")
Back to Journals » OncoTargets and Therapy » Volume 15
Overcoming Resistance to Kinase Inhibitors: The Paradigm of Chronic Myeloid Leukemia
Authors De Santis S , Monaldi C, Mancini M, Bruno S, Cavo M, Soverini S
Received 10 November 2021
Accepted for publication 5 January 2022
Published 25 January 2022 Volume 2022:15 Pages 103—116
DOI https://doi.org/10.2147/OTT.S289306
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Sara De Santis,1 Cecilia Monaldi,1 Manuela Mancini,2 Samantha Bruno,1 Michele Cavo,1,2 Simona Soverini1
1Dipartimento di Medicina Specialistica, Diagnostica e Sperimentale, UO Ematologia ‘Lorenzo e Ariosto Seràgnoli’, Università di Bologna, Bologna, Italy; 2IRCCS Azienda Ospedaliero-Universitaria di Bologna, Istituto di Ematologia “Seràgnoli”, Bologna, Italy
Correspondence: Sara De Santis
Insitute of Hematology “Lorenzo e Ariosto Seràgnoli”, Via Massarenti 9, Bologna, 40138, Italy
Tel +39 051 2143791
Fax +39 051 2144037
Email [email protected]
Abstract: Protein kinases (PKs) play crucial roles in cellular proliferation and survival, hence their deregulation is a common event in the pathogenesis of solid and hematologic malignancies. Targeting PKs has been a promising strategy in cancer treatment, and there are now a variety of approved anticancer drugs targeting PKs. However, the phenomenon of resistance remains an obstacle to be addressed and overcoming resistance is a goal to be achieved. Chronic myeloid leukemia (CML) is the first as well as one of the best examples of a cancer that can be targeted by molecular therapy; hence, it can be used as a model disease for other cancers. This review aims to summarize up-to-date knowledge on the main mechanisms implicated in resistance to PK inhibitory therapies and to outline the main strategies that are being explored to overcome resistance. The importance of molecular diagnostics and disease monitoring in counteracting resistance will also be discussed.
Keywords: protein kinases, chronic myeloid leukemia, tyrosine kinase inhibitors, synthetic lethality
Introduction
Protein kinases (PKs) are enzymes that play a pivotal role in signal transduction processes by catalyzing the transfer of phosphate from adenosine triphosphate (ATP) to specific amino acid residues (serine, threonine or tyrosine) of target proteins.1 They may be located on the cellular surface and function as receptors, or in the cytoplasm, where their activation is rather dependent on upstream signaling molecules (that may be represented by other PKs). The human kinome includes more than 500 members.2 Phosphorylation is among the most common posttranslational modifications, and is implicated in the regulation of key cellular aspects like proliferation, gene expression, metabolism, motility, membrane transport, apoptosis. Thus, it is not surprising that deregulation of PKs has been found to be one of the hallmarks of cancer. PKs are involved in the pathogenesis of solid and hematologic malignancies via a variety of mechanisms, including genomic rearrangements (eg, chromosomal translocations), gene amplification or deletions, and point mutations. Most frequently, the role of PKs as cancer drivers is due to their aberrant or constitutive activation, making them ideal targets of therapy with selective kinase inhibitors (KIs).1 The first KI to receive approval for clinical use, as early as in 2001, was the ABL1 inhibitor imatinib mesylate.3 It was acclaimed as a breakthrough in anticancer therapy and made the cover of Time Magazine on May 28th, 2001. Since then, many additional PKs known or identified as cancer drivers have been explored as therapeutic targets (they were approximately 110 as of May 2021).4 The PubChem section of the US National Library of Medicine lists more than 200 KI compounds, although just 71 of these have so far made it all to way to Food and Drug Administration (FDA) approval.4
Besides the issues of toxicity and off-target effects, which may be more or less relevant depending on the degree of specificity of the individual KI, the greatest, nearly universal hurdle that anticancer targeted therapy has to cope with is the development of drug resistance. Many cancers display a high degree of heterogeneity, with multiple coexisting subclones that are difficult to be kept in check by a single targeting approach. Moreover, once hit, cancer cells know well how to strike back. They may activate a series of protective adaptive strategies enabling them to escape therapy, and their inherent genetic instability fuels the rapid acquisition of additional alterations that may increase their fitness under the “novel” environmental conditions shaped by the targeted therapeutics. As a result, new clones may be selected and become rapidly dominant. The idea that cancer populations follow Darwinian evolutionary trajectories was formulated by David Nowell as early as in 1976,5 thus it was not a surprise when the first resistant cases were observed in clinical trials with imatinib in advanced phase chronic myeloid leukemia (CML), in 2000–2001.6 Since then, the mechanisms of resistance to imatinib have been extensively investigated, laying the foundation for subsequent-generation molecules to be developed and introduced in the clinic (reviewed in7). Since neoplastic cells follow strikingly similar strategies to evade targeted therapies, the CML experience has served as a model in many other cancers. Here, we will use CML as a paradigmatic example of how resistance to KIs may develop and how it may be counteracted, or even better prevented, by reviewing the advances made over the last two decades in CML biological characterization, treatment and monitoring, in a “bench-to-bedside and back” fashion.
CML: The First and Still the Most Successful Example of Targeted Therapy
CML is indeed a paradigm under several points of view. It is one of the first neoplasms whose molecular pathogenesis could be dissected. CML is caused by the t(9;22)(q34;q11) reciprocal translocation,8 which results in a derivative chromosome 22 called “Philadelphia” (Ph) chromosome9 where BCR and ABL1 genes are fused.10,11 Both BCR and ABL1 code for cytoplasmic PKs. The resulting chimeric protein retains a fully functional kinase domain (KD) from ABL1 but loses the N-terminal myristoylated portion, physiologically involved in regulation of kinase activity – that is replaced by the oligomerization domain of BCR.12 Thus, BCR-ABL1 is constitutively active in CML cells and behaves as an oncogene, altering the proliferation rates, survival signalling, immunological interactions, and cytoskeleton dynamics of hematopoietic cells.13 Three clinical stages are defined for CML: an initial and relatively indolent chronic phase (CP), an accelerated phase (AP), and a terminal acute phase called blast crisis (BC).14 Without therapeutic intervention, the disease follows a natural, inexorable progression. Clinical and laboratory studies indicate that BCR-ABL1 is necessary and sufficient for initiation and maintenance of CP CML,15–17 which is unusual for a cancer. In contrast, the transformation of CML from CP to BC requires additional genetic and/or epigenetic alterations, and oncogene addiction (hence, therapeutic efficacy) is greatly reduced.18,19
At least in CP CML, BCR-ABL1 was recognized since the late 80s to be an ideal therapeutic target. Imatinib mesylate stood out among hundreds of ATP-competitive compounds screened for their inhibitory activity against ABL1, and its clinical development, from Phase 1 studies to FDA approval, was extraordinarily rapid and seamless.3 Imatinib was found to combine good efficacy (although is a more potent inhibitor of other kinases, like platelet-derived growth factor receptor [PDGFR], than of ABL1) with satisfactory tolerability (its most frequent side effects being fluid retention, oedema, muscle cramps, fatigue, skin rash, nausea, diarrhea).3 However, the problem of resistance was soon observed.
Mechanisms of Resistance to Tyrosine Kinase Inhibitors (TKIs) in CML
Resistance to targeted therapies may either be primary or acquired. Primary (or upfront) resistance is defined as the lack of satisfactory response to treatment, whereas secondary (or acquired) resistance is defined as the relapse after an initial response to therapy. The meaning of “satisfactory” response depends on the disease, on the treatment endpoints and on the monitoring tools. Usually, acquired resistance occurs more frequently than primary resistance.
Resistance to TKI therapy in CML has been found to be sustained by a variety of mechanisms that recapitulate key general strategies adopted by tumor cells to evade therapy – although some have more extensively been investigated and/or are more clinically relevant than others.
TKI resistance in CML can result from two fundamentally different categories of mechanisms: BCR-ABL1-dependent (“on target”) resistance mechanisms and BCR-ABL1-independent resistance mechanisms.
“On Target” Resistance (or BCR-ABL1-Dependent Resistance)
“On target” resistance develops through direct modifications of the treatment target, so that it cannot any longer be successfully inhibited. BCR-ABL1-dependent mechanisms were the first to be identified, as early as during Phase 2 studies, in a handful of AP and BC CML patients who had relapsed after an initial response to imatinib and in whose white blood cells reactivation of BCR-ABL1 kinase activity despite continued treatment could be documented.6 BCR-ABL1-dependent mechanisms are thus the best characterized mechanisms of resistance to TKI therapy in CML. They include either qualitative or quantitative modification of the target, ie, point mutations in the KD and BCR-ABL1 overexpression, respectively (Figure 1). The exchange of the amino acid threonine (T) with isoleucine (I) at position 315 (T315I) of the BCR-ABL1 protein was the first mutation detected in imatinib-resistant CML patients (and has long been the most problematic);6,20 this substitution was predicted to fully abrogate the affinity for the drug. Structural studies have indeed shown that T315 stabilizes imatinib binding through hydrogen-bond interactions and control access of the inhibitors to a hydrophobic pocket deep in the active site that is not contacted by ATP.21 Substitution of T with the bulkier and more hydrophobic I creates a steric hindrance impeding imatinib (and second-generation ATP-competitive TKI) binding.6,22 Mutations at a “gatekeeper” residue are a “leitmotif” in resistance to anticancer target therapies.23 For example, a strikingly identical T to I amino acid substitution has been observed at homologous positions in the kinase domain of KIT (T670I) and PDGFRα (T674I) in imatinib-resistant gastrointestinal stromal tumors and hypereosinophilias, respectively. The multitude of studies that over the years have investigated BCR-ABL1-dependent mechanisms through sequencing experiments in CML patients with imatinib resistance have observed, besides T315I, a spectrum of additional mutations leading to amino acid changes in or near the phosphate-binding loop (P loop; positions M244, G250, Q252, L248, Y253, and E255), at other key contact-binding sites between imatinib and BCR-ABL1 (T315 and F317), in the C-lobe (M351, F359), and in the activation loop (H396).24 These mutations are thought to contribute to resistance by inducing local or global conformational changes that reduce imatinib-binding affinity. Since conformational changes may be more or less permissive to binding, different mutations may confer different degrees of resistance. This is usually estimated by assessing the “IC50” of the mutant, ie, the intracellular concentration of drug required to inhibit by 50% its kinase activity in vitro or the growth of a cell line engineered to express the given mutant oncoprotein (the latter is the most frequently used method).25 Very recently, however, it has been postulated that some mutations may act by altering the dissociation kinetics rather than drug affinity.26 Starting from the puzzling observation that some TKI-resistant mutants experimentally displayed similar or even higher imatinib binding affinity than the unmutated protein, it was uncovered that such mutants dissociate considerably faster, thus reducing the residence time, which is critical for drug efficacy.26 This interestingly points toward a novel mechanism of mutation-induced resistance to KIs, worth to be explored further in CML and other contexts. Importantly, such “kinetic resistance” mechanisms may be addressable by altering the treatment from a single daily dose to multiple doses.
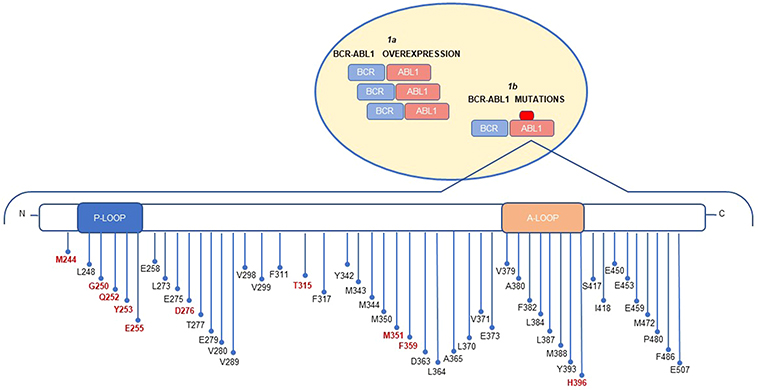 |
Figure 1 BCR-ABL1 dependent mechanisms inducing imatinib resistance include: BCR-ABL1 overexpression (1a) and BCR-ABL1 mutations (1b). The schematic diagram of ABL1 kinase domain mutations shows the distribution of mutations. The 10 most frequent mutations are highlighted in red. |
BCR-ABL1 KD mutations are a frequent finding in advanced phase patients and in heavily pretreated patients, where they account for up to 70% of resistant cases, but do not exceed 30% of the cases of resistance when CP patients treated first-line are considered.
Amplification of oncogenes is frequently observed in the pathogenesis of cancer as well as in resistance to anticancer drugs. Overexpression of the BCR-ABL1 transcript and protein due to amplification of the BCR-ABL1 fusion gene6,27–33 was indeed identified since initial studies in imatinib-resistant CML cell lines, and suggested that the overexpression of BCR-ABL1 might be an early phenomenon in the establishment of resistance and disease evolution in CML. Acquisition of a second Ph chromosome or of multiple copies of the BCR-ABL1 gene were later observed in some AP and BC patients at the time of imatinib resistance.34,35 Increase in BCR-ABL1 expression level has been associated with disease evolution in CML, and double Ph is among the so-called “major route” additional cytogenetic abnormalities that identify patients at high risk of poor response to TKI therapy and disease progression.36 BCR-ABL1 overexpression may be associated with resistance because protein levels overcome imatinib inhibitory capacity, and/or because it heralds progression of the disease from CP to BC, where genetic instability is greater and oncogenic addiction is reduced, thus impairing the therapeutic efficacy of the sole BCR-ABL1 inhibition. The extent of BCR-ABL1 transcript overexpression associated with imatinib resistance has been estimated in cell lines and patients,27–33,37 and it has been reported to range from a few folds to one log or even more. In cell lines, this has been reported to translate into two to ten-fold increase in BCR-ABL1 target protein expression.27–29,31–33 A few studies have compared the relative frequency of BCR-ABL1 KD mutations and BCR-ABL1 amplification/overexpression in the same cohort of imatinib-resistant patients. Gorre et al had observed gene amplification in 3/9 patients and mutations in 6/9 patients (it must be noted that all 9 patients were in advanced phase).6 Hochhaus et al had observed mutations in 23/66 patients (all phases of the disease), BCR-ABL1 transcript overexpression in 7/55 patients and genomic amplification in 2/32 patients with available fluorescence in situ hybridization (FISH) data.30
BCR-ABL1-Independent Resistance
Alternatively, resistance may develop via mechanisms that are independent of the target oncoprotein. In CP patients, BCR-ABL1-independent mechanisms are indeed the most frequent ones, since 60–70% of patients with unsatisfactory response to TKI therapy are negative for mutations or transcript overexpression. Several different mechanisms, not necessarily mutually exclusive, have been observed in cell lines and patients. They are outlined below.
Downstream Pathway Activation or Bypass by Parallel Pathway Activation
There is evidence that, in some cases, TKI resistance can develop despite effective target inhibition either through downstream reactivation of the signaling pathways or through the de novo activation of bypass parallel pathways that take over to sustain the oncogenic processes. Malignant growth occurs when key cellular pathways – eg, the RAS/RAF/MEK/ERK pathway, the JAK2/STAT pathway, the PI3K/AKT/mTOR pathway – are hijacked by oncogenes, or when components or upstream activators of these cascades are directly activated. Alterations in these pathways and upstream signaling molecules can thus influence sensitivity to small molecule inhibitors used for cancer therapy. In CML, a series of evidences indeed indicate a role for altered activation of the AKT and ERK pathways, elevated Bcl-2 or Bcl-xL, diminished Bim or Bad, overexpression of Src family kinases or Aurora A in BCR-ABL1-independent resistance.38–51
JAK family members are key pathogenetic players in the induction and evolution of CML. The JAK/STAT pathway mediates signaling from cytokine receptors to the nucleus: activation of JAK2 by cytokines and growth factors leads to phosphorylation and thus activation of STAT3 and STAT5, promoting their dimerization and nuclear translocation where they regulate transcription of STAT-dependent genes. JAK2 has been shown to be a major downstream signaling molecule in CML. JAK2 is routinely tyrosine-phosphorylated in the K562 cell line and in CML patient cells, and activation of JAK2 contributes to BCR-ABL1 oncogenicity via activation of PI3K/AKT and increased expression of c-Myc.39–41,43 In line with this, Samanta A et al showed that the treatment with a dual JAK2 and BCR-ABL1 inhibitor could induce apoptosis in imatinib-resistant cells.42 mTORC1 is a downstream molecule of AKT and is activated by phosphorylated AKT. Burchert et al have demonstrated that PI3K/AKT/mTOR signaling is activated in response to imatinib and facilitates imatinib resistance.44 Moreover, PI3K/AKT pathway leads to inhibition of FOXO transcriptional activity. FOXO members play a role at the G0-G1, G1-S, and G2-M checkpoints via transcriptional modulation of proteins that regulate these transitions, thereby inducing cell cycle arrest. FOXOs function downstream of the PI3K signalling pathway and are direct phosphorylation targets of the protein kinase AKT. Phosphorylation of FOXOs by AKT result in their nuclear exclusion and prevent transactivation of target genes important for cell cycle regulation. Elevated FOXO levels have also been associated with BCR-ABL1-independent resistance (Figure 2).45
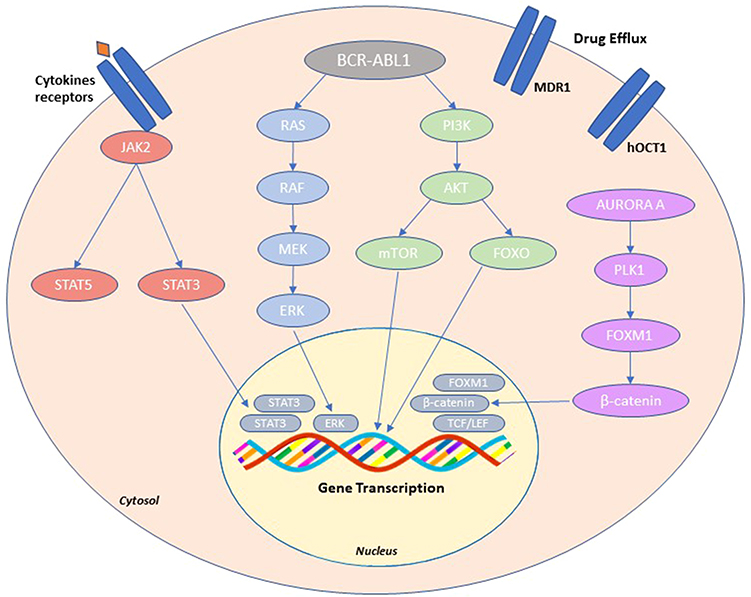 |
Figure 2 BCR-ABL1-independent resistance. Schematic overview of the main resistance mechanism such as: activation of alternative signaling pathways (PI3K/AKT, JAK/STAT, and RAS/MAPK); activation of Aurora A-PLK1-FOXM1 axis; changes in drug transporter activity (MDR1, hOCT1). |
Src family kinases play a key role in BCR-ABL1 signaling axis in that they are both substrates and activators of BCR-ABL1. Studies by Donato et al demonstrated that the src family kinase LYN, which acts downstream of BCR-ABL1, is highly expressed and persistently activated in imatinib-resistant cell lines and in primary samples from imatinib-resistant CML patients negative for BCR-ABL1 KD mutations.46–48
Finally, a recent study by Mancini et al has reported that another pathway implicated in BCR-ABL1-independent resistance is the Aurora kinase A/Polo-like 1 (PLK1) kinase/FOXM1 axis.49–51 The overexpression and hyper-activation of this axis, observed in resistant cell lines and primary patient cells, was found to induce imatinib resistance by activating multiple pathways implicated in proliferation and survival advantage of leukemic hematopoiesis. Aurora A is a serine/threonine kinase overexpressed in several cancers and it regulates centrosome maturation, entry into mitosis, formation and function of the bipolar spindle, and cytokinesis. PLK1, another serine/threonine kinase, is activated in G2 phase of cell cycle by Aurora A which phosphorylates it on T210 residues. FOXM1 is a transcription factor of the Forkhead family and it is a substrate of PLK1 which hyper-phosphorylates FOXM1 on residues Thr596 Ser678 and activates it in the G2/M phase. FOXM1 binds free β-catenin in the cytosol driving it into the nucleus where it interacts with T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription activation complex, supporting leukemic cell proliferation (Figure 2).
Pharmacokinetic Resistance
Altered intracellular drug availability modulated by drug influx and efflux transporters is another mechanism that has frequently been implicated in anticancer drug resistance. Multidrug efflux transporters of the ATP-binding cassette (ABC) transporter family, which include the multidrug resistance gene product P-glycoprotein (MDR1 or Pgp; encoded by the ABCB1 gene), and the breast cancer resistance protein (BCRP; encoded by ABCG2), may have a significant effect on restricting drug uptake from tumor cells through active efflux. In addition to high expression on hematopoietic primitive cells, both ABCB1 and ABCG2 show tissue localization in the small bowel, brain, testes, and canalicular membrane of hepatocytes and may contribute to resistance by causing drug efflux from cells at these sites. Overexpression of MDR1 has been observed in imatinib-resistant cell lines and patients.52,53 Cells from BP patients are characterized by higher expression of ABCB1 than CP patients.54 More controversial is the role of BCRP in imatinib resistance, since contrasting data have been reported about its role in imatinib transport.55–57 hOCT1 is another membrane transporter that is responsible for the intracellular transport of imatinib. Reduced expression and/or activity of hOCT1 has been found to correlate with poorer response to imatinib and worse outcomes (Figure 2).58–63
Stem Cell (SC) Resistance
It has been demonstrated that many if not all cancers arise from oncogenic transformation at the SC level. Most therapies are effective in eliminating progenitors and terminally differentiated cells, but leave SCs unaffected. SCs represent a dangerous reservoir that may feed disease recurrence at any time. The fact that very low levels of residual disease may still be detected after many years of therapy in a not negligible proportion of CML patients, and that even when molecularly undetectable, the disease may often re-emerge after TKI discontinuation is attributed to SC persistence. SCs have long been known to up- or down-regulate drug transporters to protect themselves from chemotherapeutics and targeted therapies, but other mechanisms have also been implicated. In CML, for example, it is now understood that BCR-ABL1 kinase-independent mechanisms are responsible for persistence of leukemic SC (LSC) subpopulations,64,65 and both cell intrinsic and microenvironmental mechanisms have been found to contribute to LSC maintenance. Self-renewal is an essential property of SCs, and the aberrant activation of self-renewal-related signaling pathways has been recognized as a hallmark of cancer. A number of BCR-ABL1-independent signaling pathways, including Wnt/b-catenin and Hedgehog signalling, together with other factors like epigenetic alterations, enhanced autophagy, autocrine production of growth factors, etc. (recently reviewed by Soverini et al66) have been found to contribute to CML LSC persistence and TKI resistance. The interaction between SCs and cells from the microenvironment is also known to nurture SCs and protect them from therapeutics, thus favoring the development of resistance. The LSC niche as well as its contribution to LSC persistence via cell contact as well as secreted signalling molecules have been extensively investigated and are reviewed elsewhere.66
Strategies to Overcome Resistance to TKIs
More Potent and/or Selective Second- and Third-Generation ATP-Competitive TKIs
In many cancers, especially those sustained by nonreceptor PKs, ATP-competitive inhibition has represented the first targeted therapeutic strategy. ATP-competitive inhibitors compete with ATP for binding to the KD in the cleft between the N- and C-terminal lobes.22 On the basis of the molecular mechanisms of action, two major classes of kinase inhibitors can be distinguished: types 1 and 2.67 Type 1 inhibitors target the catalytically competent active (“DFG-in”) conformation of the KD, while type 2 inhibitors recognize the inactive (“DFG-out”) kinase conformation. Imatinib is a type 2 inhibitor. Thus, even mutations at residues distant from the binding pocket may induce resistance if they that destabilize or prevent the adoption of the inactive conformation of the kinase. On the other hand, it has long been assumed that type 1 inhibitors are less specific (hence, may have more undesired, off-target effects) than type 2 inhibitors because the active conformation is very similar in most kinases. The problem of resistance to imatinib prompted the development of further ATP-competitive TKIs. While imatinib serendipitously emerged from a massive screening of a huge library of inhibitory molecules modeled over the ATP structure,68 subsequent generation-TKIs were more rationally developed to be more potent and/or more selective.69 The knowledge accumulated from the extensive characterization of imatinib-binding mode to ABL1 and of its ‘Achilles heels’, especially in terms of resistant mutations, played a key role in the development of more effective TKIs. Each imatinib-resistant point mutation has indeed been studied in detail by a conformational point of view to investigate how it may compromise imatinib binding through steric clash, elimination of direct contacts, and/or favoring an active conformation of the ABL1 kinase domain.
Out of a multitude of molecules evaluated pre-clinically or in phase 1 clinical studies, four ATP-competitive TKIs have so far been approved for clinical use based on a favorable balance between efficacy and safety: dasatinib, nilotinib, bosutinib, and ponatinib.70 They have become an important resource in the treatment of those patients who fail therapy. They have different features, binding modes and target profiles (as well as different side effects). Nilotinib is a type 2 inhibitor as well, but was obtained through a rational design aimed to modify imatinib chemical backbone in order to enhance potency against ABL1.71,72 Dasatinib and bosutinib are type 1 inhibitors; hence, they were expected not to lose efficacy in case of mutations destabilizing the inactive conformation of the kinase.73,74 Moreover, they both have the SRC kinases among their targets, which was presumed to be an added value in light of the role of SRC kinase activation in some cases of imatinib resistance. Dasatinib, nilotinib and bosutinib indeed have a much smaller spectrum of resistant mutations, mainly clustering at key contact residues: T315, Y253, E255, and F359 for nilotinib; T315, V299, and F317 for dasatinib; T315, V299, and E255 for bosutinib.75 Ponatinib is a type 2 inhibitor like imatinib and nilotinib but has more recently been obtained from a thorough structure-guided drug design specifically aimed to create a compound capable to bind the KD irrespective of any mutation, including the T315I.76
Currently, first-line options for the treatment of CML are either imatinib, dasatinib, nilotinib or bosutinib.70 In case of therapeutic failure, the TKI is switched, and the choice is between dasatinib, nilotinib, bosutinib or ponatinib (the latter in case of T315I-positive patients or if no other TKI is indicated).70 Choice is generally based weighing previous treatment history, patient comorbidities and TKI safety profile, and considering BCR-ABL1 KD mutation status.77 CML patients who experience multiple failures of sequential TKI therapy may develop polyclonal or compound mutations (CMs).78,79 The former are when two mutations have been acquired by two distinct BCR-ABL1 molecules, hence they are in two different clones; the latter are when two mutations are acquired by the same BCR-ABL1 molecule, hence they are in a single clone. A number of T315I-inclusive CMs have recently been shown to be resistant not only to imatinib and second-generation TKIs, but also to ponatinib, thus representing a great concern.80–83
Allosteric TKIs
More recently, a novel class of ABL1 inhibitors has been explored in CML. Allosteric inhibitors do not compete with ATP for binding to the same binding site. They recognize other sites that are critically implicated in the regulation of kinase activity.84 As a consequence, they tend to be much more specific. Moreover, by binding to completely different and far distant regions of the kinase, they can be expected not to be affected by mutations impairing ATP-competitive TKI fitting. Allosteric enzymes are defined as K-type or V-type depending on whether they act via a change in affinity (measured by the KM constant) or in maximum activity (defined as Vmax).
Asciminib (ABL001) is the first allosteric BCR-ABL1 inhibitor that has entered clinical evaluation.85 By binding the myristoyl site normally occupied by the myristoylated N-terminal of ABL1, which serves as an allosteric negative regulatory element but is lost upon fusion of ABL1 to BCR, asciminib mimics myristate and restores the inhibition of kinase activity. Since the myristate pocket of ABL1 is located at the very bottom of the C-terminal lobe, asciminib is expected to maintain activity against all TKI-resistant ATP-site mutations – although a very recent in vitro study has suggested that this may not entirely hold true. Eide et al86 have indeed shown that among ten BCR-ABL1 single mutants tested, asciminib potently inhibited five (G250E, Y253H, E255V, T315I, andH396R) with IC50 values below 30 nM. By contrast, some variants (C/I/V) of F359 were insensitive to asciminib (IC50 > 2500 nM). Moreover, preclinical evaluation of asciminib has predicted that resistance may still develop due to mutations in or around the myristate pocket: A344P, A337V, P465S, V468F, I502L, G671R.87 As always, however, the integration of in vitro IC50 and in vivo observations in patients who will not respond or will develop resistance will definitively clarify the actual Achilles heels of asciminib – thus more data from the currently ongoing clinical trials are eagerly awaited.
Combinations of ATP-Competitive and Allosteric TKIs
The presence of a second inhibitory site also offers the potential for simultaneous targeting of both the myristoyl-binding and ATP-binding sites, not only for enhanced kinase inhibition but also in an attempt to prevent the emergence of any mutation whatsoever. Indeed, preclinical studies have shown that the combination of the ATP-competitive inhibitors imatinib or nilotinib with allosteric inhibitors suppressed the emergence of resistance mutations in vitro or in mouse xenografts.87,88 Phase 1/2 studies exploring the combination of asciminib with imatinib, nilotinib and dasatinib are ongoing, and have so far demonstrated a good overall safety/tolerability profile. The yet clinically unexplored combination of asciminib with ponatinib, however, appears to be the most intriguing. Indeed, Eide et al86 have recently shown that the asciminib/ponatinib combo not only suppressed the outgrowth of resistant clones carrying single mutations but was also effective in tackling clinically challenging compound mutants that were resistant to either agent alone. Interestingly, the combination was effective even at very low doses of ponatinib, which would reduce the concerns deriving from ponatinib-associated toxicity.
Synthetic Lethal Combinations
Synthetic lethality results from disruption of two independent pathways, which together perform an essential function. Two genes or two pathways are said to be “synthetic lethal” if loss of either one alone has little effect on cell viability, but the simultaneous loss of both results in cell death. Packer et al first proposed that synthetic lethality could provide an approach to block the emergence of drug resistance in CML patients.89 In vitro and ex vivo studies have shown that several TKIs (eg imatinib, nilotinib and dasatinib), while inhibiting BCR-ABL1, lead to paradoxical activation of RAF, MEK and ERK. Consequently, the combined use of first and second generation TKIs associated with a MEK inhibitor (PD184352) induces apoptotic death of the clone.89 In addition, Eiring et al showed that STAT3 signaling is critical in BCR-ABL1 kinase-independent TKI resistance and, as a consequence, the combined inhibition of both BCR-ABL1 and STAT3 could kill CML stem and progenitor cells with kinase-independent TKI resistance, while inhibition of only BCR-ABL1 or only STAT3 has very limited effects.90
Moreover, it has been shown that BCR-ABL1-positive cells are deficient in both BRCA-dependent homologous recombination and DNA-dependent protein kinase (DNA-PK)-mediated nonhomologous end-joining (NHEJ). In these cells, repair of DNA double strand breaks, the most lethal DNA lesions, thus relies only on Poly(ADP)ribose polymerase 1 (PARP1)-dependent back-up NHEJ. Based on these premises, Nieborowska-Skorska et al91 successfully used PARP1 inhibitors to induce synthetic lethality in primary cells from CML patients. They also interestingly demonstrated that BRCA and DNA-PK deficiency is a common vulnerability and may be exploited for selective targeting with PARP1 inhibitors in other contexts like acute myeloid leukemia and acute lymphoblastic leukemia.
Combinations of ABL1 Inhibitors and LSC-Targeting Drugs
As discussed above, SCs are considered to be the main drivers of therapeutic resistance across all cancers: cancer SCs often survive the treatments, causing recurrence of the diseases. In CML, evidences that TKIs eliminate progenitors and more differentiated cells but fail to eradicate quiescent LSCs date back to 2002. Since CML LSCs have been shown not to depend upon BCR-ABL1 tyrosine kinase activity for their survival, the combination of TKIs and LSC-targeting drugs represents the only approach capable of completely eradicating the disease. The molecules or pathways implicated in CML LSC persistence have been extensively investigated,66 and a wide variety of agents targeting such pathways have been tested in cell lines and mouse models in order to sensitize LSCs to TKI-induced killing or eliminate LSCs by synthetic lethality, showing very promising results in many cases.66 However, these preclinical data have so far led to very few clinical trials.
An overview of TKIs and targeted combinations that are currently approved or are being actively evaluated in clinical trials is presented in Table 1.
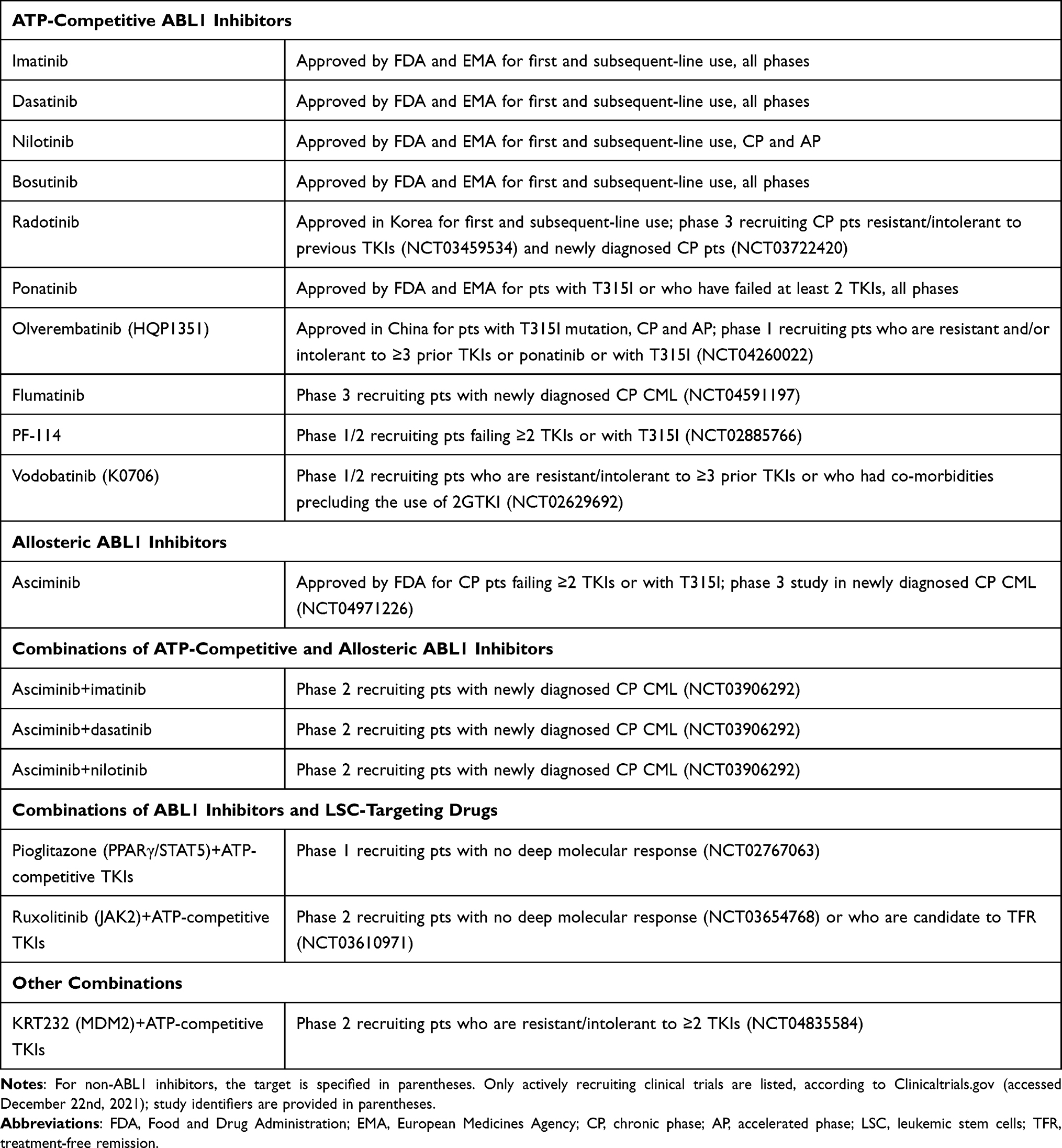 |
Table 1 Overview of the TKIs and Targeted Combinations Approved or Currently Explored in Clinical Trials for the Treatment of CML |
The Importance of Molecular Testing for Early Identification and Management of TKI Resistance
Response monitoring is essential to assess the efficacy of therapy and to intervene quickly in case of failure.75 Given its well established role as a dynamic predictor of relapse risk and overall survival, minimal residual disease (MRD) monitoring has been incorporated in routine follow-up and clinical decision-making algorithms for many hematologic malignancies.92 Once again, CML has been a forerunner and has led the way for the application of sensitive, quantitative approaches for MRD detection relying on real-time quantitative reverse transcription-polymerase chain reaction (RQ-PCR). Based on logarithmic reduction in BCR-ABL1 transcript levels from a standardized baseline, molecular response (MR) milestones have been defined that should be achieved at critical checkpoints during therapy: Early Molecular Response (EMR), corresponding to BCR-ABL1≤10% at 3 months and BCR-ABL1≤1% at 6 months; Major Molecular Response (MMR), corresponding to BCR-ABL1≤0.1% and Deep Molecular Response (DMR), corresponding to BCR-ABL1≤0.01% (down to 0.001%, the lowest MR level that RQ-PCR can quantitate).75 International guidelines (like those by the National Comprehensive Cancer Network [NCCN] and by the European LeukemiaNet [ELN])70,93 base the evaluation of response on the stepwise achievement of such MR milestones. ELN recommendations, for example, define 3 categories of response to treatment with TKIs: “optimal”, “warning” and “failure”. “Optimal” means that the response is satisfactory and the current treatment should be continued. “Failure” means that the patient should receive a different treatment to limit the risk of progression (and death). “Warning” is a grey zone in between. Continuation or change should be carefully considered, depending on patient’s characteristics, comorbidities and tolerance as well as on therapeutic endpoints.70 When response is classified as “failure” or as a “warning” (the expected MR milestone has not been achieved) or when a previously achieved milestone is lost, careful investigation into the underlying mechanisms should be undertaken. An unsatisfactory response may be due to reduced patient compliance to treatment (not infrequent with therapies that have to be administered chronically), to drug interactions (especially in elderly patients taking many concomitant medications) or to true resistance. Once nonadherence and interactions with other medications have been excluded, testing for BCR-ABL1 KD mutations is recommended.77 At the present time, mutations are indeed the only actionable mechanism of resistance.94 The presence of any mutation identifies those subjects who have a greater genetic instability, hence a higher probability of relapse once they are switched to another TKI; such patients will therefore have to be monitored more closely. Moreover, the type of mutation detected can direct, in some cases, the choice of the second- or subsequent-line TKI more likely to be effective. Mutational analysis has therefore become part of the panel of molecular tests to be performed in patients with CML and according to the ELN recommendations, it should be performed in case of failure and warning.70,77 For long time, the gold standard for performing mutational analysis of BCR-ABL1 has been Sanger sequencing.77 Sanger sequencing enables to scan the entire KD for mutations, but it has some limitations: it only allows identification of mutations if they are present in ≥20% of BCR-ABL1 transcripts and often does not allow to discriminate between compound and polyclonal mutations. More recently, several groups have developed next generation sequencing (NGS)-based targeted assays and have demonstrated that they are superior to Sanger sequencing in that they are more sensitive (with a detection limit as low as 1–3%) and enable straightforward clonal analysis whenever two mutations map within approximately 400 base pairs.95–100 The greater sensitivity of NGS enables more accurate mutation assessment (in some cases, mutations detectable by Sanger are just “the tip” of the iceberg, and additional low level mutations may be detected by NGS) and earlier identification of emerging drug-resistant clones, calling for a timely treatment change before overt resistance (that might also lead to disease progression) manifests.
Conclusions
Being the first human malignancy in whom kinase inhibition was accomplished, CML has been a precious study model for the characterization of mechanisms enabling therapeutic evasion and for the development of alternative inhibitory strategies aimed at circumventing resistance. CML is a paradigm of precision medicine not only because of the success of TKI therapies, but also because of the high standards achieved in response monitoring and dynamic patient management on the basis of molecular information deriving from highly standardized and sensitive MRD quantitation and testing for resistance-associated mutations. CML thus exemplifies how preclinical and clinical research efforts and technological advances may concur to transform patients’ outcomes and quality of life by continuous improvement of KI therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Roskoski R
2. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi:10.1126/science.1075762
3. Soverini S, Martinelli G, Iacobucci I, Baccarani M. Imatinib mesylate for the treatment of chronic myeloid leukemia. Expert Rev Anticancer Ther. 2008;8(6):853–864. doi:10.1586/14737140.8.6.853
4. Attwood MM, Fabbro D, Sokolov AV, Knapp S, Schioth HB. Trends in kinase drug discovery: targets, indications and inhibitor design. Nat Rev Drug Discov. 2021;20(11):839–861. doi:10.1038/s41573-021-00252-y
5. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–28. doi:10.1126/science.959840
6. Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–880. doi:10.1126/science.1062538
7. Soverini S, Mancini M, Bavaro L, Cavo M, Martinelli G. Chronic myeloid leukemia: the paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol Cancer. 2018;17(1):49. doi:10.1186/s12943-018-0780-6
8. Rowley JD. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–293. doi:10.1038/243290a0
9. Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst. 1960;25:85–109.
10. Heisterkamp N, Stephenson JR, Groffen J, et al. Localization of the c-ab1 oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature. 1983;306(5940):239–242. doi:10.1038/306239a0
11. Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36(1):93–99. doi:10.1016/0092-8674(84)90077-1
12. Groffen J, Stephenson JR, Heisterkamp N, Bartram C, de Klein A, Grosveld G. The human c-abl oncogene in the Philadelphia translocation. J Cell Physiol Suppl. 1984;3:179–191. doi:10.1002/jcp.1041210421
13. Sattler M, Griffin JD. Molecular mechanisms of transformation by the BCR-ABL oncogene. Semin Hematol. 2003;40(2 Suppl 2):4–10. doi:10.1053/shem.2003.50034
14. Chereda B, Melo JV. Natural course and biology of CML. Ann Hematol. 2015;94(Suppl 2):S107–21. doi:10.1007/s00277-015-2325-z
15. Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247(4944):824–830. doi:10.1126/science.2406902
16. Kelliher MA, McLaughlin J, Witte ON, Rosenberg N. Induction of a chronic myelogenous leukemia-like syndrome in mice with v-abl and BCR/ABL. Proc Natl Acad Sci U S A. 1990;87(17):6649–6653. doi:10.1073/pnas.87.17.6649
17. Elefanty AG, Hariharan IK, Cory S. bcr-abl, the hallmark of chronic myeloid leukaemia in man, induces multiple haemopoietic neoplasms in mice. EMBO J. 1990;9(4):1069–1078. doi:10.1002/j.1460-2075.1990.tb08212.x
18. Bavaro L, Martelli M, Cavo M, Soverini S. Mechanisms of Disease Progression and Resistance to Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukemia: an Update. Int J Mol Sci. 2019;20:24. doi:10.3390/ijms20246141
19. Soverini S, de Benedittis C, Mancini M, Martinelli G. Mutations in the BCR-ABL1 Kinase Domain and Elsewhere in Chronic Myeloid Leukemia. Clin Lymphoma Myeloma Leuk. 2015;15(Suppl):S120–8. doi:10.1016/j.clml.2015.02.035
20. Soverini S, Iacobucci I, Baccarani M, Martinelli G. Targeted therapy and the T315I mutation in Philadelphia-positive leukemias. Haematologica. 2007;92(4):437–439. doi:10.3324/haematol.11248
21. Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008;15(10):1109–1118. doi:10.1038/nsmb.1486
22. Nagar B, Bornmann WG, Pellicena P, et al. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 2002;62(15):4236–4243.
23. Roskoski R
24. Soverini S, Branford S, Nicolini FE, et al. Implications of BCR-ABL1 kinase domain-mediated resistance in chronic myeloid leukemia. Leuk Res. 2014;38(1):10–20. doi:10.1016/j.leukres.2013.09.011
25. Soverini S, Rosti G, Iacobucci I, Baccarani M, Martinelli G. Choosing the best second-line tyrosine kinase inhibitor in imatinib-resistant chronic myeloid leukemia patients harboring Bcr-Abl kinase domain mutations: how reliable is the IC(5)(0)? Oncologist. 2011;16(6):868–876. doi:10.1634/theoncologist.2010-0388
26. Lyczek A, Berger BT, Rangwala AM, et al. Mutation in Abl kinase with altered drug-binding kinetics indicates a novel mechanism of imatinib resistance. Proc Natl Acad Sci U S A. 2021;118:46. doi:10.1073/pnas.2111451118
27. Mahon FX, Deininger MW, Schultheis B, et al. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: diverse mechanisms of resistance. Blood. 2000;96(3):1070–1079. doi:10.1182/blood.V96.3.1070
28. Weisberg E, Griffin JD. Mechanism of resistance to the ABL tyrosine kinase inhibitor STI571 in BCR/ABL-transformed hematopoietic cell lines. Blood. 2000;95(11):3498–3505. doi:10.1182/blood.V95.11.3498
29. Campbell LJ, Patsouris C, Rayeroux KC, Somana K, Januszewicz EH, Szer J. BCR/ABL amplification in chronic myelocytic leukemia blast crisis following imatinib mesylate administration. Cancer Genet Cytogenet. 2002;139(1):30–33. doi:10.1016/S0165-4608(02)00615-5
30. Hochhaus A, Kreil S, Corbin AS, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16(11):2190–2196. doi:10.1038/sj.leu.2402741
31. Barnes DJ, Palaiologou D, Panousopoulou E, et al. Bcr-Abl expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res. 2005;65(19):8912–8919. doi:10.1158/0008-5472.CAN-05-0076
32. Mahon FX, Hayette S, Lagarde V, et al. Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res. 2008;68(23):9809–9816. doi:10.1158/0008-5472.CAN-08-1008
33. Tang C, Schafranek L, Watkins DB, et al. Tyrosine kinase inhibitor resistance in chronic myeloid leukemia cell lines: investigating resistance pathways. Leuk Lymphoma. 2011;52(11):2139–2147. doi:10.3109/10428194.2011.591013
34. Mitelman F, Levan G, Nilsson PG, Brandt L. Non-random karyotypic evolution in chronic myeloid leukemia. Int J Cancer. 1976;18(1):24–30. doi:10.1002/ijc.2910180105
35. Johansson B, Fioretos T, Mitelman F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol. 2002;107(2):76–94. doi:10.1159/000046636
36. Fabarius A, Leitner A, Hochhaus A, et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML Study IV. Blood. 2011;118(26):6760–6768. doi:10.1182/blood-2011-08-373902
37. Bianchini M, De Brasi C, Gargallo P, Gonzalez M, Bengio R, Larripa I. Specific assessment of BCR-ABL transcript overexpression and imatinib resistance in chronic myeloid leukemia patients. Eur J Haematol. 2009;82(4):292–300. doi:10.1111/j.1600-0609.2008.01199.x
38. Kuroda J, Puthalakath H, Cragg MS, et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A. 2006;103(40):14907–14912. doi:10.1073/pnas.0606176103
39. Xie S, Wang Y, Liu J, et al. Involvement of Jak2 tyrosine phosphorylation in Bcr-Abl transformation. Oncogene. 2001;20(43):6188–6195. doi:10.1038/sj.onc.1204834
40. Xie S, Lin H, Sun T, Arlinghaus RB. Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene. 2002;21(47):7137–7146. doi:10.1038/sj.onc.1205942
41. Samanta AK, Lin H, Sun T, Kantarjian H, Arlinghaus RB. Janus kinase 2: a critical target in chronic myelogenous leukemia. Cancer Res. 2006;66(13):6468–6472. doi:10.1158/0008-5472.CAN-06-0025
42. Samanta AK, Chakraborty SN, Wang Y, Schlette E, Reddy EP, Arlinghaus RB. Destabilization of Bcr-Abl/Jak2 Network by a Jak2/Abl Kinase Inhibitor ON044580 Overcomes Drug Resistance in Blast Crisis Chronic Myelogenous Leukemia (CML). Genes Cancer. 2010;1(4):346–359. doi:10.1177/1947601910372232
43. Samanta A, Perazzona B, Chakraborty S, et al. Janus kinase 2 regulates Bcr-Abl signaling in chronic myeloid leukemia. Leukemia. 2011;25(3):463–472. doi:10.1038/leu.2010.287
44. Burchert A, Wang Y, Cai D, et al. Compensatory PI3-kinase/Akt/mTor activation regulates imatinib resistance development. Leukemia. 2005;19(10):1774–1782. doi:10.1038/sj.leu.2403898
45. Wagle M, Eiring AM, Wongchenko M, et al. A role for FOXO1 in BCR-ABL1-independent tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Leukemia. 2016;30(7):1493–1501. doi:10.1038/leu.2016.51
46. Donato NJ, Wu JY, Stapley J, et al. BCR-ABL Independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101(2):690–698. doi:10.1182/blood.V101.2.690
47. Wu J, Meng F, Lu H, et al. Lyn regulates BCR-ABL and Gab2 tyrosine phosphorylation and c-Cbl protein stability in imatinib-resistant chronic myelogenous leukemia cells. Blood. 2008;111(7):3821–3829. doi:10.1182/blood-2007-08-109330
48. Wu J, Meng F, Kong LY, et al. Association between imatinib-resistant BCR-ABL mutation-negative leukemia and persistent activation of LYN kinase. J Natl Cancer Inst. 2008;100(13):926–939. doi:10.1093/jnci/djn188
49. Mancini M, Castagnetti F, Soverini S, et al. FOXM1 Transcription Factor: a New Component of Chronic Myeloid Leukemia Stem Cell Proliferation Advantage. J Cell Biochem. 2017;118(11):3968–3975. doi:10.1002/jcb.26052
50. Mancini M, De Santis S, Monaldi C, et al. Hyper-activation of Aurora kinase a-polo-like kinase 1-FOXM1 axis promotes chronic myeloid leukemia resistance to tyrosine kinase inhibitors. J Exp Clin Cancer Res. 2019;38(1):216. doi:10.1186/s13046-019-1197-9
51. Mancini M, De Santis S, Monaldi C, et al. Ponatinib treatment in chronic myeloid leukemia cell lines targets Aurora kinase A/FOXM1 axis. Hematol Oncol. 2020;38(2):201–203. doi:10.1002/hon.2703
52. Mahon FX, Belloc F, Lagarde V, et al. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood. 2003;101(6):2368–2373. doi:10.1182/blood.V101.6.2368
53. Eadie LN, Dang P, Saunders VA, et al. The clinical significance of ABCB1 overexpression in predicting outcome of CML patients undergoing first-line imatinib treatment. Leukemia. 2017;31(1):75–82. doi:10.1038/leu.2016.179
54. Reis FR, Vasconcelos FC, Pereira DL, Moellman-Coelho A, Silva KL, Maia RC. Survivin and P-glycoprotein are associated and highly expressed in late phase chronic myeloid leukemia. Oncol Rep. 2011;26(2):471–478. doi:10.3892/or.2011.1296
55. Nakanishi T, Shiozawa K, Hassel BA, Ross DD. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood. 2006;108(2):678–684. doi:10.1182/blood-2005-10-4020
56. Shukla S, Sauna ZE, Ambudkar SV. Evidence for the interaction of imatinib at the transport-substrate site(s) of the multidrug-resistance-linked ABC drug transporters ABCB1 (P-glycoprotein) and ABCG2. Leukemia. 2008;22(2):445–447. doi:10.1038/sj.leu.2404897
57. Brendel C, Scharenberg C, Dohse M, et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia. 2007;21(6):1267–1275. doi:10.1038/sj.leu.2404638
58. White DL, Saunders VA, Dang P, et al. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): reduced OCT- 1 activity is the cause of low in vitro sensitivity to imatinib. Blood. 2006;108(2):697–704. doi:10.1182/blood-2005-11-4687
59. White DL, Saunders VA, Dang P, et al. Most CML patients who have a suboptimal response to imatinib have low OCT- 1 activity: higher doses of imatinib may overcome the negative impact of low OCT- 1 activity. Blood. 2007;110(12):4064–4072. doi:10.1182/blood-2007-06-093617
60. White DL, Saunders VA, Dang P, Engler J, Hughes TP. OCT-1 activity measurement provides a superior imatinib response predictor than screening for single-nucleotide polymorphisms of OCT-1. Leukemia. 2010;24(11):1962–1965. doi:10.1038/leu.2010.188
61. White DL, Dang P, Engler J, et al. Functional activity of the OCT- 1 protein is predictive of long-term outcome in patients with chronic-phase chronic myeloid leukemia treated with imatinib. J Clin Oncol. 2010;28(16):2761–2767. doi:10.1200/JCO.2009.26.5819
62. White DL, Radich J, Soverini S, et al. Chronic phase chronic myeloid leukemia patients with low OCT- 1 activity randomized to high-dose imatinib achieve better responses and have lower failure rates than those randomized to standard-dose imatinib. Haematologica. 2012;97(6):907–914. doi:10.3324/haematol.2011.056457
63. Eadie LN, Hughes TP, White DL. Patients with low OCT- 1 activity and high ABCB1 fold rise have poor long-term outcomes in response to tyrosine kinase inhibitor therapy. Leukemia. 2018;32(10):2288–2291. doi:10.1038/s41375-018-0101-5
64. Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121(1):396–409. doi:10.1172/JCI35721
65. Hamilton A, Helgason GV, Schemionek M, et al. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood. 2012;119(6):1501–1510. doi:10.1182/blood-2010-12-326843
66. Soverini S, De Santis S, Monaldi C, Bruno S, Mancini M. Targeting Leukemic Stem Cells in Chronic Myeloid Leukemia: is It Worth the Effort? Int J Mol Sci. 2021;22:13. doi:10.3390/ijms22137093
67. Roskoski R
68. Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105(7):2640–2653. doi:10.1182/blood-2004-08-3097
69. Rossari F, Minutolo F, Orciuolo E. Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. J Hematol Oncol. 2018;11(1):84. doi:10.1186/s13045-018-0624-2
70. Hochhaus A, Baccarani M, Silver RT, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34(4):966–984. doi:10.1038/s41375-020-0776-2
71. Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7(2):129–141. doi:10.1016/j.ccr.2005.01.007
72. Golemovic M, Verstovsek S, Giles F, et al. AMN107, a novel aminopyrimidine inhibitor of Bcr-Abl, has in vitro activity against imatinib-resistant chronic myeloid leukemia. Clin Cancer Res. 2005;11(13):4941–4947. doi:10.1158/1078-0432.CCR-04-2601
73. Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305(5682):399–401. doi:10.1126/science.1099480
74. Puttini M, Coluccia AM, Boschelli F, et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006;66(23):11314–11322. doi:10.1158/0008-5472.CAN-06-1199
75. Soverini S, De Benedittis C, Mancini M, Martinelli G. Best Practices in Chronic Myeloid Leukemia Monitoring and Management. Oncologist. 2016;21(5):626–633. doi:10.1634/theoncologist.2015-0337
76. O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–412. doi:10.1016/j.ccr.2009.09.028
77. Soverini S, Hochhaus A, Nicolini FE, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118(5):1208–1215. doi:10.1182/blood-2010-12-326405
78. Khorashad JS, Kelley TW, Szankasi P, et al. BCR-ABL1 compound mutations in tyrosine kinase inhibitor-resistant CML: frequency and clonal relationships. Blood. 2013;121(3):489–498. doi:10.1182/blood-2012-05-431379
79. Shah NP, Skaggs BJ, Branford S, et al. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117(9):2562–2569. doi:10.1172/JCI30890
80. Zabriskie MS, Eide CA, Tantravahi SK, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26(3):428–442. doi:10.1016/j.ccr.2014.07.006
81. Gibbons DL, Pricl S, Posocco P, et al. Molecular dynamics reveal BCR-ABL1 polymutants as a unique mechanism of resistance to PAN-BCR-ABL1 kinase inhibitor therapy. Proc Natl Acad Sci U S A. 2014;111(9):3550–3555. doi:10.1073/pnas.1321173111
82. Byrgazov K, Lucini CB, Valent P, Hantschel O, Lion T. BCR-ABL1 compound mutants display differential and dose-dependent responses to ponatinib. Haematologica. 2018;103(1):e10–e12. doi:10.3324/haematol.2017.176347
83. Soverini S, Martelli M, Bavaro L, et al. BCR-ABL1 compound mutants: prevalence, spectrum and correlation with tyrosine kinase inhibitor resistance in a consecutive series of Philadelphia chromosome-positive leukemia patients analyzed by NGS. Leukemia. 2021;35(7):2102–2107. doi:10.1038/s41375-020-01098-w
84. Garuti L, Roberti M, Bottegoni G. Non-ATP competitive protein kinase inhibitors. Curr Med Chem. 2010;17(25):2804–2821. doi:10.2174/092986710791859333
85. Schoepfer J, Jahnke W, Berellini G, et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J Med Chem. 2018;61(18):8120–8135. doi:10.1021/acs.jmedchem.8b01040
86. Eide CA, Zabriskie MS, Savage Stevens SL, et al. Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants. Cancer Cell. 2019;36(4):431–443 e5. doi:10.1016/j.ccell.2019.08.004
87. Wylie AA, Schoepfer J, Jahnke W, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature. 2017;543(7647):733–737. doi:10.1038/nature21702
88. Zhang J, Adrian FJ, Jahnke W, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463(7280):501–506. doi:10.1038/nature08675
89. Packer LM, Rana S, Hayward R, et al. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell. 2011;20(6):715–727. doi:10.1016/j.ccr.2011.11.004
90. Eiring AM, Page BDG, Kraft IL, et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia. 2017;31(5):1253–1254. doi:10.1038/leu.2017.14
91. Nieborowska-Skorska M, Sullivan K, Dasgupta Y, et al. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J Clin Invest. 2017;127(6):2392–2406. doi:10.1172/JCI90825
92. Soverini S, Bassan R, Lion T. Treatment and monitoring of Philadelphia chromosome-positive leukemia patients: recent advances and remaining challenges. J Hematol Oncol. 2019;12(1):39. doi:10.1186/s13045-019-0729-2
93. Deininger MW, Shah NP, Altman JK, et al. Chronic Myeloid Leukemia, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2020;18(10):1385–1415. doi:10.6004/jnccn.2020.0047
94. Soverini S, Martinelli G, Rosti G, Iacobucci I, Baccarani M. Advances in treatment of chronic myeloid leukemia with tyrosine kinase inhibitors: the evolving role of Bcr-Abl mutations and mutational analysis. Pharmacogenomics. 2012;13(11):1271–1284. doi:10.2217/pgs.12.103
95. Soverini S, De Benedittis C, Machova Polakova K, et al. Unraveling the complexity of tyrosine kinase inhibitor-resistant populations by ultra-deep sequencing of the BCR-ABL kinase domain. Blood. 2013;122(9):1634–1648. doi:10.1182/blood-2013-03-487728
96. Soverini S, De Benedittis C, Castagnetti F, et al. In chronic myeloid leukemia patients on second-line tyrosine kinase inhibitor therapy, deep sequencing of BCR-ABL1 at the time of warning may allow sensitive detection of emerging drug-resistant mutants. BMC Cancer. 2016;16:572. doi:10.1186/s12885-016-2635-0
97. Soverini S, De Benedittis C, Polakova KM, et al. Next-generation sequencing for sensitive detection of BCR-ABL1 mutations relevant to tyrosine kinase inhibitor choice in imatinib-resistant patients. Oncotarget. 2016;7(16):21982–21990. doi:10.18632/oncotarget.8010
98. Soverini S, De Benedittis C, Papayannidis C, et al. Clinical impact of low-burden BCR-ABL1 mutations detectable by amplicon deep sequencing in Philadelphia-positive acute lymphoblastic leukemia patients. Leukemia. 2016;30(7):1615–1619. doi:10.1038/leu.2016.17
99. Baer C, Kern W, Koch S, et al. Ultra-deep sequencing leads to earlier and more sensitive detection of the tyrosine kinase inhibitor resistance mutation T315I in chronic myeloid leukemia. Haematologica. 2016;101(7):830–838. doi:10.3324/haematol.2016.145888
100. Machova Polakova K, Kulvait V, Benesova A, et al. Next-generation deep sequencing improves detection of BCR-ABL1 kinase domain mutations emerging under tyrosine kinase inhibitor treatment of chronic myeloid leukemia patients in chronic phase. J Cancer Res Clin Oncol. 2015;141(5):887–899. doi:10.1007/s00432-014-1845-6
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.