall survival. Therefore, we conducted a comprehensive analysis to reveal the correlation between TASP1 expression and immune infiltration levels in HCC based on TIMER. The most six frequently immune infiltrating cells, including CD4 + T cell, CD8 + T cell, neutrophil, macrophage, B cell and Dendritic cell, were selected and we found that the expression of TASP1 had a correlation with tumor purity (Cor = 0.099, p = 6.6E-02) in HCC. Additionally, the mRNA level of TASP1 had a remarkably positively correlation with the infiltration of B cell (Cor = 0.287, p = 6.31E-08), CD8 + T cell (Cor = 0.251, p =2.65E-06), CD4 + T cell (Cor = 0.406, p = 4.76E-15), macrophage (Cor = 0.409, p = 3.38E-15), neutrophil (Cor = 0.446, p = 2.69E-18) and dendritic cell (Cor = 0.407, p = 5.27E-15) (Figure 11). These findings suggested that TASP1 may play an important role in the tumor microenvironment by affecting the infiltration of immune cells. In addition, the top 3 positive or negative genes of TASP1 showed the significant correlations with varying degrees with immune cells (Figure S5).
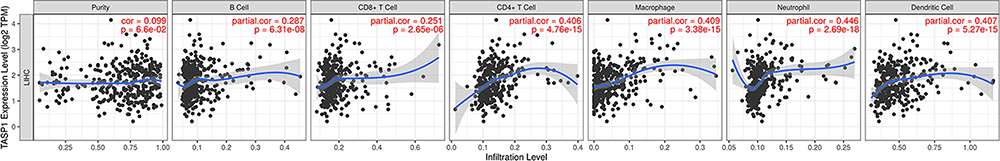 |
Figure 11 Correlation of TASP1 expression with immune cell infiltration in HCC (TIMER). Notes: TASP1 is related with tumor purity and TASP1 expression has positive correlations with the infiltrating levels of B cells, CD8 + T cells, CD4 + T cells, macrophages, neutrophils and dendritic cells. |
The Correlations of TASP1 Expression with Immune Factors
Some studies have indicated that chemokines and their receptor families are important mediators for recruiting immune cells, thus providing an ideal microenvironment for tumor biology. Moreover, different chemokines have different roles in cancers: some chemokines stimulate the development and progression of cancers, whereas some chemokines show opposite functions. Thus, TISIDB database was utilized for analyzing the correlation between TASP1 expression and chemokines. The heatmap of chemokines in various cancers suggested that TASP1 acted a key role in chemokines in cancers and we analyzed the correlation among them after filtering for |±rho| ≥ 0.1 and p < 0.05. In this study, the results indicated that TASP1 had a strong negatively correlation with CCL2, CCL5, CCL8, CCL11, CCL13, CCL14, CCL15, CCL16, CCL19, CCL21, CXCL2, CXCL16 and XCL2, and the relationship between TASP1 and CCL20 was positive (Figure 12A). The results showed that TASP1 may be involved in regulating the tumor immunology via chemokines.
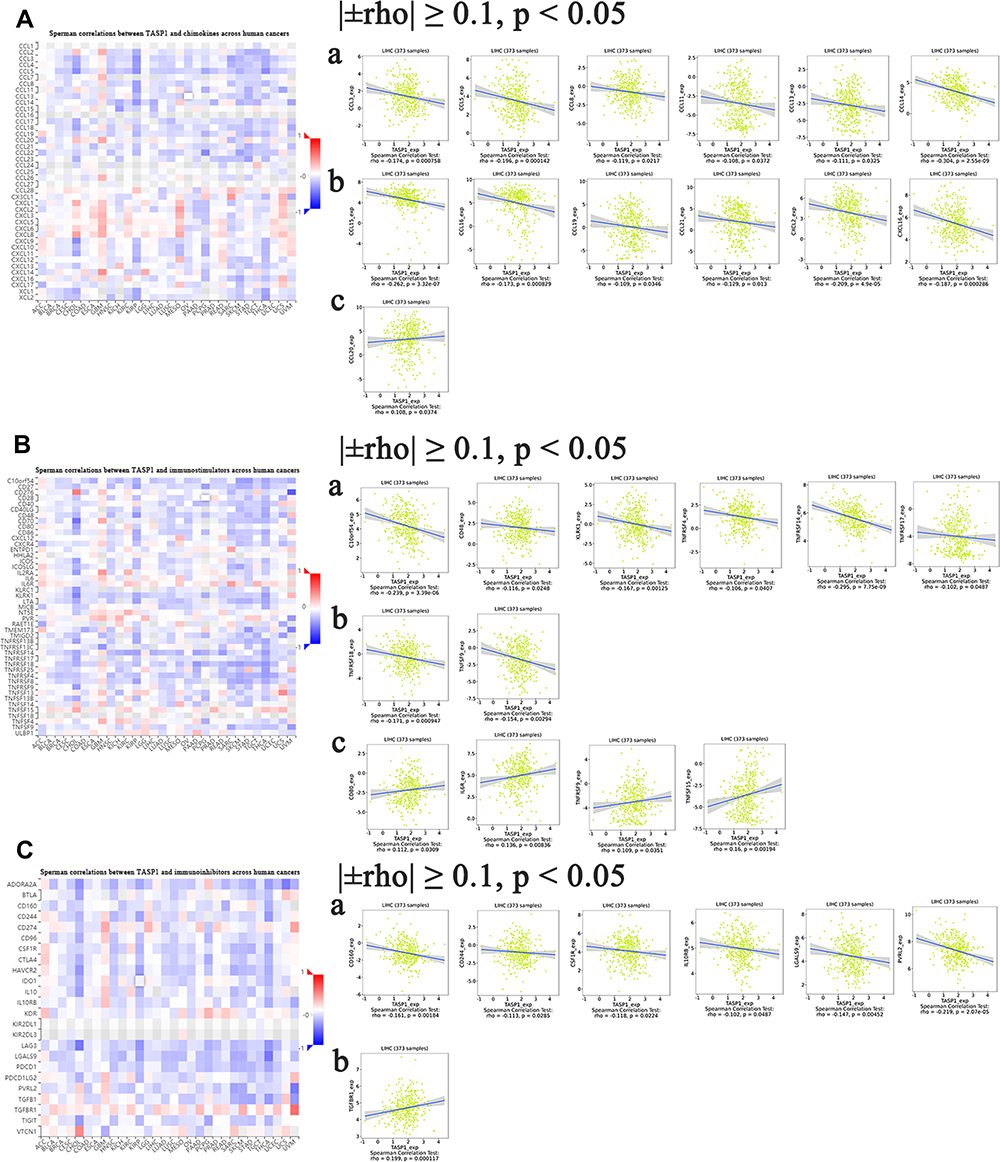 |
Figure 12 Correlation analyses between dysregulated TASP1 and cancer-related immune factor in HCC (TISIDB). Notes: (A) The association between TASP1 and chemokines was assessed in HCC. (B) The association between TASP1 and immunostimulators was assessed in HCC. (C) The association between TASP1 and immunoinhibitors was assessed in HCC. |
Except for chemokines, immune checkpoints, also known as coinhibitory molecules, modulate the tumor microenvironment and help tumor cells evade immune surveillance in many studies. Furthermore, antibodies targeting coinhibitory molecules have become popular and promising. To determine whether TASP1 may play a role in cancers via immune checkpoints, we investigated the relationship between TASP1 expression and immune checkpoints through TISIDB database. Surprisingly, as shown in the heatmaps, TASP1 was significantly correlated with immune checkpoint genes including immunoinhibitory genes and immunostimulatory genes. We found that TASP1 had a negative correlation with C10orf5, CD48, KLRK1, TNFRSF4, TNFRSF14, TNFRSF17, TNFRSF18 and TNFSF9, and a positive correlation with CD80, IL6R, TNFRSF9 and TNFSF15 of immunostimulants (Figure 12B); In addition, TASP1 was negatively related with CD160, CD244, CSF1R, IL10RB, LGALS9 and PVRL2, and positively related with TGFBR1 of immunoinhibitors (Figure 12C). These findings revealed that TASP1 may affect tumor biology via the regulation of immune checkpoints.
Discussion
Due to a difficulty in detecting HCC at an early stage, HCC, one of the most common form of cancers, is a big threat to human health. Because the molecular and cellular heterogeneity in HCC is complex, the underlying mechanisms of HCC are still unknown. Site-specific proteolysis has been reported to play an important role in regulating numerous physiological and pathological conditions caspases activation and cancers.27–29 TASP1, a highly conserved protease, mediates proteolytic cleavage occurs, then leading to the development and progression of various cancers.8,13 However, the role of TASP1 in HCC has not been understood well. To better understand the potential functions and the regulatory network of TASP1 in HCC, we collect HCC cells lines, clinical specimens and conduct bioinformatics analysis to provide novel insights for further research.
In this study, we examined the mRNA and protein expression of TASP1 in HCC cell lines and normal liver cell line, finding that the mRNA and protein expression of TASP1 in HCC cell lines was higher than that in the normal liver cell line. And based on clinical samples and public databases, the expression levels of TASP1 in HCC tissues were significantly higher than those in normal liver tissues. And high TASP1 expression was strongly correlated with pathologic differentiation in our clinical patients. Additionally, the mRNA levels of TASP1 in HCC were strongly correlated with clinic pathological features under subgroups, including age, race, gender, weight, disease stage, tumor grade, nodal metastasis and TP53 mutation from public databases. Furthermore, previous studies have shown that somatic mutation is a key role in the development and progression of cancers.30,31 Copy-number variation, one of the major alterations in cancers, also significantly affects the somatic mutations, then causing the phenotypic differences via changing the original state of the genome.32,33 DNA methylation, one of the most popular epigenetic changes, is another aspect affecting tumor biological behaviors.34,35 One previous study indicated that the methylation of LY6K promoter significantly affected the glioblastoma tumorigenicity via ERK1/2 signaling pathway.36 The recurrence in HCC was strongly involved in the CpG methylation.37 Our results showed that the promoter methylation levels of TASP1 in liver tumor tissues are decreased compared with that in normal tissues. Moreover, the expression of TASP1 was strongly correlated with the expression of DNMTs, indicating that the gene TASP1 was modulated by DNA methylation. Thus, we speculate that TASP1 may be a potential marker for diagnosis of HCC.
In our study, we analyzed TASP1 expression and its relationship with prognosis using our clinical patients and public databases. Survival analysis revealed that patients with high expression of TASP1 usually had poorer survivals than those with low TASP1 expression. Thus, we speculate that TASP1 expression may act as a prognostic biomarker for HCC. To better understand the mechanism of TASP1 in HCC, we performed bioinformatic analysis, including GO and KEGG pathway enrichment analysis, and PPI analysis, to detect the function of TASP1 in HCC and TASP1-related genes and potential molecular pathways. We found that the functional network of TASP1 participated primarily in the TGF-β pathway, Thyroid hormone signaling pathway, and Hippo signaling pathway. Accumulated evidence indicated that TGF-β signaling pathway frequently occurs in various cancers, including HCC, and it was highly associated with overall survival.38,39 Also, hippo signaling pathway plays a crucial role in biological process related with tumorigenesis and metastasis, including self-renewal, proliferation, migration, and invasion.40 These findings suggest that TASP1 is a potential drug target for therapy.
Because the tumor cells have some fundamental characteristics, such as genomic instability and mutagenesis,41,42 we conducted GSEA to identify some key kinases and kinase-related signaling pathways, which may stabilize and repair genomic DNA. The results represented that TASP1 had a strong correlation with a network of kinases, including ATM, ATR, CDK1, PLK1, CHEK1. Interestingly, several studies have confirmed that these kinases modulate genomic stability, DNA replication and cell cycle.43–46 All these kinases, related with TASP1, have been focused on in various studies. For example, CDK1 regulates the mitosis, differentiation and somatic reprogramming. Interestingly, some CDK1 inhibitors have been used to perform clinical trials for the treatment of various cancers.47 Additionally, CDK1-PLK1, together with CDK1, have been found to participate in the regulation of cell cycle in HCC.48
Furthermore, we performed GSEA to detect the networks of miRNAs related with TASP1 in HCC and find miR-202, miR-493, miR-21, miR-154, miR-515-5p and miR-519e. miRNAs, post-transcriptionally regulating gene expression, are involved in the carcinogenesis. These miRNAs detected in this study display a role in the development and progression of cancers via tumor proliferation, apoptosis, cell cycle, invasion, metastasis and angiogenesis.49–52 In fact, miR-202, miR-493 and miR-21 had been reported that they could be used to diagnose and predict the prognosis of HCC.53–55 Also, miR-21 promotes the growth of HCC via TETs-PTENp1-PTEN signaling pathway.56 This indicates that TASP1 affect HCC through the miRNAs dysregulation and changes of these miRNAs may be promising targets for HCC.
Except kinases and miRNAs, we analyzed transcription factors of TASP1 in HCC. The results showed that among significant transcription factors, E2F1 acts an important role due to the dysregulation of TASP1, and modulates the cell cycle in HCC. E2F, a common oncogenic signaling pathway, drives the development and progression of HCC.57 Furthermore, a previous study represented that the copy number of E2F1 was changed, leading the progression of HCC.58
Immune components play a critical role in the regulating tumor responses and more and more studies have focused on the interactions between tumors and immune cells. Thus, we investigated whether TASP1 can regulate the immune microenvironment in HCC. Then, we analyzed the correlation between TASP1 and the infiltration of immune cells, and found that TASP1 expression was strongly associated with the immune cell infiltration, including B cells, CD8+ T cells, CD4+ T cells, macrophages, neutrophils, and dendritic cells. Our result was aligned with other studies, indicating that the immunotherapy and clinical outcomes of cancers are dependent on the immune cell infiltration.59,60 Previous studies showed that chemokines can modulate the tumor immunity and tumor biological phenotypes by mediating the infiltration of immune cells.61–63 Further study showed that TASP1 expression was correlated with some chemokines in HCC, such as CCL2, CCL5, CCL8, CCL14, et al. And a study has been found that CCL14 served as an independent prognostic biomarker of HCC.64 In addition, CCL14 regulates angiogenesis and metastasis of breast cancer via the JARID1B/LSD1/NuRD complex.65 Above the mentioned results, we speculate that TASP1 may affect the HCC biological behaviors through immune response, such as the infiltration of immune cell and chemokine expression.
Many factors take part in the tumor microenvironment and immunological checkpoints are included. The expression levels of inhibitory immune checkpoint proteins in tumor tissues are changed compared to that in normal tissues, reflecting that immune checkpoints are involved in tumor microenvironment.66 And immune checkpoints lead to the development and progression and promote cancers from evading immune response.67 To confirm that TASP1 acts as a multimodality regulator of tumor immune response, we detect the relationship between TASP1 and immunological checkpoints. And we find that different kinds of immunological checkpoints, such as C10orf5, CD48, IL-6R, CD160, CD244 and et al, are involved in TASP1, indicating that immunological checkpoints act as an important role in HCC.
Our study has some limitations. In vitro and in vivo experiments are needed to confirm the functions and mechanisms of TASP1 in HCC.
Conclusion
In conclusion, the results showed that the expression of TASP1 in HCC was increased and TASP1 may be an independent biomarker for prognosis in HCC patients. Moreover, TASP1 could play a critical role in the development and progression of HCC via numerous regulators including kinases, miRNAs and transcript factors. In addition, TASP1 affected the tumor microenvironment by the immune cell infiltration, chemokines, and immune checkpoints. To our knowledge, we found that TASP1 was an important modulator in HCC and TASP1 was a target with greatly therapeutic promise for HCC.
Acknowledgments
We appreciated the authors, who provided these data series, and the authors of the databases used in this article. They completed this research with great efforts.
Funding
This study was supported by funds from the National Natural Science Foundation of China (grant numbers: #81873567 to Wenzhuo Yang) and Municipal Health Commission of Shanghai (#201640117 to Wenzhuo Yang).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15(10):599–616.
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
3. Wang J-H, Zhong X-P, Zhang Y-F, et al. Cezanne predicts progression and adjuvant TACE response in hepatocellular carcinoma. Cell Death Dis. 2017;8(9):e3043–e3043.
4. Llovet J, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nature Rev Dis Primers. 2016;2:16018. PubMed PMID: 27158749. doi:10.1038/nrdp.2018.8
5. DiStefano JK, Davis B. Diagnostic and prognostic potential of AKR1B10 in human hepatocellular carcinoma. Cancers. 2019;11(4):486. doi:10.3390/cancers11040486
6. López-Otín C, Bond JS. Proteases: multifunctional enzymes in life and disease. J Biol Chem. 2008;283(45):30433–30437. doi:10.1074/jbc.R800035200
7. Verma R, Mohl D, Deshaies RJ. Harnessing the power of proteolysis for targeted protein inactivation. Mol Cell. 2020;77:446–460. doi:10.1016/j.molcel.2020.01.010
8. Hsieh JJ-D, Cheng EH-Y, Korsmeyer SJ. Taspase1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell. 2003;115(3):293–303. doi:10.1016/S0092-8674(03)00816-X
9. Niizuma H, Cheng EH, Hsieh JJ. Taspase 1: a protease with many biological surprises. Mol cell Oncol. 2015;2(4):e999513. doi:10.1080/23723556.2014.999513
10. Oyama T, Sasagawa S, Takeda S, et al. Cleavage of TFIIA by Taspase1 activates TRF2-specified mammalian male germ cell programs. Dev Cell. 2013;27(2):188–200. doi:10.1016/j.devcel.2013.09.025
11. Capotosti F, Hsieh JJ-D, Herr W. Species selectivity of mixed-lineage leukemia/trithorax and HCF proteolytic maturation pathways. Mol Cell Biol. 2007;27(20):7063–7072. doi:10.1128/MCB.00769-07
12. Chen DY, Liu H, Takeda S, et al. Taspase1 functions as a non-oncogene addiction protease that coordinates cancer cell proliferation and apoptosis. Cancer Res. 2010;70(13):5358–5367.
13. Chen DY, Lee Y, Van Tine BA, et al. A pharmacologic inhibitor of the protease Taspase1 effectively inhibits breast and brain tumor growth. Cancer Res. 2012;72(3):736–746. doi:10.1158/0008-5472.CAN-11-2584
14. Takeda S, Chen DY, Westergard TD, et al. Proteolysis of MLL family proteins is essential for taspase1-orchestrated cell cycle progression. Genes Dev. 2006;20(17):2397–2409. doi:10.1101/gad.1449406
15. Zhang Y, Du P, Li Y, et al. TASP1 promotes gallbladder cancer cell proliferation and metastasis by Up-regulating FAM49B via PI3K/AKT pathway. Int J Biol Sci. 2020;16(5):739.
16. Niehof M, Borlak J. EPS15R, TASP1, and PRPF3 are novel disease candidate genes targeted by HNF4α splice variants in hepatocellular carcinomas. Gastroenterology. 2008;134(4):1191–1202.
17. Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9(2):166. doi:10.1593/neo.07112
18. Lánczky A, Nagy Á, Bottai G, et al. miRpower: a web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res Treat. 2016;160(3):439–446. doi:10.1007/s10549-016-4013-7
19. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. doi:10.1093/nar/gkx247
20. Chandrashekar DS, Bashel B, Balasubramanya SAH, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–658. doi:10.1016/j.neo.2017.05.002
21. Koch A, Jeschke J, Van Criekinge W, van Engeland M, De Meyer T. MEXPRESS update 2019. Nucleic Acids Res. 2019;47(W1):W561–W565.
22. Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46(D1):D956–D963. doi:10.1093/nar/gkx1090
23. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1–pl1.
24. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(D1):D447–D452.
25. Li T, Fan J, Wang B, et al. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77(21):e108–e110. doi:10.1158/0008-5472.CAN-17-0307
26. Ru B, Wong CN, Tong Y, et al. TISIDB: an integrated repository portal for tumor–immune system interactions. Bioinformatics. 2019;35(20):4200–4202.
27. López-Otín C, Overall CM. Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol. 2002;3(7):509–519.
28. López-Otín C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7(10):800–808.
29. Dong Y, Van Tine BA, Oyama T, Wang PI, Cheng EH, Hsieh JJ. Taspase1 cleaves MLL1 to activate cyclin E for HER2/neu breast tumorigenesis. Cell Res. 2014;24(11):1354–1366.
30. Dall’Era MA, McPherson JD, Gao AC, DeVere White RW, Gregg JP, Lara PN
31. Li G, Xu W, Zhang L, et al. Development and validation of a CIMP-associated prognostic model for hepatocellular carcinoma. EBioMedicine. 2019;47:128–141.
32. Zack TI, Schumacher SE, Carter SL, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45(10):1134–1140.
33. Urrutia E, Chen H, Zhou Z, Zhang NR, Jiang Y. Integrative pipeline for profiling DNA copy number and inferring tumor phylogeny. Bioinformatics. 2018;34(12):2126–2128.
34. Li Y-S, Zou Y, Dai D-Q. MicroRNA-320a suppresses tumor progression by targeting PBX3 in gastric cancer and is downregulated by DNA methylation. World J Gastrointest Oncol. 2019;11(10):842.
35. Zhang C, Zou Y, Dai D-Q. Downregulation of microRNA-27b-3p via aberrant DNA methylation contributes to malignant behavior of gastric cancer cells by targeting GSPT1. Biomed Pharmacother. 2019;119:109417.
36. Sastry NG, Wan X, Huang T, et al. LY6K promotes glioblastoma tumorigenicity via CAV-1–mediated ERK1/2 signaling enhancement. Neuro-Oncology. 2020.
37. Qiu J, Peng B, Tang Y, et al. CpG methylation signature predicts recurrence in early-stage hepatocellular carcinoma: results from a multicenter study. J Clin Oncol. 2017;35(7):734–742. doi:10.1200/JCO.2016.68.2153
38. Dietrich P, Wormser L, Fritz V, et al. Molecular crosstalk between Y5 receptor and neuropeptide Y drives liver cancer. J Clin Invest. 2020;130(5):2509–2526. doi:10.1172/JCI131919
39. Caballero-Díaz D, Bertran E, Peñuelas-Haro I, et al. Clathrin switches transforming growth factor-β role to pro-tumorigenic in liver cancer. J Hepatol. 2020;72(1):125–134. doi:10.1016/j.jhep.2019.09.012
40. Kovar H, Bierbaumer L, Radic-Sarikas B. The YAP/TAZ pathway in osteogenesis and bone sarcoma pathogenesis. Cells. 2020;9(4):972. doi:10.3390/cells9040972
41. Yogosawa S, Yoshida K. Tumor suppressive role for kinases phosphorylating p53 in DNA damage-induced apoptosis. Cancer Sci. 2018;109(11):3376–3382. doi:10.1111/cas.13792
42. Karimian A, Ahmadi Y, Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst). 2016;42:63–71. doi:10.1016/j.dnarep.2016.04.008
43. Ha G-H, Ji J-H, Chae S, et al. Pellino1 regulates reversible ATM activation via NBS1 ubiquitination at DNA double-strand breaks. Nat Commun. 2019;10(1):1–18. doi:10.1038/s41467-019-09641-9
44. Seibert M, Krüger M, Watson NA, et al. CDK1-mediated phosphorylation at H2B serine 6 is required for mitotic chromosome segregation. J Cell Biol. 2019;218(4):1164–1181. doi:10.1083/jcb.201806057
45. de Cárcer G, Venkateswaran SV, Salgueiro L, et al. Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat Commun. 2018;9(1):1–14. doi:10.1038/s41467-018-05429-5
46. Halder S, Torrecilla I, Burkhalter MD, et al. SPRTN protease and checkpoint kinase 1 cross-activation loop safeguards DNA replication. Nat Commun. 2019;10(1):1–18. doi:10.1038/s41467-019-11095-y
47. Wang Q, Su L, Liu N, Zhang L, Xu W, Fang H. Cyclin dependent kinase 1 inhibitors: a review of recent progress. Curr Med Chem. 2011;18(13):2025–2043. doi:10.2174/092986711795590110
48. Li L, Huang K, Zhao H, Chen B, Ye Q, Yue J. CDK1-PLK1/SGOL2/ANLN pathway mediating abnormal cell division in cell cycle may be a critical process in hepatocellular carcinoma. Cell Cycle. 2020;1–17. doi:10.1080/15384101.2019.1689482
49. Zhang L, Xu J, Yang G, Li H, Guo X. miR-202 inhibits cell proliferation, migration, and invasion by targeting epidermal growth factor receptor in human bladder cancer. Oncol Res Featuring Preclinical Clin Cancer Ther. 2018;26(6):949–957. doi:10.3727/096504018X15149787144385
50. Wang G, Fang X, Han M, Wang X, Huang Q. MicroRNA-493-5p promotes apoptosis and suppresses proliferation and invasion in liver cancer cells by targeting VAMP2. Int J Mol Med. 2018;41(3):1740–1748. doi:10.3892/ijmm.2018.3358
51. Huang Y-H, Lin Y-H, Chi H-C, et al. Thyroid hormone regulation of miR-21 enhances migration and invasion of hepatoma. Cancer Res. 2013;73(8):2505–2517. doi:10.1158/0008-5472.CAN-12-2218
52. Sahraei M, Chaube B, Liu Y, et al. Suppressing miR-21 activity in tumor-associated macrophages promotes an antitumor immune response. J Clin Invest. 2019;129:12.
53. Zhuang D, Liang L, Zhang H, Feng X. MiR-202 suppresses hepatocellular carcinoma progression via down-regulating BCL2 expression. Oncol Res Featuring Preclinical Clin Cancer Ther. 2020;28:399–408. doi:10.3727/096504020X15864296270581
54. Ding W, Tan H, Li X, et al. MicroRNA-493 suppresses cell proliferation and invasion by targeting ZFX in human hepatocellular carcinoma. Cancer Biomarkers. 2018;22(3):427–434. doi:10.3233/CBM-171036
55. Wang W-Y, Zhang H-F, Wang L, et al. miR-21 expression predicts prognosis in hepatocellular carcinoma. Clin Res Hepatol Gastroenterol. 2014;38(6):715–719. doi:10.1016/j.clinre.2014.07.001
56. Cao L-Q, Yang X-W, Chen Y-B, Zhang D-W, Jiang X-F, Xue P. Exosomal miR-21 regulates the TETs/PTENp1/PTEN pathway to promote hepatocellular carcinoma growth. Mol Cancer. 2019;18(1):148. doi:10.1186/s12943-019-1075-2
57. Xing J, Bhuria V, Bui KC, et al. Haprolid inhibits tumor growth of hepatocellular carcinoma through Rb/E2F and Akt/mTOR inhibition. Cancers. 2020;12(3):615. doi:10.3390/cancers12030615
58. Kent LN, Bae S, Tsai S-Y, et al. Dosage-dependent copy number gains in E2f1 and E2f3 drive hepatocellular carcinoma. J Clin Invest. 2017;127(3):830–842. doi:10.1172/JCI87583
59. Bindea G, Mlecnik B, Tosolini M, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–795. doi:10.1016/j.immuni.2013.10.003
60. Liu X, Wu S, Yang Y, Zhao M, Zhu G, Hou Z. The prognostic landscape of tumor-infiltrating immune cell and immunomodulators in lung cancer. Biomed Pharmacother. 2017;95:55–61. doi:10.1016/j.biopha.2017.08.003
61. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4(7):540–550. doi:10.1038/nrc1388
62. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. 2013;25(2):214–221. doi:10.1016/j.coi.2012.12.003
63. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5(4):263–274. doi:10.1038/nrc1586
64. Gu Y, Li X, Bi Y, et al. CCL14 is a prognostic biomarker and correlates with immune infiltrates in hepatocellular carcinoma. Aging. 2020;12(1):784. doi:10.18632/aging.102656
65. Li Q, Shi L, Gui B, et al. Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res. 2011;71(21):6899–6908. doi:10.1158/0008-5472.CAN-11-1523
66. Borch TH, Donia M, Andersen MH, Svane IM. Reorienting the immune system in the treatment of cancer by using anti-PD-1 and anti-PD-L1 antibodies. Drug Discov Today. 2015;20(9):1127–1134. doi:10.1016/j.drudis.2015.07.003
67. Rotte A, Jin J, Lemaire V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol. 2018;29(1):71–83. doi:10.1093/annonc/mdx686
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.