")
Back to Journals » OncoTargets and Therapy » Volume 12
Overexpression of microRNA-216a inhibits autophagy by targeting regulated MAP1S in colorectal cancer
Authors Wang Y, Zhang S, Dang S, Fang X, Liu M
Received 4 December 2018
Accepted for publication 27 April 2019
Published 12 June 2019 Volume 2019:12 Pages 4621—4629
DOI https://doi.org/10.2147/OTT.S196992
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tohru Yamada
Yunfeng Wang, Songyan Zhang, Shuwei Dang, Xuan Fang, Ming Liu
Department of General Surgery, The Fourth Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang, People’s Republic of China
Background: Autophagy executes the rapid degradation of unneeded proteins and organelles through the lysosomal pathway, and is a crucial catabolic process widely conserved among eukaryotes. miRNAs can modulate autophagy by targeting genes encoding proteins involved in the process. A great deal of researchhas indicated that miR-216a was a functional miRNA related to tumorigenesis. However, the contribution of miR-216a to autophagy in colorectal cancer (CRC) remains unclear. The purpose of this study was to investigate the role of miR-216a in autophagy in CRC cells.
Methods: The expression levels of miR-216a in 67 paired CRC patients were evaluated by qRT-PCR. Direct gene targeting predicted by TargetScan and miRanda was confirmed by luciferase activity. Western blot and flow cytometry were used to identify the regulatory mechanism of miR-216a on autophagy in CRC cells.
Results: We determined that miR-216a is downregulated in CRC by screening its expression in 67 CRC tissue samples. Dual luciferase reporter assays showed that miR-216a binds the 3′-UTR of MAP1S, suggesting that MAP1S is a direct target of miR-216a. miR-216a could inhibit autophagy in HCT-116 and HT-29 CRC cells through downregulating MAP1S expression. Flow cytometry and Western blot analysis demonstrated that overexpression of miR-216a reduced MAP1S mRNA and protein levels. Moreover, we determined that miR-216a-regulated inhibition of autophagy via MAP1S regulation involves the TGF-β pathway.
Conclusion: Taken together, our findings indicate that miR-216a was a tumor-suppressor miRNA in human CRC, which can inhibit autophagy via the TGF-β/MAP1S pathway.
Keywords: miR-216a, MAP1S, autophagy, TGF-β, colorectal cancer
Introduction
Autophagy is characterized by autophagosome-dependent lysosomal degradation of long-lived proteins and unneeded organelles, which sustain cellular homeostasis.1 Elimination of pathogens and engulfment of apoptotic cells are also important functions of autophagy.2,3 In cancer cells, metabolic stress robustly induces autophagy, which is sustained when apoptosis is blocked.4 Abnormal regulation of autophagy is involved in several pathophysiological processes in numerous diseases;4 however, the correlation between autophagy and tumors remains controversial.5
microRNAs (miRs, miRNAs), which belong to a group of short, noncoding RNAs, have emerged as potential targets for anticancer therapy, as they regulate approximately 60% of protein-coding genes.6 Through binding target mRNA molecules, miRNAs generally downregulate gene expression and can influence cell proliferation, apoptosis, and the cell cycle.7–9 Dysregulated miRNA expression is a common feature in human cancers,10,11 and previous studies have demonstrated that several miRNAs are dysregulated in colorectal cancer (CRC), and associated with its progression.12–14 Aberrant expression of miRNAs is increasingly recognized as involved in the inappropriate stimulation of cellular programs, such as autophagy.15,16
miR-216a, a functional miRNA related to tumorigenesis, has recently become the subject of a great deal of research. Its expression is frequently abnormal in tumors, suggesting that it may have a significant role in cancer.17,18 Moreover, a recent study demonstrated that miR-216a enhances radiosensitivity through inhibition of autophagy in pancreatic cancer cells;19 however, the contribution of miR-216a to autophagy in CRC remains unclear. Our computational analysis predicted that the MAP1S gene is a target for regulation by miR-216a and that its encoded protein, microtubule-associated protein 1S (MAP1S), is a key regulator that enhances autophagy flux.20 Moreover, recent research has demonstrated a link between TGF-β/MAP1S pathway-mediated autophagy and suppression of tumorigenesis.21
In the present study, we show that levels of miR-216a expression are reduced in CRC tissues and cells and confirm the autophagy-mediating protein, MAP1S, as a direct transcriptional target of miR-216a in CRC. We further demonstrate that the TGF-β/MAP1S signaling pathway participates in miR-216a-mediated suppression of CRC cell autophagy.
Materials and methods
Patient and tissue samples
Pairs of fresh tumor and adjacent nontumor tissue samples (n=67) were collected during surgical resection at the Second Affiliated Hospital of Harbin Medical University (Harbin, China) between 2017 and 2018. Patients who had received prior therapy were excluded. All patients provided written informed consent, according to our institutional guidelines, and the study protocol was approved by the Institutional Review Board of Harbin Medical University, which was conducted in accordance with the Declaration of Helsinki.
Cell culture and transfection
The normal colon epithelial cell line, FHC, and the human CRC cell lines, HT-29, HCT-116, SW-480, and SW-620, were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). FHC cells were cultured in DMEM (Hyclone, Logan, UT, USA) and CRC cells were incubated in RPMI-1640 medium. RPMI-1640 and DMEM were supplemented with 10% FBS (Gibco, Grand Island, NY, USA). Cells were incubated at 37℃ in 5% CO2. Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) was used for transient transfection, according to the manufacturer’s protocol. The efficiency of transfection was assessed by RT-qPCR and Western blotting.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA and miRNAs were isolated from tissues and cells using Trizol (Thermo Fisher Scientific) and a mirVana miRNA Isolation kit (Ambion, Waltham, MA, USA), respectively, according to the manufacturer’s instructions. miRNA samples (2 μg) were used to perform reverse transcription with a High Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific). Real-time PCR was performed using SYBR Premix Ex TaqTM Ⅱ (TaKaRa, Tokyo, Japan). The sequences of the primer sets were as follows: MAP1S forward, 5′-CTCCTCACTGGTCGCTAAAACT-3′ and reverse 5′-GGGACTGGTAGAAAGGGGTATGT-3′; miR-216a forward, 5′-CTCAACGCGTGCGTCTGCTCTGGTGGCT-3′ and reverse 5′-GGCGCAAGCTTCACCTGGAGTAAGACACGACTTC-3′; TGF-β forward, 5′-ATGTTGAGACCTTCAACACC-3′ and reverse 5′-AGGTAGTCAGTCAGGTCCCGGCC-3′. The relative expression levels of MAP1S and miR-216a were, respectively, normalized to β-actin and U6.
Construction of promoter reporter plasmids and luciferase reporter assays
A fragment of the 3′-UTR of MAP1S containing miR-216a-binding sites was amplified by PCR and inserted downstream of the firefly luciferase gene in the pGL3-promoter vector (Promega, Madison, WI, USA). Mutant reporter plasmids were constructed using the Quick Change Mutagenesis kit (Stratagene, Palo Alto, CA, USA) and verified by sequencing (Agilent Technologies, Inc., Palo Alto, CA, USA). Luciferase activities were analyzed using the Dual-Luciferase Reporter Assay System (Promega Corporation), according to the manufacturer’s instructions.
Western blotting
To extract proteins, cells were treated with RIPA lysis buffer supplemented with protease inhibitor cocktail and protein phosphatase inhibitor. Protein concentrations in the resulting lysates were measured using a BCA kit (Beyotime Institute of Biotechnology, Shanghai, China). Samples were then separated by electrophoresis and transferred to PVDF membranes, which were incubated with primary antibodies overnight at 4℃. Primary antibodies used were against MAP-1S, microtubule-associated protein light chain 3 (LC3)-I, LC3-II, Beclin-I, N-cadherin, and Vimentin (Santa Cruz Biotechnology, Dallas, TX, USA). The following day, membranes were incubated with secondary antibody for 2 hrs at room temperature. Expression levels of detected proteins were determined by enhanced chemiluminescence reagent. β-actin was used as a normalization control.
Evaluation of autophagy by flow cytometry
Cell autophagy was detected by monodansylcadaverine (MDC; Sigma-Aldrich, St. Louis, MO, USA) staining. Cells were washed with PBS and then incubated with 0.05 mmol/L MDC in PBS at 37°C for 45 mins. Subsequently, cells were washed three times with PBS and immediately analyzed by flow cytometry. Cell Quest software was used to analyze 104 cells. The number of MDC-positive cells was positively correlated with autophagy status.
Statistical analysis
Results are expressed as mean ± SD. Comparisons between two groups were carried out using the Student’s t-test. Levels of correlation were examined by Spearman correlation analysis. P<0.05 was considered to indicate a statistically significant difference. Statistical analysis was performed using SPSS version 21 (IBM Corp., Armonk, NY, USA) or GraphPad Prism version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
miR-216a inhibits autophagy in CRC cells
First, we investigated miR-216a expression levels to clarify its potential role in CRC. Compared with adjacent nontumor and FHC cells, levels of miR-216a expression were decreased in 67 CRC tumor samples and four CRC cell lines (Figure 1A and B). The CRC cell lines, HT-29 and HCT-116, were selected for further investigation, as they showed intermediate levels of miR-216a expression.
 | Figure 1 miR-216a inhibits CRC cell autophagy. (A and B) miR-216a expression levels in 67 CRC tissue samples and four CRC cell lines. (C and D) HCT-116 and HT-29 cells were transfected with miR-216a mimic with or without the autophagy activator, rapamycin, or the autophagy inhibitor, 3-MA. The number of MDC-positive cells was detected by flow cytometry. Cell lysates were then examined for LC3-I, LC3-II, Beclin 1, E-cadherin, N-cadherin, and Vimentin expression by Western blotting. β-actin was used as an internal control. Data are presented as means ± SD values of 3–4 independent experiments. *p<0.05, ***p<0.001. Abbreviations: CRC, colorectal cancer; 3-MA, 3-Methyladenine; MCD, monodansylcadaverine. |
To further investigate the potential involvement of miR-216a in autophagy, we exposed cells to miR-216a mimic, the autophagy activator, rapamycin, or the autophagy inhibitor, 3-MA and examined levels of autophagy by flow cytometry and Western blot analysis. The number of MDC-positive cells (correlates positively with autophagy status) was assessed by flow cytometry, while the expression of autophagy marker proteins, such as LC3, Beclin-I, N-cadherin, E-cadherin, and Vimentin, was evaluated by Western blotting. As shown in Figure 1C and D, lower numbers of MDC-positive cells; lower levels of LC3-II, Beclin-I, N-cadherin, and Vimentin expression; and higher E-cadherin expression were detected in the miR-216a transfected group, compared with controls. These data indicate that treatment with miR-216a mimic inhibits autophagy in HCT-116 and HT-29 CRC cells; however, the inhibitory effect was slightly lower than that observed in cells treated with the autophagy inhibitor, 3-MA. The autophagy activator, rapamycin, significantly enhanced the level of autophagy, as reflected by the higher numbers of MDC-positive cells; increased expression of LC3-I, LC3-II, Beclin-I, N-cadherin, and Vimentin; and reduced levels of E-cadherin compared with controls; however transfection of miR-216a into rapamycin-treated HCT-116 and HT-29 CRC cell lines decreased the level of autophagy compared with rapamycin treatment alone. These results indicate that miR-216a partially antagonizes the autophagy-inducing effects of rapamycin, further verifying that miR-216a contributes to autophagy inhibition. Nevertheless, the inhibitory effects of treatment with both miR-216a and 3-MA simultaneously were similar to those of 3-MA transfection alone in HCT-116 and HT-29 cells. Although results in the two cells seemed inconsistent in co-transfection of miR-216a with rapamycin group, it may be the sensitivity of different cells on miR-216a and rapamycin was different. However, these results suggest that miR-216a is a tumor-suppressor miRNA that can inhibit autophagy in human CRC.
miR-216a directly targets and down-regulates MAP1S in CRC cells
In most cases, miRNAs bind to target mRNAs to regulate downstream genes and influence biological processes. To explore potential targets of miR-216a, we used TargetScan (
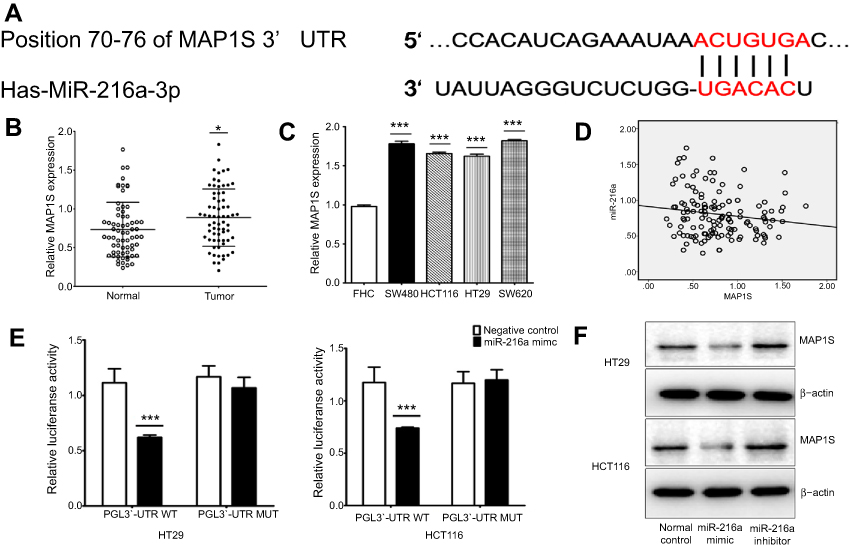 | Figure 2 miR-216a directly targets and downregulates MAP1S in CRC cells. (A) Prediction of miR-216a targets was conducted in silico and MAP1S was selected as a candidate. The bottom line shows the mutations introduced into the two putative miR-216a-binding sites. (B and C) Expression levels of MAP1S in 67 CRC tissue samples and four CRC cell lines. (D) Correlation between miR-216a and MAP1S levels in CRC tissues was analyzed by Spearman correlation analysis. (E) A fragment of the 3′-UTR of MAP1S containing miR-216a binding sites was amplified by PCR and inserted downstream of the firefly luciferase gene in the pGL3-promoter vector. Mutant reporter plasmids were constructed using the Quick Change Mutagenesis kit and verified by sequencing. Luciferase activities were analyzed using the dual-luciferase reporter assay system. (F) HCT-116 and HT-29 cells were transfected with miR-216a mimic or miR-216a inhibitor and total cell lysates subjected to Western blot analysis. β-actin was used as an internal control. Data are presented as the means ± SD values of 3–4 independent experiments. *p<0.05, ***p<0.001. |
Dual luciferase reporter assays were performed in both HT-29 and HCT-116 cells. The results showed that miR-216a could significantly decrease luciferase activity driven by the wild-type MAP1S 3ʹ-UTR sequence compared with a vector-only control (Figure 2E). Additionally, partial mutation of the perfectly complementary sites in the 3ʹ-UTR of MAP1S abolished the suppressive effect of miR-216a, due to the disruption of the interaction between miR-216a and the MAP1S 3ʹ-UTR.
To further investigate whether miR-216a could directly regulate MAP1S expression, HT-29 and HCT-116 cells were treated with either miR-216a mimic or miR-216a inhibitor and compared with untreated controls. Total cells lysates were obtained and subjected to Western blotting. Compared with controls, MAP1S expression levels were significantly decreased in CRC cells treated with miR-216a mimic, while they were increased in those treated with miR-216a inhibitor (2F). These data suggest that MAP1S is a direct target of miR-216a and is negatively regulated by miR-216a in CRC cells.
miR-216a is involved in MAP1S-regulated autophagy
We next explored whether autophagy inhibition in response to miR-216a was achieved through regulation of MAP1S. The data presented in Figure 3A and B demonstrate that treatment of HT-29 and HCT-116 cells with siRNAs targeting MAP1S induced a significant decrease in MAP1S mRNA and protein expression. Next, we transfected miR-216a mimic with or without MAP1S-targeting siRNAs and found that the number of MDC-positive cells detected by flow cytometry was significantly decreased on simultaneous transfection with miR-216a mimic and MAP1S-targeting siRNAs (Figure 3C). Western blot analysis was conducted to further verify these results and showed that MAP1S, LC3-I, LC3-II, and Beclin-I protein expression levels were downregulated in cells transfected with miR-216a mimic or MAP1S-targeting siRNAs (Figure 3D). Furthermore, treatment with a combination of miR-216a mimic and MAP1S-targeting siRNA failed to alter the inhibition of autophagy compared with treatment with MAP1S-targeting siRNAs alone in HT29 and HCT116 cells, as determined by both in flow cytometry and Western blot analyses. Overall, these results indicate that miR-216a is involved in MAP1S-regulated autophagy.
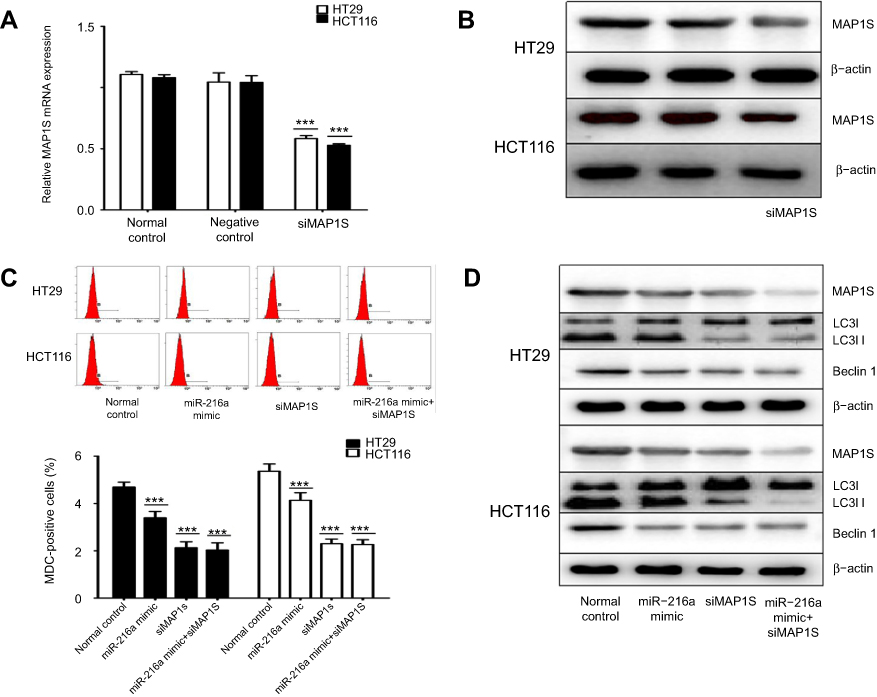 | Figure 3 miR-216a is involved in MAP1S-regulated autophagy. (A and B) The influence of MAP1S-targeting siRNAs on MAP1S expression in HCT-116 and HT-29 cells. (C and D) miR-216a mimic were transfected into HCT-116 and HT-29 cells with or without MAP1S siRNAs and the number of MDC-positive cells was detected by flow cytometry to assess the level of autophagy in each treatment condition. Western blotting was also applied to analyze the levels of MAP1S and other autophagy marker proteins in the same cells. β-actin was used as an internal control. Data are presented as means ± SD values of 3–4 independent experiments. ***p<0.001. |
Suppression of CRC cell autophagy by miR-216a involves the TGF-β/MAP1S signaling pathway
We further investigated whether the TGF-β/MAP1S signaling pathway is involved in miR-216a suppression of CRC cell autophagy. Compared with controls, an increased level of TGF-β expression was observed in both CRC tumors and CRC cell lines (Figure 4A and B). Next, both HT29 and HCT116 cells were transfected with miR-216a mimic and then treated with or without TGF-β and the level of autophagy was assessed. Compared with the control group, transfection with miR-216a mimic significantly suppressed autophagy; fewer MDC-positive cells were detected by flow cytometry (Figure 4C), while lower levels of LC3-I, LC3-II, Beclin-I, N-cadherin expression, and Vimentin, and higher levels of E-cadherin expression were determined by Western blotting (Figure 4D) in both cell lines. Levels of autophagy were elevated in both HT29 and HCT116 cells after treatment with TGF-β; however, this induction could be inhibited by combination treatment with miR-216a mimic in both HT29 and HCT116 cells, as shown by the reduced proportion of MDC-positive cells (Figure 4C); lower expression of LC3-1, LC3-II, Beclin-I, N-cadherin, and Vimentin; and higher expression of E-cadherin (Figure 4D) in the CRC cells. These results indicate that the TGF-β/MAP1S signaling pathway participates in miR-216a-mediated suppression of CRC cell autophagy.
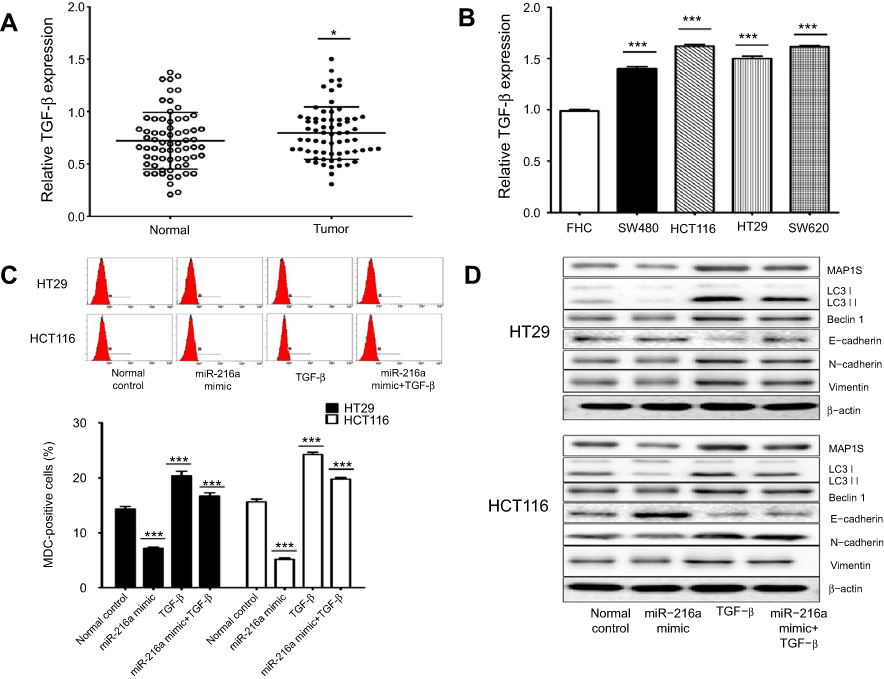 | Figure 4 The TGF-β/MAP1S signaling pathway is involved in miR-216a suppression of CRC cells autophagy. (A and B) Expression levels of TGF-β in 67 CRC tissue samples and four CRC cell lines. (C and D) Treatment of HCT-116 and HT-29 cells with miR-216a mimic, TGF-β, or miR-216a mimic combined with TGF-β; the number of MDC-positive cells was detected by flow cytometry to assess the level of autophagy in each treatment condition. Western blotting was also applied to analyze the levels of MAP1S and other autophagy marker proteins in the same cells. β-actin was used as an internal control. Data are presented as means ± SD values of 3–4 independent experiments. *p<0.05, ***p<0.001. |
Discussion
CRC is the third most common cancer and the fourth leading cause of cancer-associated death worldwide.22 Despite advances in surgical resection combined with chemotherapy and radiotherapy, median survival rates of patients with CRC remain low, since they are usually diagnosed at an advanced stage, beyond the optimal time for surgical resection.23 Exploring the molecular mechanisms underlying CRC carcinogenesis is vital, as it may provide new therapeutic targets and improve the long-term survival patients with this disease; however, the molecular mechanisms underlying the regulation of CRC progression remain incompletely understood. Increasing evidence supports roles for miRNAs as regulators of tumor progression; hence, miRNAs have emerged as anticancer targets and are the subject of intensive investigation.9–11 Among potential miRNA therapeutic targets, miR-216a is one of the most prominent tumor suppressors, which regulates a variety of target mRNAs in several cancers.17–19 miR-216a has recently received particular research attention because it targets mRNAs to inhibit cancer progression. In this study, we evaluated the relationship between miR-216a and autophagy in CRC cells.
We showed that miR-216a was expressed at a lower level in both CRC cells and CRC tissues compared with paired normal tissues and the FHC cell line (Figure 1A and B). Consistent with our results, downregulation of miR-216a has also been reported in other human cancers, including melanoma17 and pancreatic cancer,18 suggesting that miR-216a may have roles in several types of malignancy.
Using both flow cytometry and Western blot analyses, we demonstrated that upregulation of miR-216a inhibited autophagy (Figure 1). To further confirm these results we performed additional analyses using an autophagy activator (rapamycin) and an inhibitor (3-MA).24,25 Treatment of CRC cells with both miR-216a and rapamycin led to inhibition of autophagy compared with rapamycin treatment alone, providing further support for the function of miR-216a in inhibiting autophagy (Figure 1C and D); however, treatment with miR-216a and 3-MA did not result in a difference in autophagy inhibition levels compared with treatment with each individually. These results demonstrate that miR-216a acts a suppressor of CRC cell autophagy.
As miRNAs are involved in cancer pathogenesis through direct regulation of the expression of their targets, their investigation is potentially helpful to understanding of the molecular mechanisms underlying CRC progression. In our study, bioinformatics analysis indicated that MAP1S may be a miR-216a-regulated target, since there is a potential miR-216a-binding site in the MAP1S 3ʹ-UTR (Figure 2A). To verify this prediction, we conducted dual-luciferase reporter assays which confirmed MAP1S as a direct target of miR-216a (Figure 2E). Furthermore, we detected higher MAP1S expression levels in HT-29, HCT-116, SW-480, and SW-620 CRC cells, than in the normal colon epithelial cell line, FHC (Figure 2C). Meanwhile, examination of tumor and adjacent normal tissue samples from 67 CRC patients showed that MAP1S is also significantly overexpressed in CRC tumors relative to nonmalignant tissue (Figure 2D). Western blot analysis further verified the regulated effect of miR216a on MAP1S (Figure 2F).
The combination of miR-216a mimic and MAP1S-targeting siRNA treatment and treatment with MAP1S-targeting siRNAs alone failed to have any effect on CRC cell autophagy (Figure 3C), indicating that, once MAP1S is blocked, the level of autophagy did not increase on treatment with the miR-216a. This demonstrates a direct relationship between miR-216a and MAP1S in autophagy inhibition. Further results verified that miR-216a can downregulate MAP1S protein levels and those of autophagy marker proteins in HCT-116 and HT-29 CRC cells (Figure 3D). These data suggest that MAP1S is a direct target of miR-216a and confirms the role of MAP1S in the autophagy pathway, where miR-216a downregulates MAP1S expression and inhibits autophagy in HCT-116 and HT-29 CRC cells.
Among miR-216a target genes, MAP1S is a confirmed downstream regulator of the TGF-β pathway, which is a well-characterized positive mediator of autophagy.26 The TGF-β pathway is established as a promotor of the progression of several cancers, through positive regulation of various oncogenes.27 Based on our data in this study, we confirm that miR-216a can suppress CRC cell autophagy. Levels of autophagy were elevated by treatment of CRC cells with TGF-β. This induction could be attenuated by combination treatment with miR-216a mimic, compared with TGF-β treatment alone. Therefore, we supposed that TGF-β signaling at least partly participated in miR-216a-mediated suppression of CRC cell autophagy. Based on previous study that the link between TGF-β/MAP1S pathway-mediated-autophagy and suppression of tumorigenesis, we will focus on to further explore the regulatory mechanism between miR-216, TGF-β, and MAP1S in CRC, which could provide a new therapeutic target for colorectal patients.
Conclusion
In summary, our results identify miR-216a as a tumor-suppressor miRNA in human CRC, which can regulate autophagy through the inhibition of MAP1S. We also determined that miR-216a inhibits autophagy via the TGF-β/MAP1S pathway. Regulation via the miR-216a/MAP1S axis may modulate CRC cell function.
Acknowledgments
This work was supported by funds from the National Natural Science Foundation of China (Grant No. 81372612 and 81302059).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi:10.1016/j.cell.2007.12.018
2. Colombo MI. Autophagy: A pathogen driven process. IUBMB Life. 2007;59:238–242. doi:10.1080/15216540600994357
3. Qu X, Zou A, Sun Q, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi:10.1016/j.cell.2006.12.044
4. Lum JJ, Bauer DE, Kong M, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;12:237–248. doi:10.1016/j.cell.2004.11.046
5. Mathew R, Kongara S, Beaudoin B, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381. doi:10.1101/gad.1545107
6. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi:10.1038/nrc1997
7. Li Y, Wang H, Tao K, et al. miR-29b suppresses CML cell proliferation and induces apoptosis via regulation of BCR/ABL1 protein. Exp Cell Res. 2013;31:1094–1101. doi:10.1016/j.yexcr.2013.02.002
8. He X, Dong Y, Wu CW, et al. MicroRNA-218 inhibits cell cycle progression and promotes apoptosis in colon cancer by downregulating BMI1 polycomb ring finger oncogene. Mol Med. 2012;18:1491–1498. doi:10.2119/molmed.2012.00304
9. Chen D, Guo W, Qiu Z, et al. MicroRNA-30d-5p inhibits tumour cell proliferation and motility by directly targeting CCNE2 in non-small cell lung cancer. Cancer Lett. 2015;362:208–217. doi:10.1016/j.canlet.2015.03.041
10. Cheng Y, Xiang G, Meng Y, et al. MiRNA-183-5p promotes cell proliferation and inhibits apoptosis in human breast cancer by targeting the PDCD4. Reprod Biol. 2016;16:225–233. doi:10.1016/j.repbio.2016.10.005
11. Ma ZB, Kong XL, Cui G, et al. Expression and clinical significance of miRNA-34a in colorectal cancer. Asian Pac J Cancer Prev. 2014;15:9265–9270. doi:10.7314/APJCP.2014.15.21.9265
12. Zhang D, Zhao L, Shen Q, et al. Down-regulation of KIAA1199/CEMIP by miR-216a suppresses tumor invasion and metastasis in colorectal cancer. Int J Cancer. 2017;140:2298–2309. doi:10.1002/ijc.30656
13. Xiao R, Li C, Chai B. miRNA-144 suppresses proliferation and migration of colorectal cancer cells through GSPT1. Biomed Pharmacother. 2015;74:138–144. doi:10.1016/j.biopha.2015.08.006
14. Yan L, Yao J, Qiu J. miRNA-495 suppresses proliferation and migration of colorectal cancer cells by targeting FAM83D. Biomed Pharmacother. 2017;96:974–981. doi:10.1016/j.biopha.2017.11.138
15. Ren Y, Chen Y, Liang X, et al. MiRNA-638 promotes autophagy and malignant phenotypes of cancer cells via directly suppressing DACT3. Cancer Lett. 2017;390:126–136. doi:10.1016/j.canlet.2017.01.009
16. Hu J, Meng Y, Zhang Z, et al. MARCH5 RNA promotes autophagy, migration, and invasion of ovarian cancer cells. Autophagy. 2017;13:333–344. doi:10.1080/15548627.2016.1256520
17. Hu J, Guo X, Yang L. Morin inhibits proliferation and self-renewal of CD133+ melanoma cells by upregulating miR-216. J Pharmacol Sci. 2018;136:114–120. doi:10.1016/j.jphs.2018.02.003
18. Zhang Y, Tang X, Shi M, et al. MiR-216a decreases MALAT1 expression, induces G2/M arrest and apoptosis in pancreatic cancer cells. Biochem Biophys Res Commun. 2017;483:816–822.
19. Zhang X, Shi H, Lin S, et al. MicroRNA-216a enhances the radiosensitivity of pancreatic cancer cells by inhibiting beclin-1-mediated autophagy. Oncol Rep. 2015;34:1557. doi:10.3892/or.2015.4078
20. Li W, Zou J, Yue F, et al. Defects in MAP1S‐mediated autophagy cause reduction in mouse lifespans especially when fibronectin is overexpressed. Aging Cell. 2016;15:370–379. doi:10.1111/acel.12441
21. Song K, Hu W, Yue F, et al. Transforming growth factor TGF-β increase levels of microtubule-associated protein MAP1S and autophagy flux in pancreatic ductal adenocarcinomas. PLoS One. 2015;10. doi:10.1371/journal.pone.0143150
22. World Cancer Research Fund International: worldwide data; 2012. Available from:
23. Stein U, Walther W, Arlt F, et al. MACC1, a newly identified key regulator of HGF-MET signaling, predicts colon cancer metastasis. Nat Med. 2009;15:59–67. doi:10.1038/nm.1889
24. Sotthibundhu A, McDonagh K, von Kriegsheim A, et al. Rapamycin regulates autophagy and cell adhesion in induced pluripotent stem cells. Stem Cell Res Ther. 2016;15:166. doi:10.1186/s13287-016-0425-x
25. Wu Y, Wang X, Guo H, et al. Synthesis and screening of 3-MA derivatives for autophagy inhibitors. Autophagy. 2013;9:595–603. doi:10.4161/auto.23641
26. Kiyono K, Suzuki HI, Matsuyama H, et al. Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009;69:8844–8852. doi:10.1158/0008-5472.CAN-08-3660
27. Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.