")
Back to Journals » OncoTargets and Therapy » Volume 11
Overexpression of GPR35 confers drug resistance in NSCLC cells by β-arrestin/Akt signaling
Authors Wang W, Han T, Tong W, Zhao J, Qiu X
Received 29 May 2018
Accepted for publication 19 July 2018
Published 26 September 2018 Volume 2018:11 Pages 6249—6257
DOI https://doi.org/10.2147/OTT.S175606
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Takuya Aoki
Wei Wang,1 Tianci Han,1 Wei Tong,1 Jian Zhao,1 Xueshan Qiu2
1Department of Thoracic Surgery, Cancer Hospital of China Medical University, Liaoning Cancer Hospital and Institute, Shenyang 110042, China; 2Department of Pathology, The First Hospital of China Medical University, Shenyang 110001, China
Background: Non-small-cell lung cancer (NSCLC) is the major leading cause of cancer-related death around the world. The resistance to chemotherapy limits the effects of clinical treatment. The aim of this study was to identify novel mechanisms involved in NSCLC chemoresistance.
Materials and methods: We explored the public database and commercial tissue microarray to evaluate the expression of G protein-coupled receptor 35 (GPR35). We established the chemoresistant A549 cell line to further investigate the biological function of GPR35 in vitro and in vivo. Then, we measured the altered signalings that GPR35 knocking down by Western blot assay.
Results: We demonstrated that GPR35 expression was significantly elevated in NSCLC tissues and correlated with poor prognosis. GPR35 was upregulated in our in vitro chemoresistance cell model. GPR35 depletion reduced the half maximal inhibitory concentration of chemodrugs and restored the sensitivity both in vitro and in vivo. Mechanically, we found that GPR35-mediated chemoresistance occurred partially via β-arrestin-2/Akt signaling. Furthermore, inhibition of β-arrestin-2 or Akt activation could suppress the GPR35 expression and overcome chemoresistance.
Conclusion: Our results suggested that GPR35 might serve as a novel therapeutic target to enhance the chemotherapy efficacy in NSCLC.
Keywords: GPR35, NSCLC, chemoresistance, β-arrestin-2
Introduction
Non-small-cell lung cancer (NSCLC) is an aggressive and major type of lung cancer, accounting for approximately 85% of lung cancer cases.1,2 Most patients with NSCLC were first diagnosed in advanced stage, and the 5-year survival rate of NSCLC is still dismal at about 17%.3 The clinical procedures for NSCLC include surgery, chemotherapy, and radiotherapy. Among these, platinum agent is the first-line chemodrugs combined with other cytotoxic agents, such as docetaxel and paclitaxel.4,5 However, due to frequent chemoresistance, the effective treatment remains a critical challenge.
G protein-coupled receptor 35 (GPR35), also known as CXCR8, a receptor of CXCL17, is identified during the genomic DNA screen.6,7 The gene gpr35 is located in the chromosome 2q37.3, which was reported to be partially genomic loss in lung cancer.8 The expression of GPR35 is found widely, especially in pancreas and small intestine.9 However, GPR35 expression is relative low in stomach, kidney, and liver.10 Because GPR35 belongs to the orphan G protein-coupled receptor (GPCR) family, the ligands and downstream effector have been investigated previously, and the therapeutic potential is revealed in immune, nervous, and cardiovascular systems.11,12 For instance, kynurenic acid is the first reported ligand and presents in many tissues, such as brain and colon, and agonism of GPR35 by kynurenic acid resulted in interleukin-4 suppression.13,14 Moreover, activation of GPR35 could promote the recruitment of β-arrestins and therefore, initiate the signal transduction.15 Functionally, it is demonstrated that GPR35 mediates smooth muscle cell migration and endothelial cell proliferation.16 GPR35 also induces hypoxia-inducible factor-1 alpha to regulate cellular processes.17 Besides, GPR35 is identified as a susceptibility gene associated with chronic anthracycline-induced cardiotoxicity in pediatric cancer patients.18 However, the role of GPR35 in lung cancer needs to be further elucidated.
Here, we disclosed that GPR35 was a key regulator in our established chemoresistance cell model. We initially found that the expression of GPR35 was significant in NSCLC tissues compared with that in adjacent nontumor tissues. High expression of GPR35 indicated shorter overall survival and progression-free survival. Furthermore, depletion of GPR35 could remarkably overcome chemoresistance both in vitro and in vivo. Mechanically, we indicated that GPR35/β-arrestin-2/Akt loop contributed to chemoresistance, implying that depletion of GPR35 or β-arrestin-2 or pharmaceutical inhibition of Akt might be a practicable approach for chemotherapy.
Materials and methods
Cell lines and reagents
Human lung cancer cell line A549 was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were routinely maintained in DMEM supplemented with 10% FBS and antibiotics. A549 doxorubicin (Dox)-resistant cells (A549R) were obtained by the following procedure: 2 μM Dox culturing for 1 month, then 4 μM Dox culturing for 2 weeks, 6 μM Dox culturing for 2 weeks, 10 μM Dox culturing for 2 weeks, and maintained in the medium containing 8 μM Dox. Cells were cultured at 37°C in a humidified atmosphere with 5% CO2. Dox, cisplatin, and LY294002 were obtained from Selleck Inc. (Shanghai, China).
Clinical samples
The study associated with clinical samples was approved by the ethics committee of The First Hospital of China Medical University. The NSCLC tissue microarrays (TMAs) were purchased form Xi’an Alenabio Inc. (Xi’an, China). The data from Gene Expression Omnibus (GEO) database were obtained from Oncomine (www.oncomine.org).19 The GEO datasets were GSE10072, GSE32863, GSE31210, and GSE19188.20–23 The prognosis data were obtained from K-M plot (http://kmplot.com/analysis/).24 We used the latest updated vision including 1,926 cases of overall survival data and 982 cases of progression-free survival and we chose “auto select best cutoff” to analyze the prognosis value of GPR35.
siRNA transfection and lentivirus infection siRNAs targeting GPR35 and β-arrestin-2 were purchased from GenePharma (Shanghai, China). The sequences were as follows: GPR35, 5′-CCAAGCACUCAGCUCUCUAAU-3′, 5′-GCAGGUUCUGACCAAGAACAA-3′; β-arrestin-2, 5′-GGACCGCAAAGUGUUUGUG-3′, 5′-CCAACCUCAUUGAAUUUGA-3′. Cell transfection was performed using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA) according to the manufacturer’s protocol. To establish the stable GPR35 knocking down cells, cells were infected with lentivirus containing shRNA targeting GPR35 (GeneCopoeia, Guangzhou, China) combined with polybrene (5 μg/mL). Infected cells were selected by 2 μg/mL puromycin for 1 week.
Immunohistochemical staining (IHC)
IHC was performed as described previously.25 In brief, the slides were deparaffinized and rehydrated, then antigen retrieval was performed by citric acid buffer (pH 6.0) and heated by microwave. Peroxidase activity was blocked and incubated with primary antibodies against GPR35 (ab150635 from Abcam, Cambridge, UK) Ki67 overnight at 4°C. After washing with phosphate buffered solution with 0.1% Tween-20, the slides were incubated with secondary antibody for 30 minutes and reacted with diaminobenzidine under microscope and counterstained with hematoxylin. The primary antibody rabbit IgG was used as negative control.
To evaluate the expression of GPR35 in clinical samples, we performed semiquantitative scoring of the IHC staining, as described previously.26 Briefly, the score of staining was calculated by positive intensity (0, 0%–15%; 1, 15%–50%; 2, 50%–70%; 3, >70%) × ratio (0, low staining; 1, moderate staining; 2, high staining; 3, extremely high staining). A cutoff score larger than 4 was defined as high expression.
Cell viability assay
In vitro cell drug sensitivity was determined by MTT assay. To determine the half maximal inhibitory concentration (IC50) of cells, the cells (104 cells/well) were seeded into 96-wells plate, and then treated with gradient concentration of chemoagents for 48 hours. Twenty microliters of 5 mg/mL MTT was added into each well and incubated for 4 hours. The cell viability was detected by dimethyl sulfoxide (DMSO) resolving and measuring the OD at 570 nm.
Western blot
Cells were harvested and lyzed in RIPA buffer containing protease and phosphatase inhibitor cocktail (Roche, Basel, Switzerland). The total protein concentration was measured by bicinchoninic acid method. An equal amount of protein from each sample was loaded into 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes. After 5% BSA blocking, primary antibodies were incubated at 4°C overnight. Then, the membranes were incubated with secondary antibodies and reacted with enhanced chemiluminescence detection solution. Results were normalized to β-actin. The primary antibodies used were as follows: anti-GPR35 (ab76217), anti-Bcl2 (ab32124), anti-p21 (ab109520), anti-bcl-xl (ab32370), and anti-β-arrestin-2 (ab54790), which were from Abcam. Anticleaved caspase 3 (#9661), antiphosphorylated Akt (#4060), antitotal Akt (#2920), antiphosphorylated S6 (#4858), anti-S6 (#2317), and anticleaved PARP (#5625) were from Cell Signaling Technology (Beverly, MA, USA).
Flow cytometry
Cell apoptosis was double stained by Annexin V/7AAD apoptosis kit according to the manufacture’s protocol and performed on BD FACS Calibur. Cells with Annexin V positive stain were analyzed.
Mice study
The mice experiments were approved by the committee of China Medical University and conducted in accordance with the Guideline of the Care and Use of Laboratory Animals in China Medical University. To measure the in vivo chemoresistance effect, we performed A549R cells derived from xenograft tumor analysis; 3×106 indicated cells were subcutaneously injected into the fight flank of 6- to 7-week-old male nude mice. When the average tumor volume reached 50 mm3, the mice were intraperitoneally injected with cisplatin (5 mg/kg/week) or DMSO to stimulate chemotherapy. Tumor size was measured every 3 days and calculated by the formula (length×width2/2). After treatment for 3 weeks, in the day 24, the mice were sacrificed and the tumors were fixed in 10% formalin solution, paraffin-embedded, and then sectioned successively for IHC. To determine the tumor cell proliferation in vivo, staining with Ki67 was performed.
Statistical analysis
Statistical analysis was performed using SPSS 21.0 (IBM Corporation, Armonk, NY, USA) and visualized by GraphPad Prism 5. In vitro data were presented as mean±standard error of the mean (SEM) from at least three independent experiments. Difference between experimental groups was tested by Student’s t-test or one-way ANOVA with two tails. The Mann–Whitney test with two tails was used to measure the difference between normal tissues group and tumor group of GPR35 IHC staining. Survival curves were measured by Kaplan–Meier method with log-rank test. A P-value <0.05 was considered statistically significant.
Results
Expression of GPR35 in NSCLC
To investigate the expression pattern of GPR35 in NSCLC tissues, we explored the GEO database and found that GPR35 was significantly upregulated in tumor tissues compared with normal tissues in four independent cohorts (Figure 1A–D). To further confirm these findings, we purchased a commercial NSCLC TMA. Besides NSCLC tissues, 50 normal lung epithelial tissue spots were found. We performed IHC to detect the expression of GPR35 in clinical samples. As shown in Figure 1E, GPR35 was mostly located in the cytoplasm and membrane of cells. By quantification of the IHC staining, we noticed that the expression of GPR35 was significantly high in tumor tissues (Figure 1F). Furthermore, we found that high expression of GPR35 was associated with poor prognosis (Figure 1G) and shorter progression-free survival (Figure 1H), according to the K-M plot analysis. Taken together, the results indicated that GPR35 might act as an oncogene in NSCLC and associate with increased relapse.
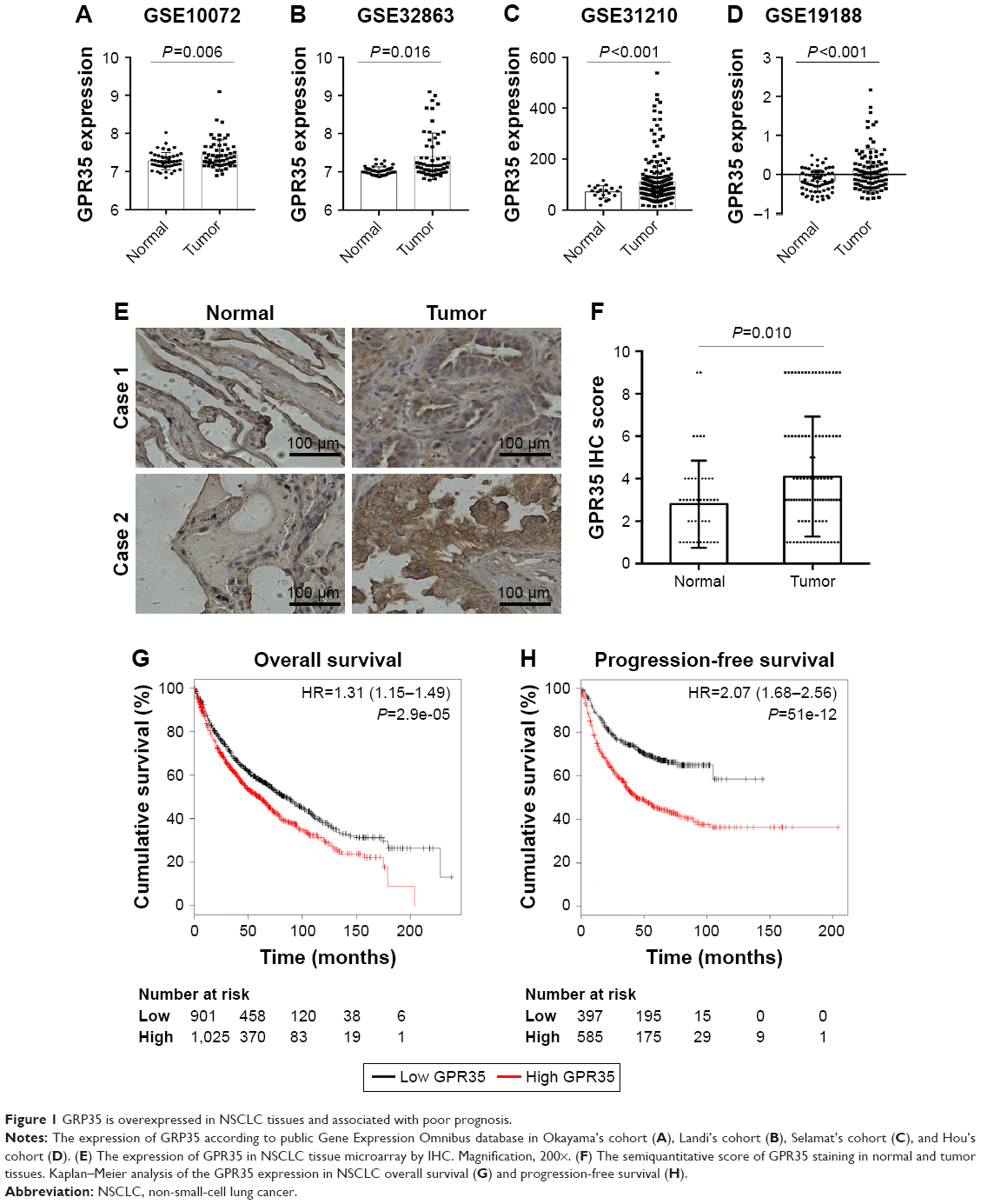 | Figure 1 GRP35 is overexpressed in NSCLC tissues and associated with poor prognosis. |
Increased GPR35 expression in chemoresistant NSCLC cells
To study the function of GPR35, we generated in vitro chemoresistant model using A549 NSCLC cell. A549-resistant cells (A549R) were established by exposing cells to increased concentration of Dox. As shown in Figure 2A, compared with parental cells, A549R cells exhibited an increased resistance to Dox. We performed colony formation assay and found that A549R cells could still grow and suffer long-term chemotreatment, while parental cells were mostly dead (Figure 2B). In view of the crucial role of GPR35, we found that under Dox treatment, expression of GPR35 was higher in A549R cells than in A549 cells, as well as the expression of Bcl-2, Bcl-xl (Figure 2C), suggesting that antiapoptotic proteins were upregulated and A549R cells were resistant to chemotherapy and the apoptosis rate of A549R cells were less than control cells. Then, we knocked down GPR35 in A549R cells and treated with Dox and Cis; the results indicated that IC50 was increased and A549R cells restored sensitivity to chemodrugs (Figure 2D and E). In addition, we detected the cell apoptosis rate and found that depletion of GPR35 increased cellular apoptosis rate, and when treated with Dox or Cis, cell apoptosis was elevated significantly (Figure 2F). The Western blot showed similar results. Knocking down of GPR35 was verified, and we noticed that cleaved caspase 3 and cleaved PARP were increased, indicating that knocking down of GPR35 elevated cell apoptosis (Figure 2G).
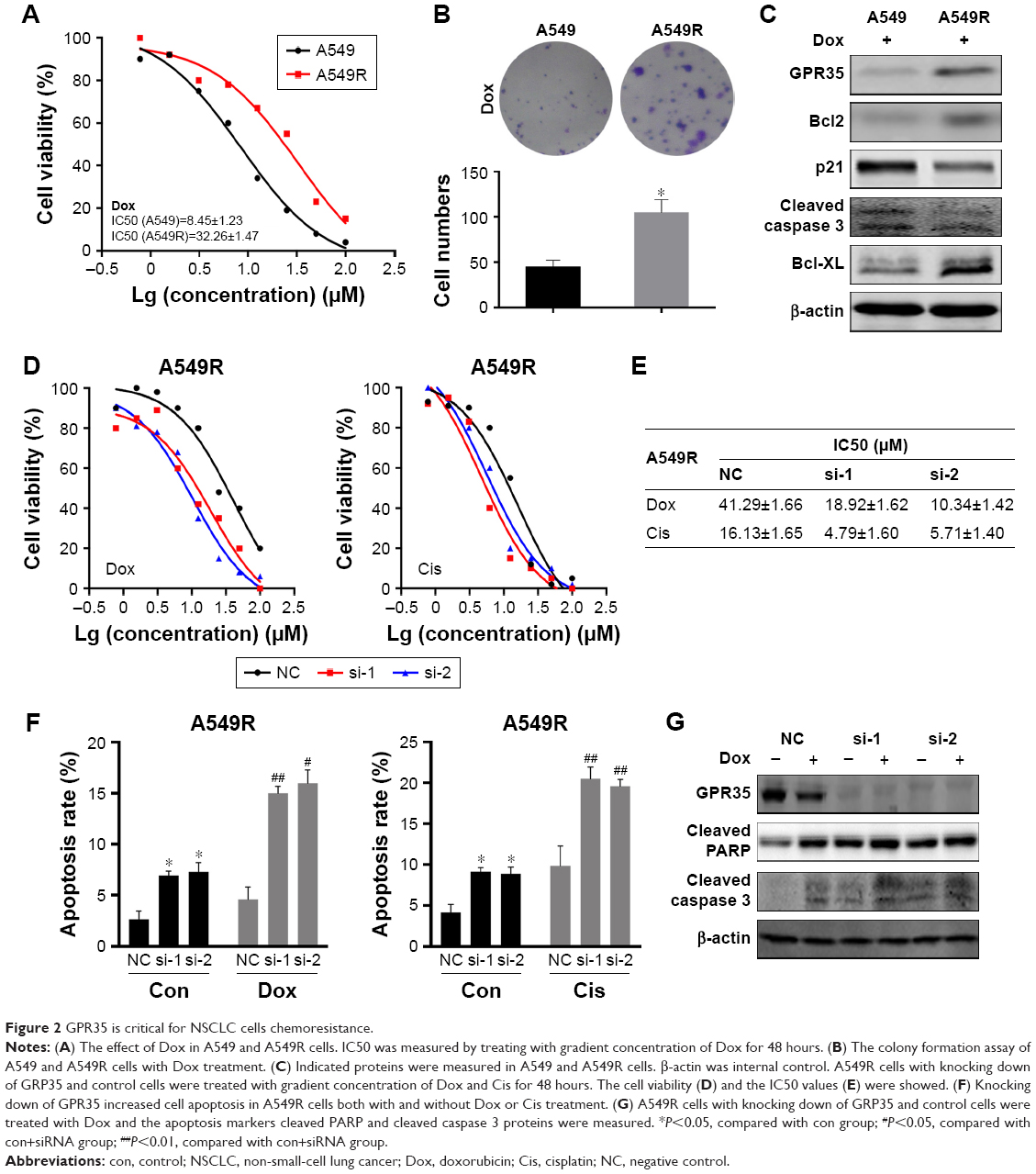 | Figure 2 GPR35 is critical for NSCLC cells chemoresistance. |
Knocking down of GPR35 abrogates cisplatin resistance in vivo
We next evaluated the effects of knocking down of GPR35 in vivo. Nude mice bearing A549R or A549-GPR35 knockdown cell xenografts were treated with cisplatin to stimulate the chemotreatment. The tumor growth of A549R xenografts followed by the cisplatin treatment showed a weak inhibition, as we demonstrated in vitro experiments. On the contrary, knocking down of GPR35 could partially suppress the tumor growth. When combined with chemotreatment, the suppression effect was significantly enhanced (Figure 3A). The tumor growth was examined every 3 days (Figure 3B) and the tumor weights were examined (Figure 3C). Of note, the IHC staining results of Ki67, a marker of cell proliferation, also indicated that combination of GPR35 depletion and cisplatin remarkably inhibited tumor cell growth (Figure 3D and E).
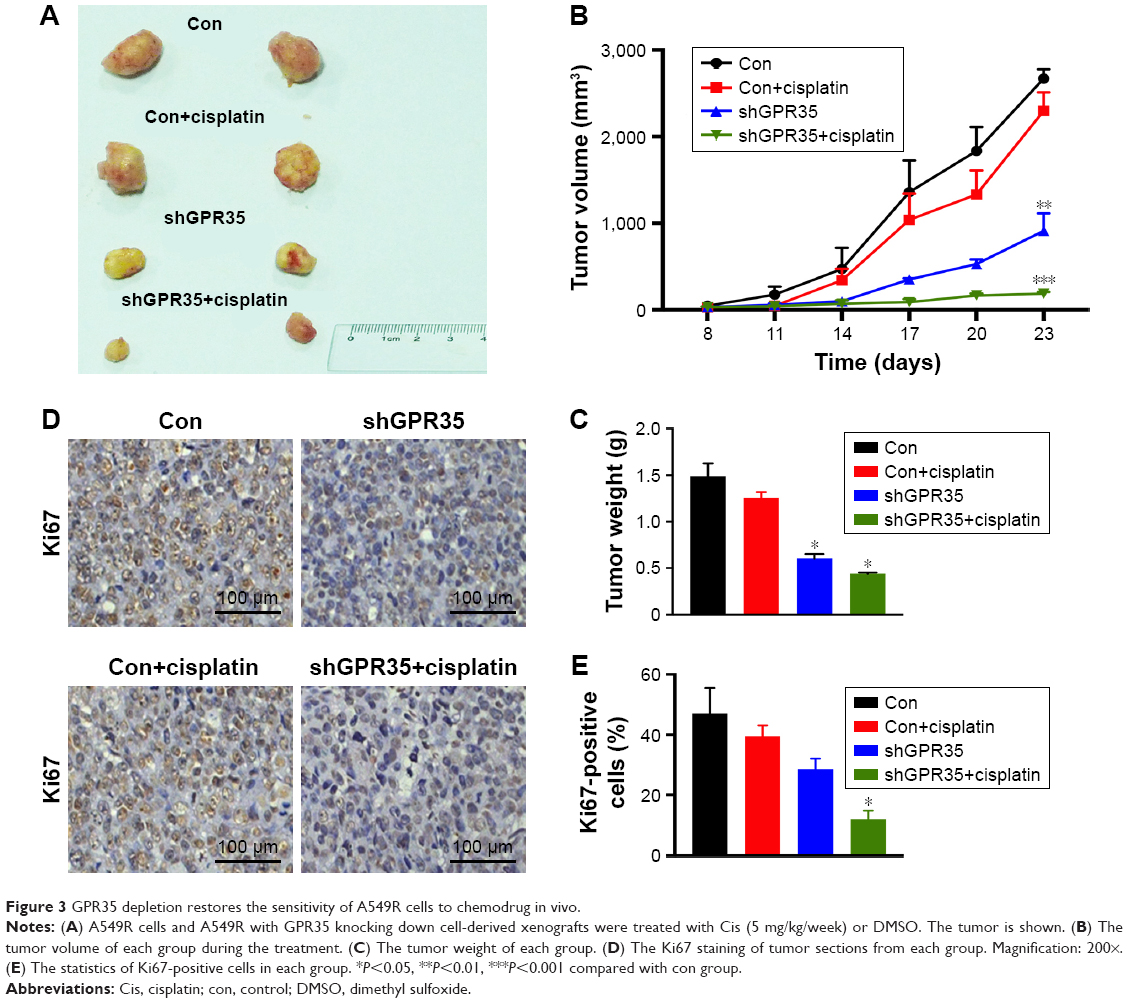 | Figure 3 GPR35 depletion restores the sensitivity of A549R cells to chemodrug in vivo. |
GPR35 regulates chemoresistance via β-arrestin-2/Akt loop
GPR35 could activate β-arrestins and has been demonstrated to have the tumorigenesis role, so we explored the mechanism of GPR35-mediated chemoresistance. GPR35 knockdown considerably reduced the expression of β-arrestin-2 and the activation state of Akt signaling (Figure 4A). Indeed, GPR35 activation could recruit β-arrestin-2. However, our results indicated that GPR35 also regulated β-arrestin-2 expression. More importantly, knocking down of β-arrestin-2 in A549R cells led to the suppression of GPR35 (Figure 4B). Furthermore, inhibition of Akt signal using LY294002 markedly decreased GPR35 and β-arrestin-2 expression (Figure 4C). We hypothesized that there was a regulation loop of GPR35/β-arrestin-2/Akt. Then, we ectopically expressed GPR35 in A549 cells, and we noticed that β-arrestin-2 as well as phosphor-AKT were elevated (Figure 4D). Subsequently, we knocked down β-arrestin-2 and treated cells with Dox and Cis and found that cell apoptosis rate was elevated (Figure 4E). Furthermore, combination of LY294002 and chemodrugs significantly induced the cell apoptosis as well (Figure 4F). Taken together, the results led to a regulatory loop mediating chemoresistance of NSCLC.
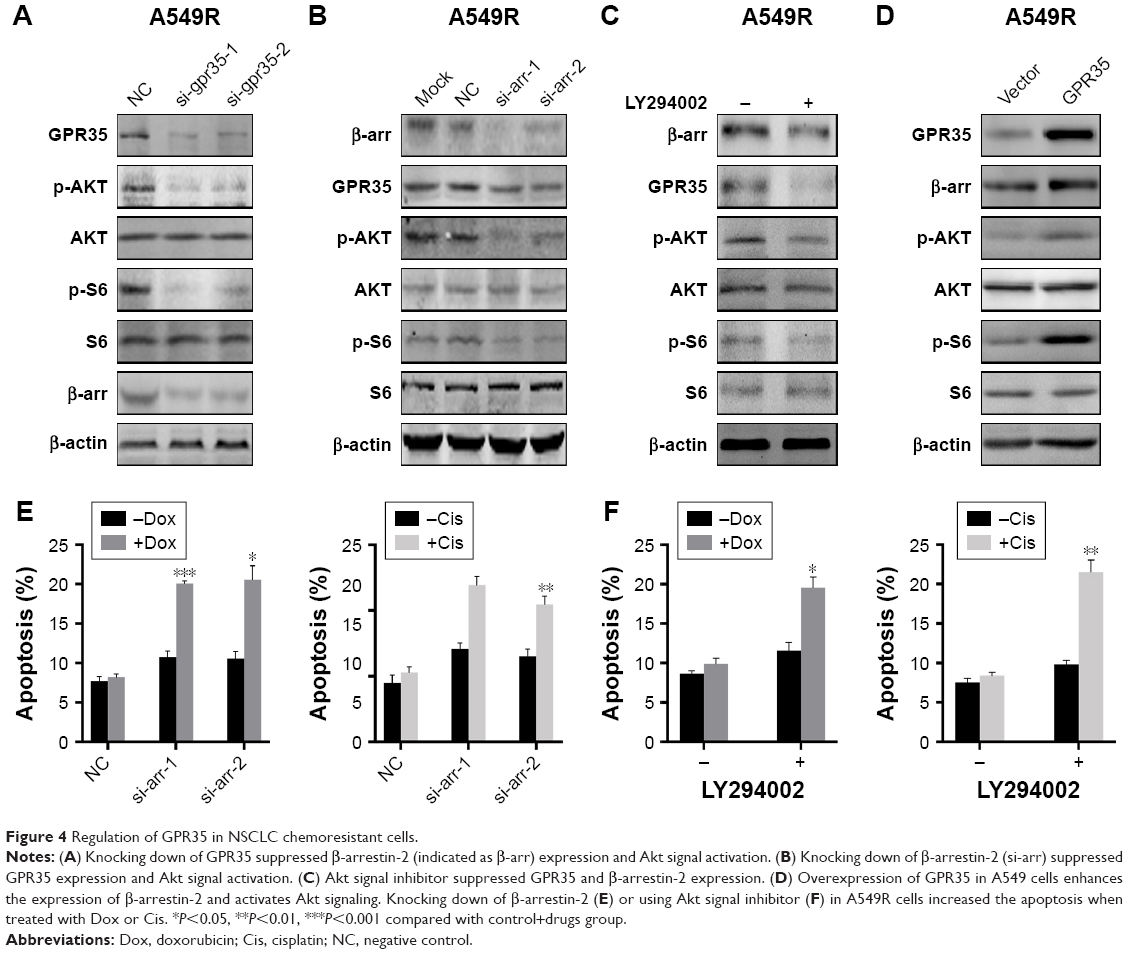 | Figure 4 Regulation of GPR35 in NSCLC chemoresistant cells. |
Discussion
The chemoresistance and relapse are the major causes of NSCLC treatment failure and contributors to poor prognosis.27 However, the mechanisms of chemoresistance in the progression of NSCLC are poorly characterized. In the present study, we showed that elevated expression of GPR35 enhanced the chemoresistant ability of NSCLC cells. In addition, interrupting GPR35/β-arrestin-2/Akt loop could partially restore the sensitivity. These results may further elucidate how GPR35 promotes the progression of NSCLC.
In the current study, we observed that GPR35 was predominantly expressed in NSCLC tissues both in mRNA and protein levels. More importantly, high expression of GPR35 was significantly associated with poor prognosis. Therefore, GPR35 might have oncogenesis role in NSCLC. Previous reports indicated that GPR35 was also overexpressed in many types of cancers, such as breast cancer and gastric cancer.28 Furthermore, we established in vitro A549 cell model of Dox toxicity resistance. In this regard, GPR35 was upregulated in the A549R cells, which is related to the high expression of β-arrestin-2 and activation of Akt signal. It has been demonstrated that β-arrestin-2 could upregulate MDR1 and related genes, leading to multidrug resistance in breast cancer cells.29 Moreover, we noticed that β-arrestin-2 also activated Akt signal, and it has been mentioned in dopamine receptor signaling before.30 Consistent with these findings, our data showed that either knocking down of β-arrestin-2 or using Akt inhibitor could suppress GPR35 expression and abrogate GPR35-mediated chemoresistance. Whether the antagonist of GPR35 has similar effect needs to be further explored.
As GPR35 is recognized as an orphan GPCR receptor, its agonists and antagonist were identified in many studies. However, their effects on cancer needed further investigation. For example, pamoic acid, an agonist of GPR35, could recruit β-arrestin-2 and activate extracellular signal-regulated kinase 1/2 (ERK1/2) signaling and promote tumorigenesis.11 Contradictorily, several agonists may also have anticancer effect. It is reported that aspirin metabolites, salicyluric acid, targets GPR35 and might contribute to the cancer-preventive effect of Aspirin.31 Kynurenic acid enhanced the expression of p21 Waf1/Cip1 in colon cancer HT-29 cells, leading to cell proliferation inhibition.32 Together with our findings, more studies should be done to demonstrate the mechanism of GPR35 in tumorigenesis and treatment.
Here, our data suggested that GPR35/β-arrestin-2/Akt loop contributed to the cancer cell chemoresistance. Recent study using genome-wide Crispr-cas9 activation screen has shown that GPR35 was involved in BRAF inhibitor resistance.33 Collectively, it suggested that GPR35 may potentiate a broad-spectrum resistance to chemodrugs. Besides, GPR35 activation also enhances cAMP production, intracellular Ca2+ concentration, and ERK phosphorylation as well,34,35 which are also related with chemoresistance.
In summary, our study revealed an important role of GPR35 in NSCLC chemoresistance. Furthermore, targeting the GPR35/β-arrestin-2/Akt signal may serve as a potential therapeutic approach to decrease chemoresistance and improve patient’s survival.
Acknowledgments
This study was supported by the Natural Science Foundation of Liaoning Province, China (20170540570), and the project of “Liaoning clinical research center for colorectal cancer” (2015225005), and “Liaoning BaiQianWan Talents Program” ([2017] No.C13).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Costa DB, Shaw AT, Ou SH, et al. Clinical Experience With Crizotinib in Patients With Advanced ALK-Rearranged Non-Small-Cell Lung Cancer and Brain Metastases. J Clin Oncol. 2015;33(17):1881–1888. | ||
Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–289. | ||
Moro-Sibilot D, Smit E, de Castro Carpeño J, et al. Outcomes and resource use of non-small cell lung cancer (NSCLC) patients treated with first-line platinum-based chemotherapy across Europe: FRAME prospective observational study. Lung Cancer. 2015;88(2):215–222. | ||
Park K, Kim JH, Cho EK, et al. East Asian Subgroup Analysis of a Randomized, Double-Blind, Phase 3 Study of Docetaxel and Ramucirumab Versus Docetaxel and Placebo in the Treatment of Stage IV Non-small Cell Lung Cancer Following Disease Progression after One Prior Platinum-Based Therapy (REVEL). Cancer Res Treat. 2016;48(4):1177–1186. | ||
O’Dowd BF, Nguyen T, Marchese A, et al. Discovery of three novel G-protein-coupled receptor genes. Genomics. 1998;47(2):310–313. | ||
Maravillas-Montero JL, Burkhardt AM, Hevezi PA, Carnevale CD, Smit MJ, Zlotnik A. Cutting edge: GPR35/CXCR8 is the receptor of the mucosal chemokine CXCL17. J Immunol. 2015;194(1):29–33. | ||
Son JW, Jeong KJ, Jean WS, et al. Genome-wide combination profiling of DNA copy number and methylation for deciphering biomarkers in non-small cell lung cancer patients. Cancer Lett. 2011;311(1):29–37. | ||
Wang J, Simonavicius N, Wu X, et al. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J Biol Chem. 2006;281(31):22021–22028. | ||
Taniguchi Y, Tonai-Kachi H, Shinjo K. Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett. 2006;580(21):5003–5008. | ||
Zhao P, Sharir H, Kapur A, et al. Targeting of the orphan receptor GPR35 by pamoic acid: a potent activator of extracellular signal-regulated kinase and β-arrestin2 with antinociceptive activity. Mol Pharmacol. 2010;78(4):560–568. | ||
Shore DM, Reggio PH. The therapeutic potential of orphan GPCRs, GPR35 and GPR55. Front Pharmacol. 2015;6:69. | ||
Stone TW. Kynurenic acid antagonists and kynurenine pathway inhibitors. Expert Opin Investig Drugs. 2001;10(4):633–645. | ||
Fallarini S, Magliulo L, Paoletti T, de Lalla C, Lombardi G. Expression of functional GPR35 in human iNKT cells. Biochem Biophys Res Commun. 2010;398(3):420–425. | ||
Jenkins L, Brea J, Smith NJ, et al. Identification of novel species-selective agonists of the G-protein-coupled receptor GPR35 that promote recruitment of β-arrestin-2 and activate Gα13. Biochem J. 2010;432(3):451–459. | ||
Mccallum JE, Mackenzie AE, Divorty N, et al. G-Protein-Coupled Receptor 35 Mediates Human Saphenous Vein Vascular Smooth Muscle Cell Migration and Endothelial Cell Proliferation. J Vasc Res. 2015;52(6):383–395. | ||
Hu HH, Deng H, Ling S, et al. Chemical genomic analysis of GPR35 signaling. Integr Biol. 2017;9(5):451–463. | ||
Ruiz-Pinto S, Pita G, Patiño-García A, et al. Exome array analysis identifies GPR35 as a novel susceptibility gene for anthracycline-induced cardiotoxicity in childhood cancer. Pharmacogenet Genomics. 2017;27(12):445–453. | ||
Rhodes DR, Yu J, Shanker K, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6. | ||
Landi MT, Dracheva T, Rotunno M, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008;3(2):e1651. | ||
Selamat SA, Chung BS, Girard L, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012;22(7):1197–1211. | ||
Okayama H, Kohno T, Ishii Y, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72(1):100–111. | ||
Hou J, Aerts J, den Hamer B, et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010;5(4):e10312. | ||
Győrffy B, Surowiak P, Budczies J, Lánczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013;8(12):e82241. | ||
Guo YJ, Zhou YJ, Yang XL, Shao ZM, Ou ZL, Zl O. The role and clinical significance of the CXCL17-CXCR8 (GPR35) axis in breast cancer. Biochem Biophys Res Commun. 2017;493(3):1159–1167. | ||
Qian Y, Xia S, Feng Z. Sox9 mediated transcriptional activation of FOXK2 is critical for colorectal cancer cells proliferation. Biochem Biophys Res Commun. 2017;483(1):475–481. | ||
Fang L, Cai J, Chen B, et al. Aberrantly expressed miR-582-3p maintains lung cancer stem cell-like traits by activating Wnt/β-catenin signalling. Nat Commun. 2015;6:8640. | ||
Okumura S, Baba H, Kumada T, et al. Cloning of a G-protein-coupled receptor that shows an activity to transform NIH3T3 cells and is expressed in gastric cancer cells. Cancer Sci. 2004;95(2):131–135. | ||
Jing X, Zhang H, Hu J, et al. β-arrestin 2 is associated with multidrug resistance in breast cancer cells through regulating MDR1 gene expression. Int J Clin Exp Pathol. 2015;8(2):1354–1363. | ||
Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122(2):261–273. | ||
Deng H, Fang Y. Aspirin metabolites are GPR35 agonists. Naunyn Schmiedebergs Arch Pharmacol. 2012;385(7):729–737. | ||
Walczak K, Turski WA, Rzeski W. Kynurenic acid enhances expression of p21 Waf1/Cip1 in colon cancer HT-29 cells. Pharmacol Rep. 2012;64(3):745–750. | ||
Konermann S, Brigham MD, Trevino AE, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517(7536):583–588. | ||
Berlinguer-Palmini R, Masi A, Narducci R, et al. GPR35 activation reduces Ca2+ transients and contributes to the kynurenic acid-dependent reduction of synaptic activity at CA3-CA1 synapses. PLoS One. 2013;8(11):e82180. | ||
Deng H, Hu H, Fang Y. Multiple tyrosine metabolites are GPR35 agonists. Sci Rep. 2012;2:373. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.