")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Overexpression of SOX9 alleviates the progression of human osteoarthritis in vitro and in vivo
Authors Ouyang Y, Wang W, Tu B, Zhu Y, Fan C, Li Y
Received 2 February 2019
Accepted for publication 29 June 2019
Published 13 August 2019 Volume 2019:13 Pages 2833—2842
DOI https://doi.org/10.2147/DDDT.S203974
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Cristiana Tanase
Yuanming Ouyang,1,2 Wei Wang,1,2 Bing Tu,1,2 Yi Zhu,1,2 Cunyi Fan,1,2 Yanfeng Li1,2
1Department of Orthopedics, Shanghai Sixth People’s Hospital East Campus Shanghai University of Medicine and Health, Shanghai 201306, People’s Republic of China; 2Department of Orthopedics, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai 200233, People’s Republic of China
Purpose: Recent findings have identified that SOX9 served as a key role during the pathogenesis of osteoarthritis (OA). This study aimed to investigate the mechanisms by which SOX9 regulated the formation of OA in vitro and in vivo.
Materials and methods: The relative expressions of SOX9 in patients with OA and normal fracture of thighbone were analyzed by real-time-PCR. In vitro, IL-1β induced inflammatory response in human chondrocytes was used to evaluate the function of SOX9. The recombinant SOX9 lentivirus vector (Lenti-SOX9) was used to upregulate the expression of SOX9 in cells. ELISA was used to measure the concentration of tumornecrosis factor-α (TNF-α). The protein expressions of SOX9, matrixmetalloproteinase-13 (MMP13), Collagen II, Aggrecan and Smad3 were analyzed by Western blot. Cell proliferation and cell apoptosis were detected by CCK-8 assay and flow cytometry, respectively. In vivo, the effect of SOX9 on surgically induced OA mice was evaluated.
Results: The gene level of SOX9 was remarkably downregulated in patients with OA compared with normal people, while the concentration of TNF-α was upregulated. In addition, IL-1β reduced the expressions of SOX9, Collagen II and Aggrecan and increased the level of MMP13 in chondrocytes. Moreover, Lenti-SOX9 notably inhibited IL-1β-induced growth inhibition and apoptosis in chondrocytes via increasing the expression of Smad3. Finally, Lenti-SOX9 markedly alleviated the symptoms of OA mice in vivo.
Conclusion: Upregulation of SOX9 inhibited IL-1β-induced inflammatory response via increasing the level Smad3 in human chondrocytes and exhibited therapeutic effect on surgically induced OA mice in vivo. Therefore, SOX9 may serve as a potential target in the treatment of OA in the future.
Keywords: SOX9, osteoarthritis, collagen, inflammation, IL-1β
Introduction
Arthritis is considered to be one of the most common clinical diseases. In addition, osteoarthritis (OA) is the commonest chronic arthritis in aging people or animal.1,2 OA involves synovitis, subchondral bone sclerosis and progressive articular cartilage damage, which will induce severe ache in the joint part and severe disability in final.1,2 Damage of articular cartilage caused the changes in extracellular matrix of cartilage.3 Collagen II and aggrecan are the primary composition of the extracellular matrix in cartilage, and matrix metalloproteinase (MMPs) are very important for maintaining integrity of cartilage.4 Inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and IL-1β were considered to be important in inducing damage of articular cartilage.5,6 IL-1β could regulate the differentiation and function of chondrocytes, which promotes the expression of MMPs; IL-1β then breaks down the cartilage matrix via inducing cell apoptosis.4,7 Therefore, inhibiting IL-1β-induced inflammation might be an effective therapeutic strategy for treating OA.
The SOX9 gene was reported to be cloned in the early ’90s via using a PCR-based screening, which was close relative of SRY.8 The SRY is a new gene identified as the sex-determining region on the Y chromosome.8 Recent studies have shown that the SOX (SRY-related HMG box)-containing genes are now well known as master transcription factors in cell fate determination and differentiation in a variety of lineages.8 In mesenchymal condensations, SOX9 is found to be early expressed and takes a great effect on the subsequent differentiation of prechondrogenic precursor cells into chondrocytes.9 Furthermore, Dy et al confirmed that SOX9 was important for normal cartilage formation.10 SOX9 is required to form a growth plate and prevent chondrocytes from transitioning from a proliferative state to a hypertrophic state and an osteoblastic state.10,11 Moreover, SOX9 could inhibit the proliferation and hypertrophy of chondrocyte.10
Recent findings have identified that SOX9 serves as a key role during the pathogenesis of OA. This study aimed to investigate the mechanisms by which SOX9 regulated the formation of OA in vitro and in vivo.
Materials and methods
Patients and specimen collection
This study was approved by the Medical Ethical Committee of the Shanghai Jiao Tong University Affiliated Sixth People’s Hospital. The guidelines of the Declaration of Helsinki and Tokyo were also obeyed during this process. OA human cartilage tissues were obtained from patients with OA (n=20, 9 women; age range from 53 to 65 years) who were diagnosed with OA and received total knee arthroplasty in Shanghai Jiao Tong University Affiliated Sixth People’s Hospital. The American College of Rheumatology (ACR) classification criteria was used for the diagnosis of patients with OA.12 All patients have signed the informed consents.
Cell culture and transfection
Human chondrocytes CHON-001 (American Type Culture Collection, Manassas, VA, USA) were cultivated in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) with 10% of FBS (Thermo Fisher Scientific) in a humidified incubator with 5% CO2 at 37°C.
The lentiviral vector and pLv-CMV-SOX9 lentiviral plasmid were obtained from GenePharma (Shanghai, China). The SOX9 lentivirus and control lentivirus were transfected into 293T cells for 72 hrs. Then, the SOX9 lentivirus supernatant was collected. CHON-001 cells (4×105 cells per well) were plated into 60-mm cell plates overnight at 37°C. Then, cells were transfected with control lentivirus and SOX9 lentivirus supernatant for 72 hrs.
Real time quantitative PCR (qPCR)
The mRNA expression of SOX9, MMP13, collagen II and aggrecan was quantified by using real-time quantitative PCR (qPCR). Based on the instructions of the manufacturer, total RNA was isolated from human OA cartilage tissues or CHON-001 cells by TRIzol reagent, respectively. 1 µg of total RNA was used to synthesize the first-strand cDNA. The QuantiTect Reverse Transcription kit from QIAGEN (Venlo, the Netherlands) was used in this procedure. In addition, the process of qPCR was performed according to the following conditions: 10 mins 95°C, followed by 40 cycles of 15 s 95°C and 1 min 60°C. 5 µL SYBR Green Master Mix, 0.25 µL of reverse primer and forward primer, respectively, and 4.5 µL diluted cDNA were included in the reaction system with a total volume of 10 µL. Fold change (2−ΔΔCt) was used to analyze the relative expression of target gene.13 GAPDH was used to be the inner standard to normalize the gene concentration of target mRNAs. Each gene analysis was performed in triplicate. The real-time qPCR primers were as following: SOX9, forward primer 5ʹ-GGTCTGGTTGGGGTAGAGGT-3ʹ and reverse primer 5ʹ-TGAAGATGGCGTTAGGAGAGA-3ʹ; MMP13, forward primer 5ʹ-CCAGAACTTCCCAACCAT-3ʹ and reverse primer 5ʹ-ACCCTCCATAATGTCATACC-3ʹ; collagen II, forward primer 5ʹ-CTCAAGTCGCTGAACAACCA-3ʹ and reverse primer 5ʹ-GTCTCCGCTCTTCCACTCTG-3ʹ; aggrecan, forward primer 5ʹ-AAGTGCTATGCTGGCTGGTT-3ʹ and reverse primer 5ʹ-GGTCTGGTTGGGGTAGAGGT-3ʹ; GAPDH, forward primer 5ʹ-TCTCCTCTGACTTCAACAGCGAC-3ʹ and reverse primer 5ʹ-CCCTGTTGCTGAGCCAAATTC-3ʹ.
TNF-α activity measurement
TNF-α activity was measured as described previously.14 Briefly, the level of TNF-α in human OA cartilage tissues or in CHON-001 cells was measured by using commercial ELISA kits R&D (Minneapolis, MN, USA). The absorbance of each sample was measured at 450 nm (ELISA) with a microplate reader (Thermo Scientific, Waltham, MA, USA).
Cell viability determination
Cell viability was assessed using the CCK-8 according to the manufacturer’s instructions (Sigma Aldrich, St. Louis, MO, USA). CHON-001 cells (5×103 cells per well) were cultured in 96-well plate for 24 hrs. Then, CHON-001 cells were treated with IL-1β for 24 hrs. After that, 10 µL CCK-8 was added to each well and the cells were incubated at 37°C for another 4 hrs. The optical density was read at a wavelength of 450 nm with a microplate reader.
Western blot assay
Total protein from CHON-001 cells was extracted with RIPA lysis buffer. After sonicating on ice, lysates were centrifuged at 12,000× g for 30 mins at 4°C. BCA protein assay kit purchased from Thermo Fisher Scientific was used to determine the protein concentration. Equal amount of protein (40 µg) was separated by 12% SDS-PAGE, and transferred into PVDF membranes. And then, membranes were incubated with blocking buffer (5% nonfat milk in PBS containing 0.1% Tween-20 (PBST)) for 2 hrs at room temperature and incubated with the primary antibodies against SOX9, MMP13, collagen II, aggrecan, smad3 and GAPDH (Cell Signaling Technology, Danvers, MA, USA) overnight at 4°C. PBST was used to wash the membranes for three times (5 mins for each time). Then, the membranes were incubated with HRP-conjugated secondary antibodies (Cell Signaling Technologies) for 2 hrs. Finally, the membranes were detected by enhanced chemiluminescence kit (Thermo Fisher Scientific) and quantified by the Image J (version 2.0, National Institutes of Health, Bethesda, MD, USA) software.
Apoptosis analysis
An Annexin V-FITC and propidium iodide (PI) staining kit (Thermo Fisher Scientific) was used to detect the cell apoptosis according to the manufacturer’s protocol. Briefly, CHON-001 cells were treated with IL-1β for 72 hrs. Then, cells were stained with Annexin-FITC and PI for 5 mins at room temperature in the dark, and the cells were subjected to flow cytometric analysis.
Surgically induced OA mice models
A total of 12 male C57BL/6N mice (8-week old, n=4/group) weighing 20–25 g were purchased from the Vital River (Beijing, China). Food and water were available ad libitum. Mice were housed under a constant temperature of 20±2°C, a relative humidity of 50±10% and a 12 hrs light/dark cycle. The experimental protocol was approved by the Animal Care and Use Committee of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital. A total of 12 mice were randomly divided into three groups: sham (control) group, OA model group, OA model with the treatment of SOX9-expressing lentivirus (OA + lenti-SOX9). The medial meniscus (DMM) of the experimental mice was destabilized surgically to simulate OA, as previously described.15 In brief, after anesthesia with peritoneal injection of Zoletil 50, the attachment of the medial meniscus to the tibial plateau (medial meniscotibial ligament) of the right knee was transected with a microsurgical knife. The lateral meniscotibial ligament was identified and protected during the surgery. A sham operation, consisting of an arthrotomy without the transaction of medial meniscotibial ligament, was also performed in the right knee joint of mice in the sham control group. SOX9-expressing lentivirus (5 nM) was injected into the articular cavity of mice once a week after surgery. All animals were sacrificed 8 weeks after surgery. Knee joint tissues were collected for further evaluation.
Histological analysis
After fixing in 4% paraformaldehyde for 24 hrs at 4°C, the knee joint samples were decalcified in 10% EDTA solution at 4°C for 2 weeks. Then, the knee joint samples were dehydrated through a gradient concentration of alcohol. Next, the knee joint samples were cleared and embedded in paraffin blocks. 5 µm thick of frontal serial sections across entire joints were obtained. Ten slides per joint at every 50 µm were selected. And then, the slides were stained with Safranin O/Fast Green to show cartilage destruction. In order to determine the extent of subchondral bone thickness, synovitis and cartilage degeneration, the stained sections were photographed digitally under a microscope. The destruction of articular cartilage was graded using the Osteoarthritis Research Society International (OARSI) scoring system for medial femoral condyle and medial tibial plateau.16 The section was assigned as follows (six OA grades): 0= surface intact, cartilage intact; 1= surface intact; 2= surface discontinuity; 3= vertical fissures; 4= erosion; 5= denudation; and 6= deformation. Furthermore, a summed OARSI score (0–12) from medial femoral condyle and medial tibial plateau was used to evaluate the degree of articular cartilage destruction according to the previous reference.16 The severity of synovitis was graded using a scoring system which was previously described:17 enlargement of the synovial lining cell layer on a scale of 0–3 (0=1–2 cells, 1=2–4 cells, 2=4–9 cells and 3=10 or more cells) and density of cells in the synovial stroma on a scale of 0–3 (0= normal cellularity, 1= slightly increased cellularity, 2= moderately increased cellularity and 3= greatly increased cellularity). Axio Vision software (Carl Zeiss, Oberkochen, Germany) was chosen to measure the thickness of the medial subchondral bone plate according to Safranin-O-stained sections.
Statistical analysis
All experiments were performed independently at least three times. Values are shown as mean ± SD. GraphPad Prism software (version 7.0, La Jolla, CA, USA) was used to perform all analysis. The comparison between two groups was analyzed by Student’s t-test. One-way ANOVA followed by Tukey’s test was used to calculate the statistical significance among multiple groups. P<0.05 was considered statistically significant.
Results
Expression of the SOX9 in the patients with OA
The relative expressions of SOX9 in fracture of thighbone were illustrated by real-time qPCR firstly. And, the production of TNF-α in OA tissues was measured by ELISA. The data indicated that the expression of SOX9 in healthy tissues were much higher than that in OA tissues (Figure 1A, P<0.01). In addition, the production of TNF-α was increased significantly in patients with OA compared with healthy controls (Figure 1B). Thus, SOX9 appeared to be downregulated in OA tissues.
 |
Figure 1 Expression of the SOX9 in patients with osteoarthritis (OA). (A) Relative gene expression of SOX9 in normal articular cartilage of patients with femur and articular cartilage of OA patients was detected by using real-time qPCR. **P<0.01 compared with normal group, n=3. (B) The concentration of TNF-α in normal synovial fluid of patients with femur and synovial fluid of OA patients was detected by using ELISA. **P<0.01 compared with normal group, n=3. |
IL-1β induced inflammatory response in CHON-001 cells
IL-1β is considered one of the key cytokines involved in the pathogenesis of OA, which was used to induce inflammatory response in human chondrocytes.18 As shown in Figure 2A, compared to the control group, significant downregulation of collagen II, aggrecan and SOX9 mRNA levels were observed in IL-1β-treated CHON-001 cells, whereas the mRNA level of MMP13 was upregulated. Consistently, IL-1β markedly reduced protein expressions of collagen II, aggrecan and SOX9 and increased MMP13 protein (Figure 2B and C). In addition, the production of TNF-α in cell medium was increased significantly after IL-1β treatment (Figure 2D). All these data suggested IL-1β induced significantly inflammatory response in CHON-001 cells.
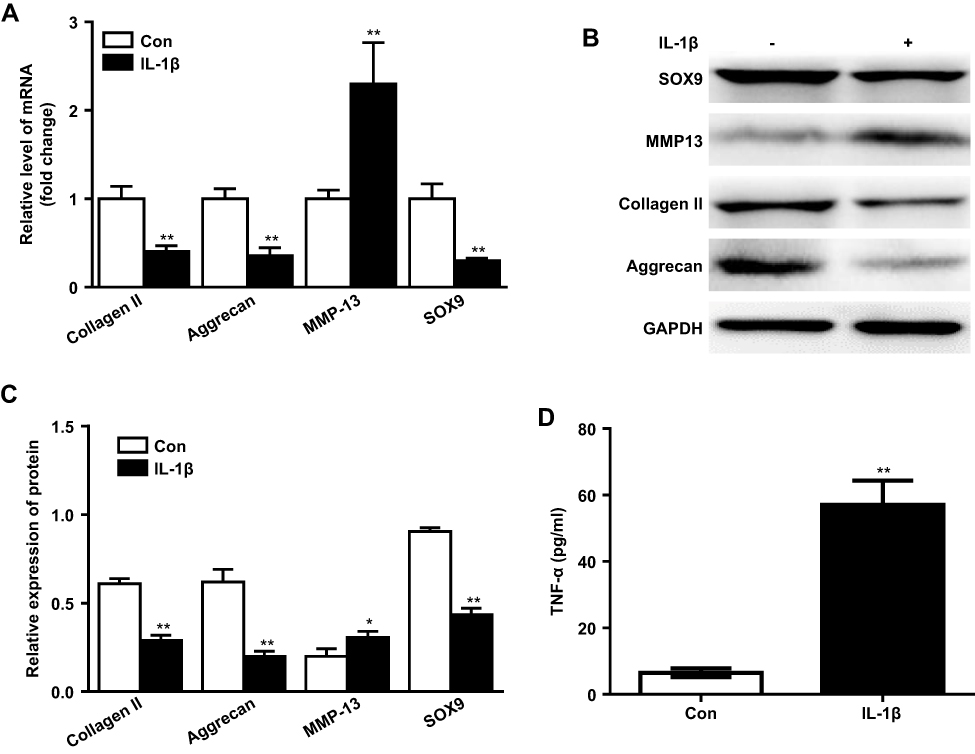 |
Figure 2 IL-1β induced inflammatory response in human chondrocytes. (A) CHON-001 cells were stimulated with IL-1 β (10 ng/mL) for 24 hrs. Relative gene expressions of collagen II, aggrecan, MMP13 and SOX9 in human cells were detected with real-time qPCR. **P<0.01 compared with control group, n=3. (B) Protein expressions of collagen II, aggrecan, MMP13 and SOX9 in CHON-001 cells after treating with IL-1β for 72 hrs were detected with Western blot. (C) Relative protein expressions of collagen II, aggrecan, MMP13 and SOX9 in cell were quantified. *P<0.05, **P<0.01 compared with control group, n=3. (D) CHON-001 cells were treated with IL-1β for 72 hrs, and the concentrations of TNF-α in cell medium were evaluated with ELISA. **P<0.01 compared with control group, n=3. |
Overexpression of SOX9 in CHON-001 cells
CHON-001 cells were treated with various concentrations of IL-1β. The results indicated that IL-1β dose-dependently inhibited mRNA and protein levels of SOX9 (Figure 3A–C). Next, chondrocytes were transfected with a lentiviral vector containing cytomegalovirus (CMV) driven pLv-SOX9-GFP or one reporter vector pLv-GFP for 6 hrs. As shown in Figure 3D–F, Lenti-SOX9 significantly upregulated the mRNA and protein levels of SOX9 in CHON-001 cells.
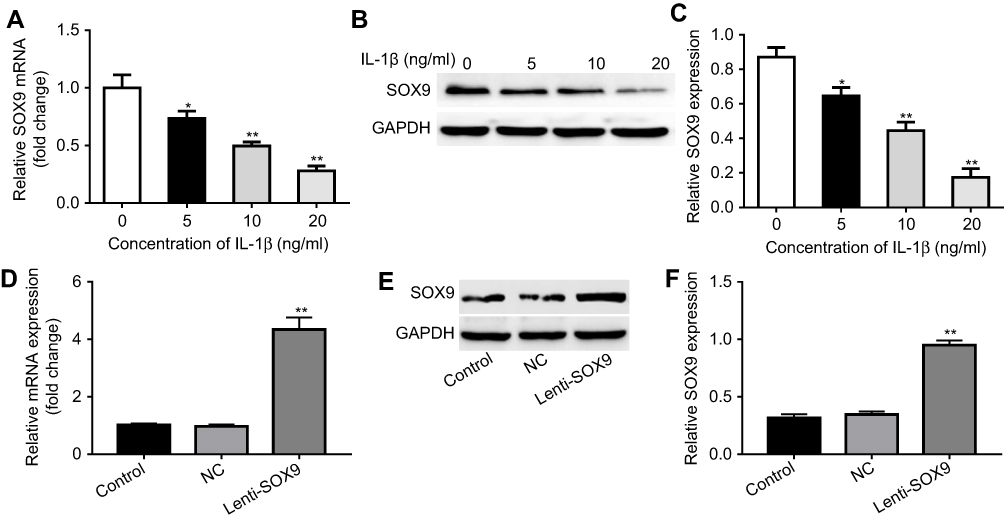 |
Figure 3 Overexpression of SOX9 in human chondrocytes. (A) CHON-001 cells were treated with 5, 10 or 20 ng/mL IL-1β for 24 hrs. Relative gene expression of SOX9 in cells was detected with real-time qPCR. *P<0.05, **P<0.01 compared with control group, n=3. (B) CHON-001 cells were treated with 5, 10 or 20 ng/mL IL-1β for 72 hrs. Protein expression of SOX9 in cells was measured by Western blot. (C) Quantification of SOX9 protein. *P<0.05, **P<0.01compared with control group, n=3. (D) CHON-001 cells were transfected with control lentivirus vector or Lenti-SOX9 for 6 hrs. Relative gene expression of SOX9 in CHON-001 cells was detected with real-time qPCR. **P<0.01 compared with control group, n=3. (E) Protein expression of SOX9 in cells was detected by Western blot. (F) Quantification of SOX9 protein. **P<0.01 compared with control group, n=3. |
Overexpression of SOX9 suppressed IL-1β-induced apoptosis in CHON-001 cells
In order to evaluate the effect of Lenti-SOX9 on the viability of chondrocytes, CCK-8 assay was used. As shown in Figure 4A, the viability of CHON-001 reduced markedly in IL-1β group compared with the control group. However, this effect was notably reversed by Lenti-SOX9. In addition, the result of cell apoptosis analysis demonstrated that Lenti-SOX9 significantly inhibited in IL-1β-induced apoptosis of CHON-001 cells (Figure 4B and C). The cell apoptotic rates were increased to 34% in IL-1β-treated CHON-001 cells, while the apoptotic rates were decreased to 16% following transfection with Lenti-SOX9 (Figure 4B and C). Furthermore, the level of TNF-α was significantly increased by IL-1β treatment (Figure 4D). However, the production of TNF-α was markedly reduced in the presence of Lenti-SOX9 (Figure 4D).
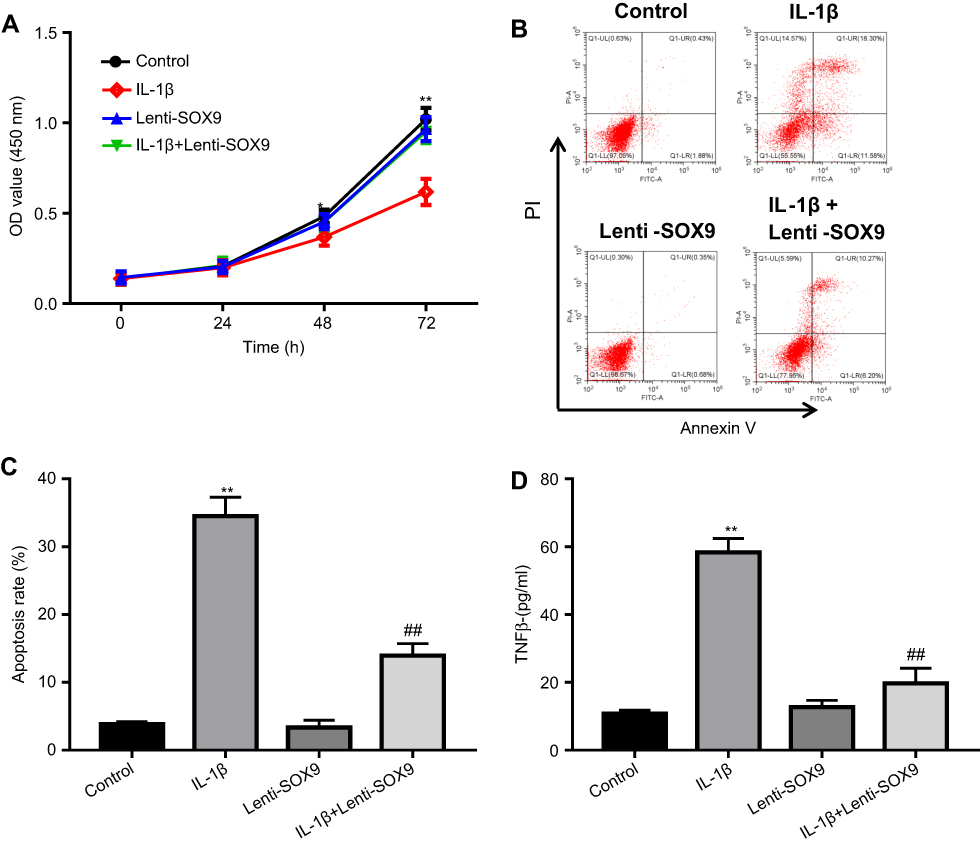 |
Figure 4 Overexpression of SOX9 suppressed IL-1β-induced apoptosis in human chondrocytes. (A) CHON-001 cells were transfected with Lenti-SOX9 for 6 hrs and then stimulated with IL-1β (10 ng/mL) for 24 hrs. Cell viability was measured by CCK-8 assay. *P<0.05, **P<0.01 compared with control group, n=3. (B) CHON-001 cells were transfected with Lenti-SOX9 for 6 hrs and then stimulated with IL-1β (10 ng/mL) for 72 hrs. Cell apoptosis of cells was measured by flow cytometry. (C) Quantification analysis of apoptosis. **P<0.01 compared with control group. ##P<0.01 compared with group treated with IL-1β, n=3. (D) The concentration of TNF-α in cell medium of different groups was measured with ELISA . **P<0.01 compared with control group. ##P<0.01 compared with group treated with IL-1β, n=3. |
Effect of Lenti-SOX9 on the expression of collagen II and smad3 in CHON-001 cells
Next, we investigated the mechanism underlying the protective effect of Lenti-SOX9 in IL-1β-treated human chondrocytes. The mRNA level of smad3 and the protein expressions of collagen II and smad3 were measured by real-time qPCR and Western blot, respectively. As shown in Figure 5A and B, notable downregulation of the mRNA expression of smad3 was observed in IL-1β-stimulated chondrocytes. Nevertheless, Lenti-SOX9 obviously inhibited the downregulation of smad3 by IL-1β in cells. In addition, Lenti-SOX9 distinctly decreased the protein degradation of collagen II and smad3 (Figure 5B–D). Furthermore, the expressions of smad3 in healthy articular cartilage were much higher than that in OA tissues (Figure 5E, P<0.01). A significant positive relationship between smad3 and SOX9 was demonstrated by correlation analyses (Figure 5F). All these data suggested Lenti-SOX9 significantly inhibited IL-1β-induced inflammatory response in human chondrocytes.
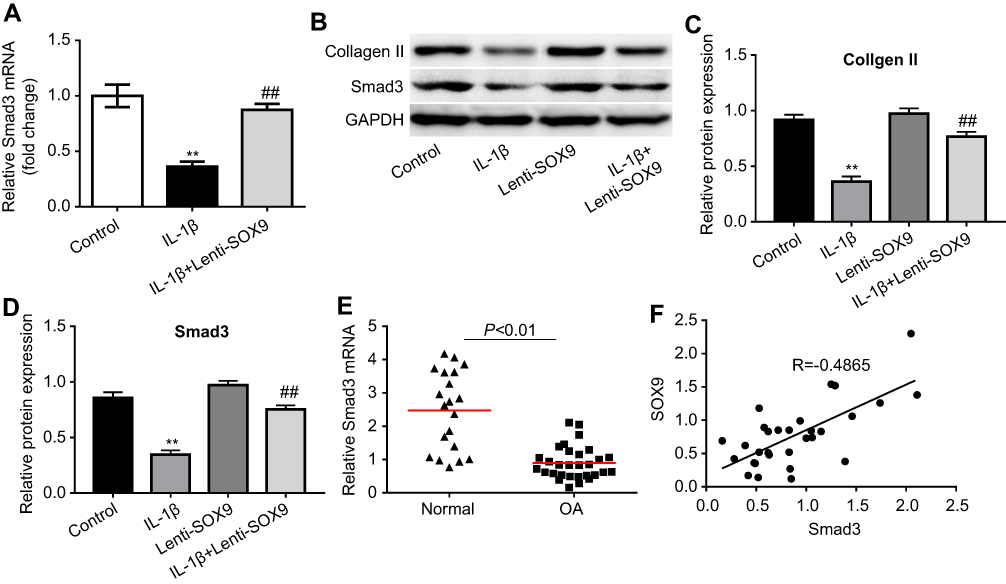 |
Figure 5 Effect of Lenti-SOX9 on the expressions of collagen II and smad3 in human chondrocytes. (A) CHON-001 cells were transfected with Lenti-SOX9 for 6 hrs following stimulation with IL-1β (10 ng/mL) for 24 hrs. Relative gene level of smad3 in cells was detected with real-time qPCR. **P<0.01 compared with control group. ##P<0.01 compared with group treated with IL-1β, n=3. (B) CHON-001 cells were transfected with Lenti-SOX9 for 6 hrs following stimulation with IL-1β (10 ng/mL) for 72 hrs. The protein expressions of collagen II and smad3 were measured by Western blot. (C, D) Quantification analysis of collagen II and smad3. **P<0.01 compared with control group. ##P<0.01 compared with group treated with IL-1β, n=3. (E) Relative gene expression of smad3 in normal articular cartilage and in patients with OA was measured with real-time qPCR. (F) The relationship between SOX9 and smad3 was evaluated by correlation analysis. |
SOX9 alleviated the progression of OA in mice
To assess the effect of SOX9 on the progression of OA in vivo, surgical-induced OA mice model was established. As presented in Figure 6A, the surface of the cartilage in control group was integrated according to the result of Safranin O staining. Compared with the control group, cartilage superficial destruction, extensive proteoglycan loss, cartilage erosion and obvious hypocellularity were observed in OA group. However, Lenti-SOX9 notably inhibited the proteoglycan loss and cartilage destruction as compared to OA group. Consistent with the results of Safranin O staining, the OASI score (Figure 6B) of OA group was apparently higher than that in control group. As expected, Lenti-SOX9 markedly lower OARSI score, compared with OA group. In addition, evidently relieved synovitis and the decreases in subchondral bone plate thickness were observed in Lenti-SOX9-treated group (Figure 6C and D). Taken together, these results demonstrated that upregulation of SOX9 could alleviate the progression of OA in mice.
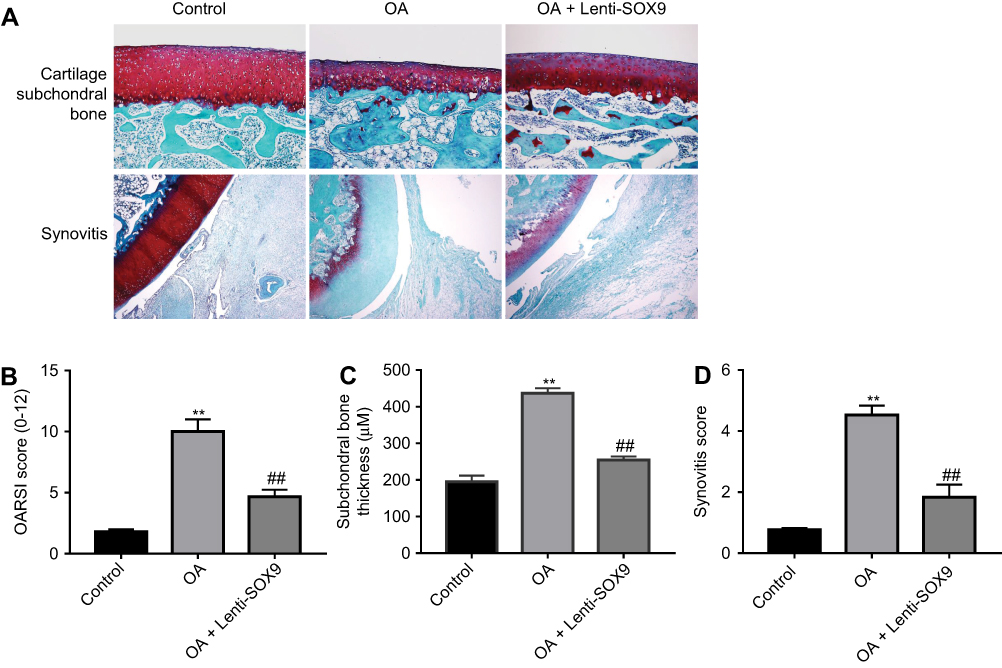 |
Figure 6 SOX9 alleviated the progression of osteoarthritis (OA) in mice. Mice were randomly divided into three groups: control group, OA group and Lenti-SOX9 group. The control group underwent sham operation, and the OA and the Lenti-SOX9 groups were subjected to DMM surgery for establishing OA models. After surgery, the Lenti-SOX9 group received an intra-articular injection of Lenti-SOX9 weekly for 8 weeks. (A) Histological morphology of cartilage was determined by Safranin O staining. (B, C, D) OA damage in mice was evaluated with OARSI score, subchondral bone plate thickness and synovitis scores. **P<0.01 compared with sham control group. ##P<0.01 compared with the OA group, n=5. |
Discussion
As a pivotal transcription factor, SOX9 is known for its function in different tissue processes including in the cartilage development and its maintenance in adults.11 In addition, SOX9 has been indicated to regulate the differentiation and hypertrophy of chondrocyte.19 Though there are many studies about SOX9, the mechanisms by which SOX9 regulates the pathogenesis of OA remain unclear. In this study, we investigated the effect of SOX9 in human chondrocytes in vitro and the protective effects of SOX9 in OA mice in vivo. Our results indicated that overexpression of SOX9 inhibited IL-1β-induced cell apoptosis and reduced the degradation of collagen-II in human chondrocytes. Furthermore, we found that overexpression of SOX9 suppressed IL-1β-stimulated smad3 decrease. Besides, treatment of SOX9 lentivirus attenuated the progression of OA in mice in vivo.
In OA cartilage, it has been detected that cell death showed the morphological and molecular features of apoptosis. The detected fate suggested that chondrocyte death/survival may play a vital role in the pathogenesis of OA.20 In addition, apoptotic cell death in OA cartilage was associated with matrix degradation and calcification. In turn, many studies also suggested the important role of cell death/survival in OA pathogenesis. Furthermore, in apoptosis, it has been reported that there was a close and positive relationship between the severity of cartilage destruction and matrix depletion in human osteoarthritic tissue specimens.21 Our results showed that SOX9 inhibited IL-1-induced cell apoptosis in human chondrocytes.
It has been well known that the major structure of the ECM constituted collagen II and aggrecan. Moreover, the normal physiological function of the cartilage was also maintained by collagen II and aggrecan. Loss of collagen II and aggrecan contributes a lot to accelerate the progress of OA. According to previous studies, retroviral expression of SOX9 in human OA articular chondrocytes was demonstrated to induce an upregulation in expression level of collagen II.22,23 These data corresponded well with our results. We speculated that SOX9 transduction may be able to renew the capacity of late passage human OA articular chondrocytes to form cartilage matrix.
Ferguson et al have proved that smad3 is one of the key mediators which could regulate the inhibitory effect of TGF-β on terminal differentiation of chondrocyte.24 Without smad3 signaling, chondrocytes may be able to break their quiescent state and undergo anomalous terminal differentiation in further. Spontaneous maturation in smad3-deficient chondrocytes will be blocked if smad3 were overexpressed.25,26 Moreover, an increased risked hip and knee OA was reported to be closely linked with a single nucleotide polymorphism in the smad3 gene.27 Surprisingly, retroviral transduction with SOX9 inhibited IL-1β-induced degradation of smad3 in human chondrocytes in the current study.
Articular cartilage destruction with changes in subchondral bone and synovitis is supposed to be key factors in OA formation.28 Based on this, suffering patients will benefit a lot from the agents which were designed to be specifically interdict mechanisms involved in synovitis, destruction of articular cartilage and subchondral bone remodeling. In addition, these agents will provide exciting new hope for the treatment of OA.29 In our study, OA mice models induced by DMM were used to evaluate the effect of SOX9 in vivo. OARSI grade, synovitis scoring system and subchondral bone plate thickness were used to assess the severity of cartilage destruction, synovitis and subchondral bone thickening, respectively. Interestingly, overexpression of SOX9 apparently decreased the OARSI scores as well as subchondral bone plate thickness and relieved the severity of synovitis in mice OA models, which suggested that overexpression of SOX9 alleviated the progression of OA.
Conclusion
In summary, we have shown that SOX9 played a key role in regulating the formatting of OA. Furthermore, overexpression of SOX9 decreased cartilage destruction and subchondral bone plate thickness and relieved synovitis in OA mice. Taken together, these findings suggested that overexpression of SOX9 might be a potential therapeutic approach for the treatment of OA.
Acknowledgment
The study was supported by the Projects of National Science Foundation of China (Grant Nos. 81570992 and 81571261), the Projects of National Science Foundation of Shanghai, China (Grant No. 17401901000), SUMHS Seed Foundation Project (Grant No. HMSF-16-21-010), Science and Technology Development Foundation of Pudong New District, Shanghai, China (Grant Nos. PKJ2016-Y55 and PWZxq2017-03), and the study was also partly sponsored by the Interdisciplinary Program of Shanghai JiaoTong University (No. YG2017MS22, YG2017QN56 and YG2016QN22), the Program of Shanghai Sixth People’s Hospital East Campus Foundation (No. 2019YY001), the Program of Shanghai Sixth People’s Hospital Foundation (No. LY2Y0272), the Shanghai Three-year Action plan to Further Accelerate the Development of Traditional Chinese Medicine (2018-2020) No. ZY 2018-2020-FWTX-8009).
Disclosure
The authors declare that they have no competing financial interests in this work.
References
1. Castaneda S, Roman-Blas JA, Largo R, Herrero-Beaumont G. Subchondral bone as a key target for osteoarthritis treatment. Biochem Pharmacol. 2012;83(3):315–323. doi:10.1016/j.bcp.2012.01.018
2. Kraus VB, Nevitt M, Sandell LJ. Summary of the OA biomarkers workshop 2009 – biochemical biomarkers: biology, validation, and clinical studies. Osteoarthritis Cartilage. 2010;18(6):742–745. doi:10.1016/j.joca.2009.11.014
3. Liu Q, Zhang X, Dai L, et al. Long noncoding RNA related to cartilage injury promotes chondrocyte extracellular matrix degradation in osteoarthritis. Arthritis Rheumatol. 2014;66(4):969–978. doi:10.1002/art.38309
4. Luo Z, Zheng B, Jiang B, et al. Peiminine inhibits the IL-1beta induced inflammatory response in mouse articular chondrocytes and ameliorates murine osteoarthritis. Food Funct. 2019;10(4):2198–2208. doi:10.1039/C9FO00307J
5. Zheng W, Feng Z, You S, et al. Fisetin inhibits IL-1beta-induced inflammatory response in human osteoarthritis chondrocytes through activating SIRT1 and attenuates the progression of osteoarthritis in mice. Int Immunopharmacol. 2017;45:135–147. doi:10.1016/j.intimp.2017.02.009
6. Zheng W, Tao Z, Chen C, et al. Plumbagin prevents IL-1beta-induced inflammatory response in human osteoarthritis chondrocytes and prevents the progression of osteoarthritis in mice. Inflammation. 2017;40(3):849–860. doi:10.1007/s10753-017-0530-8
7. Gao B, Gao W, Wu Z, et al. Melatonin rescued interleukin 1beta-impaired chondrogenesis of human mesenchymal stem cells. Stem Cell Res Ther. 2018;9(1):162. doi:10.1186/s13287-018-0892-3
8. Lovell-Badge R. The early history of the Sox genes. Int J Biochem Cell Biol. 2010;42(3):378–380. doi:10.1016/j.biocel.2009.12.003
9. Haag J, Gebhard PM, Aigner T. SOX gene expression in human osteoarthritic cartilage. Pathobiology. 2008;75(3):195–199. doi:10.1159/000124980
10. Dy P, Wang W, Bhattaram P, et al. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev Cell. 2012;22(3):597–609. doi:10.1016/j.devcel.2011.12.024
11. Lefebvre V, Dvir-Ginzberg M. SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect Tissue Res. 2017;58(1):2–14. doi:10.1080/03008207.2016.1235566
12. Palmieri B, Lodi D, Capone S. Osteoarthritis and degenerative joint disease: local treatment options update. Acta Bio-medica. 2010;81(2):94–100.
13. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi:10.1093/nar/29.9.e45
14. Lu Z, Luo M, Huang Y. lncRNA-CIR regulates cell apoptosis of chondrocytes in osteoarthritis. J Cell Biochem. 2018. In press.
15. Vasheghani F, Zhang Y, Li YH, et al. PPARgamma deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Ann Rheum Dis. 2015;74(3):569–578. doi:10.1136/annrheumdis-2014-205743
16. Pritzker KP, Gay S, Jimenez SA, et al. Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage. 2006;14(1):13–29. doi:10.1016/j.joca.2005.07.014
17. Lewis JS, Hembree WC, Furman BD, et al. Acute joint pathology and synovial inflammation is associated with increased intra-articular fracture severity in the mouse knee. Osteoarthritis Cartilage. 2011;19(7):864–873. doi:10.1016/j.joca.2011.04.011
18. Wojdasiewicz P, Poniatowski LA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014;2014:561459. doi:10.1155/2014/561459
19. Hattori T, Muller C, Gebhard S, et al. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development. 2010;137(6):901–911. doi:10.1242/dev.045203
20. Musumeci G, Castrogiovanni P, Trovato FM, et al. Biomarkers of chondrocyte apoptosis and autophagy in osteoarthritis. Int J Mol Sci. 2015;16(9):20560–20575. doi:10.3390/ijms160920560
21. Musumeci G, Aiello FC, Szychlinska MA, et al. Osteoarthritis in the XXIst century: risk factors and behaviours that influence disease onset and progression. Int J Mol Sci. 2015;16(3):6093–6112. doi:10.3390/ijms16036093
22. Tew SR, Li Y, Pothacharoen P, et al. Retroviral transduction with SOX9 enhances re-expression of the chondrocyte phenotype in passaged osteoarthritic human articular chondrocytes. Osteoarthritis Cartilage. 2005;13(1):80–89. doi:10.1016/j.joca.2004.10.011
23. Li Y, Tew SR, Russell AM, et al. Transduction of passaged human articular chondrocytes with adenoviral, retroviral, and lentiviral vectors and the effects of enhanced expression of SOX9. Tissue Eng. 2004;10(3–4):575–584. doi:10.1089/107632704323061933
24. Ferguson CM, Schwarz EM, Reynolds PR, et al. Smad2 and 3 mediate transforming growth factor-beta1-induced inhibition of chondrocyte maturation. Endocrinology. 2000;141(12):4728–4735. doi:10.1210/endo.141.12.7848
25. Li TF, Darowish M, Zuscik MJ, et al. Smad3-deficient chondrocytes have enhanced BMP signaling and accelerated differentiation. J Bone Miner Res. 2006;21(1):4–16. doi:10.1359/JBMR.050911
26. Yang X, Chen L, Xu X, et al. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J Cell Biol. 2001;153(1):35–46. doi:10.1083/jcb.153.1.35
27. Valdes AM, Spector TD, Tamm A, et al. Genetic variation in the SMAD3 gene is associated with hip and knee osteoarthritis. Arthritis Rheum. 2010;62(8):2347–2352. doi:10.1002/art.27631
28. Zheng W, Zhang H, Jin Y, et al. Butein inhibits IL-1beta-induced inflammatory response in human osteoarthritis chondrocytes and slows the progression of osteoarthritis in mice. Int Immunopharmacol. 2017;42:1–10. doi:10.1016/j.intimp.2016.11.009
29. Wei Y, Bai L. Recent advances in the understanding of molecular mechanisms of cartilage degeneration, synovitis and subchondral bone changes in osteoarthritis. Connect Tissue Res. 2016;57(4):245–261. doi:10.3109/03008207.2015.1113271
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.