")
Back to Journals » Cancer Management and Research » Volume 13
Overexpression of 15-Hydroxyprostaglandin Dehydrogenase Inhibits A549 Lung Adenocarcinoma Cell Growth via Inducing Cell Cycle Arrest and Inhibiting Epithelial-Mesenchymal Transition
Authors Wang W, Yang C, Deng H
Received 26 July 2021
Accepted for publication 20 October 2021
Published 30 November 2021 Volume 2021:13 Pages 8887—8900
DOI https://doi.org/10.2147/CMAR.S331222
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Kattesh Katti
Weixuan Wang,1,* Changmei Yang,2,* Haiteng Deng2
1Institute of Chinese Medicine, Guangdong Pharmaceutical University, Guangzhou, People’s Republic of China; 2MOE Key Laboratory of Bioinformatics, Center for Synthetic and Systematic Biology, School of Life Sciences, Tsinghua University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Haiteng Deng
MOE Key Laboratory of Bioinformatics, Center for Synthetic and Systematic Biology, School of Life Sciences, Tsinghua University, Beijing, People’s Republic of China
Tel +86-10-62790498
Email [email protected]
Purpose: Lung cancer is one of the most commonly diagnosed cancer as well as the leading cause of cancer-related mortality worldwide, among which lung adenocarcinoma (LUAD) is the most frequent form of lung cancer. Previous studies have shown that 15-hydroxyprostaglandin dehydrogenase (15-PGDH) catalyzes the oxidation of prostaglandins to reduce their biological activities and behaves as a tumor suppressor in various cancers. Thus, we aimed to systematically examine the effects of 15-PGDH overexpression on cellular processes in lung adenocarcinoma cells.
Methods: The stable 15-PGDH-overexpressing A549 cell line was constructed using lentivirus particles. CCK-8 assay was used to determine the cell proliferation rate and sensitivity to cisplatin. Tandem mass tag (TMT)-based quantitative proteomic analysis was used to identify differentially expressed proteins between control and 15-PGDH-overexpression cells. The cell cycle was determined by a flow cytometer. The expression levels of mesenchymal and epithelial markers were measured using Western blotting. Wound healing and transwell assays were used to detect the cell migration and cell invasion ability, respectively.
Results: Analysis of datasets in The Cancer Genome Atlas revealed that the PGDH gene expression level in the lung adenocarcinoma tissues was significantly lower than that in the pericarcinous tissues. 15-PGDH overexpression in A549 cells reduced cell proliferation rate. Quantitative proteomics revealed that 15-PGDH overexpression inhibited PI3K/AKT/mTOR signaling pathway, which is a signaling pathway driving tumor cell growth and epithelial-mesenchymal transition (EMT) process. In addition, both cell cycle and DNA repair-related proteins were down-regulated in 15-PGDH overexpressed cells. 15-PGDH overexpression induced G1/S cell cycle arrest and increased susceptibility to DNA damaging reagent cisplatin. Importantly, overexpression of 15-PGDH inhibited EMT process with the downregulation of β-catenin and Snail-1 as well as upregulation of E-cadherin and ZO-1.
Conclusion: 15-PGDH is a tumor suppressor in lung cancer and may serve as a potential therapeutic target to prevent lung adenocarcinoma.
Keywords: 15-PGDH, proteomics, lung adenocarcinoma, cell proliferation, cell cycle arrest, epithelial-mesenchymal transition
Introduction
Lung cancer is one of the most commonly diagnosed cancers over the world.1 In 2020, new cases of lung cancer account for 11.4% of total cancer cases.1 Lung adenocarcinoma (LUAD) is the most common subtype of lung cancer and the only form of lung cancer, which non-smokers develop.2,3 The 5-year survival rate of lung cancer is still low, varying from 4% to 17% depending on the stage and regional differences.4 Nearly 70% of lung cancer patients are diagnosed with locally advanced or metastatic diseases, which emphasizes the necessity of early diagnosis for lung cancer.5 What is worse, many patients died of recurrent lung cancer even with early diagnosis and optimal treatment.6–8 Thus, further investigation of therapeutic targets that are associated with the tumorigenesis and progression of lung cancer is urgently required.
Previous studies have shown that 15-hydroxyprostaglandin dehydrogenase (15-PGDH) is a NAD+-dependent dehydrogenase that catalyzes the oxidation of prostaglandins and lipoxins, generating 15-keto derivatives with much reduced biological activities.9,10 Recently, accumulating attentions have been drawn to the vital role of 15-PGDH in tumorigenesis and progression. Emerging evidence has indicated that 15-PGDH was down-regulated in various cancers, including lung cancer, breast cancer, colorectal cancer, etc.11–13 Moreover, in non-small cell lung cancer and pancreatic ductal adenocarcinoma, a low expression level of 15-PGDH is associated with poor prognoses.14,15 The low expression of 15-PGDH correlated with high microvessel density, a standard quantification of angiogenesis, which indicated that 15-PGDH may improve the prognosis of non-small cell lung cancer through an anti-angiogenic mechanism.14 For breast cancer and gastric cancer patients, the low expression of 15-PGDH is associated with reduced overall survival.16,17 In addition, the expression level of 15-PGDH is associated with drug resistance.16 The down-regulated expression level of 15-PGDH is related to tamoxifen resistance and decreases overall survival in ERα-positive breast cancer patients.16 15-PGDH knockout markedly increased colon tumorigenesis and sensitized C57BL/6J mice to carcinogen azoxymethane.12 Accordingly, mice injected with lung adenocarcinoma A549 cells expressing wild-type 15-PGDH prevented lung cancer through down-regulating the expression of CD44 and Bcl-2.11 Similar researches were conducted in colon carcinomas and breast cancer, which showed inhibition of tumor growth by 15-PGDH overexpression.13,18 These findings indicate the important role of 15-PGDH in tumor progression and treatment. However, few studies have systematically examined the effects of 15-PGDH overexpression on cellular processes, including cell cycle and epithelial-mesenchymal transition (EMT), especially in lung adenocarcinoma.
In the present study, we established a stable cell line, in which 15-PGDH was overexpressed for comprehensively determining the effects of 15-PGDH on protein expression and biological processes. Our data showed that 15-PGDH was a key regulator in lung adenocarcinoma and may serve as a potential therapeutic target in lung cancer prevention.
Materials and Methods
Cell Culture
The human lung adenocarcinoma cell line, A549, and human embryonic kidney cell line, 293T, were obtained from the cell bank of the Chinese Academy of Sciences (Shanghai, China). Cells were grown in RPMI-1640 medium or Dulbecco’s Modified Eagle Medium (Wisent, Montreal, QC, Canada) and incubated at 37°C with 5% CO2. The medium was supplemented with 10% FBS (Wisent) and 1% penicillin/streptomycin (Wisent).
Establishment of Stable 15-PGDH Overexpression Cell Line
Human PGDH cDNA was obtained from the A549 cell line. A Flag-tag was added at the C-terminus and the recombinant human PGDH DNA was cloned into PLVX-IRES-ZsGreen1 lentiviral vector. 293T cells were transfected with PLVX-IRES-ZsGreen1 or PLVX-PGDH-IRES-ZsGreen1, respectively. The supernatants were concentrated using polyethylene glycol (PEG) 6000 and precipitated lentiviral particles were resuspended in PBS. A549 cells were then infected by lentiviral particles along with 5 μg/mL polybrene (Sigma, St Louis, MO, USA) and sorted by a flow cytometer. A single green fluorescent protein (GFP)-positive cell was seeded into a 96-well plate. A clone with intense and uniform GFP expression was chosen for further experiments.
Western Blotting Analysis
Empty vector (EV) and 15-PGDH overexpression (PGDH(+)) cells were harvested and lysed for 30 min on ice with RIPA lysis buffer (Solarbio, Beijing, China), supplemented with 1% Protease Inhibitor Cocktail (Thermo Fisher Scientific, Rockford, IL). Supernatants were collected after centrifugation at 12,000 rpm for 20 min at 4°C. After concentration determined with BCA protein assay kit (Solarbio), proteins were separated by 12% SDS-PAGE gels and transferred onto PVDF membranes. Western blotting analysis followed a standard procedure. β-Actin antibody was purchased from Cell Signaling Technology (Danvers, MA, USA) and used as an internal control. Flag antibody was purchased from Sigma. Primary antibodies against β-catenin, E-cadherin, and Snail1 were purchased from Cell Signaling Technology. ZO-1 antibody was obtained from Proteintech (Chicago, IL, USA). DTL antibody was purchased from Bioss (Beijing, China). KIAA0101 antibody was purchased from Solarbio. EXO1 antibody was purchased from cloud-clone (Wuhan, China).
Quantitative Real-Time PCR (qPCR)
Total RNA was extracted from EV and PGDH(+) cells using the RNAprep Pure Cell Kit (Tiangen, Beijing, China). cDNA was then synthesized by the TIANScript RT kit (Tiangen). qPCR was performed using the Roche LightCycler® 480II Detection System with SYBR green (Tiangen), and β-ACTIN was used as an internal control. All primers were acquired from Primer Bank (http://pga.mgh.harvard.edu/primerbank/) and listed in Supplementary Table 1.
Cell Proliferation Rates Determined by CCK-8
Cells were seeded in 96-well plates with 2000 cells/well. Cell proliferation rates were determined using Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Kumamoto, Japan). CCK-8 reagent was added to each well and incubated at 37°C for 2 h. Optical density (OD) was measured at 450 nm with a microplate reader (Bio-Rad, Hercules, USA).
Tandem Mass Tag (TMT)-Based Quantitative Proteomic Analysis
Equal amounts of protein (200 μg) were extracted from EV and PGDH(+) cells and reduced by dithiothreitol (Merck, Whitehouse Station, NJ) at 5 mM for 60 min at room temperature and alkylated with 12.5 mM iodoacetamide (Sigma, St Louis, MO) for 45 min in the dark at room temperature. In-solution digestion was then carried out with trypsin (Promega, Fitchburg, WI) for 14 h at 37 °C and peptides were desalted using Sep-Pak C18 cartridges, and labeled by TMT 6-plex reagents (Thermo Fisher Scientific) according to the manufacturer’s instructions. The TMT labeled peptides were mixed and desalted with Sep-Pak C18 cartridges (Waters, Milford, MA) followed by HPLC separation. The collected eluents were concatenated in 12 fractions and all the fractions were dried down using SpeedVac and dissolved in 0.1% formic acid for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
For LC-MS/MS analysis, TMT-labeled peptides were separated at a flow rate of 0.3 μL/min with a Thermo-Dionex Ultimate 3000 HPLC system that was directly interfaced with a Thermo Orbitrap Fusion Lumos mass spectrometer. The analytical column was a homemade fused silica capillary column (75 μm inner-diameter, 150 mm length; Upchurch, Oak Harbor, WA, USA) and packed with C-18 resin (300 Å, 5 μm; Varian, Lexington, MA, USA). Mobile phase A consisted of 0.1% formic acid, and mobile phase B consisted of 100% acetonitrile and 0.1% formic acid. The Thermo Orbitrap Fusion Lumos mass spectrometer was operated in the data-dependent acquisition mode using Xcalibur 4.0.27.10 software.
Peak lists from LC-MS/MS analysis were generated with the SEQUEST searching algorithm using Proteome Discoverer software (version 1.4; Thermo Fisher Scientific). Spectra were searched against the UniProt human reference proteome (released on March 17, 2017, containing 21,042 entries) using an in-house Proteome Discoverer Searching Algorithm (version 1.4; Thermo Fisher Scientific). The search criteria were as follows: full tryptic specificity was required; two missed cleavage sites were allowed; oxidation of methionine, deamidation at asparagine and glutamine were set as variable modifications; carbamidomethylation of cysteine, TMT 6-plex at lysine or protein N-terminus were set as the fixed modifications; precursor ion mass tolerance was 20 ppm and fragment ion mass tolerance was 20 mmu. Peptide false discovery rate (FDR) was estimated using Proteome Discoverer, with a cutoff score of 1%. Relative protein quantification was performed with Proteome Discoverer 1.4 software based on the reporter ion intensities per peptide. Protein ratios were calculated as the median of all peptide hits belonging to a protein. Proteins with fold-change >1.5 or <0.67 were considered differentially expressed.
Immunoprecipitation Followed by LC-MS/MS Analysis
Equal amounts of proteins from EV and PGDH(+) cells were incubated with Anti-FLAG M2 Affinity Gel (Sigma, St Louis, MO) at 4 °C in a rotary wheel. After 6 h incubation, the beads were washed and boiled with 4× loading buffer (Solarbio) followed by 1D SDS-PAGE separation. In-gel digestion was then carried out following a standard procedure. Briefly, the gel was excised, reduced with 25 mM dithiothreitol and alkylated with 55 mM iodoacetamide followed by trypsin digestion for 14 h at 37 °C. The peptides were extracted twice with 0.1% formic acid in 50% acetonitrile aqueous solution and the volume was reduced using speedvac. LC-MS/MS was used for analysis. Protein identification was carried out using Proteome Discoverer Searching Algorithm (Version 1.4). The label-free quantitation method was used to determine 15-PGDH-interacting proteins. When a protein was only identified in PGDH(+) cells with spectra counts >5 or the ratio of the spectra counts for a protein in immunoprecipitated samples from PGDH(+) and EV cells was higher than 5, the protein was considered as 15-PGDH-interacting proteins.
Cell Cycle Analysis for EV and PGDH(+) Cells
EV and PGDH(+) cells were seeded into 6-well plates. After 36 h incubation, cells were trypsinized, washed twice with PBS, and fixed with 70% ethanol at 4°C overnight. The cells were centrifuged and resuspended in PBS, treated with RNase and stained with Propidium Iodide (Solarbio). Cell cycle was measured with a BD FACSAria II Flow Cytometer (BD Biosciences, San Jose, CA, USA). The percentage of cells in each cell cycle phase was assessed using Modfit software.
Susceptibility of EV and PGDH(+) Cells to Cisplatin
EV and PGDH(+) cells were seeded into 96-well plates with 4000 cells/well. After 36 h incubation, cells were treated with cisplatin (Selleck, Houston, TX) in triplicate for 30 h. CCK-8 reagent was added to each well and incubated at 37 °C for 2 h. OD was measured at 450 nm with a microplate reader (Bio-Rad). Cell viability was represented as the percentage of viable cells compared to untreated cells.
Wound Healing Assay
EV and PGDH(+) cells were seeded into 6-well plates. When the cell confluence reached 95%, a sterile 200 µL pipette tip was used to scratch the cells to generate a wound. Cells were then cultured in RPMI-1640 supplemented with 2% FBS. Closures of these wounds were imaged at 0 and 48 h with a Nikon microscope (Olympus Corporation, Tokyo, Japan). The wound area was evaluated by ImageJ software version 1.52k (National Institutes of Health, Bethesda, MD, USA).
Transwell Invasion Assay
Transwell invasion assay was performed using 8-μm-pore transwell chambers (Corning, New York, NY). Briefly, EV and PGDH(+) cells were starved for 12 h. Then, cells were trypsinized, washed twice with serum-free medium, and resuspended in serum-free medium. Cells were counted and seeded into the upper insert of the transwell chamber while RPMI-1640 medium containing 10% FBS were added to the lower chamber. Cells were then incubated at 37°C with 5% CO2. Cells that invaded through the Matrigel (Solarbio, Beijing, China)-coated membrane were trypsinized and CCK-8 reagent was added to treated cells and incubated at 37°C for 2 h. Optical density (OD) was measured at 450 nm with a microplate reader.
Statistical Method
GraphPad Prism 7.0 software (La Jolla, CA, USA) was used for statistical analysis. Significant differences were determined using Student’s t-test. p-value <0.05 was considered significant.
Results
PGDH is Downregulated in Multiple Tumors Compared with Normal Tissues
Analyzing datasets from The Cancer Genome Atlas (TCGA) with GEPIA website (http://gepia.cancer-pku.cn),19 we found that PGDH was significantly lower in the majority of tumor tissues compared with paired normal tissues (Supplementary Figure 1A). We next analyzed PGDH gene expression in LUAD using TCGA datasets and found that the gene expression level of PGDH was decreased approximately 2-fold in the LUAD tissues compared with normal tissues (Figure 1A). Considering that the number of normal samples in the TCGA database is rather small, the Genotype-Tissue Expression (GTEx) normal sample database was added to the analysis, and a similar result was obtained (Figure 1B), suggesting that 15-PGDH is a tumor suppressor in lung adenocarcinoma initiation and progression.
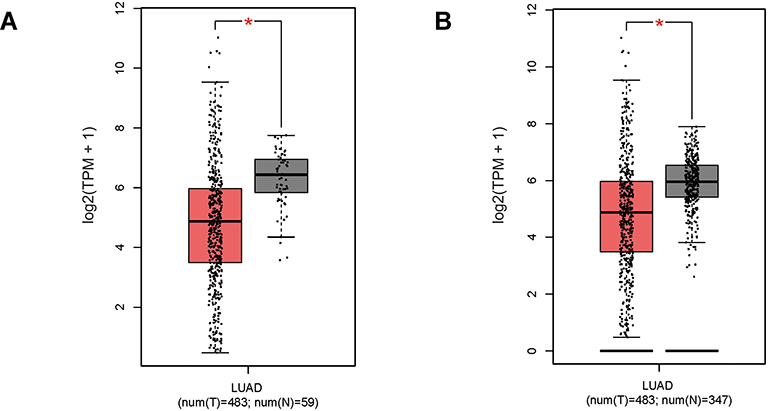 |
Figure 1 Gene expression level of PGDH was lower in Lung adenocarcinoma (LUAD) tissues compared with normal tissues. (A) PGDH gene expression level analysis between TCGA tumor samples and TCGA normal samples. (B) PGDH gene expression level analysis between TCGA tumor samples and TCGA normal + GTEx normal samples. TPM, Transcripts Per Million. The expression data were log2(TPM+1) transformed for differential analysis. *p<0.05. |
15-PGDH Overexpression Inhibits Cell Growth of A549 Cells
To study the effects of 15-PGDH overexpression on cellular processes, we established a stable cell line in which 15-PGDH was overexpressed in A549 cells. Lentiviral particles containing 15-PGDH vector were transfected into A549 cells and sorted by a flow cytometer to produce stable 15-PGDH-overexpressing cell line (named PGDH(+) cells) (Figure 2A and B). The control cells were transfected with pLVX-IRES-ZsGreen1 empty vector (named EV cells). CCK-8 assay showed that there was nearly no difference in cell growth between wild-type and EV A549 cells (Supplementary Figure 2A). 15-PGDH overexpression in A549 cells was confirmed by Western blotting and qPCR (Figure 2C and D), showing an approximately 10-fold increase of PGDH mRNA expression level. Proliferation rates of EV and PGDH(+) cells were determined by CCK-8 assay, showing that PGDH(+) cells grew slower than EV cells observably from 36 h. At 96 h, the number of PGDH(+) cells was approximately 45% less than that of EV cells (Figure 2E). In addition, SW033291,9 an inhibitor of 15-PGDH enzyme activity, was used to treat PGDH(+) cells and found an increased cell proliferation rate compared with the untreated PGDH(+) cells (Supplementary Figure 2B).
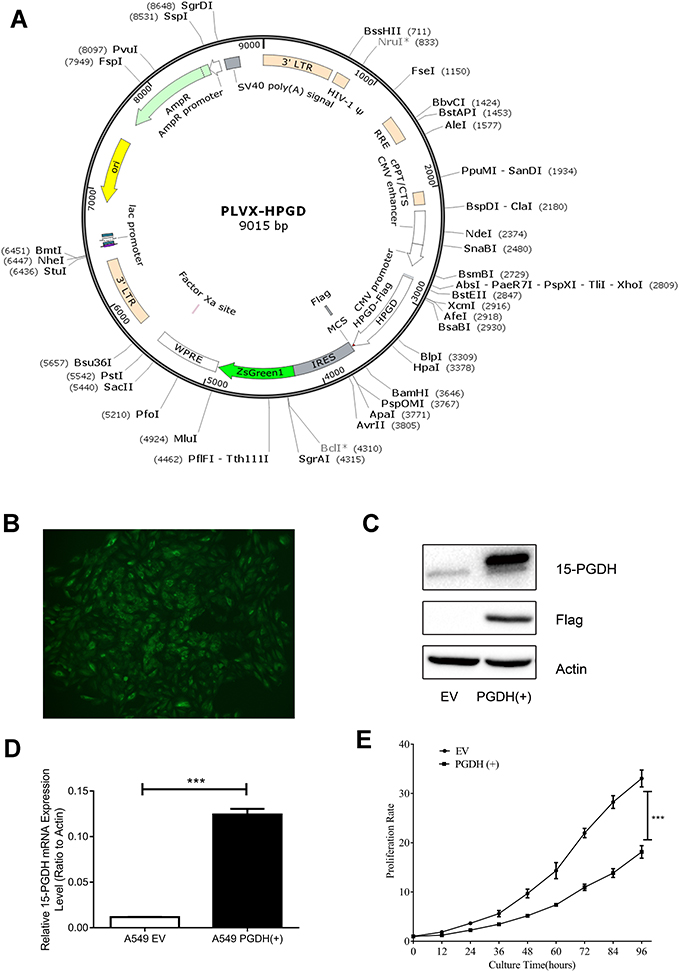 |
Figure 2 15-PGDH overexpression in A549 cells suppressed cell proliferation. (A) PLVX-PGDH-IRES-ZsGreen1 plasmid map visualization was performed with SnapGene software. (B) A 15-PGDH overexpression single clone cell line with intense and uniform GFP expression (400×). (C) Western blotting analysis confirmed that 15-PGDH is overexpressed successfully in A549 cells. (D) qPCR analysis confirmed overexpressed PGDH mRNA levels in A549 cells. (E) Growth curves of EV and PGDH(+) A549 cells determined by CCK-8 assay. Cell proliferation rates were represented as the ratio between OD450 at a particular time point and 0 hour. All values represent mean from three biological replicates ± SEM. Data were analyzed using Student’s t-test. ***p< 0.001. |
TMT-Based Quantitative Proteomic Analysis of EV and PGDH(+) A549 Cells
We next carried out the TMT-based quantitative proteomic analysis to identify differentially expressed proteins between EV and PGDH(+) A549 cells. Equal amounts of protein from EV and PGDH(+) cells were digested in solution, labeled with TMT reagents and subsequently fractionated by HPLC. Twelve fractions were analyzed by nano-LC-MS/MS to identify differentially expressed proteins (Figure 3A). We identified approximately 6000 proteins and the quantitation values were determined based on TMT ratios. The false-positive rate threshold was set to be 1%. Based on TMT ratios (>1.5 or <0.67), 404 proteins were differentially expressed between EV and PGDH(+) A549 cells, with 97 proteins down-regulated and 307 up-regulated (Supplementary Tables 2 and 3). Gene list annotations were summarized in a pie chart using the PANTHER bioinformatics platform (http://www.pantherdb.org/), showing that more than a quarter of down-regulated proteins were associated with cellular metabolism, including nucleotide-containing compound metabolic process, lipid metabolic process, and amino acid metabolic process (Figure 3B). These results suggest that 15-PGDH overexpression decreased cellular metabolism, pointing out the important role of 15-PGDH in the regulation of cellular metabolic homeostasis.
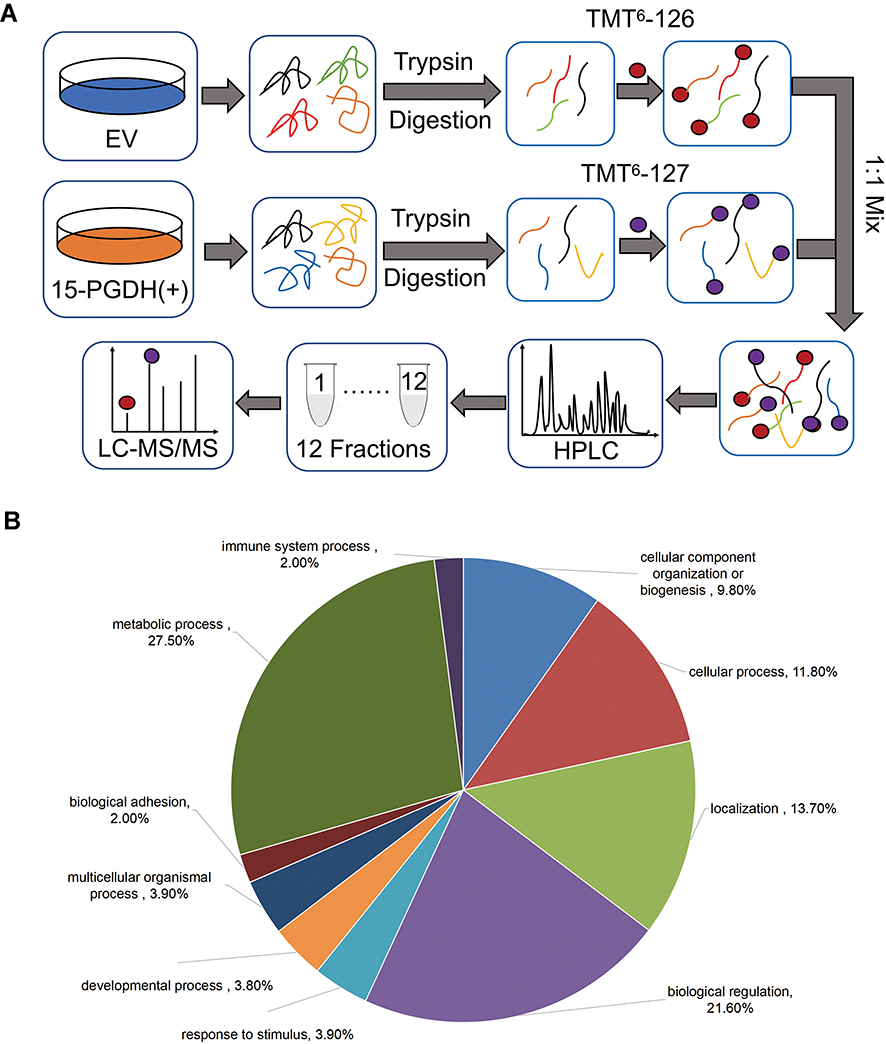 |
Figure 3 Quantitative proteomic study of EV and PGDH(+) A549 cells. (A) The schematic diagram of TMT-based quantitative proteomic analysis of EV and PGDH(+) A549 cells. (B) Functional classification of down-regulated proteins between EV and PGDH(+) A549 cells by PANTHER bioinformatics platform (http://www.pantherdb.org). |
Among differentially expressed proteins, seven proteins involved in cell growth and proliferation regulation were downregulated in PGDH(+) cells, including rapamycin-insensitive companion of mTOR (RICTOR), ribosomal protein S6 kinase beta-1 (RPS6KB1), protein NDRG1 (NDRG1), serine/threonine-protein kinase 26 (STK26), progranulin (GRN), proliferation-associated nucleolar protein p120 (NOP2), and protein S100-P (S100P) (Figure 4A). Also, the expression level of three DNA repair-related proteins including exonuclease 1 (EXO1), PCNA-associated factor (KIAA0101), and denticleless protein homolog (DTL) and seven cell cycle regulation-related proteins including 3ʹ-5ʹ RNA helicase YTHDC2 (YTHDC2), cell division cycle protein 20 homolog (CDC20), protein PIMREG (FAM64A), borealin (CDCA8), zinc finger protein 318 (ZNF318), NOP2, and RPS6KB1 were lower in PGDH(+) cells compared with EV cells (Figure 4B). The downregulation of these DNA repair-related proteins (EXO1, KIAA0101, and DTL) in PGDH(+) cells was verified using Western blotting (Supplementary Figure 3A).
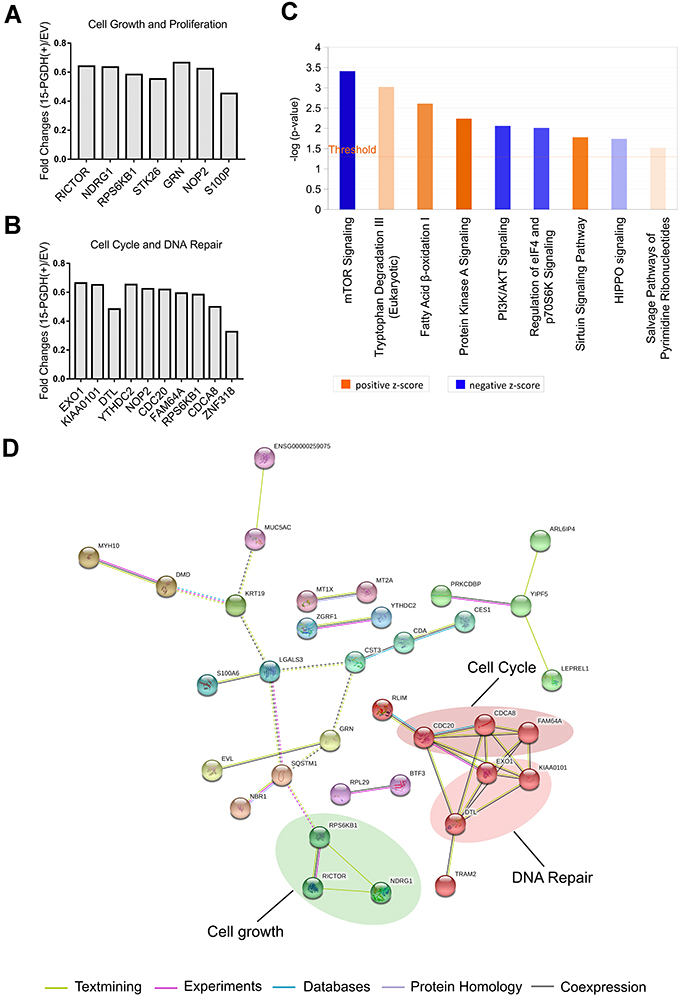 |
Figure 4 Proteomic analysis of EV and PGDH(+) A549 cells using IPA software and String database. (A) TMT ratios of down-regulated proteins related to cell growth and proliferation. (B) TMT ratios of down-regulated proteins related to cell cycle and DNA repair. (C) The enriched canonical pathways of differentially expressed proteins by IPA analysis. (D) Interaction network of down-regulated proteins between PGDH(+) and EV cells was analyzed by String software. |
Further, Ingenuity Pathway Analysis (IPA) software was used to identify significantly changed canonical pathways associated with differentially expressed proteins between EV and 15-PGDH(+) cells. The significantly repressed canonical pathways included mTOR signaling, PI3K/AKT signaling, and regulation of eIF4 and p70S6K signaling (Figure 4C), which indicated that 15-PGDH overexpression may suppress tumor cell proliferation via inhibiting PI3K/AKT/mTOR signaling pathway. Down-regulated proteins in PGDH(+) cells were further analyzed by STRING database to create protein interaction network.20 We found that these interacting proteins can be divided into three categories, including cell growth (RICTOR, RPS6KB1, and NDRG1), cell cycle (CDC20, FAM64A, and CDCA8), and DNA repair (EXO1, KIAA0101, and DTL) (Figure 4D), indicating the vital roles of these proteins in 15-PGDH-mediated cellular processes and suggesting 15-PGDH overexpression affects cell cycle regulation and sensitivity to DNA damaging chemotherapy drugs. Further, we immunoprecipitated 15-PGDH complexes from EV and PGDH(+) cells to identify binding partners of 15-PGDH. 15-PGDH interacting proteins can be divided into eight categories, including cell cycle, DNA repair, cytoskeleton, RNA binding, tRNA ligase, chaperon, and protein, amino acid, ion, electron transport (Figure 5). The immunoprecipitation results suggest that 15-PGDH regulates EMT process, cell cycle, and cell susceptibility to DNA damaging drugs by interacting with cytoskeleton, cell cycle- and DNA repair-related proteins.
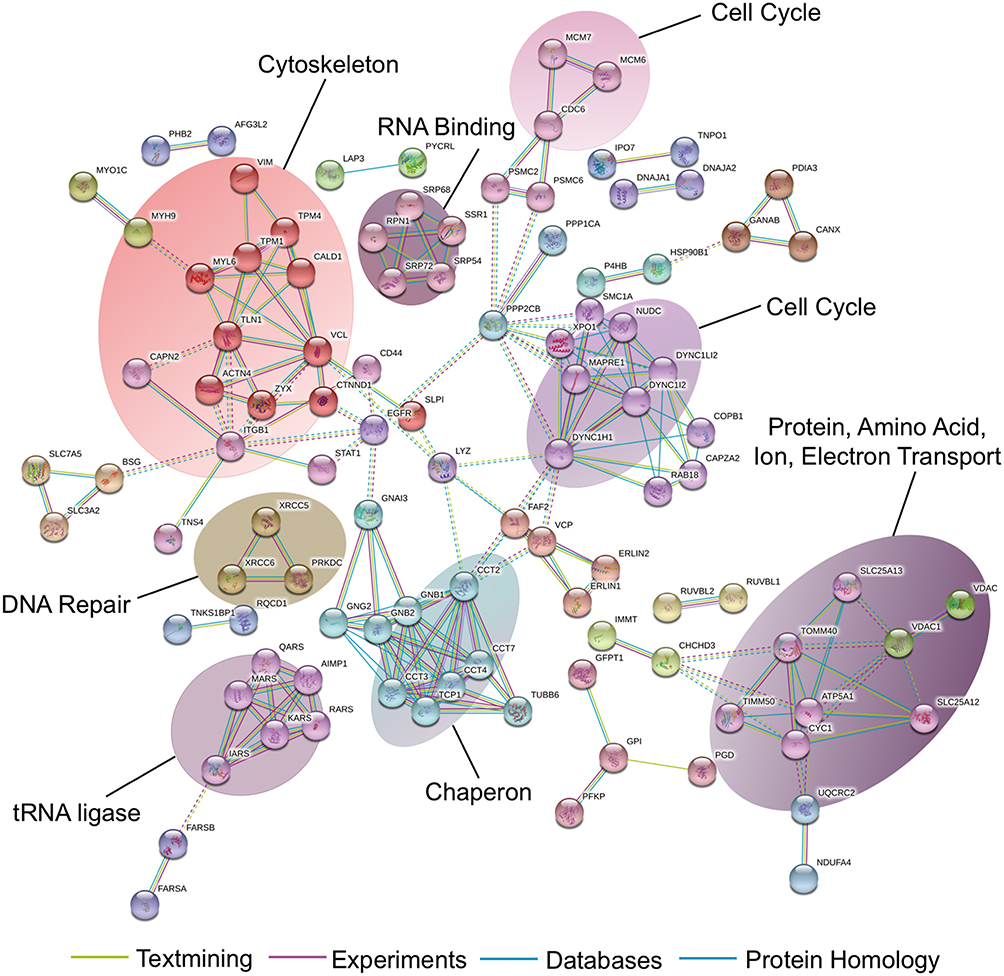 |
Figure 5 Identification of 15-PGDH-interacting proteins. Analysis of 15-PGDH-interacting proteins using String database. |
Overexpression of 15-PGDH Induces G1/S Arrest, Increases Cell Sensitivity to Cisplatin, and Inhibits EMT Process
Down-regulation of cell cycle-related proteins (YTHDC2, NOP2, CDC20, FAM64A, RPS6KB1, CDCA8, and ZNF318) suggests that 15-PGDH overexpression affects cell cycle. To explore the effects of 15-PGDH overexpression on cell cycle, we performed cell cycle detection for EV and PGDH(+) cells. The result showed a significant increase in the percentage of PGDH(+) cells at G1 phase compared to EV cells, with a concurrent decrease in the proportion of cells at S and G2/M phases (Figure 6A and B). The aforementioned experiment indicated that 15-PGDH overexpression induced G1/S arrest, which contributes to the suppressed cell growth in A549 cells.
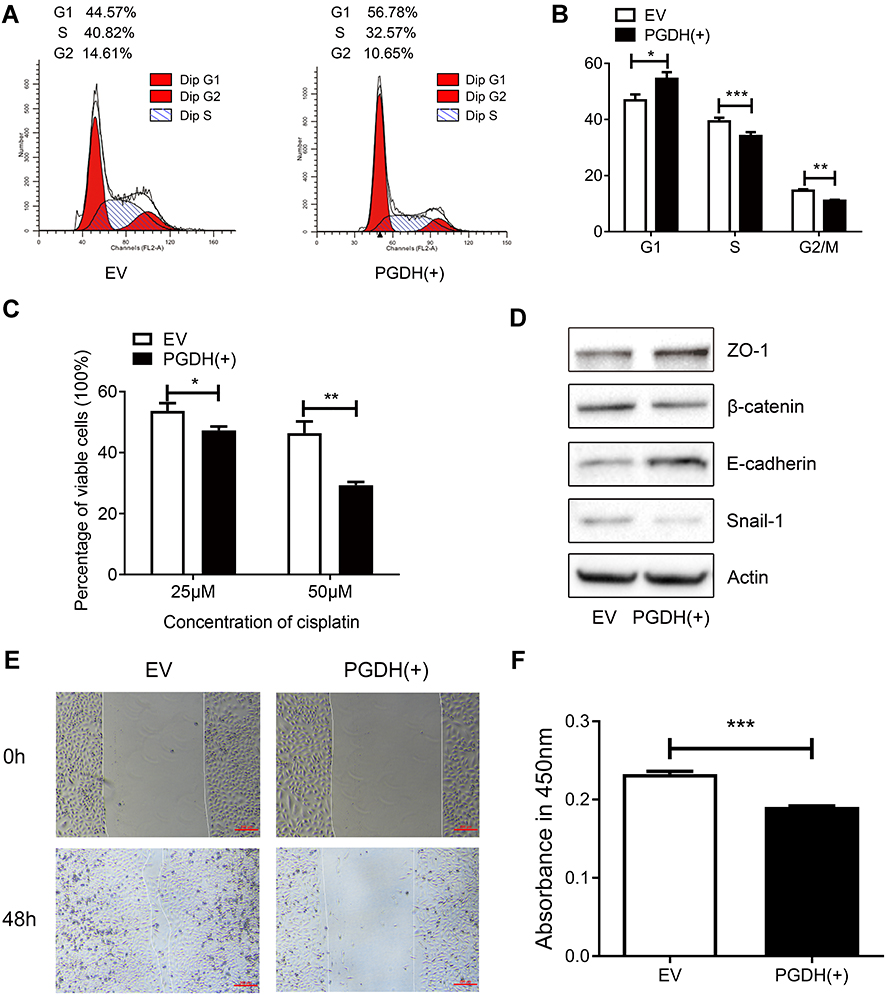 |
Figure 6 15-PGDH overexpression leads to G1/S arrest, increases cell susceptibility to cisplatin, and inhibits EMT process. (A) A representative cell cycle analysis determined by flow cytometry of EV and PGDH(+) cells. (B) Cell cycle analysis of EV and PGDH(+) cells. (C) Percentage of viable cells between EV and PGDH(+) cells treated with different concentrations of cisplatin for 30 h. (D) Western blotting of expression levels of ZO-1, β-catenin, E-cadherin, Snail1, and β-actin in EV and PGDH(+) cells. (E) Cell migration capacity of EV and PGDH(+) cells were determined by wound healing assay. Scale bar, 200 μm. (F) Cell invasion capacity of EV and PGDH(+) cells were determined using transwell assay. All values represent mean from three biological replicates ± SEM. Data were analyzed using Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001. |
Down-regulation of DNA repair-related proteins (EXO1, KIAA0101, and DTL) suggests that 15-PGDH overexpression may render cancer cell more sensitive to DNA damaging drugs. Cisplatin, a DNA damaging chemotherapy drug, is an effective anti-cancer agent that is widely used in the treatment of multiple solid tumors including LUAD.21,22 The way cells respond to DNA damage plays a key role in cisplatin sensitivity. To examine the susceptibility of EV and PGDH(+) cells to cisplatin, both cells were treated with different concentrations of cisplatin. Dose-dependent effects of cisplatin were measured by CCK-8 assay and represented as the percentage of viable cells after cisplatin treatment. The percentage of viable cells was 53.70 ± 1.45% and 47.26 ± 0.75% for EV and PGDH(+) cells, respectively, after treatment with 25 µM cisplatin for 30 h. The percentage of viable cells decreased to 29.26% when PGDH(+) cells were treated with 50 µM cisplatin for 30 h (Figure 6C), much lower than the percentage of viable EV cells (46.37%) and it demonstrated that PGDH(+) cells are extremely sensitive to cisplatin treatment compared with EV cells. Taken together, 15-PGDH can sensitize A549 cells to chemotherapy drug cisplatin, which may be caused by the downregulated proteins associated with DNA repair.
As aforementioned, the enriched downregulated signaling pathways in 15-PGDH overexpression cells included PI3K/AKT/mTOR, which was reported to play essential roles in EMT.23,24 Thus, we further tested whether overexpression of 15-PGDH inhibited the EMT process. Western blotting analysis showed that PGDH(+) cells exhibited significantly lower expression of the mesenchymal markers (Snail1 and β-catenin) and higher expression of the epithelial markers (ZO-1 and E-cadherin) (Figure 6D). Wound healing and transwell assay showed that 15-PGDH overexpression significantly decreased the cell migration and cell invasion ability (Figure 6E and F). These results suggest that 15-PGDH overexpression can significantly inhibit the EMT process, which contributed to the suppressed cell growth in A549 cells.
Discussion
Previous studies have shown that 15-PGDH is downregulated and behaves as a tumor suppressor in various cancers, such as lung cancer, breast cancer, and colon cancer.11–13 In the present study, we found that the gene expression level of PGDH was significantly lower in lung adenocarcinoma when compared with paired normal tissues based on TCGA data analysis, pointing out the key role of 15-PGDH in lung cancer initiation and development. To characterize the effects of 15-PGDH overexpression on cellular processes, we established a 15-PGDH-expressing cell line, PGDH(+) cells. 15-PGDH overexpression in A549 cells was confirmed by Western blotting and qPCR. By measuring the proliferation rates of EV and 15-PGDH(+) cells, we found that PGDH(+) cells grew much slower, demonstrating that 15-PGDH overexpression inhibited lung cancer cell growth and proliferation.
Chemotherapy is commonly used for lung adenocarcinoma treatment. Among chemotherapeutic drugs, platinum-based antineoplastic agents, a class of DNA damage drugs, are the primary therapeutics in lung adenocarcinoma treatment.25 However, the overall survival rate of patient remains low due to drug resistance. The increased DNA repair pathway activity may be the reason for drug resistance in cancers.26 In our study, we found that PGDH(+) cells were more sensitive to cisplatin, and the expression levels of DNA repair-related proteins, EXO1, KIAA0101, and DTL, were down-regulated in PGDH(+) cells. Researches have shown that EXO1 interacts with Ku70 and affects DNA damage repair. EXO1 knockdown sensitizes ovarian cancer cells to cisplatin and doxorubicin.27,28 KIAA0101 is overexpressed in various human malignancies, such as lung cancer, esophageal cancer, and hepatocellular carcinoma,29–33 and the increased KIAA0101 expression level is associated with poor survival and drug resistance.31,32 KIAA0101 induced cisplatin resistance of esophageal cancer cells by decreasing apoptosis.32 KIAA0101 knockdown increased the sensitivity of ovarian cancer cells to cisplatin treatment through inhibiting Wnt/β-catenin signaling pathway.31 These results indicate that 15-PGDH activation may be a potent strategy when facing drug resistance, mainly due to its ability in regulating DNA damage repair pathway.
EMT is a process known as the transition of epithelial cells to motile mesenchymal cells, which plays a vital role in cancer progression.34 In our study, we found that 15-PGDH overexpression inhibited EMT and PI3K/AKT/mTOR signaling pathway in PGDH(+) cells. The PI3K/AKT/mTOR pathway is activated in the majority of cancers and plays an essential role in numerous biological processes including EMT.23,35,36 AKT is activated by phosphatidylinositol 3,4,5-triphosphate (PIP3), a lipid second messenger generated by PI3K, and leads to the phosphorylation of mTOR that participates in the initiation of translation of proteins essential for cell metabolism, cell growth, cell cycle, and EMT through its downstream targets, 4EBP1 and p70S6K.35,37 These results demonstrate that 15-PGDH plays a vital role in the prevention of lung cancer progression by inhibiting PI3K/AKT/mTOR signaling pathway.
There are many factors that can affect cell proliferation, among which cell cycle arrest is an important factor. In our present study, 15-PGDH overexpression is associated with the decreased expression levels of several DNA repair-related proteins (EXO1, KIAA0101, and DTL), and 15-PGDH overexpression induces G1/S cell cycle arrest. Researches have shown that EXO1 knockdown inhibits DNA repair, induces cell cycle arrest, and inhibits cell proliferation in astrocytoma.38 EXO1 interacts with cell cycle regulation protein PCNA and 14-3-3 isoforms, which may therefore affect cell cycle.39,40 EXO1 knockdown decreases cell proliferation and tumorigenicity of hepatocellular carcinoma cells while EXO1 overexpression increases cell proliferation.41 KIAA0101 knockdown inhibits cell growth and causes G1/S cell cycle arrest in breast cancer by promoting the interaction between p53 and Sp1, which results in a decreased level of free Sp1.42 Sp1 can bind to the promoter and positively regulate the expression levels of CCNE2, CDK6, and CDKN1A, which are important regulators in cell proliferation and cell cycle progression.42 Overexpression of KIAA0101 increases cell proliferation and reduces the proportion of cells in G1 phase in esophageal cancer cells by upregulating the expression levels of cyclin A and cyclin B1.32 DTL knockdown reduces cell proliferation through inducing G2 arrest.43 DTL inactivation can prevent the ubiquitination of p53, which increases the levels of p53 and its downstream target p21, leading to G2 arrest.43 These results indicate that 15-PGDH overexpression inhibits cell proliferation by inducing cell cycle arrest via decreasing the expression levels of DNA repair-related proteins.
In addition, EMT plays a vital role in the regulation of cell proliferation. In our present study, we found that overexpression of 15-PGDH downregulated the expression of the mesenchymal marker Snail1 and upregulated the expression of the epithelial marker E-cadherin. Snail1, a vital positive regulator in EMT and cell proliferation, induces EMT by repressing the expression of E-cadherin and upregulating the expression of mesenchymal genes.44 Researches have shown that in lung adenocarcinoma cells, Snail1 overexpression promotes cell proliferation by upregulating the expression levels of Cyclin D1 and PCNA, and vice versa.45 β-Catenin is also a positive regulator in EMT and cell proliferation. β-Catenin knockdown inhibits tumor growth of kidney cancer in nude mice by inhibiting PCNA and Ki-67.46 In colon cancer cells, β-catenin is abnormally activated and promotes cell proliferation through its downstream targets, such as c-Myc, CCND1, and survivin.47–49 These results indicate that 15-PGDH overexpression inhibits EMT, decreases the expression levels of EMT regulator Snail1 and β-catenin, which subsequently inhibits cell proliferation.
Conclusions
Taken together, our findings provide a comprehensive view of the effects of 15-PGDH overexpression on cellular processes, demonstrating that 15-PGDH overexpression suppresses cell growth, induces G1/S arrest, increases tumor cell sensitivity to chemotherapy drug cisplatin, and inhibits EMT process in lung adenocarcinoma A549 cells. Our data suggest that 15-PGDH plays an essential role in lung cancer development and indicate that chemicals like hyaluronidase,11 which upregulate 15-PGDH expression level would be a potential strategy for cancer treatment and cancer prevention.
Data Sharing Statement
The data used to support the findings of this study are included in the article.
Funding
This work was supported by Basic and Applied Basic Research fund of Guangdong Province (Grant no. 2021A1515012553), Basic and Applied Basic Research Fund of Guangdong Province (Grant no. 2019A1515110123), Innovative Strong School Project of Guangdong Pharmaceutical University (Grant no. 2018KQNCX130), The Medical Science and Technology Research Fund of Guangdong Province (Grant no. A2019531).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Aisner DL, Sholl LM, Berry LD, et al. The impact of smoking and TP53 mutations in lung adenocarcinoma patients with targetable mutations-the lung cancer mutation consortium (LCMC2). Clin Cancer Res. 2018;24(5):1038–1047. doi:10.1158/1078-0432.CCR-17-2289
3. Gazdar AF, Linnoila RI. The pathology of lung cancer–changing concepts and newer diagnostic techniques. Semin Oncol. 1988;15(3):215–225.
4. Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389(10066):299–311. doi:10.1016/S0140-6736(16)30958-8
5. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584–594. doi:10.4065/83.5.584
6. Brock MV, Hooker CM, Ota-Machida E, et al. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med. 2008;358(11):1118–1128. doi:10.1056/NEJMoa0706550
7. Sporn JC, Kustatscher G, Hothorn T, et al. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene. 2009;28(38):3423–3428. doi:10.1038/onc.2009.26
8. Hoffman PC, Mauer AM, Vokes EE. Lung cancer. Lancet. 2000;355(9202):479–485. doi:10.1016/s0140-6736(00)82038-3
9. Zhang Y, Desai A, Yang SY, et al. TISSUE REGENERATION. Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science. 2015;348(6240):aaa2340. doi:10.1126/science.aaa2340
10. Tai HH, Cho H, Tong M, Ding Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: structure and biological functions. Curr Pharm Des. 2006;12(8):955–962. doi:10.2174/138161206776055958
11. Ding Y, Tong M, Liu S, Moscow JA, Tai HH. NAD+-linked 15-hydroxyprostaglandin dehydrogenase (15-PGDH) behaves as a tumor suppressor in lung cancer. Carcinogenesis. 2005;26(1):65–72. doi:10.1093/carcin/bgh277
12. Myung SJ, Rerko RM, Yan M, et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci U S A. 2006;103(32):12098–12102. doi:10.1073/pnas.0603235103
13. Wolf I, O’Kelly J, Rubinek T, et al. 15-hydroxyprostaglandin dehydrogenase is a tumor suppressor of human breast cancer. Cancer Res. 2006;66(15):7818–7823. doi:10.1158/0008-5472.can-05-4368
14. Li Y, Li S, Sun D, Song L, Liu X. Expression of 15-hydroxyprostaglandin dehydrogenase and cyclooxygenase-2 in non-small cell lung cancer: correlations with angiogenesis and prognosis. Oncol Lett. 2014;8(4):1589–1594. doi:10.3892/ol.2014.2371
15. Arima K, Komohara Y, Bu L, et al. Downregulation of 15-hydroxyprostaglandin dehydrogenase by interleukin-1β from activated macrophages leads to poor prognosis in pancreatic cancer. Cancer Sci. 2018;109(2):462–470. doi:10.1111/cas.13467
16. Volpato M, Cummings M, Shaaban AM, et al. Downregulation of 15-hydroxyprostaglandin dehydrogenase during acquired tamoxifen resistance and association with poor prognosis in ERalpha-positive breast cancer. Explor Target Antitumor Ther. 2020;1:355–371. doi:10.37349/etat.2020.00021
17. Li Y, Li J, Dong J, et al. 15-PGDH expression in gastric cancer: a potential role in anti-tumor immunity. Cancer Manag Res. 2020;12:7419–7426. doi:10.2147/CMAR.S245726
18. Yan M, Rerko RM, Platzer P, et al. 15-Hydroxyprostaglandin dehydrogenase, a COX-2 oncogene antagonist, is a TGF-beta-induced suppressor of human gastrointestinal cancers. Proc Natl Acad Sci U S A. 2004;101(50):17468–17473. doi:10.1073/pnas.0406142101
19. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–W102. doi:10.1093/nar/gkx247
20. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi:10.1093/nar/gky1131
21. Basu A, Krishnamurthy S. Cellular responses to Cisplatin-induced DNA damage. J Nucleic Acids. 2010;2010:201367. doi:10.4061/2010/201367
22. Zamble DB, Lippard SJ. Cisplatin and DNA repair in cancer chemotherapy. Trends Biochem Sci. 1995;20(10):435–439. doi:10.1016/s0968-0004(00)89095-7
23. Karimi Roshan M, Soltani A, Soleimani A, Rezaie Kahkhaie K, Afshari AR, Soukhtanloo M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie. 2019;165:229–234. doi:10.1016/j.biochi.2019.08.003
24. Chen Y, Wang BC, Xiao Y. PI3K: a potential therapeutic target for cancer. J Cell Physiol. 2012;227(7):2818–2821. doi:10.1002/jcp.23038
25. Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5(3):288–300. doi:10.21037/tlcr.2016.06.07
26. Basourakos SP, Li L, Aparicio AM, Corn PG, Kim J, Thompson TC. Combination platinum-based and DNA damage response-targeting cancer therapy: evolution and future directions. Curr Med Chem. 2017;24(15):1586–1606. doi:10.2174/0929867323666161214114948
27. He D, Li T, Sheng M, Yang B. Exonuclease 1 (Exo1) participates in mammalian non-homologous end joining and contributes to drug resistance in ovarian cancer. Med Sci Monit. 2020;26:e918751. doi:10.12659/MSM.918751
28. Zhou J, Wang Y, Wang Y, et al. FOXM1 modulates cisplatin sensitivity by regulating EXO1 in ovarian cancer. PLoS One. 2014;9(5):e96989. doi:10.1371/journal.pone.0096989
29. Yu P, Huang B, Shen M, et al. p15(PAF), a novel PCNA associated factor with increased expression in tumor tissues. Oncogene. 2001;20(4):484–489. doi:10.1038/sj.onc.1204113
30. Hu S, Zeng W, Zhang W, et al. KIAA0101 as a new diagnostic and prognostic marker, and its correlation with gene regulatory networks and immune infiltrates in lung adenocarcinoma. Aging. 2020;13(1):301–339. doi:10.18632/aging.104144
31. Chen H, Xia B, Liu T, Lin M, Lou G. KIAA0101, a target gene of miR-429, enhances migration and chemoresistance of epithelial ovarian cancer cells. Cancer Cell Int. 2016;16:74. doi:10.1186/s12935-016-0353-y
32. Cheng Y, Li K, Diao D, et al. Expression of KIAA0101 protein is associated with poor survival of esophageal cancer patients and resistance to cisplatin treatment in vitro. Lab Invest. 2013;93(12):1276–1287. doi:10.1038/labinvest.2013.124
33. Liu L, Chen X, Xie S, Zhang C, Qiu Z, Zhu F. Variant 1 of KIAA0101, overexpressed in hepatocellular carcinoma, prevents doxorubicin-induced apoptosis by inhibiting p53 activation. Hepatology. 2012;56(5):1760–1769. doi:10.1002/hep.25834
34. Mamuya FA, Duncan MK. aV integrins and TGF-β-induced EMT: a circle of regulation. J Cell Mol Med. 2012;16(3):445–455. doi:10.1111/j.1582-4934.2011.01419.x
35. Karar J, Maity A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front Mol Neurosci. 2011;4:51. doi:10.3389/fnmol.2011.00051
36. Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer. 2005;5(12):921–929. doi:10.1038/nrc1753
37. Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. Semin Cancer Biol. 2019;59:125–132. doi:10.1016/j.semcancer.2019.07.009
38. Dai Y, Tang Z, Yang Z, et al. EXO1 overexpression is associated with poor prognosis of hepatocellular carcinoma patients. Cell Cycle. 2018;17(19–20):2386–2397. doi:10.1080/15384101.2018.1534511
39. Nielsen FC, Jager AC, Lutzen A, Bundgaard JR, Rasmussen LJ. Characterization of human exonuclease 1 in complex with mismatch repair proteins, subcellular localization and association with PCNA. Oncogene. 2004;23(7):1457–1468. doi:10.1038/sj.onc.1207265
40. Engels K, Giannattasio M, Muzi-Falconi M, Lopes M, Ferrari S. 14-3-3 proteins regulate exonuclease 1-dependent processing of stalled replication forks. PLoS Genet. 2011;7(4):e1001367. doi:10.1371/journal.pgen.1001367
41. Yang G, Dong K, Zhang Z, et al. EXO1 Plays a Carcinogenic Role in Hepatocellular Carcinoma and is related to the regulation of FOXP3. J Cancer. 2020;11(16):4917–4932. doi:10.7150/jca.40673
42. Lv W, Su B, Li Y, Geng C, Chen N. KIAA0101 inhibition suppresses cell proliferation and cell cycle progression by promoting the interaction between p53 and Sp1 in breast cancer. Biochem Biophys Res Commun. 2018;503(2):600–606. doi:10.1016/j.bbrc.2018.06.046
43. Song B, Wang Y, Titmus MA, et al. Molecular mechanism of chemoresistance by miR-215 in osteosarcoma and colon cancer cells. Mol Cancer. 2010;9:96. doi:10.1186/1476-4598-9-96
44. Stemmer V, de Craene B, Berx G, Behrens J. Snail promotes Wnt target gene expression and interacts with beta-catenin. Oncogene. 2008;27(37):5075–5080. doi:10.1038/onc.2008.140
45. Lin Z, Lin X, Zhu L, Huang J, Huang Y. TRIM2 directly deubiquitinates and stabilizes Snail1 protein, mediating proliferation and metastasis of lung adenocarcinoma. Cancer Cell Int. 2020;20:228. doi:10.1186/s12935-020-01316-6
46. Yang CM, Ji S, Li Y, Fu LY, Jiang T, Meng FD. β-Catenin promotes cell proliferation, migration, and invasion but induces apoptosis in renal cell carcinoma. Onco Targets Ther. 2017;10:711–724. doi:10.2147/ott.s117933
47. He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–1512. doi:10.1126/science.281.5382.1509
48. Tetsu O, McCormick F. β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398(6726):422–426. doi:10.1038/18884
49. Kim PJ, Plescia J, Clevers H, Fearon ER, Altieri DC. Survivin and molecular pathogenesis of colorectal cancer. Lancet. 2003;362(9379):205–209. doi:10.1016/S0140-6736(03)13910-4
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.