")
Back to Journals » OncoTargets and Therapy » Volume 11
Overexpression of NELFCD promotes colorectal cancer cells proliferation, migration, and invasion
Authors Song S, Li D, Yang C, Yan P, Bai Y, Zhang Y, Hu G, Lin C, Li X
Received 3 September 2018
Accepted for publication 30 October 2018
Published 5 December 2018 Volume 2018:11 Pages 8741—8750
DOI https://doi.org/10.2147/OTT.S186266
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Shenglei Song, Daojiang Li, Chunxing Yang, Peicheng Yan, Yang Bai, Yi Zhang, Gui Hu, Changwei Lin, Xiaorong Li
Department of Gastrointestinal Surgery, The Third Xiang Ya Hospital of Central South University, Changsha, Hunan 410013, China
Purpose: Negative elongation factor complex member C/D (NELFCD), mapped to chromosome 20q13.32, has been found to be significantly overexpressed in colorectal cancer (CRC) by our previous research. However, whether its overexpression contributes to CRC development is unknown. We aimed to explore the biological and clinical roles of NELFCD in CRC.
Materials and methods: The expression of NELFCD was detected by qRT-PCR and Western blot. The biological function of NELFCD on CRC cell proliferation, migration, invasion, and apoptosis was detected by cell counting kit-8, plate colony formation assay, transwell migration and invasion assays, and flow cytometry in vitro and by murine xenograft tumor growth in vivo. Moreover, we evaluated the correction between its expression level and clinicopathologic parameters.
Results: We found NELFCD was overexpressed in 50 pairs of CRC tissues in comparison to the adjacent nontumor tissues (P<0.05). Knockdown of NELFCD significantly impaired cell proliferation, migration and invasion abilities, facilitated cell apoptosis in vitro, and inhibited tumorigenesis of CRC cells in vivo. NELFCD levels were remarkably connected with tumor location in CRC patients.
Conclusion: NELFCD is overexpressed and plays an oncogenic role in CRC. Targeting NELFCD may provide a potential therapeutic option for NELFCD-amplified tumors.
Keywords: NELFCD, colorectal carcinoma, CRC, oncogene
Introduction
Colorectal cancer (CRC) is the third most common cancer in men (100,6000 cases, 10.6% of the total) and the second most common cancer in women (79,4900 cases, 9.2% of the total) worldwide.1 Although the increased use of screening and removal of precancerous adenomas has decreased the CRC incidence in the United States,2,3 the incidence and mortality of CRC are still increased in economically developing countries, especially in China.4,5 Novel diagnostic, prognostic, and treatment biomarkers still need to be explored and applied to improve tumor therapies and patient survival.
Emerging evidence indicates that CRC is a heterogeneous disease, arising through the accumulation of genetic and epigenetic alterations, with three major molecular pathways involved: chromosomal instability, microsatellite instability (MSI), and the CpG island methylator phenotype.6–8 Chromosomal instability is a common sign of CRC, resulting in copy number alterations (CNAs) and gain or loss in copies of DNA sections, which confers growth advantage and metastatic competence to tumor cells, thus playing a crucial role in CRC initiation and progression.9–11 Multiple putative oncogenes at the chromosome 20q amplicon contribute to the progression from colorectal adenoma to carcinoma.12 Our previous research found that negative elongation factor complex member C/D (NELFCD), which mapped to chromosome 20q, is markedly overexpression in CRC, which may be the result of CNA of NELFCD.13 Therefore, in this study, we aimed to know more about the role of NELFCD in CRC.
NELFCD is mapped to chromosome 20q13.32, and its official full name is negative elongation factor complex member C/D. The negative elongation factor (NELF) complex of proteins interacts with the DSIF protein complex to repress transcriptional elongation by RNA polymerase II. The protein encoded by NELFCD is an essential part of the NELF complex.14–18 As a subunit of NELF, NELFCD has rarely been studied. The biological roles of NELFCD in CRC remain unclear. Thus, based on our previous study, we explored whether NELFCD, which was overexpressed by CNA, was involved in CRC development.
Materials and methods
Clinical samples
Two cohorts of CRC patients were included. For The Cancer Genome Atlas (TCGA) cohort, mRNA expression data of NELFCD in CRC were acquired from Gene Expression Profiling Interactive Analysis.19 For Changsha cohort, 50 CRC cancer tissues and matched adjacent normal tissues (more than 5 cm away from the tumor) were obtained from patients who were pathologically and clinically diagnosed with CRC. Elective surgery was carried out on these patients at Third Xiangya Hospital of Central South University (Changsha, China) from 2016 to 2017. All samples were immediately frozen in liquid nitrogen after surgery and stored at −80°C until DNA and RNA extraction. Clinical information of these patients was collected including age, sex, smoking history, tumor location, tumor size (maximum diameter), tumor differentiation, TNM stage, tumor grade, lymph node metastasis, and MSI status. Tumor location, tumor size, tumor differentiation, and TNM staging were based on pathology reports. The use of tissues for this study was approved by the ethics committee of Third Xiangya Hospital of Central South University and conducted in accordance with the Declaration of Helsinki. Before using these clinical materials for research purposes, all the patients signed an informed consent. None of these patients received any preoperative chemotherapy or radiotherapy.
Extraction of total RNA, cDNA synthesis, and quantitative real-time (qRT)-PCR
Total RNA were extracted from tissues and cell lines using Trizol (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s protocol. cDNA was synthesized using the PrimeScript RT reagent Kit (TOYOBO, Osaka, Japan) following the manufacturer’s protocol. For qRT-PCR, the NELFCD primer sequences were sense, 5′-ATGCTGAACTTCACCGTTAAGC-3′ and antisense, 5′-TGGGCAAACAGGTACGTGTG-3′. To normalize for RNA concentration, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. The sequences of the GAPDH primers were sense, 5′-ACCTGACCTGCCGTCTAGAA-3′ and antisense, 5′-TCCACCACCCTGTTGCTGTA-3′. The 2−ΔΔCT method was used to calculate the relative amount of NELFCD compared with GAPDH expression. qRT-PCR was performed using the KOD SYBR qPCR Mix kit (TOYOBO). The amplification protocol included an initial denaturation step at 98°C for 2 minutes, followed by 40 cycles of 98°C for 10 seconds, 60°C for 10 seconds and 68°C for 30 seconds. qRT-PCR was performed in a Light Cycler 480 II instrument (Hoffman-La Roche Ltd., Basel, Switzerland).
Western blotting
Total protein was extracted from tissues and cell lines using RIPA (KeyGen Biotech, Nanjing, China) following the manufacturer’s protocol. The total protein lysates were incubated with SDS-PAGE Sample Loading Buffer (Biosharp, Hefei, China) for 5 minutes at 100°C and then were kept at −80°C until use. The following primary antibodies were used: mouse anti-NELFCD (1:500; Santa Cruz Biotechnology, Dallas, TX, USA) and rabbit anti-GAPDH (1:1,000; Santa Cruz). The protein extracts (40 μg) were then subjected to 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA) electrophoretically. After blocking with 5% degreased milk and washing with PBS for 10 minutes three constitutive times, the membranes were incubated overnight at 4°C with mouse anti-NELFCD or rabbit anti-GAPDH. The membranes were then washed with PBS for 10 minutes three constitutive times and incubated with secondary antibodies for 1 hour. Finally, the membranes were washed with PBS. The protein bands’ signals were detected with the Odyssey Clx (Gene Company Ltd, Chai Wan, Hong Kong). ImageJ (1.48V; NIH, USA) analysis was used to quantify the signal intensities of Western blots of NELFCD taking GAPDH as the control protein.
Cell culture
Human SW480 and HCT116 CRC cell lines were obtained from the Cell Center of Xiangya School of Medicine, Central South University (Hunan, China). SW480 and HCT116 were maintained in L-15 and McCoy’s 5A Medium (KeyGen Biotech, Nanjing, China) with 10% FBS (Biological Industries, Beit Haemek, Israel). All cells were maintained in 1% penicillin/streptomycin and cultured at 37°C in a humidified incubator with 5% CO2.
Establishment of stable cell lines
Short hairpin RNA (sh-RNA) was targeted at the mRNA of NELFCD gene. The sh-RNA sequence was cloned into the lentivirus (synthesized by Genepharma, Suzhou, China) expression vector to knockdown the expression of NELFCD. A sh-RNA duplex (sh-NELFCD: sh-NELFCD, 5′-GGAACAGATGATTGCACATAC-3′) corresponding to the NELFCD RNA sequences and a negative control (sh-NC, 5′-TTCTCCGAACGTGTCACGT-3′) were transfected into SW480 and HCT-116 following the manufacturer’s protocol, and infected cells expressing each vector were selected with 5 μg/mL puromycin (Biofroxx, Berlin, Germany) for ten constitutive days. NELFCD expression levels were examined via qRT-PCR and Western blotting.
Plate colony formation assays
SW480 and HCT116 cells (1×103) were counted and seeded into six-well plates, then cultured at 37°C in a humidified incubator with 5% CO2 for 10–14 d until the colonies were visible. The cells were washed with PBS and fixed with methanol for 30 minutes. The colonies were stained with Crystal Violet solution for 30 minutes and washed with PBS. After air-drying, the colonies with more than 50 cells were counted.
Cell counting kit-8 (CCK-8)
SW480 and HCT-116 cells were plated in flat-bottom 96-well plates (1×104 and 5×103 cells/well) and supplemented with 100 mL L15 and Mc medium per well. After incubation at 37°C in a humidified incubator with 5% CO2 for 24, 48, 72, and 96 hours, respectively, 10 μL of CCK-8 (Dojindo, Kumamoto, Japan) was added to each well. Then, after 3 hours of culture, colorimetric analysis was performed on a microplate reader (BioTek, Winooski, VT, USA) at a wavelength of 450 nm. The assay was performed using six replicates. All experiments were performed in triplicate.
Transwell migration and invasion assays
For migration assays, infected SW480 cells (1×105 in 200 μL of serum-free L15 medium and 0.1% BSA) and HCT-116 cells (1×105 in 200 μL of serum-free Mc medium and 0.1% BSA) were seeded into the upper chamber of transwell plates in a 24-well format with 8 mm diameters (Corning Costar, USA). Then, 600 μL of medium containing 10% FBS was added to the bottom chamber as a chemoattractant. After 24–48 hours of culture, cells were fixed with methanol and stained with Crystal Violet solution. The remaining cells were removed from the top of the permeable membrane using a cotton swab. Then, cells that migrated through the upper chamber were counted in five random fields (×200) under a light microscope (Olympus Corporation, Tokyo, Japan).
For invasion assays, the top chambers were coated with basement membrane Matrigel (BD Biosciences, San Jose, CA, USA) at 37°C for 30 minutes. Infected SW480 and HCT116 cells (2×105) in serum-free medium were added into the top chambers; the bottom chambers were filled with medium containing 10% FBS. After 24–48 hours of culture, cells were fixed with methanol and were stained with Crystal Violet solution. The remaining cells were removed from the top of the permeable membrane using a cotton swab. Then, cells that migrated through the upper chamber were counted in five random fields (200×) under a light microscope (Olympus Corporation, Tokyo, Japan).
Flow cytometry
Apoptosis analysis: HCT116 and SW480 cells transfected with sh-NC or sh-NELFCD were trypsinized and resuspended in 1× binding buffer at 2.5×106 cells/mL. Then, 100 μL of this cell suspension was incubated with 5 μL of 7-aminoactinomycin D (BD Pharmingen, San Diego, CA, USA) and 5 μL APC Annexin (BD Pharmingen) for 15 minutes in the dark. The reaction was terminated with the addition of 400 μL 1× binding buffer and analyzed with flow cytometer (BD Pharmingen) using the Flowjo software.
Cell-cycle analysis: The cells were fixed in 70% ethanol, washed twice with PBS, and then labeled with PI-RNase Staining Buffer (BD Pharmingen) for 30 minutes in the dark. Samples were run on a flow cytometer, and the percentages of cells within each phase of the cell cycle were analyzed using Modfit software (Verity Software House, Topsham, ME, USA).
In vivo tumor growth
Four- to six-week-old female BALB/c nude mice were purchased from Hunan Silaikejingda Experimental Animal Co., Ltd. (Changsha, China). The mice were randomly divided into the test and control groups. There were six mice in each group. For the establishment of human tumors in the BALB/c nude mice, stable, inducible SW480-shNC or SW480-shNELFCD cells (4×106 cells/mouse) were subcutaneously injected into the right abdominal flanks in test and control mice, respectively. The tumor sizes were measured every 3 days, and the tumor volumes were calculated as length × width2 × 0.5. After 4 weeks, all of the mice were sacrificed, and all of the tumors were weighed, sectioned, and stained with H&E staining. All animal experimental procedures used in this study were approved by the Animal Ethics Committee of Central South University and conducted in accordance with the Guideline of the Care and Use of Laboratory Animals in Central South University.
Statistical analysis
All results were expressed as the mean ± SD unless otherwise indicated. To compare the difference between two groups, the Students t-test (two tailed) was performed. Pearson chi-squared test or Fisher’s exact test was used for analysis of the associations between patient clinicopathologic characteristics and NELFCD expression. All statistical tests were performed using SPSS, version 19.0 (IBM Corporation, Armonk, NY, USA), and a two-tailed P-value of 0.05 was considered statistically significant.
Results
NELFCD is upregulated in CRC tissues
The mRNA expression of NELFCD in 50 pairs of CRC (tumor) and adjacent normal tissue (normal) (Figure 1A), and protein expression of NELFCD in six pairs of CRC (tumor) and adjacent normal tissue (normal) (Figure 1B) from Changsha cohort were detected. The mRNA expression data of NELFCD in CRC were acquired from TCGA cohort (Figure 1C). In both Changsha cohort and TCGA cohort, the expression of NELFCD in cancer tissues was obviously higher than those in the noncancerous tissues. These results confirmed that upregulated expression of NELFCD might be crucial in CRC tumorigenesis. Based on the median NELFCD mRNA expression level (Changsha cohort) in 50 pairs of tissues, we divided the patients into two subgroups, with high or low tumor NELFCD expression levels (Figure 1D). Clinicopathologic factors were compared between the two NELFCD expression groups (Table 1). NELFCD expression was significantly correlated with tumor location (P=0.045). However, NELFCD expression was not associated with any other clinicopathologic factors in our study.
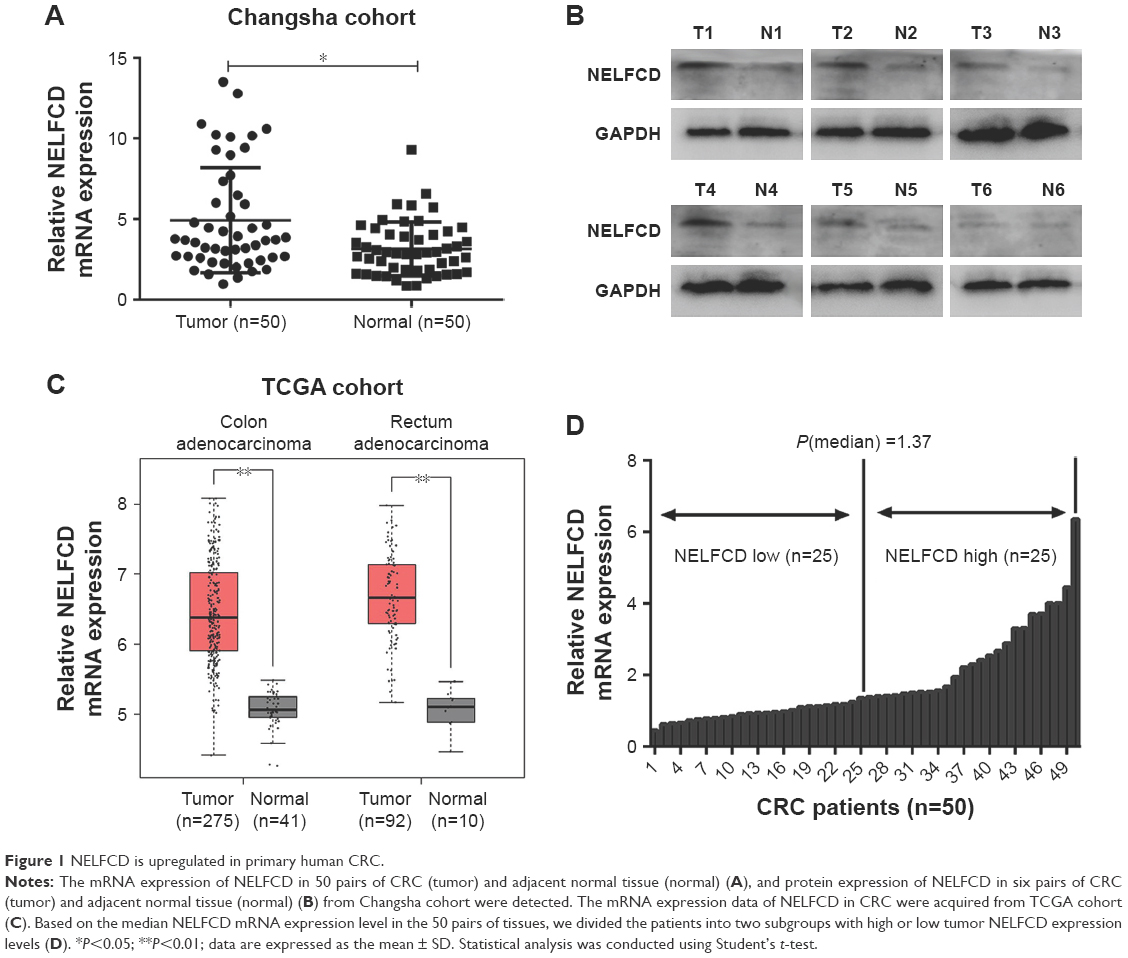 | Figure 1 NELFCD is upregulated in primary human CRC. |
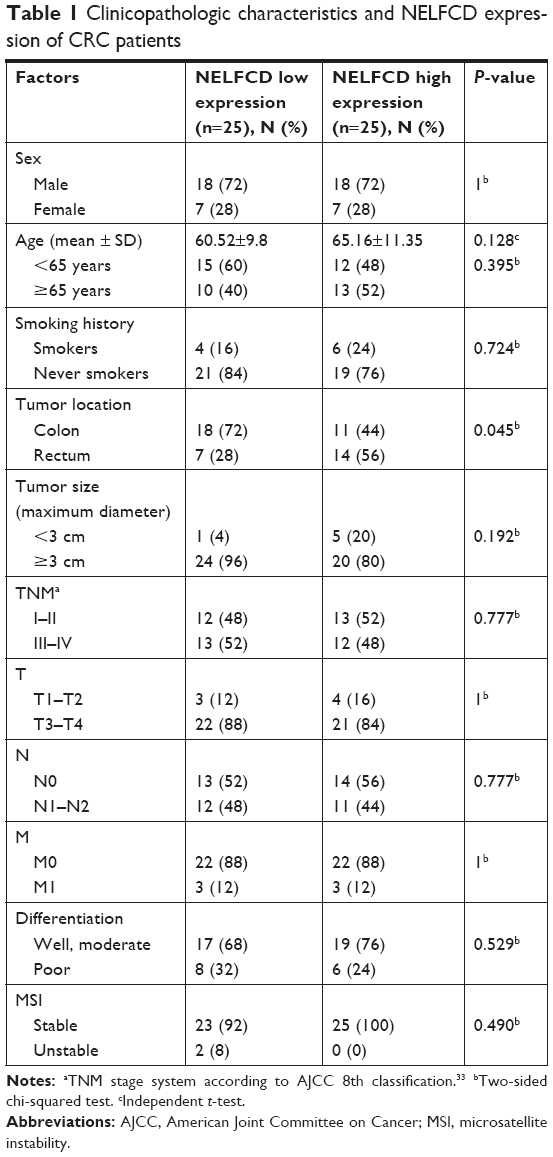 | Table 1 Clinicopathologic characteristics and NELFCD expression of CRC patients |
Knockdown of NELFCD represses CRC cell proliferation in vitro
We conducted NELFCD knockdown assays using sh-RNA to investigate the biological function of NELFCD in CRC cells. Using qRT-PCR and Western blotting, we confirmed that NELFCD expression in SW480 and HCT116 cells transfected with sh-NELFCD was significantly lower than that in cells transfected with sh-NC (Figure 2A and B).
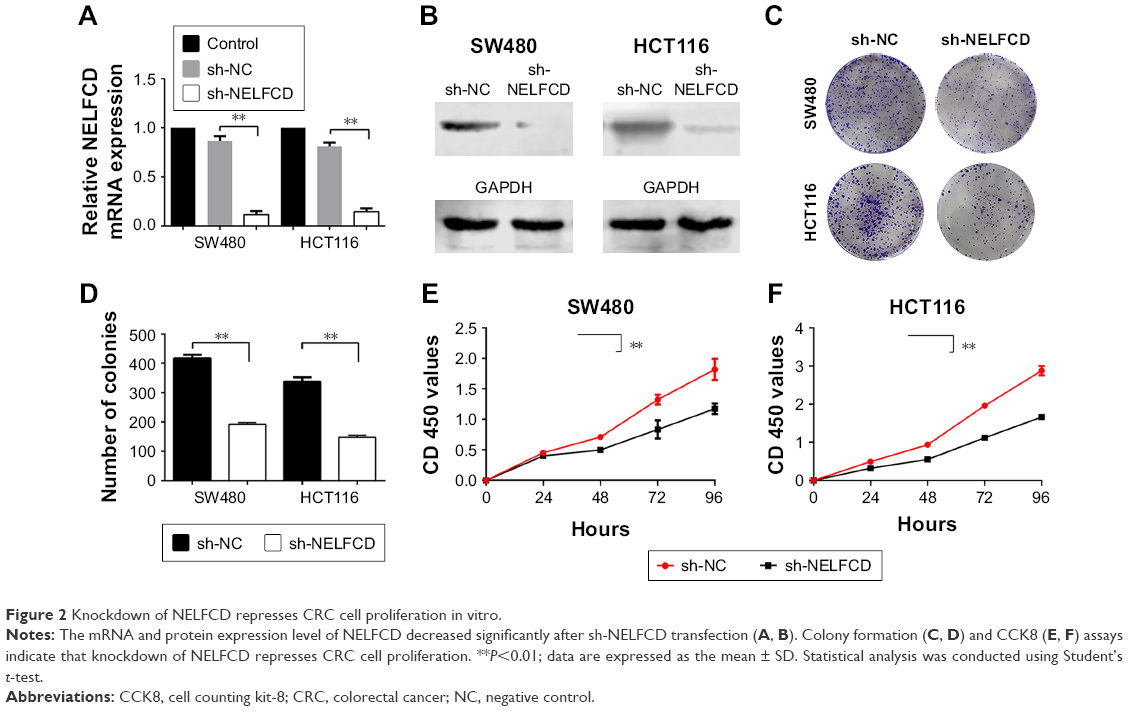 | Figure 2 Knockdown of NELFCD represses CRC cell proliferation in vitro. |
To determine whether knockdown of NELFCD represses cell proliferation in SW480 and HCT116 cells, plate colony formation assays were performed. The data of plate colony formation assays, compared with SW480 and HCT116 cells infected with sh-NC, showed that SW480 and HCT116 cells infected with sh-NELFCD resulted in 53.8% and 56.2% growth inhibition, respectively (P<0.01) (Figure 2C and D).
We next sought to determine whether NELFCD knockdown affected cell proliferation by CCK-8 assay. The data, compared with SW480 and HCT-116 cells infected with sh-NC, showed that the proliferation of SW480 and HCT-116 cells infected with sh-NELFCD was inhibited to 11.8%–35% (P<0.01), 29.6%–41.2% (P<0.01), 36.9%–43% (P<0.01), and 35.4%–42.4% (P<0.01) at 24, 48, 72, and 96 hours, respectively (Figure 2E and F). Knockdown of NELFCD remarkably inhibited cell proliferation.
Knockdown of NELFCD represses CRC cell migration and invasion, retards the G2/M transition, and facilitates cell apoptosis in vitro
We explored the effect of NELFCD on migration and invasion. Cells that migrated and invasived through the transwell plates were counted in five random fields (200×), and the average value of the five fields was expressed. The results showed that knockdown of NELFCD suppressed 52.3%–66.1% of the cells’ migratory activity in SW480 and HCT116 cells (Figure 3A and B) and inhibited 52%–86.5% of the cells’ invasive activity in SW480 and HCT116 cells (Figure 3C and D). These results, taken together, clearly indicated that knockdown of NELFCD expression markedly retarded cell migration and invasion in CRC cells.
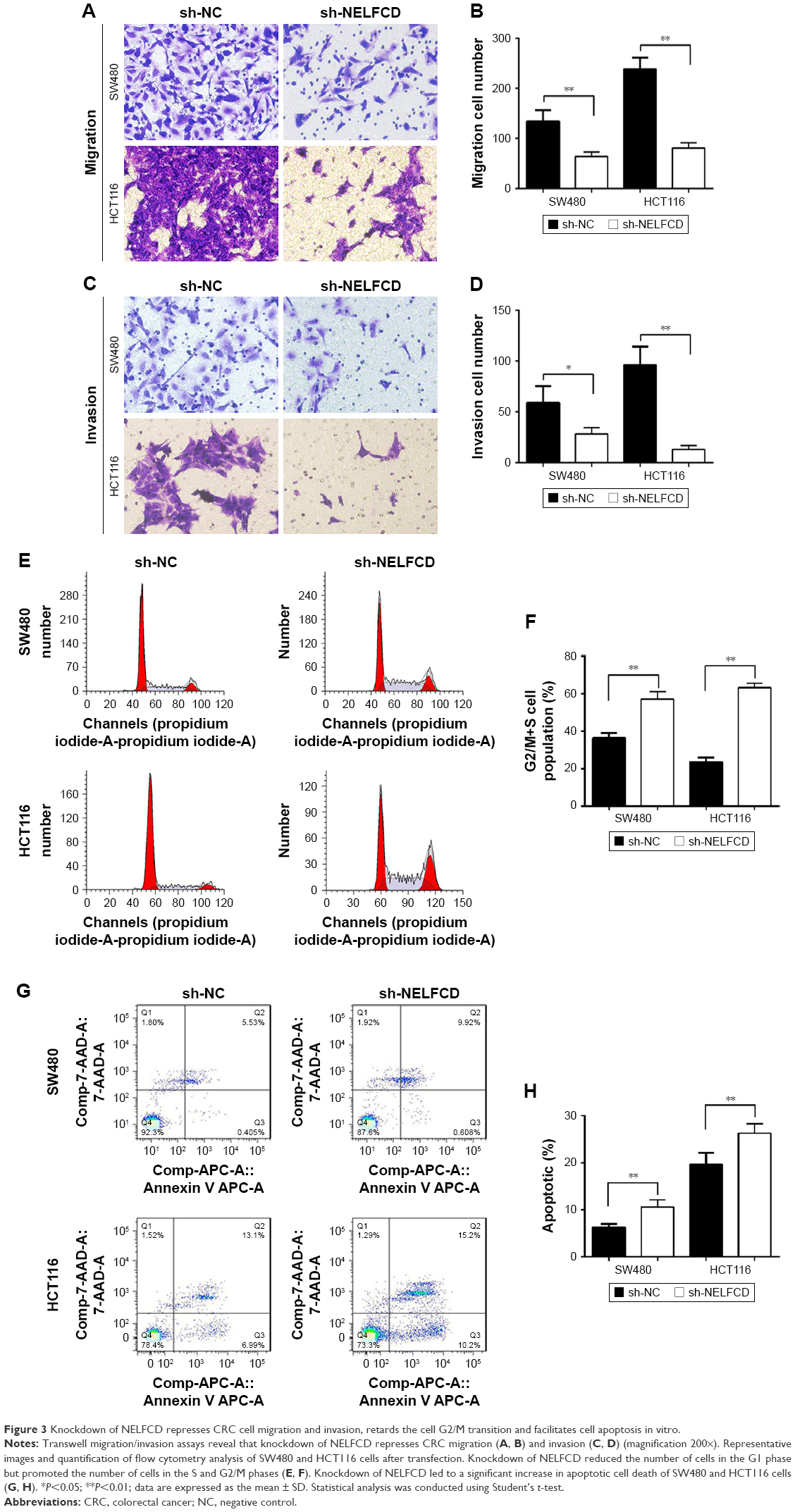 | Figure 3 Knockdown of NELFCD represses CRC cell migration and invasion, retards the cell G2/M transition and facilitates cell apoptosis in vitro. |
Next, flow cytometry was conducted to analyze the role of NELFCD on the cell cycle in CRC cells. Results indicated that knockdown of NELFCD reduced the number of cells in the G1 phase but promoted the number of cells in the S and G2/M phases in comparison to sh-NC group (Figure 3E and F).
We further assessed the effect of NELFCD on cell apoptosis. Knockdown of NELFCD led to a significant increase in apoptotic cell death of SW480 and HCT116 cells, as analyzed by flow cytometric analysis (Figure 3G and H). These data revealed that knockdown of NELFCD actually facilitated cell apoptosis in SW480 and HCT116 cells. These results indicate that knockdown of NELFCD represses CRC cell migration and invasion, retards the G2/M transition and facilitates cell apoptosis in vitro.
Knockdown of NELFCD represses tumor growth in vivo
To further investigate the tumorigenic ability of NELFCD, SW480-shNC and SW480-shNELFCD cells were subcutaneously injected into the right abdominal flanks of nude mice. The results demonstrated that the volume and weight of tumors in nude mice injected with SW480-shNELFCD were markedly suppressed (64.6% decrease in tumor weight) relative to that of mice injected with SW480-shNC (Figure 4A–D). These data indicated that knockdown of NELFCD markedly inhibited the tumorigenicity of SW480 cells in the nude mouse xenograft model.
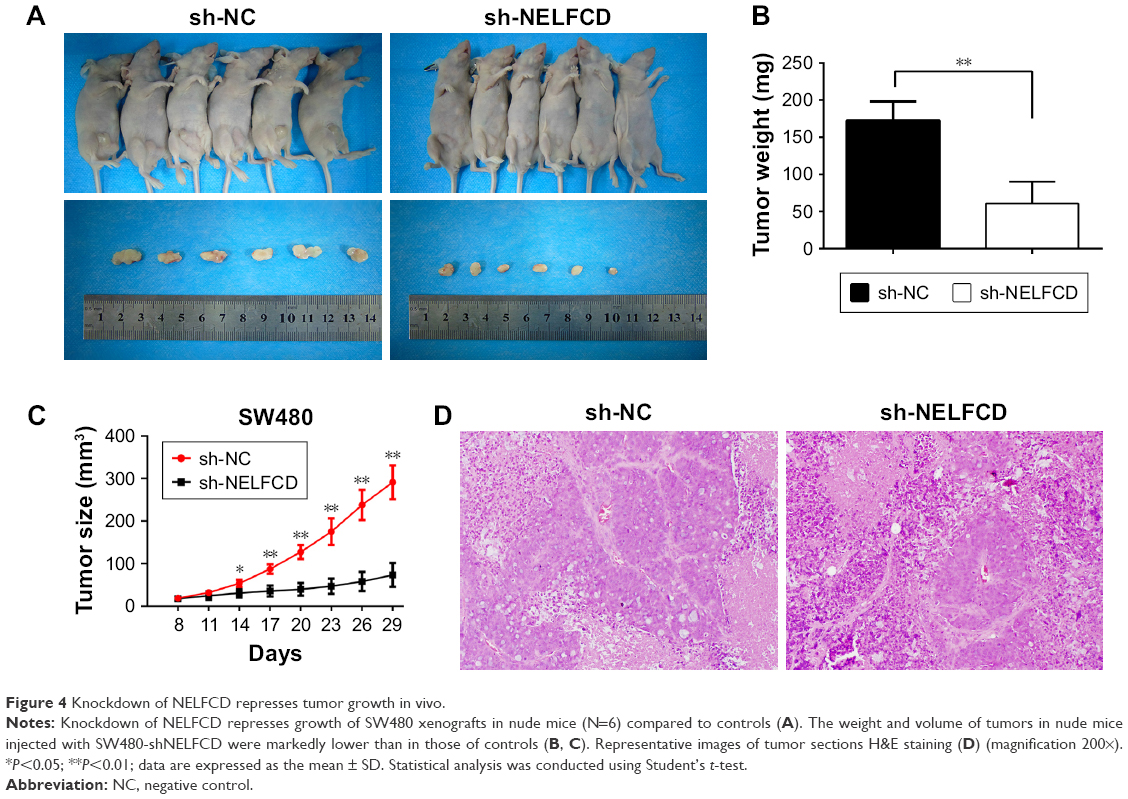 | Figure 4 Knockdown of NELFCD represses tumor growth in vivo. |
Discussion
Multiple putative oncogenes at the chromosome 20q amplicon contribute to the progression of colorectal adenoma to carcinoma. Gain of the long arm of chromosome 20 is one of the most common chromosomal alterations in colorectal carcinogenesis. This genetic alteration has also been found to be related to worse clinical outcomes in CRC patients. Therefore, these findings suggest that chromosome 20q harbors oncogenes that are particularly relevant to CRC.12 DDX27 (20q13.13) has been identified to be highly amplified in both TCGA CRC and primary CRC, and this amplification was positively correlated with its mRNA overexpression. Ectopic expression of DDX27 increased CRC cell proliferation, migration, and invasion, but it suppressed apoptosis. Silencing DDX27 exerted the opposite effects in vitro and significantly inhibited murine xenograft tumor growth and lung metastasis in vivo.20 TM9SF4 (20q11.21) has been found to modulate tumor pH alterations, which are associated with drug resistance and invasiveness of colon cancer cells.21 PALGL2 (20q11.21) has also been reported to serve as a tumor oncogene in the development and progression of CRC.22
Here, we reported for the first time that NELFCD (20q13.32) exerted an oncogenic role in CRC cells. Knockdown of NELFCD significantly impaired cell proliferation, migration, and invasion abilities; it also facilitated cell apoptosis in vitro and inhibited the tumorigenesis of CRC cells in vivo. However, an intensive understanding of the underlying molecular mechanisms of NELFCD, including its transcriptional target genes and co-factors or downstream effectors, is still lacking. Some reports indicate that NELFCD inhibited PAK kinase activity and negatively regulated MAPK signal transduction.23–27 There are four major MAPK pathways in mammals: the extracellular signal-regulated kinase (ERK), extracellular signal-regulated kinase 5 (ERK5), c-Jun N- terminal kinase (JNK), and p38 pathways. JNK and p38 MAPK signalling is associated with cancers in humans and may control cancer development.28–32 Therefore, we hypothesize that NELFCD acts as an oncogene by negatively regulating MAPK signal transduction. Further studies should be performed to verify the potential mechanism of NELFCD.
In this study, the data revealed that NELFCD expression levels in CRC tissues were significantly higher than those in matched adjacent normal tissues, which is consistent with TCGA database. Moreover, the overexpression of NELFCD was associated with tumor location among clinicopathologic factors (P,0.05). However, NELFCD expression was not associated with any other clinicopathologic factors in our study. We believe that this is because the number of patients with CRC was not large.
Conclusion
We first investigated the potential role of NELFCD in CRC. Our research provided evidence to support the oncogenic role of NELFCD in CRC tumorigenesis. However, the mechanism underlying the oncogenic role of NELFCD in CRC remains unclear, and further work is needed. We conclude that NELFCD is overexpressed and plays an oncogenic role in CRC. Targeting NELFCD may provide a potential therapeutic option for NELFCD-amplified tumors.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (No 81773130, No 81602568) and China Postdoctoral Science Foundation (No 2018M643009).
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. Epub 2018 Oct 23. | ||
Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116(3):544–573. | ||
Siegel RL, Miller KD, Jemal A, Statistics C. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. | ||
Center MM, Jemal A, Smith RA, Ward E. Worldwide variations in colorectal cancer. Dis Colon Rectum. 2010;53(7):1099. | ||
Chen W, Sun K, Zheng R, et al. Cancer incidence and mortality in China, 2014. Chin J Cancer Res. 2018;30(1):1–12. | ||
Issa JP. Colon Cancer: it’s CIN or CIMP. Clin Cancer Res. 2008;14(19):5939–5941. | ||
Al-Sohaily S, Biankin A, Leong R, Kohonen-Corish M, Warusavitarne J. Molecular pathways in colorectal cancer. J Gastroenterol Hepatol. 2012;27(9):1423–1431. | ||
Carethers JM, Jung BH. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology. 2015;149(5):1177–1190. | ||
Camps J, Nguyen QT, Padilla-Nash HM, et al. Integrative genomics reveals mechanisms of copy number alterations responsible for transcriptional deregulation in colorectal cancer. Genes, chromosomes & cancer. 2009;48(11):1002–1017. | ||
Redon R, Ishikawa S, Fitch KR, et al. Europe PMC Funders Group Global variation in copy number in the human genome. Nature. 2009;444(7118):444–454. | ||
Wang H, Liang L, Fang J, Xu J. Somatic gene copy number alterations in colorectal cancer: new quest for cancer drivers and biomarkers. Oncogene. 2016;35(16):2011–2019. | ||
Carvalho B, Postma C, Mongera S, et al. Multiple putative oncogenes at the chromosome 20q amplicon contribute to colorectal adenoma to carcinoma progression. Gut. 2009;58(1):79–89. | ||
Li D, Lin C, Chen M, et al. Comprehensive bioinformatics analysis of the characterization and determination underlying mechanisms of over-expression and co-expression of genes residing on 20q in colorectal cancer. Oncotarget. 2017;8(45):78642–78659. | ||
Yamaguchi Y, Takagi T, Wada T, et al. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell. 1999;97(1):41–51. | ||
Bonthron DT, Hayward BE, Moran V, Strain L. Characterization of TH1 and CTSZ, two non-imprinted genes downstream of GNAS1 in chromosome 20q13. Hum Genet. 2000;107(2):165–175. | ||
Narita T, Yamaguchi Y, Yano K. Human transcription elongation factor NELF: identification of novel subunits and reconstitution of the functionally active complex human transcription elongation factor NELF: identification of novel subunits and reconstitution of the functionally active. Mol Cell Biol. 2003;23(6):1863–1873. | ||
Narita T, Yung TM, Yamamoto J, et al. NELF interacts with CBC and participates in 3′ end processing of replication-dependent histone mRNAs. Mol Cell. 2007;26(3):349–365. | ||
Vos SM, Pöllmann D, Caizzi L, et al. Architecture and RNA binding of the human negative elongation factor. Elife. 2016;5(5):1–27. | ||
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. | ||
Tang J, Chen H, Wong CC, et al. DEAD-box helicase 27 promotes colorectal cancer growth and metastasis and predicts poor survival in CRC patients. Oncogene. 2018;37(22):3006–3021. | ||
Lozupone F, Borghi M, Marzoli F, et al. TM9SF4 is a novel V-ATPase-interacting protein that modulates tumor pH alterations associated with drug resistance and invasiveness of colon cancer cells. Oncogene. 2015;34(40):5163–5174. | ||
Liu B, Lu C, Song Y, et al. The role of pleomorphic adenoma gene-like 2 in gastrointestinal cancer development, progression, and prognosis. Int J Clin Exp Pathol. 2014;7(6):3089–3100. | ||
Salojin KV, Zhang J, Delovitch TL. TCR and CD28 are coupled via ZAP-70 to the activation of the Vav/Rac-1-/PAK-1/p38 MAPK signaling pathway. J Immunol. 1999;163(2):844–853. | ||
Slack-Davis JK, Eblen ST, Zecevic M, et al. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J Cell Biol. 2003;162(2):281–291. | ||
Jin S, Zhuo Y, Guo W, Field J. p21-activated Kinase 1 (Pak1)-dependent phosphorylation of Raf-1 regulates its mitochondrial localization, phosphorylation of BAD, and Bcl-2 association. J Biol Chem. 2005;280(26):24698–24705. | ||
Chan PM, Lim L, Manser E. PAK is regulated by PI3K, PIX, CDC42, and PP2Calpha and mediates focal adhesion turnover in the hyperosmotic stress-induced p38 pathway. J Biol Chem. 2008;283(36):24949–24961. | ||
Cheng C, Kong X, Wang H, et al. Trihydrophobin 1 interacts with PAK1 and regulates ERK/MAPK activation and cell migration. J Biol Chem. 2009;284(13):8786–8796. | ||
Yang SH, Sharrocks AD, Whitmarsh AJ. MAP kinase signalling cascades and transcriptional regulation. Gene. 2013;513(1):1–13. | ||
Bulavin DV, Fornace AJ. p38 MAP kinase’s emerging role as a tumor suppressor. Adv Cancer Res. 2004;92:95–118. | ||
Hui L, Bakiri L, Stepniak E, Wagner EF. p38alpha: a suppressor of cell proliferation and tumorigenesis. Cell Cycle. 2007;6(20):2429–2433. | ||
Wagner EF, Nebreda AR, Ár N. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–549. | ||
Urosevic J, Garcia-Albéniz X, Planet E, et al. Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH. Nat Cell Biol. 2014;16(7):685–694. | ||
Weiser MR. AJCC 8th Edition: Colorectal Cancer. Ann Surg Oncol. 2018;25(6):1454–1455. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.