")
Back to Journals » OncoTargets and Therapy » Volume 9
Overexpression of p53 activated by small activating RNA suppresses the growth of human prostate cancer cells
Authors Ge Q, Wang C, Ruan Y, Chen Z, Liu J, Ye Z
Received 21 September 2015
Accepted for publication 21 November 2015
Published 8 January 2016 Volume 2016:9 Pages 231—241
DOI https://doi.org/10.2147/OTT.S96710
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Qiangqiang Ge,1,* Chenghe Wang,2,* Yajun Ruan,1,* Zhong Chen,1 Jihong Liu,1 Zhangqun Ye1
1Department of Urology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, 2Department of Urology, Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: Previous research has reported that a particular double-stranded RNA, named dsP53-285, has the capacity to induce expression of the tumor suppressor gene TP53 in chimpanzee cells by targeting its promoter. Usually, it is the wild-type p53 protein, rather than mutants, which exhibits potent cancer-inhibiting effects. In addition, nonhuman primates, such as chimpanzees, share almost identical genome sequences with humans. This prompted us to speculate whether dsP53-285 can trigger wild-type p53 protein expression in human prostate cancer (PCa) cells and consequently suppress cell growth. The human PCa cell lines LNCaP and DU145 were transfected with dsP53-285 for 72 hours. Compared with the dsControl and mock transfection groups, expression of both p53 messenger RNA and p53 protein was significantly enhanced after dsP53-285 transfection, and this enhancement was followed by upregulation of p21, which indirectly indicated that dsP53-285 induced wild-type p53 expression. Moreover, overexpression of wild-type p53 mediated by dsP53-285 downregulated the expression of Cyclin D1 and cyclin-dependent kinase 4/6, thereby inducing PCa cell cycle arrest in G0/G1 phase and then inhibiting cell proliferation and clonogenicity. More importantly, dsP53-285 suppressed PCa cells mainly by modulating wild-type p53 expression. In conclusion, our study provides evidence that dsP53-285 can significantly stimulate wild-type p53 expression in the human PCa cell lines LNCaP and DU145 and can exert potent antitumor effects.
Keywords: p53, small activating RNA, prostate cancer
Introduction
Prostate cancer (PCa) represents the second most common cause of cancer-related deaths in males in the USA, with a reported 29,480 deaths in 2014 and an estimated annual incidence of 233,000 new cases.1 As a transcription factor, wild-type p53, encoded by the human TP53 gene, is a well-known tumor suppressor protein, which prevents cells from DNA damage and tumorigenesis by inducing DNA repair, cell cycle arrest, senescence, apoptosis, or autophagy.2–4 Reduction in wild-type p53 is clearly associated with the development of PCa cells,5,6 thus overexpression of wild-type TP53 should contribute to inhibition of the growth and progression of PCa cells.
RNA activation (RNAa) is a lately discovered phenomenon that small double-stranded RNAs (dsRNAs) can activate specific gene expression by targeting complementary sequences on the gene promoter, and such dsRNA molecules are termed as small activating RNAs (saRNAs).7 A previous study reported that a candidate saRNA, dsP53-285, can induce wild-type p53 expression by targeting its promoter in chimpanzee cells, and the overexpression of wild-type p53 triggered by such RNAa has a potent capacity to induce upregulation of p21WAF1/CIP1 (p21), which suppresses the growth of the chimpanzee cells. Moreover, this RNAa phenomenon is conserved in mammalian species.8 This prompted us to investigate whether this saRNA can also activate the expression of wild-type p53 and inhibit the growth of human PCa cells.
In this study, dsP53-285 was transfected via liposomes into the human PCa cell lines LNCaP and DU145. We found that expression of wild-type p53 and its downstream gene, p21, was significantly upregulated at 72 hours following transfection. Enhanced p53 can inhibit proliferation and clonogenicity of PCa cells by modulating the expression of Cyclin D1 and cyclin-dependent kinase (CDK).
Materials and methods
Design of dsRNAs
The dsRNAs used in this study possess two-nucleotide 3′ overhangs and were chemically synthesized by RiboBio Co., Ltd. (Guangzhou, People’s Republic of China). The p53-targeted small interfering RNA siP53 was used to silence p53 expression, and a control dsRNA (dsControl) lacking significant homology to all known human sequences was designed as a negative control.9,10 The sequences of all tested RNAs are listed in Table S1.
Cell culture and transfection
The human PCa cell lines LNCaP and DU145 (American Type Culture Collection [ATCC], Manassas, VA, USA) were cultured in RPMI 1640 medium (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA) in a humidified atmosphere with 5% CO2 at 37°C. LNCaP cells are known to harbor wild-type p53, whereas DU145 cells are known to contain two mutations in the p53 gene, specifically in codon 223 (proline to leucine) and codon 274 (valine to phenylalanine).11
The day before transfection, cells were treated with trypsin and seeded into a new six-well plate at a density of 50%–60%, then cultured without antibiotics. All dsRNAs were transfected at a final concentration of 50 nM with Lipofectamine RNAiMax (Thermo Fisher Scientific) according to the manufacturer’s instructions. Additionally, dsRNA was replaced by opti-MEM (Invitrogen/Gibco, Carlsbad, CA, USA), a medium in which liposomes is combined with dsRNA, for mock transfection. The Institutional Review Board of Tongji Hospital approved the use of cells in this study.
RNA extraction and real-time quantitative polymerase chain reaction
Extraction of total cellular RNA from PCa cells was carried out using Trizol reagent (Thermo Fisher Scientific). RNA (500 ng) was reverse transcribed using a reverse transcription kit (TaKaRa, Dalian, People’s Republic of China) according to the manufacturer’s instructions. Real-time polymerase chain reaction (PCR) was performed on a Mx3000P system (Stratagene, Cedar Creek, TX, USA) with SYBR Premix Ex Taq II (TaKaRa), and the program was set for an initial denaturation step of 5 minutes at 95°C, followed by 40 cycles at 95°C for 15 seconds, 60°C for 30 seconds, and 72°C for 30 seconds, with a final extension at 72°C for 5 minutes. Glyceraldehyde 3-phosphate dehydrogenase was used as an internal control. Relative expression of the targeted genes was calculated by the 2−ΔΔCt method. The sequences of all primers, provided commercially by Thermo Fisher Scientific, are listed in Table S2.
Protein isolation and immunoblot analysis
PCa cells were harvested at 72 hours after transfection, and total proteins were isolated by RIPA lysis buffer (Beyotime Biotech, Haimen, People’s Republic of China) supplemented with protease and phosphatase inhibitor cocktail (Hoffman-La Roche Ltd., Basel, Switzerland). Detection of total cellular protein concentration was conducted using a BCA protein assay kit (Beyotime). Proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (Boster, Wuhan, People’s Republic of China), then transferred to polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). The final blots were blocked in 5% nonfat dried milk for 1 hour at room temperature and then incubated overnight with the primary antibodies at 4°C. The primary antibodies were p53 (1:1,000), p21 (1:1,000) (both Cell Signaling Technology, Denver, MA, USA), Cyclin D1 (1:1,000), CDK4 (1:1,000), CDK6 (1:500) (all Affinity, Cincinnati, OH, USA), glyceraldehyde 3-phosphate dehydrogenase (1:1,000), and α-tubulin (1:500) (both Boster). The membranes were then incubated with the appropriate secondary antibodies for 1 hour at room temperature, before detection with an enhanced chemiluminescence assay kit (EMD Millipore).
Cell cycle analysis
Cells were treated with trypsin at 72 hours after transfection and fixed in 70% ethanol for 2 hours at 4°C. After being washed twice with phosphate-buffered saline, the cells were incubated with 0.1 mg/mL RNase A (Sigma-Aldrich Co., St Louis, MO, USA) at 37°C for 30 minutes. The cells were then resuspended in 0.05 mg/mL of propidium iodide (Keygen Biotech, Nanjing, People’s Republic of China) at 4°C for 30 minutes while being protected from light. Finally, the processed cells were analyzed in a FACSort flow cytometer (BD Biosciences, San Jose, CA, USA). Evaluation of the data was performed by CellQuest software (BD Biosciences).
Cell proliferation assay
Cell proliferation ability was assessed by the CellTiter 96® AQueous One Solution Cell Proliferation Assay kit (Promega Corporation, Fitchburg, WI, USA). Cells transfected with dsRNA were treated with trypsin and grown in a 96-well plate at a density of 1,000 cells/well. Cell growth was assessed daily for 5 days. At each measured time point, the medium of each well was substituted with 100 μL of fresh medium premixed with 10 μL of CellTiter 96® AQueous One Solution. After incubation at 37°C for 2 hours, the absorbance of each well at 490 nm was detected by a microplate reader (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Colony formation assay
At 24 hours after transfecting with indicated dsRNAs, cells were treated with trypsin and reseeded into new six-well plates with 1,000 cells/well. The culture medium was changed every 3 days to maintain cell growth. On day 10, the colonies were fixed with methanol and stained with 0.5% crystal violet (Sigma-Aldrich Co.) for 30 minutes at room temperature. Colony formation rate was calculated by the following equation: colony formation rate = number of colonies/number of seeded cells ×100%.
Statistical analysis
All values are presented as mean ± standard deviation. Statistical significance was evaluated using Student’s t-test. Analyses were carried out using SPSS software (Version 13.0; SPSS Inc., Chicago, IL, USA). P<0.05 was considered statistically significant.
Results
dsP53-285 activates p53 expression by targeting its promoter
The results verified that dsP53-285 can induce p53 expression by targeting its promoter sequences at positions -285/-267 relative to the transcription start site in chimpanzee cells. As the sequences of the -285/-267 site are completely identical between human and chimpanzee (Figure 1A),8 it is probable that dsP53-285 also has the ability to activate p53 expression in human PCa cells.
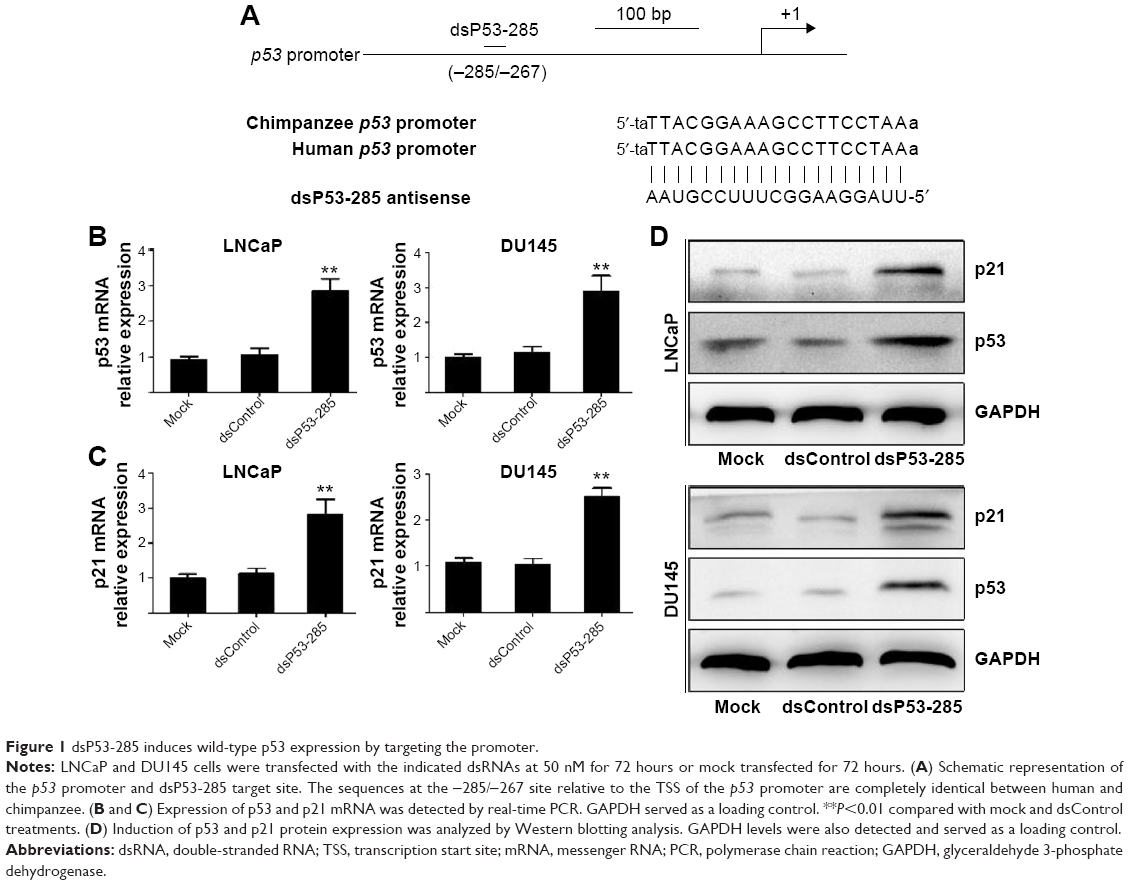 | Figure 1 dsP53-285 induces wild-type p53 expression by targeting the promoter. |
We transfected synthetic dsP53-285 into LNCaP and DU145 cells and analyzed p53 expression after 72 hours. Compared with the mock and dsControl groups, dsP53-285 caused significant upregulation of p53 messenger RNA (mRNA) and protein (Figure 1B and D). As a downstream gene of TP53, p21 is directly regulated only by wild-type TP53.
Real-time polymerase chain reaction revealed that p21 mRNA was overexpressed in both cells after dsP53-285 transfection (Figure 1C), and Western blotting further confirmed this (Figure 1D). These results indicated that dsP53-285 can activate wild-type p53 expression by targeting its promoter.
dsP53-285 facilitates PCa cell cycle arrest and inhibits cell growth
To further evaluate the effect of dsP53-285 on PCa cell growth, genes that directly control cell cycle progression,12 such as Cyclin D1 and CDK4/6, were assessed. Compared with the mock and dsControl treatments, Cyclin D1 and CDK4/6 mRNA levels were significantly reduced at 72 hours after dsP53-285 transfection in LNCaP and DU145 cells (Figure 2A). Furthermore, immunoblotting analysis also confirmed the inhibitory effects mediated by dsP53-285 (Figure 2B).
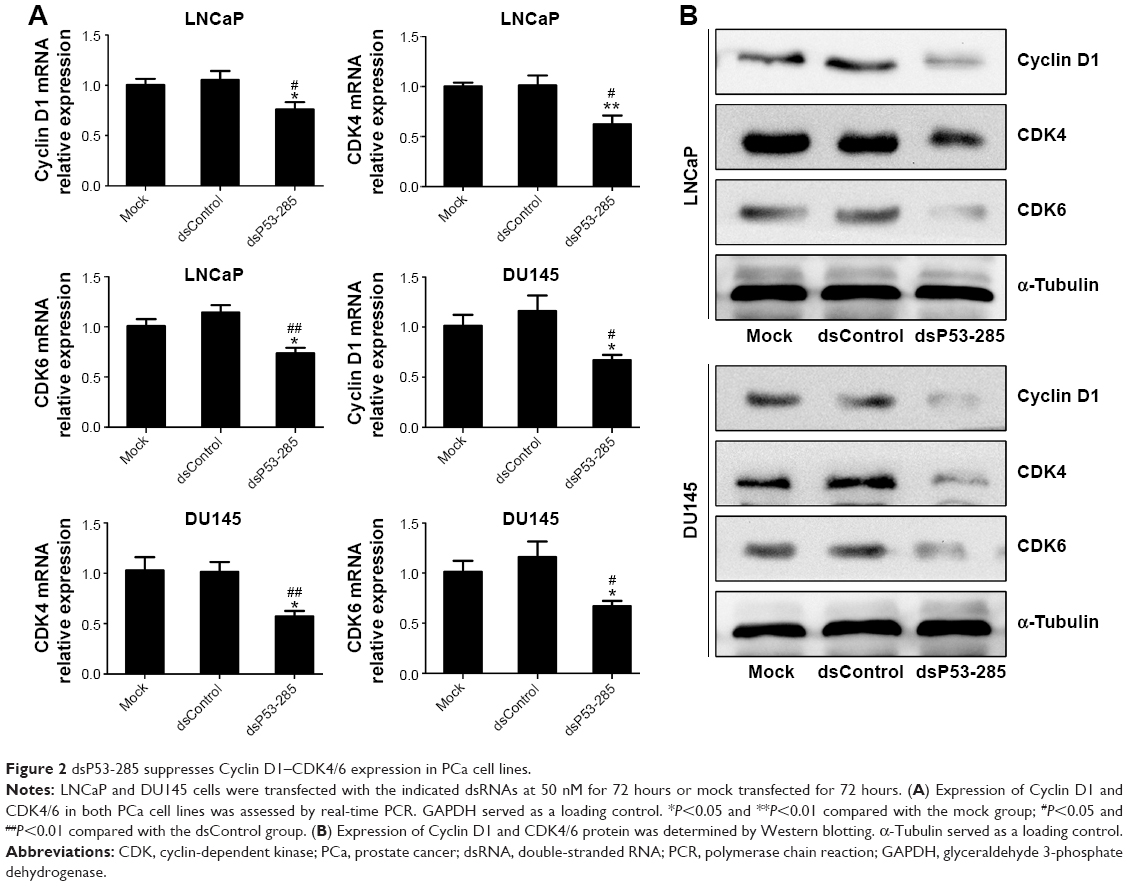 | Figure 2 dsP53-285 suppresses Cyclin D1–CDK4/6 expression in PCa cell lines. |
Flow cytometry was conducted to assess the cell cycle distribution after transfection with the indicated dsRNAs. As shown in Figure 3A, compared with the mock and dsControl groups, cells transfected with dsP53-285 presented a prominent accumulation of cells in the G0/G1 phase and a decrease in the S phase.
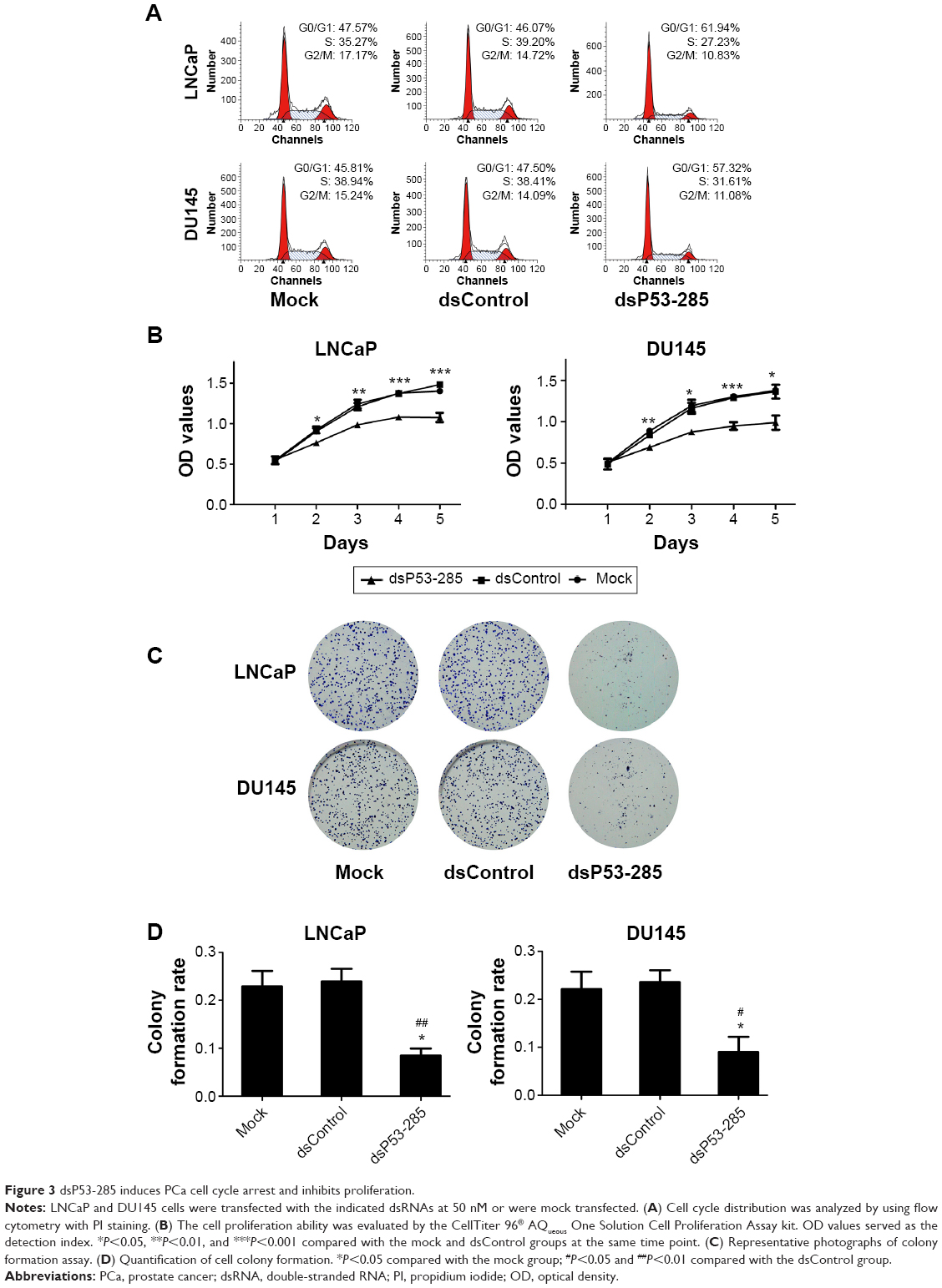 | Figure 3 dsP53-285 induces PCa cell cycle arrest and inhibits proliferation. |
Moreover, a cell proliferation assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay) was performed daily for 5 days to analyze the viability of PCa cells. Compared with the mock and dsControl treatments, the proliferative ability of PCa cells transfected with dsP53-285 was distinctly decreased from the second day (Figure 3B).
Colony formation assay was performed to identify differences in growth modes between the groups. The colony formation ability of PCa cells transfected with dsP53-285 appeared to be suppressed as the number and size of colonies were obviously less than those of the mock and dsControl groups (Figure 3C and D). These data showed that dsP53-285 can induce cell cycle arrest and inhibit the cell proliferation and colony formation abilities of PCa cell lines LNCaP and DU145.
dsP53-285 inhibits Cyclin D1–CDK4/6 expression mainly by manipulating p53
To verify that dsP53-285 inhibits PCa cell proliferation and clonogenicity mainly by regulating p53 expression, we knocked down p53 expression by transfecting a small interfering RNA (siP53) into LNCaP and DU145 cells. As seen in Figure 4A and C, compared with dsP53-285 transfection, cotransfection of dsP53-285 and siP53 for 72 hours significantly decreased the expression levels of p53 mRNA and protein in both cell types. Based on that p21 is directly modulated by wild-type p53, the p21 mRNA and protein expression were correspondingly inhibited after silencing of p53 (Figure 4B and C). We then examined Cyclin D1–CDK4/6 expression after cotreatment with dsP53-285 and siP53. Consistent with our expectations, the inhibitory effects of dsP53-285 on Cyclin D1–CDK4/6 mRNA and protein expression were downregulated after cotransfection of siP53 in LNCaP and DU145 cells (Figure 5A and B).
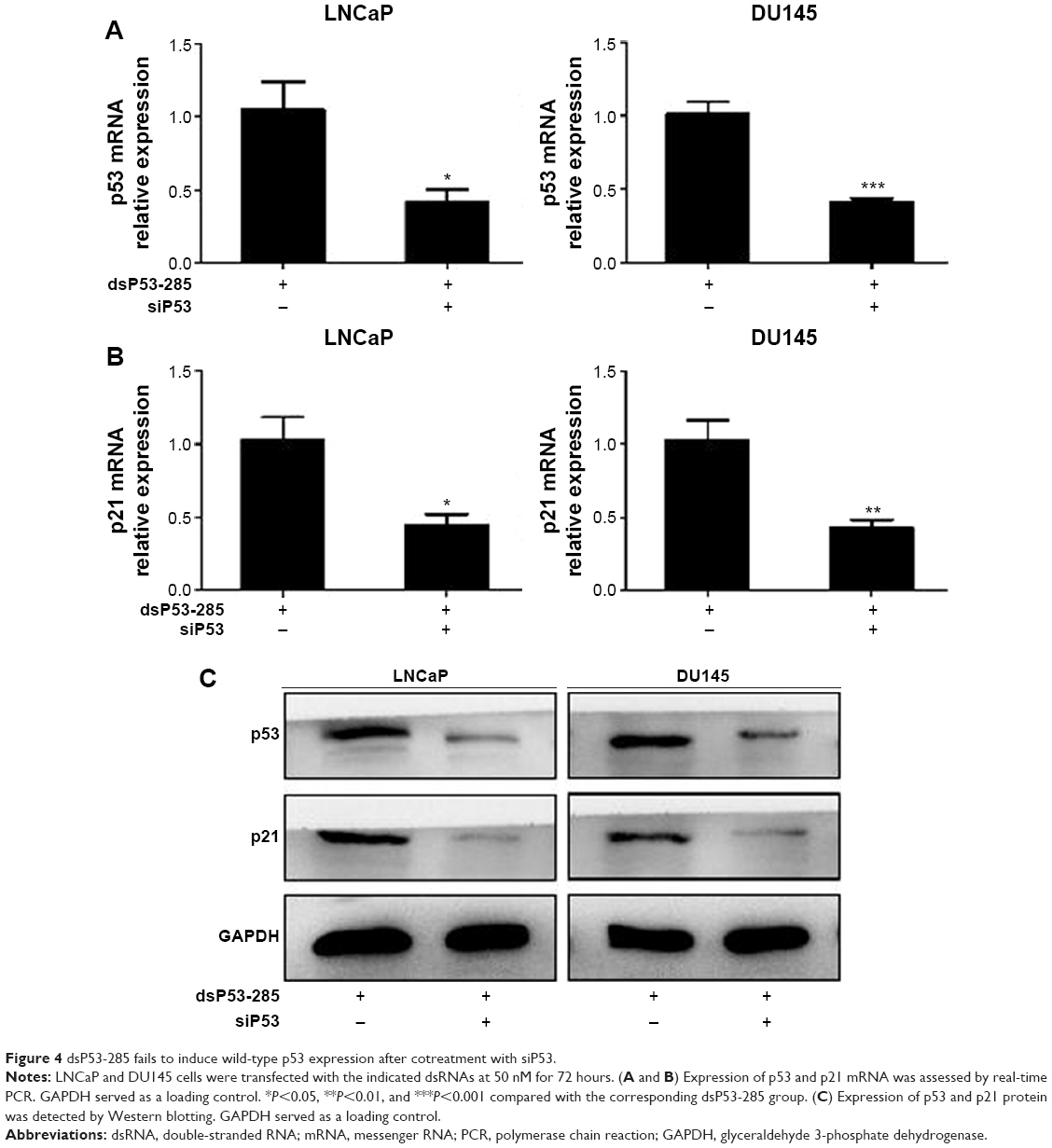 | Figure 4 dsP53-285 fails to induce wild-type p53 expression after cotreatment with siP53. |
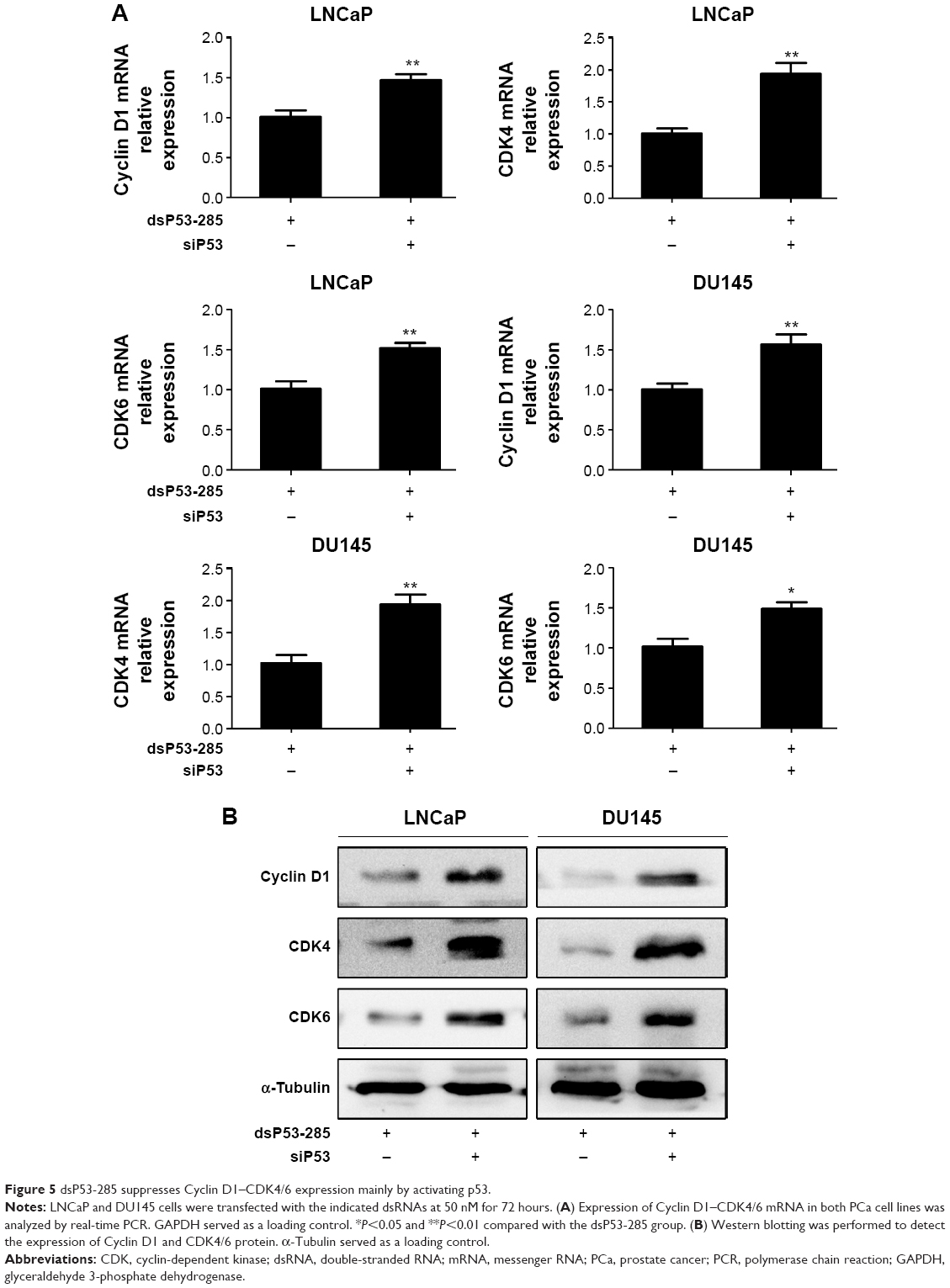 | Figure 5 dsP53-285 suppresses Cyclin D1–CDK4/6 expression mainly by activating p53. |
These findings indicate that dsP53-285 inhibits Cyclin D1–CDK4/6 complex largely by modulating p53 expression.
dsP53-285 suppresses PCa cells by upregulating p53
We next investigated whether p53 was mainly responsible for inhibiting the proliferation and clonogenicity of PCa cells. We cotransfected dsP53-285 and siP53 into LNCaP and DU145 cells. Flow cytometry showed a significant decrease in the G0/G1 population and a corresponding increase in the S population in both cell lines, which indicated that depletion of p53 markedly reduced G0/G1 cycle arrest mediated by dsP53-285 (Figure 6A). Additionally, the cell proliferation ability was also upregulated after knockout of p53 (Figure 6B), and the colonies formed by dsP53-285 and siP53 cotransfected cells were greater in number and larger in size than those of the single dsP53-285-transfected cells (Figure 6C and D). These data together strongly indicate that dsP53-285 suppresses PCa cell growth primarily by regulating p53 expression.
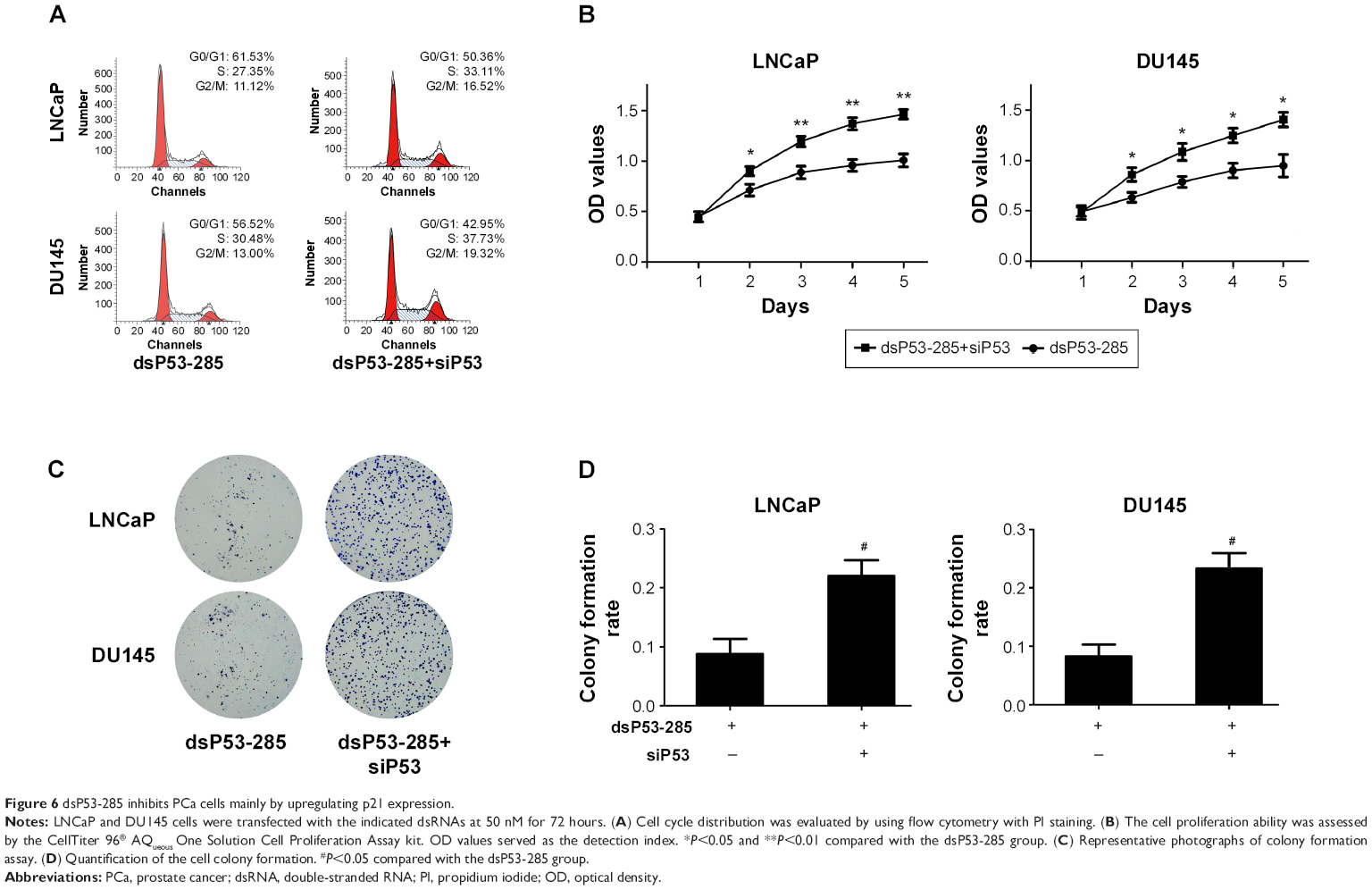 | Figure 6 dsP53-285 inhibits PCa cells mainly by upregulating p21 expression. |
Discussion
In the present study, we demonstrated that an exogenous synthetic dsP53-285, which is completely complementary to sequences at positions -285/-267 relative to the transcription start site of the p53 gene promoter, had a robust capacity to activate wild-type p53 expression in human PCa cells. As a downstream gene of TP53, p21 expression was accordingly upregulated. Moreover, genes (Cyclin D1 and CDK4/6) mediating the cell cycle were downregulated, resulting in cycle arrest of PCa cells. The cell proliferation and colony formation abilities were also inhibited. In addition, the antitumor effects of dsP53-285 were caused by modulating wild-type p53 expression.
p53 proteins in cancer cells are divided into two forms: wild-type p53 and TP53 mutants. TP53 mutants occur at high frequencies in all human cancers and usually exhibit cancer-promoting effects by interfering with the sequence-specific DNA-binding ability of wild-type p53.13–16 Thus, we aimed to confirm the type of p53 proteins activated by dsP53-285. As a tumor suppressor gene, p21 is positively regulated by wild-type p53 and participates in the p53-mediated DNA-damaging response. However, TP53 mutants have been shown to suppress the expression of wild-type p53 targeting p21 which functions as a vital cell cycle inhibitor and causes alteration in the Cyclin–CDK complex.17 In this study, we demonstrated that the expression levels of p21 were positively correlated with upregulation of p53. Thus, we conclude that the upregulated expression of TP53 proteins mediated by dsP53-285 was wild type rather than mutant.
Persistent proliferation, mediated by abnormalities in cell cycle regulation, is one prominent characteristic of cancer cells. Therefore, cell cycle arrest is considered to be a potential pathway for cancer therapy.18 The cell cycle is modulated by Cyclin–CDK complexes. Reduction in Cyclin D1–CDK4/6 complexes effectively restrains the progression of G0/G1 to S phase. Our results showed that dsP53-285 could induce wild-type p53 expression, thus promoting p21 expression. It is known that p21 is a broad-acting CDK inhibitor and can prompt cell cycle arrest in the G1 phase by binding to and downregulating Cyclin–CDK complexes.19,20 Correspondingly, the cell proliferation ability is suppressed. Earlier studies have shown that p21 is frequently expressed at low levels in PCa cells and is associated with prognosis.21 Therefore, upregulation of p21 induced by dsP53-285 may be a promising strategy to suppress cancer cell growth.
As a gene silencing mechanism, RNA interference mainly functions at the posttranscriptional level, efficiently directing specific degradation of homologous mRNA or repressing translation,22,23 whereas RNAa is a transcriptional gene-activating mechanism that involves transcriptional and epigenetic alterations. The activating function appears at around 24–48 hours after transfection, peaks at 72 hours, and lasts for ~14 days.7,24 Previous studies have shown that, both in vitro and in vivo, specific gene activation by RNAa can effectively inhibit cancer cell growth and progression.25,26 As saRNA does not alter the expression of nonspecific genomic loci while activating gene expression, it should not pose a potential threat to host genome integrity.7 Therefore, RNAa should offer a safe method for cancer therapy via upregulation of specific tumor suppressors.
Conclusion
In summary, we have provided proof of principle that dsP53-285 can significantly stimulate wild-type p53 expression and consequently lead to inhibition of the proliferation and clonogenicity of PCa cells. More importantly, the antitumor effect of dsP53-285 on PCa cells is mainly achieved by manipulating wild-type p53 expression. To the best of our knowledge, our study is the first to identify that dsP53-285 can induce p53 expression in PCa cells LNCaP and DU145 cells and inhibit cell progression. However, further studies are needed to explore the exact mechanism of RNAa and to develop effective methods for saRNA delivery in vivo for clinical cancer therapy.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no 81372759), People’s Republic of China. The funders had no role in study design, data collection or analysis, decision to publish, or preparation of the paper.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, et al. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | ||
Liu J, Zhang C, Hu W, et al. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015;356(2 pt A):197–203. | ||
Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137(3):413–431. | ||
Chen F, Wang W, El-Deiry WS. Current strategies to target p53 in cancer. Biochem Pharmacol. 2010;80(5):724–730. | ||
Burchardt M, Burchardt T, Shabsigh A, et al. Reduction of wild type p53 function confers a hormone resistant phenotype on LNCaP prostate cancer cells. Prostate. 2001;48(4):225–230. | ||
Rokhlin OW, Gudkov AV, Kwek S, et al. p53 is involved in tumor necrosis factor-alpha-induced apoptosis in the human prostatic carcinoma cell line LNCaP. Oncogene. 2000;19(15):1959–1968. | ||
Li LC, Okino ST, Zhao H, et al. Small dsRNAs induce transcriptional activation in human cells. Proc Natl Acad Sci U S A. 2006;103(46):17337–17342. | ||
Huang V, Qin Y, Wang J, et al. RNAa is conserved in mammalian cells. PLoS One. 2010;5(1):e8848. | ||
Srsen V, Gnadt N, Dammermann A, et al. Inhibition of centrosome protein assembly leads to p53-dependent exit from the cell cycle. J Cell Biol. 2006;174(5):625–630. | ||
Miyazaki M, Sakaguchi M, Akiyama I, et al. Involvement of interferon regulatory factor 1 and S100C/A11 in growth inhibition by transforming growth factor beta 1 in human hepatocellular carcinoma cells. Cancer Res. 2004;64(12):4155–4161. | ||
Isaacs WB, Carter BS, Ewing CM. Wild-type p53 suppresses growth of human prostate cancer cells containing mutant p53 alleles. Cancer Res. 1991;51(17):4716–4720. | ||
Wang C, Ge Q, Chen Z, et al. A new double stranded RNA suppresses bladder cancer development by upregulating p21 (Waf1/CIP1) expression. Biomed Res Int. 2015;2015:304753. | ||
Hollstein M, Sidransky D, Vogelstein B, et al. p53 mutations in human cancers. Science. 1991;253(5015):49–53. | ||
Milner J, Medcalf EA, Cook AC. Tumor suppressor p53: analysis of wild-type and mutant p53 complexes. Mol Cell Biol. 1991;11(1):12–19. | ||
Kato S, Han SY, Liu W, et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A. 2003;100(14):8424–8429. | ||
Schlomm T, Iwers L, Kirstein P, et al. Clinical significance of p53 alterations in surgically treated prostate cancers. Modern Pathol. 2008;21(11):1371–1378. | ||
Vikhanskaya F, Lee MK, Mazzoletti M, et al. Cancer-derived p53 mutants suppress p53-target gene expression – potential mechanism for gain of function of mutant p53. Nucleic Acids Res. 2007;35(6):2093–2104. | ||
Sperka T, Wang J, Rudolph KL. DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol. 2012;13(9):579–590. | ||
Stivala LA, Cazzalini O, Prosperi E. The cyclin-dependent kinase inhibitor p21CDKN1A as a target of anti-cancer drugs. Curr Cancer Drug Targets. 2012;12(2):85–96. | ||
Starostina NG, Kipreos ET. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol. 2012;22(1):33–41. | ||
Sánchez-González C, Ciudad CJ, Izquierdo-Pulido M, et al. Urolithin A causes p21 up-regulation in prostate cancer cells. Eur J Nutr. Epub 2015 May 12. | ||
Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. | ||
Wang Z, Rao DD, Senzer N, et al. RNA interference and cancer therapy. Pharm Res. 2011;28(12):2983–2995. | ||
Zheng L, Wang L, Gan J, et al. RNA activation: promise as a new weapon against cancer. Cancer Lett. 2014;355(1):18–24. | ||
Kosaka M, Kang MR, Yang G, et al. Targeted p21WAF1/CIP1 activation by RNAa inhibits hepatocellular carcinoma cells. Nucl Acid Ther. 2012;22(5):335–343. | ||
Ren S, Kang MR, Wang J, et al. Targeted induction of endogenous NKX3-1 by small activating RNA inhibits prostate tumor growth. Prostate. 2013;73(14):1591–1601. |
Supplementary materials
 | Table S1 Sequences of dsRNAs used in the present study |
 | Table S2 Sequences of real time quantitative PCR primers used in the present study |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.