")
Back to Journals » OncoTargets and Therapy » Volume 11
Overexpressing lncRNA SNHG16 inhibited HCC proliferation and chemoresistance by functionally sponging hsa-miR-93
Authors Xu F, Zha G, Wu Y, Cai W, Ao J
Received 31 July 2018
Accepted for publication 31 August 2018
Published 7 December 2018 Volume 2018:11 Pages 8855—8863
DOI https://doi.org/10.2147/OTT.S182005
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Takuya Aoki
Fengfeng Xu,1 Guoqing Zha,2 Yanpeng Wu,1 Weilong Cai,1 Jian Ao1
1Department of General Surgery, The Fifth Affiliated Hospital of Guangzhou Medical University, Guangzhou, People’s Republic of China; 2The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, People’s Republic of China
Background: Long noncoding RNAs (lncRNAs) have been identified as prognostic biomarkers and functional regulators in human cancers. The present study aimed to determine the expressions and functions of an lncRNA, Small Nucleolar RNA Host Gene 16 (SNHG16), in hepatocellular carcinoma (HCC).
Patients and methods: SNHG16 expressions were tested by quantitative real-time PCR (qRT-PCR) in HCC cell lines, as well as 43 pairs of HCC tissues and pair-matched healthy hepatic tissues. It was overexpressed in Hep3B and HuH7 cells. The effects of SNHG16 overexpression in HCC in vitro proliferation, 5-fluorouracil (5-FU) chemoresistance, and in vivo tumor growth were tested. A potential microRNA (miRNA) sponge target of SNHG16, hsa-miR-93, was tested by luciferase reporter assay and qRT-PCR. In addition, hsa-miR-93 was upregulated in SNHG16-overexpressed HCC cells to examine its effect on SNHG16-mediated cancer cell functional regulation in HCC.
Results: SNHG16 levels were markedly downregulated in both HCC cell lines and HCC tissues. Lentivirus-mediated SNHG16 overexpression inhibited HCC cell proliferation, 5-FU chemoresistance, and in vivo tumor growth. Hsa-miR-93 was confirmed to be directly sponging on SNHG16. Its upregulation in HCC cells reversed SNHG16 overexpression and induced tumor-suppressing effects in HCC cells.
Conclusion: Our data demonstrate that SNHG16 plays a critical role in HCC development via functionally sponging hsa-miR-93.
Keywords: lncRNA, SNHG16, miRNA, hsa-miR-93, proliferation
Introduction
Hepatocellular carcinoma (HCC) is one of the most commonly diagnosed human cancers in the world.1,2 In developed countries, HCC is the sixth leading cause of cancer-related death in men.1 However, in less developed countries, including China, HCC has become the second leading cause of cancer-related death in men.1 Every year, nearly 1 million new HCC cases and more than ¾ million HCC-associated deaths occur worldwide, with China accounting for >50% of the total number of cases and deaths.1,2 The underlying pathological mechanisms of HCC may be very complicated, though most HCC cases may be attributed to chronic infection with hepatitis B or C virus.3,4 Unfortunately, despite recent advances in HCC early diagnosis and specialized HCC treatments, cancer patients still suffer from poor prognosis and high recurrence rate.5–7 Therefore, it is of crucial importance to understand the pathophysiological conditions contributing to HCC carcinogenesis and development, in order to seek new diagnosis and treatment strategies to improve the overall prognosis of HCC patients.
Long noncoding RNAs (lncRNAs) are families of noncoding molecular transcripts with more than 200 nucleotides in length, which have been identified as having significant functional roles in various human diseases.8–10 Specifically, lncRNAs have been shown to be deregulated in many types of human cancers, and the deregulation of lncRNAs was demonstrated to induce aberrant gene expression, thus contributing to the development, progression, or apoptosis of human cancers, including HCC.11–15 Notably, our recent study revealed that a novel lncRNA colon cancer associated transcript-1 (CCAT1) was aberrantly upregulated in human HCC, and its overexpression significantly promoted HCC cell progression and migration.11
MicroRNAs (miRNAs) are families of small-length (8–22 nt long) noncoding RNAs that may modulate gene and protein expressions in various cancer types via either oncogenic or tumor-suppressive activities.16–18 Recently, it has been demonstrated that many RNA transcripts, including lncRNAs, may function as competing endogenous RNAs (ceRNAs) by binding miRNAs and preventing their targeted transcripts from being degraded.19,20 In human HCC specifically, our recent study demonstrated that CCAT1 could competitively sponge on miRNA let-7 to affect its downstream cancer-related signaling pathways.11 Yet, whether lncRNAs other than CCAT1 may also be dysregulated and functioning as miRNA-sponging epigenetic regulators in human HCC is not clear.
It was recently identified that another lncRNA, Small Nucleolar RNA Host Gene 16 (SNHG16), might act as a potential cancer biomarker or functional cancer development modulator in various human cancers.21–25 Thus in this study, we explored the expression and functions of SNHG16 in HCC. We hypothesized that SNHG16 may be dysregulated in HCC cell lines and human HCC tumors. Also, we hypothesized that it may have functional roles in regulating HCC cell biological function, possibly through competitive sponging on certain miRNA candidates.
Patients and methods
Ethics statement
This study was approved by the Ethics Committees of the First and Fifth Affiliated Hospitals of Guangzhou Medical University (#903-GD-452-21) in Guangzhou, Guangdong Province, China. Written informed consents were acquired from each participating patient. For animal experiments, all procedures were reviewed and approved by the Animal Study Committee of the First and Fifth Affiliated Hospitals of Guangzhou Medical University in Guangzhou, Guangdong Province, China, and in compliance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and Animal Welfare Act. In addition, all protocols were conducted in accordance with the principles of the Declaration of Helsinki.
Cell culture and clinical samples
HCC cell lines (Hep3B, HuH7, SNU398, SNU423, SNU429, Hep3G2, SK-HEP-1, and PLC/PRF/5) were all purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). In addition, two healthy liver cell lines, THLE2 and THLE3, were purchased from the American Type Culture Collection, Manassas, VA, USA. All cells were maintained in a 6-well plate in RPMI-1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Thermo Fisher Scientific), 100 μg/mL streptomycin (Thermo Fisher Scientific), and 100 U/mL penicillin (Thermo Fisher Scientific) in a humidified 37°C incubator with 5% CO2.
Clinical samples, including HCC tissues and adjacent noncancerous hepatic tissues, were collected from 43 patients with HCC during surgeries from June 2010 to January 2018. Participating patients did not receive any chemotherapy or radiation treatment prior to surgery. Upon surgical removal, all tissues were preserved in liquid nitrogen prior to further processing.
RNA extraction and quantitative real-time PCR (qRT-PCR)
RNA extraction and qRT-PCR were performed according to the methods in our previous publications.11,26 Briefly, total RNAs were extracted from HCC cell lines or clinical samples using a TRIzol reagent (Takara Bio Inc., Shiga Prefecture, Japan) according to the manufacturer’s instruction. First-strand cDNA was generated using the Reverse Transcription System Kit (Thermo Fisher Scientific) according to the manufacturer’s instruction. For SNHG16, qRT-PCR was performed using the SYBR Green PCR kit protocol in a StepOne Plus system (Thermo Fisher Scientific). The 18S rRNA was used as an endogenous control. For hsa-miR-93, qRT-PCR was performed using a mirVana™ qRT-PCR miRNA Detection Kit (Thermo Fisher Scientific). U6 snRNA was used as an endogenous control. All qRT-PCRs were performed on a Biosystems 7300 Real-Time PCR system (Thermo Fisher Scientific). Relative gene expression levels were calculated using the 2(−ΔΔCt) method.
SNHG16 upregulation assay
A lentiviral vector containing human lncRNA SNHG16, SNHG16(L), was purchased from GenePharma (Shanghai, China). In addition, a nonspecific lentiviral vector, NS(L), was also purchased from GenePharma. Hep3B and HuH7 cells were transduced with NS(L) or SNHG16(L) in the presence of polybrene (8 μg/mL; GenePharma) for 48 hours. After a selection procedure using blasticidin (10 μg/mL; GenePharma) for 72 hours, surviving cells were collected and reseeded in six-well plates. After 3–6 passages, qRT-PCR was performed to examine endogenous SNHG16 expression in Hep3B and HuH7 cells.
Cell proliferation assay
HCC in vitro proliferation was measured according to the method in our previous publication.11 Briefly, ~5×103 Hep3B or HuH7 cells were plated in 96-well plates for 96 hours. Every 24 hours, cell proliferation was assessed using the cell counting kit-8 ([CCK-8]; Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the manufacturer’s protocol. The cell proliferation curves were plotted using the absorbance (570 nm) reading at each time point.
Chemoresistance assay
Approximately 5×103 Hep3B or HuH7 cells were plated in 96-well plates and treated with 5-fluorouracil (5-FU) at concentrations of 0, 2, 10, and 20 μM for 48 hours. Then, a CCK-8 assay was performed according to the manufacturer’s protocol. At each 5-FU concentration, viable cells were measured using the absorbance (570 nm) and then normalized to the absorbance at 0 μM 5-FU.
In vivo tumorigenicity assay
HCC in vivo tumor growth was evaluated according to the method in our previous publication.26 Briefly, 100 μL, 1×106 HuH7 cells were inoculated subcutaneously into the dorsal flanks of 5-week-old female athymic nude mice (n=8 for each group). In vivo tumor volume (V) was monitored each week by measuring the subcutaneous length (L) and width (W) and then calculated according to the formula V=(L×W2/2). All nude mice were killed after 5 weeks. HuH7 tumors were extracted and examined under a broad-light microscope.
Luciferase assay
A luciferase assay was performed according to the method in our previous publications.11,26 Briefly, a pmirGLO dual-luciferase plasmid with the insertion of wild-type (WT) SNHG16 (SNHG16[WT]) was purchased from GenePharma. The putative hsa-miR-93 binding sites on SNHG16 were mutated. The mutant (MU) SNHG16 (SNHG16[MU]) was also inserted in a pmirGLO plasmid, also from GenePharma. A total of 5×104 293T cells/well were seeded into 24-well plates and co-transfected with SNHG16(WT) or SNHG16(MU) and a synthetic hsa-miR-93 mimics ([miR93-mimics]; GenePharma) or a nonspecific miRNA mimics ([NS-mimics]; GenePharma) using lipofectamine 2000 (Thermo Fisher Scientific). After 48 hours, relative luciferase activities were measured using a dual luciferase reporter assay system (Promega Corporation, Fitchburg, WI, USA), and normalized to the luciferase activity measured in cells co-transfected with miR93-mimics and SNHG16(WT).
Statistical analysis
All experiments were performed in at least three biological repeats. For comparisons, Student’s t-test or Wilcoxon signed-rank test was performed. All P-values were two-sided and were obtained using the SPSS 18.0 software package (SPSS Inc., Chicago, IL, USA). Differences were defined as statistically significant for P-values <0.05.
Results
Downregulation of SNHG16 and inhibition of HCC in vitro proliferation on upregulation
To investigate the role of SNHG16 in HCC, we first examined the endogenous expression of SNHG16 in eight human HCC cell lines (Hep3B, HuH7, SNU398, SNU423, SNU429, Hep3G2, SK-HEP-1, and PLC/PRF/5) and two normal liver cell lines (THLE2 and THLE3). Downregulated expression of SNHG16 was observed in all eight HCC cell lines compared with that in THLE2 cells (Figure 1A; *P<0.05, ΔP>0.05). We then measured SNHG16 expression level in 43 pairs of HCC tissues and pair-matched noncancerous (healthy) hepatic tissues. This demonstrated that SNHG16 expression was significantly decreased in HCC tissues as compared with that in pair-matched healthy hepatic tissues (Figure 1B; *P<0.005, Wilcoxon signed-rank test).
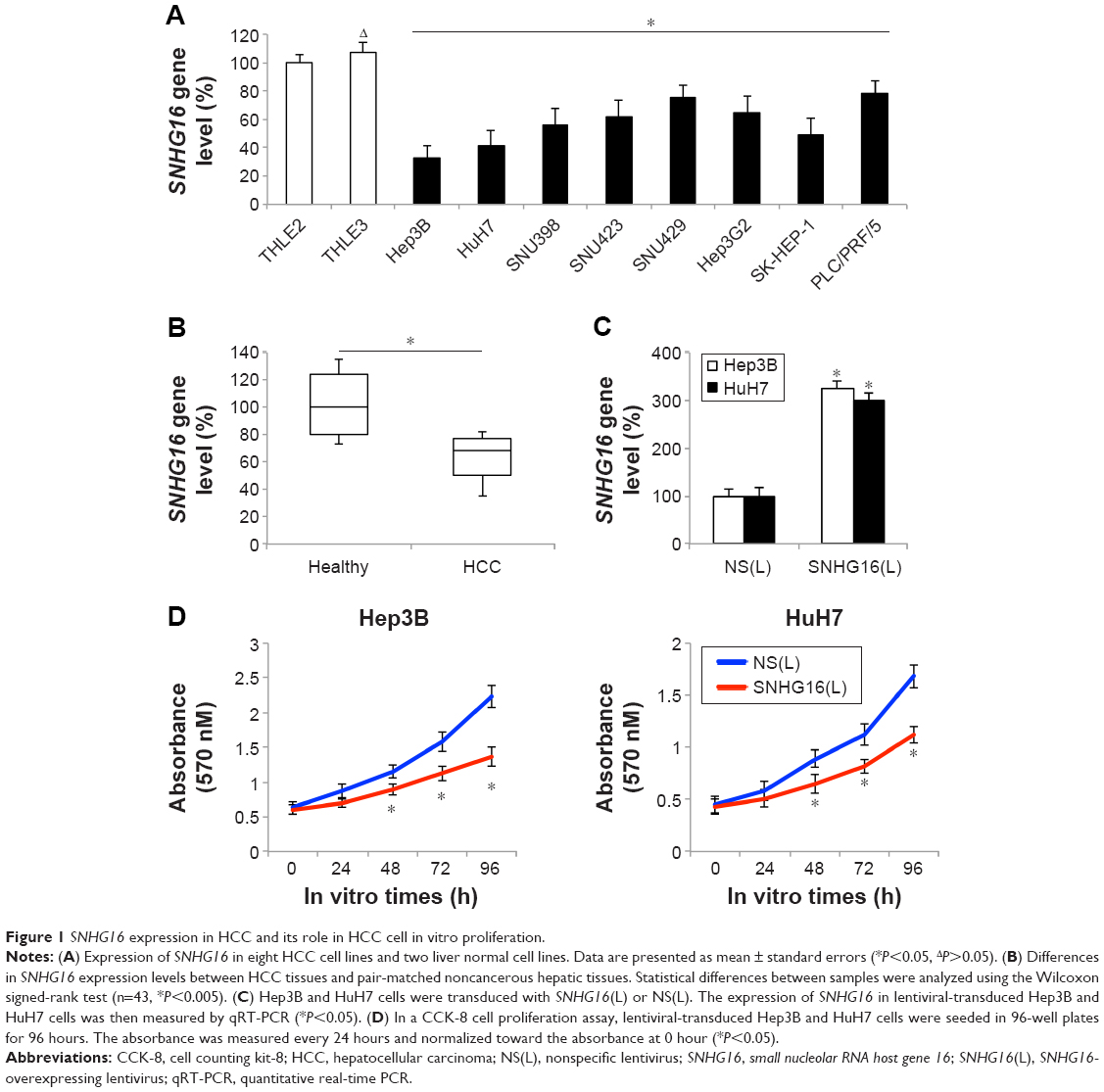 | Figure 1 SNHG16 expression in HCC and its role in HCC cell in vitro proliferation. |
Then, we stably overexpressed SNHG16 by transducing lentiviruses of SNHG16(L) or NS(L) into the HCC cell lines, Hep3B, and HuH7. Analysis of qRT-PCR showed that SNHG16 gene levels significantly increased in HCC cells transduced with SNHG16(L) than in HCC cells transduced with NS(L) (Figure 1C, *P<0.05). Then, a CCK-8 assay indicated that SNHG16 upregulation significantly inhibited cell proliferation in Hep3B and HuH7 cells (Figure 1D, *P<0.05).
SNHG16 upregulation inhibited HCC chemoresistance and in vivo tumorigenicity
To further investigate the functional roles of SNHG16 in HCC, we examined chemoresistance to 5-FU in lentiviral-transduced Hep3B and HuH7 cells. The results showed that SNHG16 upregulation significantly inhibited 5-FU chemoresistance (between 2 and 20 μM) in Hep3B and HuH7 cells (Figure 2A, *P<0.05).
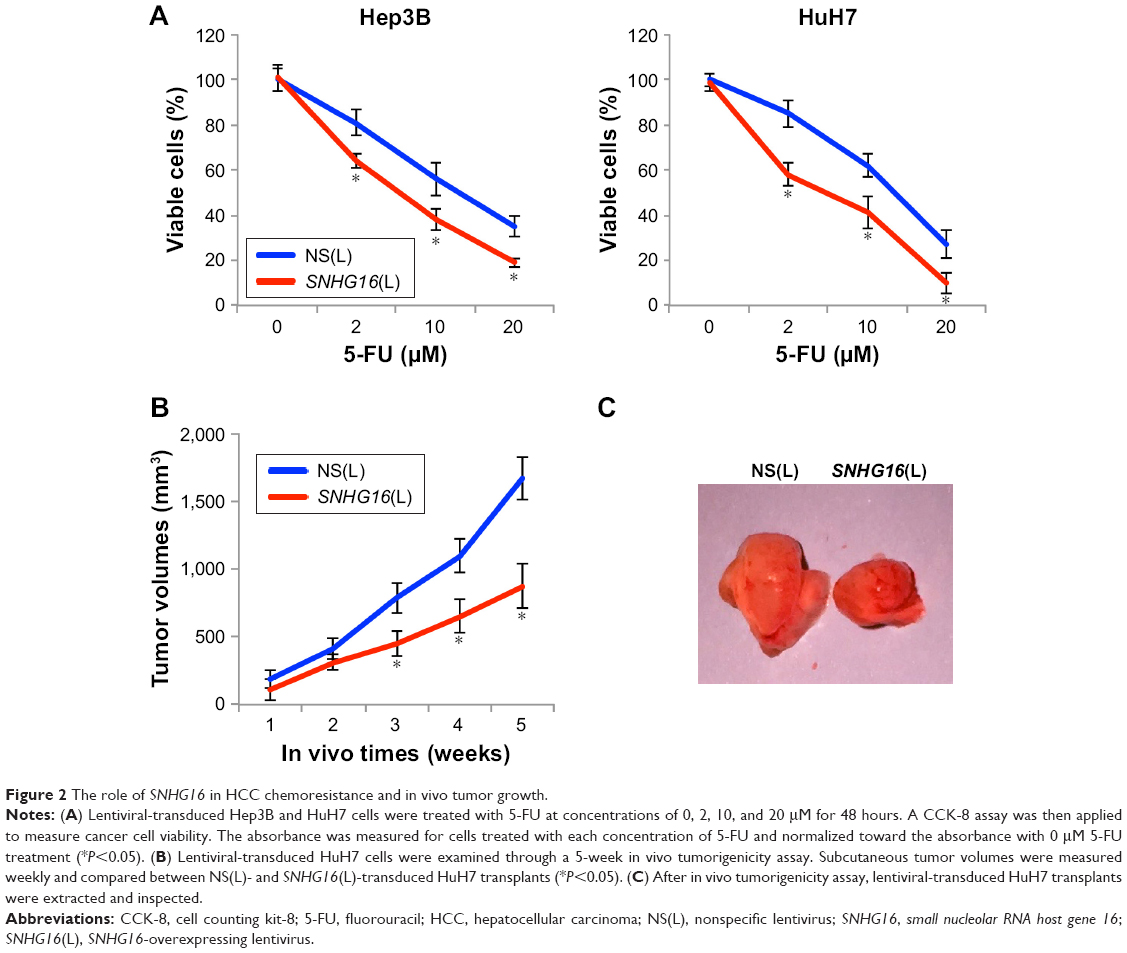 | Figure 2 The role of SNHG16 in HCC chemoresistance and in vivo tumor growth. |
We also examined in vivo tumorigenicity using lentiviral-transduced HuH7 cells. By measuring weekly subcutaneous tumor volumes, we discovered that 3–5 weeks after lentiviral-transduced HuH7 cells were transplanted, SNHG16 upregulation significantly inhibited the in vivo growth of transplanted HCC tumors (Figure 2B, *P<0.05). At the end of 5-week in vivo tumorigenicity assay, HuH7 transplants were extracted and directly compared under a microscope. This confirmed our in vivo discovery, showing that SNHG16-overexpressed HuH7 transplants were significantly smaller than control HuH7 transplants (Figure 2C).
hsa-miR-93 sponges SNHG16
To investigate the mechanisms responsible for SNHG16 upregulation-induced tumor suppression in HCC, bioinformatics analyses were performed to search for SNHG16-associated molecular regulators. By searching through online miRNA–lncRNA database of StarBase v2.0,27,28 we noticed that human mature miRNA, hsa-miR-93, has a very strong potential to bind SNHG16 (Figure 3A). To verify this hypothesis, we constructed two luciferase plasmids, one containing the DNA sequence of SNHG16(WT) and the other containing the DNA sequence of SNHG16(MU) with eliminated hsa-miR-93 binding site. Then, a luciferase assay demonstrated that only in 293T cells co-transfected with SNHG16(WT) and miR93-mimics, the relative luciferase activity was significantly suppressed, thus confirming that hsa-miR-93 could directly be sponged by SNHG16 (Figure 3B, *P<0.05).
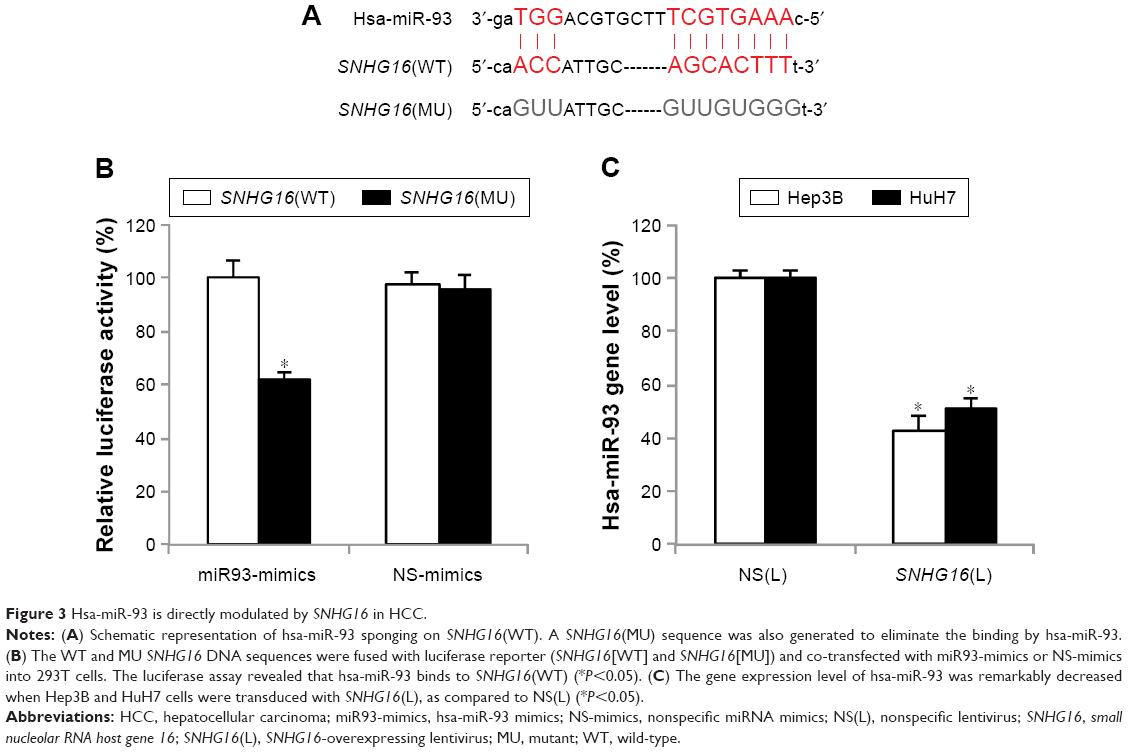 | Figure 3 Hsa-miR-93 is directly modulated by SNHG16 in HCC. |
In addition, we examined whether the expression of hsa-miR-93 was regulated by SNHG16 in HCC cells. qRT-PCR then showed that SNHG16 upregulation significantly decreased the mRNA level of endogenous hsa-miR-93 in both Hep3B and HuH7 cells (Figure 3C, *P<0.05).
SNHG16 inhibits HCC functions by competitively binding hsa-miR-93
To explore whether hsa-miR-93 can functionally reverse the roles of SNHG16 in HCC cells, we overexpressed hsa-miR-93 in Hep3B and HuH7 cells with upregulated SNHG16 expression (Figure 4A, *P<0.05).
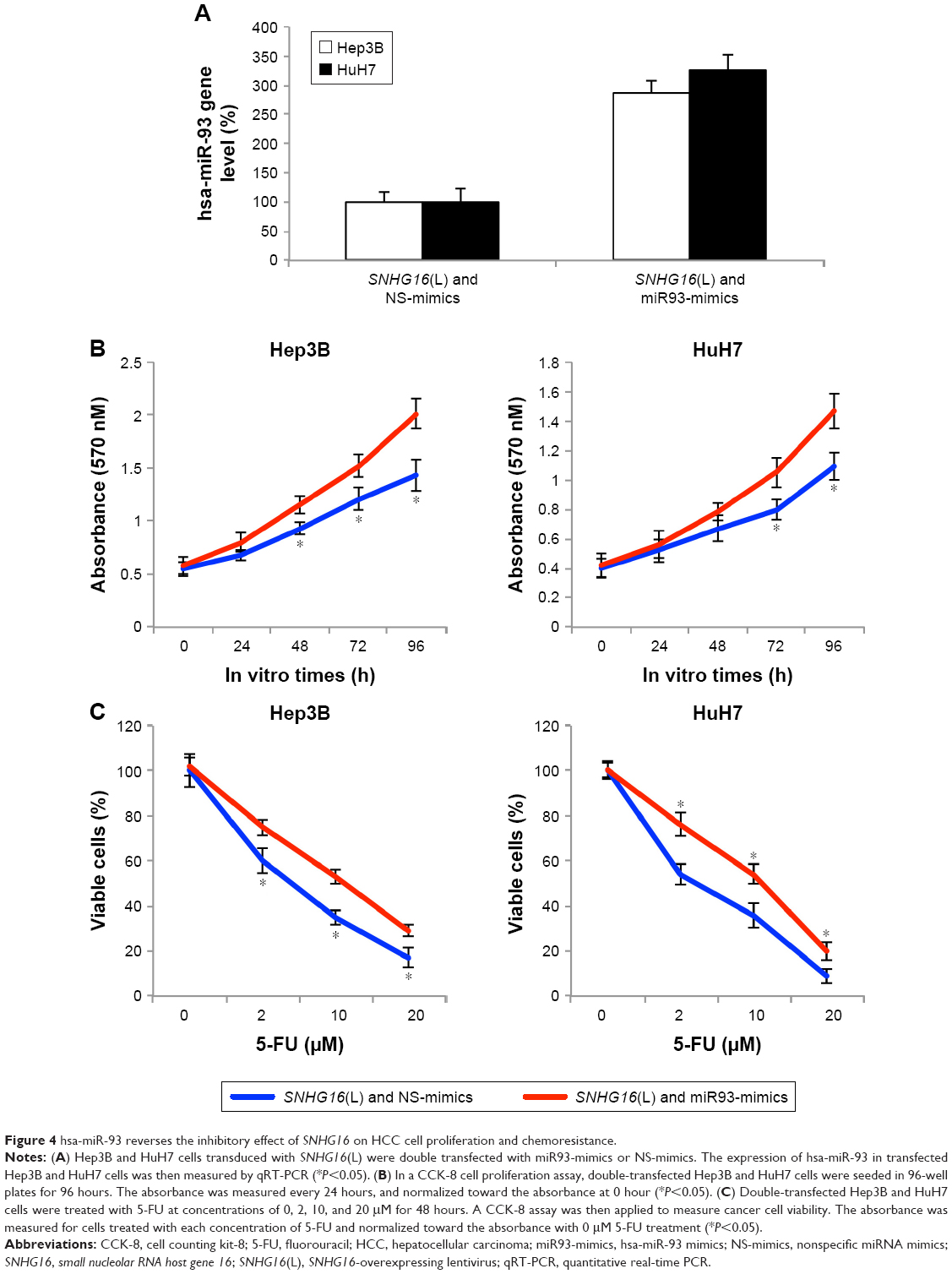 | Figure 4 hsa-miR-93 reverses the inhibitory effect of SNHG16 on HCC cell proliferation and chemoresistance. |
Then, functional assays were used to clarify the role of hsa-miR-93 binding in SNHG16-mediated HCC regulation. CCK-8 assays indicated that in SNHG16-upregulated Hep3B and HuH7 cells, overexpressing hsa-miR-93 significantly promoted HCC proliferation (Figure 4B, *P<0.05). Chemoresistance assay also indicated that in SNHG16-upregulated Hep3B and HuH7 cells, overexpressing hsa-miR-93 significantly increased 5-FU resistance (Figure 4C, *P<0.05). Thus, these results showed that SNHG16 inhibited HCC functions via competitively binding hsa-miR-93.
Discussion
In this study, we evaluated the expression of lncRNA SNHG16 in HCC and discovered that SNHG16 was markedly downregulated in both HCC cancer cell lines and human tumors. Interestingly, our findings seem to be different from several previous studies in human cancers, which demonstrated that SNHG16 was upregulated in colorectal cancer, gastric cancer, or breast cancer.22,24,29 Based on those previous findings and ours, it seems like the deregulating pattern of SNHG16 in human cancers could be either upregulation or downregulation, possibly determined by the sites and microenvironments of specific cancer types.
We also evaluated the biological functions of SNHG16 in HCC cells. Through lentiviral transduction, we successfully overexpressed SNHG16 in HCC cell lines. Then, through several in vitro and in vivo cancer function assays, we identified that SNHG16 overexpression had significant tumor-suppressing effects by inhibiting HCC cancer cell in vitro proliferation, 5-FU chemoresistance, and in vivo tumor growth, indicating that SNHG16 might function as a tumor suppressor in HCC. Interestingly, SNHG16 was demonstrated to function as oncogene in human breast cancer, gastric cancer, or bladder cancer.22–24 The findings in our study thus presented new evidence that SNHG16 may exert both oncogenic and tumor-suppressing effects on human cancer development or progression. Furthermore, our recent study identified another lncRNA, CCAT1, to function as oncogene in HCC.11 Therefore, the epigenetic regulations of lncRNAs may be very complex, involving both oncogenic and tumor-suppressing ones in HCC.
In studies on human cancers, it has been demonstrated that SNHG16 might act as ceRNAs to sponge on another group of epigenetic regulators, miRNAs. In human esophagus cancer, SNHG16 was shown to directly sponge on miR-140-5 p, thus inversely upregulating ZEB1 gene to promote cancer cell progression.21 In addition, SNHG16 was demonstrated to induce breast cancer cell migration by competitively binding miR-98 with E2F5.24 In this study, we presented several lines of evidence to demonstrate that SNHG16 acted as a direct molecular sponge of hsa-miR-93, a known oncogenic transcript in HCC.30 First, our luciferase assay provided convincing evidence that SNHG16 may directly bind with hsa-miR-93. Second, qRT-PCR assay demonstrated that hsa-miR-93 was inversely downregulated in SNHG16-overexpressed HCC cell lines. Third and most importantly, in double-transfected HCC cells, upregulating hsa-miR-93 was able to reverse the tumor-suppressing effects induced by SNHG16 upregulation on cancer cell proliferation and 5-FU chemoresistance. Therefore, our results support the notion that tumor-suppressing functions of SNHG16 may very possibly act through competitively binding on hsa-miR-93. Intriguingly, it was demonstrated that hsa-miR-93 exerted oncogenic regulations on HCC development by activating c-Met/PI3K/Akt pathway in HCC.30 The possibility of SNHG16 sponging on hsa-miR-93 to inversely inhibit c-Met/PI3K/Akt pathway would certainly require further investigations.
Conclusion
Overall, our studies indicate that SNHG16 is markedly downregulated in HCC and may act as a tumor suppressor to inhibit HCC development, possibly by functioning as a competitive sponge of hsa-miR-93. Furthermore, based on the findings in our study, future clinical studies focusing on the correlation between SNHG16 and HCC patients’ clinicopathological characteristics may help to further define the translational implication of SNHG16 as it may be developed as a novel prognostic marker or therapeutic target to benefit HCC diagnosis and therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
de Martel C, Ferlay J, Franceschi S, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13(6):607–615. | ||
Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006;45(4):529–538. | ||
Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. | ||
Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 2010;7(8):448–458. | ||
Pascual S, Herrera I, Irurzun J. New advances in hepatocellular carcinoma. World J Hepatol. 2016;8(9):421–438. | ||
Kwok ZH, Tay Y. Long noncoding RNAs: lincs between human health and disease. Biochem Soc Trans. 2017;45(3):805–812. | ||
Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21(6):354–361. | ||
Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett. 2013;339(2):159–166. | ||
Deng L, Yang SB, Xu FF, Zhang JH. Long noncoding RNA CCAT1 promotes hepatocellular carcinoma progression by functioning as let-7 sponge. J Exp Clin Cancer Res. 2015;34:18. | ||
Gibb EA, Vucic EA, Enfield KS, et al. Human cancer long non-coding RNA transcriptomes. PLoS One. 2011;6(10):e25915. | ||
Du Z, Fei T, Verhaak RG, et al. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat Struct Mol Biol. 2013;20(7):908–913. | ||
Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21(11):1253–1261. | ||
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–874. | ||
Lujambio A, Ropero S, Ballestar E, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67(4):1424–1429. | ||
Tong AW, Nemunaitis J. Modulation of miRNA activity in human cancer: a new paradigm for cancer gene therapy? Cancer Gene Ther. 2008;15(6):341–355. | ||
Saito Y, Jones PA. Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle. 2006;5(19):2220–2222. | ||
Wang Y, Xu Z, Jiang J, et al. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev Cell. 2013;25(1):69–80. | ||
Sumazin P, Yang X, Chiu HS, et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011;147(2):370–381. | ||
Zhang K, Chen J, Song H, Chen LB. SNHG16/miR-140-5p axis promotes esophagus cancer cell proliferation, migration and EMT formation through regulating ZEB1. Oncotarget. 2018;9(1):1028–1040. | ||
Lian D, Amin B, Du D, Yan W. Enhanced expression of the long non-coding RNA SNHG16 contributes to gastric cancer progression and metastasis. Cancer Biomark. 2017;21(1):151–160. | ||
Cao X, Xu J, Yue D. LncRNA-SNHG16 predicts poor prognosis and promotes tumor proliferation through epigenetically silencing p21 in bladder cancer. Cancer Gene Ther. 2018;25(1–2):10–17. | ||
Cai C, Huo Q, Wang X, Chen B, Yang Q. SNHG16 contributes to breast cancer cell migration by competitively binding miR-98 with E2F5. Biochem Biophys Res Commun. 2017;485(2):272–278. | ||
Yu M, Ohira M, Li Y, et al. High expression of ncRAN, a novel non-coding RNA mapped to chromosome 17q25.1, is associated with poor prognosis in neuroblastoma. Int J Oncol. 2009;34(4):931–938. | ||
Xu FF, Xie WF, Zha GQ, Chen HW, Deng L. MiR-520f promotes cell aggressiveness by regulating fibroblast growth factor 16 in hepatocellular carcinoma. Oncotarget. 2017;8(65):109546–109558. | ||
Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011;39(Database issue):D202–D209. | ||
Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(Database issue):D92–D97. | ||
Christensen LL, True K, Hamilton MP, et al. SNHG16 is regulated by the Wnt pathway in colorectal cancer and affects genes involved in lipid metabolism. Mol Oncol. 2016;10(8):1266–1282. | ||
Ohta K, Hoshino H, Wang J, et al. MicroRNA-93 activates c-Met/PI3K/Akt pathway activity in hepatocellular carcinoma by directly inhibiting PTEN and CDKN1A. Oncotarget. 2015;6(5):3211–3224. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.