")
Back to Journals » OncoTargets and Therapy » Volume 15
Ovarian Cancer Therapy: Homologous Recombination Deficiency as a Predictive Biomarker of Response to PARP Inhibitors
Authors Miller RE, Elyashiv O , El-Shakankery KH , Ledermann JA
Received 23 June 2022
Accepted for publication 13 September 2022
Published 4 October 2022 Volume 2022:15 Pages 1105—1117
DOI https://doi.org/10.2147/OTT.S272199
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
Rowan E Miller,1,2 Osnat Elyashiv,1 Karim H El-Shakankery,1 Jonathan A Ledermann1,3
1Department of Medical Oncology, University College London Hospital, London, UK; 2Department of Medical Oncology, St Bartholomew’s Hospital, London, UK; 3UCL Cancer Institute, University College London, London, UK
Correspondence: Jonathan A Ledermann, UCL Cancer Institute, 72 Huntley Street, London, WC1E 6DD, UK, Email [email protected]
Abstract: Poly(adenosine diphosphate-ribose) polymerase (PARP) inhibitors have revolutionised the management of patients with high-grade serous and endometrioid ovarian cancer demonstrating significant improvements in progression-free survival. Whilst the greatest benefit is seen with BRCA1/2 mutant cancers, it is clear that the benefit extends beyond this group. This sensitivity is thought to be due to homologous recombination deficiency (HRD), which is present in up to 50% of the high-grade serous cancers. Several different HRD assays exist, which fall into one of three main categories: homologous recombination repair (HRR)-related gene analysis, genomic “scars” and/or mutational signatures, and real-time HRD functional assessment. We review the emerging data on HRD as a predictive biomarker for PARP inhibitors and discuss the merits and disadvantages of different HRD assays.
Keywords: ovarian cancer, PARP inhibitors, BRCA mutations, homologous recombination deficiency, maintenance therapy
Introduction
Maintenance therapy with poly(adenosine diphosphate-ribose) polymerase (PARP) inhibitors has transformed the treatment of the most common and lethal forms of ovarian cancer (high-grade serous and endometrioid, HGOC). The introduction of PARP inhibitor (PARPi) maintenance therapy has resulted in significant improvements in progression-free survival (PFS) in both relapsed and first-line settings.1–6 Whilst PARPi were initially only licensed for BRCA1 or BRCA2 (BRCA1/2) mutant cancers,7,8 it soon became apparent that the benefit extended beyond this group. The basis of this sensitivity is thought to be due to homologous recombination deficiency (HRD) which can be identified in up to 50% of high-grade serous ovarian cancer (HGSC).9,10 Most frequently, this is due to the lack of a functional copy of either BRCA1 or BRCA2. However, BRCA1/2 genes can be inactivated by non-mutational process such as DNA methylation, and there are other proteins involved in homologous recombination repair (HRR) whose loss results in an HRD phenotype, similar to that observed with BRCA1/2 loss.11
Maintenance therapy with PARPi is approved for patients with HGOC in recurrent platinum-sensitive disease regardless of biomarker status, and this also applies to niraparib in the first-line setting.1,4,6,12,13 Despite this broad approval, a common theme is apparent across most PARPi studies; maximal benefit is observed in those cancers characterised by a BRCA1/2 mutation, followed by HRD/BRCA1/2 wild-type cancers with minimal benefit seen in tumors that are negative on HRD testing.11 Reflecting this, the most recent PAPRi approval for olaparib in combination with bevacizumab was only granted for HRD cancers (defined by the presence of a BRCA1/2 mutation and/or a high genomic instability score; GIS).14,15 In this review, we discuss the pivotal studies of PARPi used in HGOC. Specifically, we consider the relevance of HRD in ovarian cancer from a molecular and clinical perspective.
Homologous Recombination Repair (HRR) and PARP Inhibitors
Environmental exposure and endogenous toxins lead to constant DNA damage that is repaired by complex pathways to ensure genomic integrity, progression through the cell cycle and error-free replication.16 DNA damage results in single-strand DNA (ssDNA) and double-strand DNA (dsDNA) breaks and repair is essential for maintaining genomic integrity. dsDNA breaks are the most serious lesions and these are repaired mainly by homologous recombination repair (HRR). Complex pathways are involved in this process and BRCA1 and BRCA2 proteins play a key role in the repair process. In the absence of HRR (as occurs with deleterious BRCA1/2 and other HRR gene mutations), the repair of dsDNA breaks is reliant on other processes such as non-homologous end-joining (NHEJ) and ssDNA repair mechanisms; however, these mechanisms are more error-prone than HRR.17 Several of these alternative ssDNA repair pathways are modulated by PARP, leaving the cells sensitive to PARP inhibition.18 The selective killing of BRCA1/2 deficient cells by PARP inhibitors exploits the process of synthetic lethality,19,20 now used for cancer treatment. PARP proteins are trapped onto DNA at sites of ssDNA breaks by PARPi. When trapped PARP is encountered by the DNA replication machinery, it leads to stalling of the replication fork, collapse and the generation of a dsDNA break leading to genetic disarray and ultimately cell death, which is particularly marked in cells with HRD.21 Almost half of HGSCs exhibit defects with the HRR pathway, through a variety of underlying mechanism, some of which are yet unexplained.9 Most frequently, this is due to loss of function mutations and epigenetic modification in BRCA1/2 or another HRR genes including RAD51C/D, BRIP1, PALB2, ATM and Fanconi anaemia genes.9 These cancers are more reliant on error prone forms of DNA repair and exhibit a distinct clinical phenotype including greater response to platinum-based chemotherapy and PARPi.10,22
Testing for Homologous Recombination Deficiency (HRD)
The value of HRD status as a predictive and prognostic biomarker is becoming increasingly apparent and some international guidelines now recommend HRD testing in addition to BRCA1/2 testing in all patients with newly diagnosed high-grade epithelial ovarian cancer.23 Three main categories of HRD assays have been developed (Figure 1): 1) HRR pathway-related genes that identify specific causes of HRD, 2) genomic “scars” or mutational signatures that measure the patterns of somatic mutations that accumulate in HRD cancers irrespective of the underlying cause and 3) functional assays that have the potential to reflect the current HRD status. To date, the only HRD assays that have been validated in clinical trials are those based on next-generation sequencing of DNA from tumor tissue. The key component of the assay is to detect genomic “scars” and measure levels of loss of heterozygosity (LOH); additional measurements such as telomeric allelic imbalance (TAI) and large-scale transition (LST) are applied to one assay.11
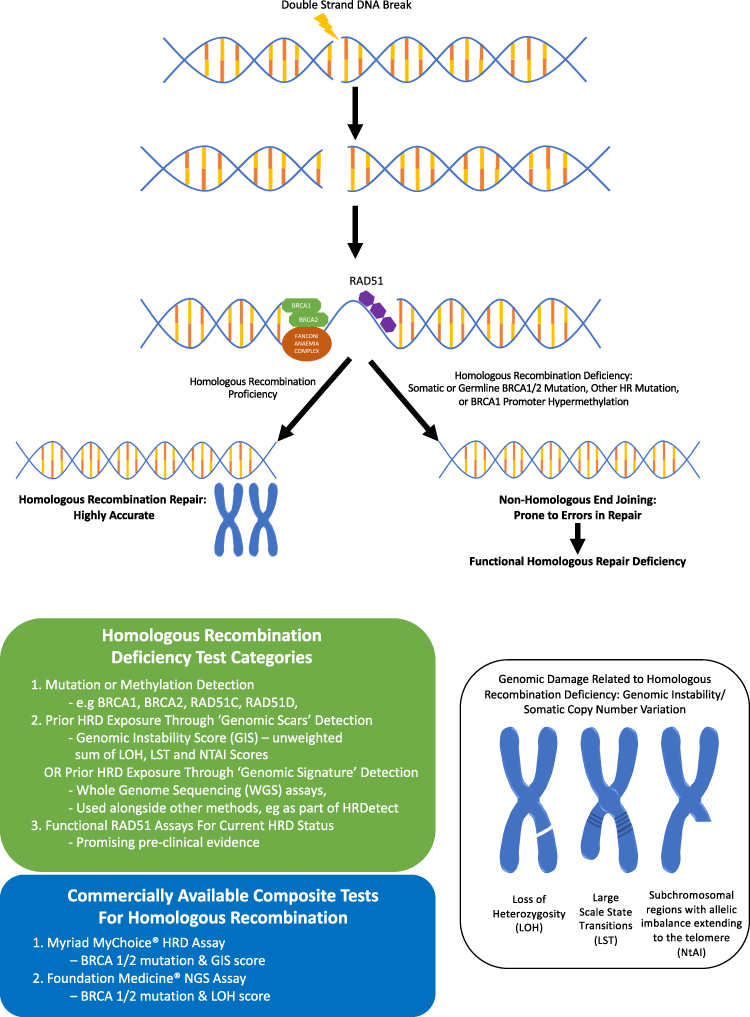 |
Figure 1 Homologous recombination deficiency (HRD) and types of HRD testing. |
HRR Gene Tests
BRCA1/2
Deleterious germline (inherited) BRCA1/2 mutations are present in between 13% and 15% of HGSC.9,24 Cancers that develop in patients with germline BRCA1/2 mutations frequently contain a somatic loss of function mutation in the corresponding wild-type BRCA1/2 allele resulting in defective HRR. Germline genetic testing by either direct sequencing or panel testing is relatively cheap and quick. However, it will fail to identify epigenetic BRCA1/2 modifications and has a limited scope in identifying HRD cancers in the whole ovarian cancer population. An additional 5–7% of HGSC harbour somatic BRCA1/2 mutations that have arisen during tumorigenesis.9 Data from clinical trial samples suggest that there is biallelic inactivation in >80% of the somatic BRCA1/2 tumors, with mutations predominately clonal, suggesting that somatic BRCA1/2 mutations arise early in tumor development.25 Clinical outcomes with PARPi treatment in patients with somatic BRCA1/2 mutations are comparable to those with germline BRCA1/2 mutations with similar response rates and progression-free survival across multiple Phase 3 trials.2,26,27
Non BRCA1/2 HRR Genes
Germline mutations in HRR genes such as RAD51C, RAD51D, PALB2 and BRIP1 are associated with an increased lifetime risk of HGSC.28–30 Together with somatic aberrations in these genes and other key HRR pathway genes including CHK1/2, ATM, CDK12 and Fanconi anaemia pathway, these are present in approximately 30% of HGSC and thought to confer an HRD phenotype.31 This is supported by preclinical studies, which have established that mutations in these genes result in sensitivity to DNA repair inhibition.10,32,33 Clinical studies have shown that somatic mutations in non-BRCA1/2 HRR genes result in a PFS and overall survival advantage, similar to that seen with BRCA1/2 mutations in patients treated with platinum chemotherapy and PARPi compared to those that are BRCA1/2 and HRR wild-type.22,34 For example, in a retrospective analysis from Study 19 (see below), a cohort of BRCA1/2 wild-type patients were identified with HRR mutations (from tumor tissue). This cohort derived a similar benefit from olaparib as those with a BRCA1/2 mutation (hazard ratio [HR] 0.21 and HR 0.18, respectively), which was of a greater magnitude than those without an HRR gene mutation (HR 0.71).34 However, a major limitation is that because of the relatively rarity of non-BRCA1/2 HRR mutations, these studies usually group all HRR mutations together making it difficult to draw conclusion on the relevance of an individual mutation. Evidence for individual HRR genes are often anecdotal and emerging data suggests that mutations in different HRR genes, such as ATM and BRCA1/2, can be associated with different sensitivities to PARPi.35 Therefore, adopting a one-size fits all approach when using individual HRR mutations to predict PARPi response is discouraged.
Determining the clinical significance of variants of uncertain significance (VUS) remains a challenge. VUS are usually rarer missense mutations with unknown consequences on the function of the gene product.36 The rate of germline VUS reporting varies from different laboratories and depends on testing prevalence and the ancestry of the population tested.36 The frequency of VUS is often elevated in underrepresented populations such as in African American and Asiatic groups.37,38 Somatic VUS may be more frequent and diverse than germline variants, having arisen in the context of an elevated mutation rate or genomic instability. Considering the difficulty of predicting the functional relevance of an individual mutation and the variation in the assignment of VUS, corroborating evidence from a phenotypic assay should ideally be required.11
HRR Gene Hypermethylation
Clinical studies provide conflicting evidence for the accuracy of HRR gene promoter methylation as a biomarker for predicting PARPi (or platinum) responses in HGSC.39–42 Defective methylation of cytosine residues of cytosine-phosphate-guanine (CpG) dinucleotides within promoter regions, results in decreased gene expression.43 Epigenetic silencing of BRCA1 and other HRR genes such as RAD51C are associated with high HRD scores and account for up to 15% of HRD HGSC.44,45 Whilst some studies have reported that BRCA1/RAD51C methylation is associated with better prognosis39,46 other studies have described inconsistent findings and poor reliability of this as a biomarker for PARPi response.39,47,48 Subsequently it became clear that prior studies were affected by technical factors and that the zygosity of BRCA1 methylation is a key factor in determining PARPi sensitivity. For example, Kondrashova et al showed that both copies of BRCA1 must be methylated for PARPi response and that losing methylation of a single BRCA1 copy was enough to restore HRR DNA repair and lead to platinum/PARPi resistance.49 However, it remains to be confirmed if the same requirements for methylation zygosity are applicable to RAD51C methylated cases.11 Chemotherapy exposure has also been shown to result in demethylation of previously methylated BRCA1 copies, which may occur more easily than resistance mechanisms such as reversion mutations, described with pathogenic BRCA1/2 mutations.50–52 Caution is required when assigning methylation status of HRR genes and gene copy number is critical for accurate HRD assessment. Furthermore, epigenetic modifications are not detected using present-day next-generation sequencing, which limits the use of this method to identify HRD cancers.
Genomic Signatures and Scars
Copy Number Scar Assays
HRD cancers display genomic instability illustrated by abnormal copy-number profiles and thousands of somatic mutations. The resulting genomic “scars” serve as an indirect measure of HRD as they represent a permanent footprint of genomic changes induced by historical DNA repair deficiency regardless of underlying aetiology.11 To date, two commercial assays have been prospectively validated in ovarian cancer PARPi clinical trials: Foundation One CDx (Foundation Medicine) which evaluates for the percentage of genomic regions with LOH determined through tumor single-nucleotide polymorphism (SNP) sequencing and myChoice CDx (Myriad Genetics) which generates a GIS score by combining measurements of LOH, TAI, and LSTs obtained from allele-specific copy number profiles. LOH, LST, and TAI are highly associated with each other and indicate increasing genomic instability. Prior studies have reported that SNP-based copy-number variant assays reliable predict BRCA1/2 status by quantification of LST, LOH and TAI.44,53,54 Further studies demonstrated that combining information from two or more of these assays further improved the ability to distinguish between HRR proficient and deficient tumors.55 Many of the pivotal clinical trials exploring PARPi in ovarian cancer discussed below (see also Table 1) incorporated either the myChoice CDx or Foundation Medicine LOH assay to determine tumor HRD. However, it should be noted that subgroup analyses based on HRD status were often performed as predefined exploratory endpoints which were not sufficiently powered or adjusted, preventing definitive analysis. Despite this limitation, these assays consistently identify a subgroup of BRCA1/2 wild-type cancers which derive a greater magnitude of benefit from PARPi therapy.11 There have been no direct comparisons of these assays within clinical trials. However, a retrospective analysis from two clinical datasets demonstrated positive agreement between MyChoice GIS and %LOH of only 65–83% suggesting these assays should not be considered interchangeable in predicting PARPi response in clinical practice.56 Furthermore, these tests are expensive and not affordable for all healthcare systems. As such there is a need to develop more accessible, cheaper assays. Currently, an international academic effort is underway to identify more reliable, cost-effective HRD assays using samples from the PAOLA1 trial.13,57
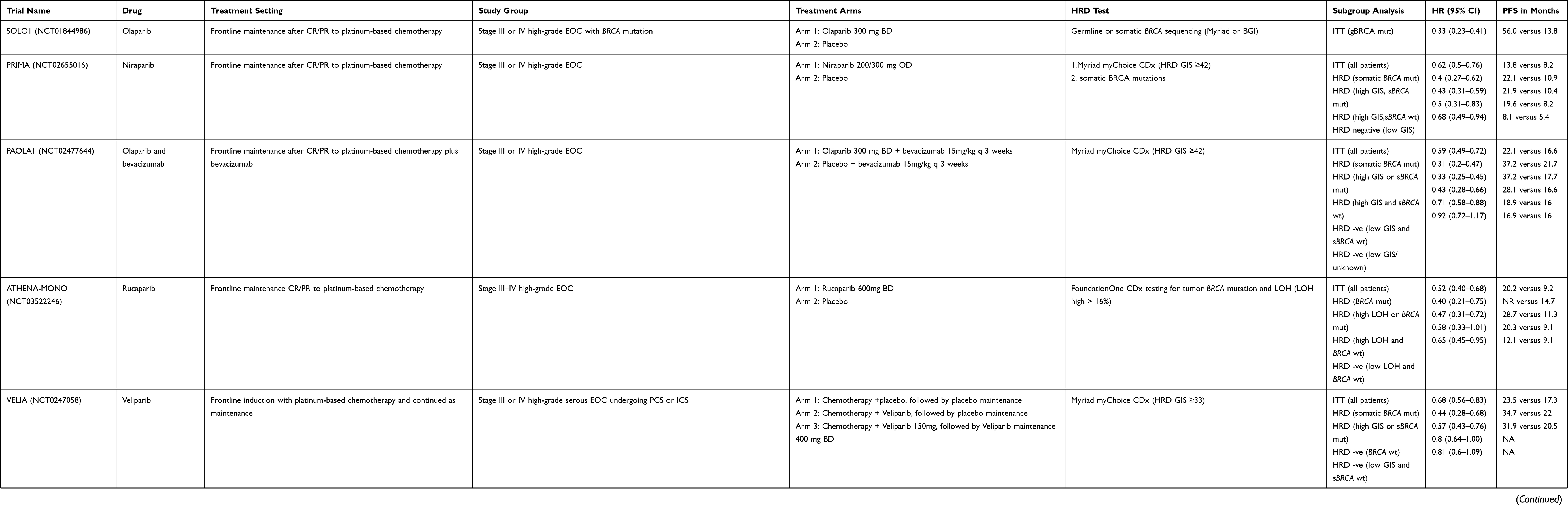 |
Table 1 Pivotal PARP Inhibitor Trials |
Mutational Signatures
Mutational signatures are alternative means of assessing the impact of HRD on the genome by quantifying the type of mutations found and the patterns of nucleotide transitions.58 Each mutational process contains components of DNA damage, repair and replication and generates a characteristic mutational pattern or signature.58 In HGSC, mutational signatures have been associated with response to platinum and overall survival.59,60 Signature 3 is a mutational signature based on single base substitutions and is characterized by a large number of bigger deletions with overlapping microhomology at breakpoint junctions. Signature 3 has been associated with BRCA1/2 mutation and BRCA1 promoter methylation in ovarian, breast, pancreatic and stomach cancers and has been proposed as an HRD biomarker.11,61,62 HRDect in another mutational signature-based assays which may provide better sensitivity and specificity. This assay utilises whole-genome sequencing (WGS), and an algorithm incorporates a weighted aggregate of six HRD-associated signatures predictive of BRCA1/2 deficiency into a single score (rearrangement signature3, base-substitution signature3, rearrangement signature5, HRD index, base-substitution signature8, microhomology-mediated deletions).63 This assay was developed using WGS data from BRCA1/2 mutant and control (BRCA1/2 wild-type) breast cancer samples and validated in further breast, ovarian and pancreatic cancer cohorts. HRDdetect significantly outperforms current genomic scar assays in predicting BRCA1/2 deficiency.63,64 However, the role of HRDdetect and other mutational signature assays in predicting PARPi sensitivity is unknown, and they have not been utilised in prospective clinical trials.
The major limitation of genomic scar and signature assays is that by definition they reflect prior existence of HRD and do not reflect current HRR status, which can be restored by a variety of mechanisms. For example, a BRCA1/2 mutation may have imprinted a genomic HRD scar, but upon gene reversion, the tumor may regain HRR function. Functional assays could overcome this limitation as they provide a dynamic evaluation of the current HRR level.
Functional Assays
Considering the limitations of scar and signature-based assays, measuring RNA, protein expression or a functional assay may provide a better assessment of the current HRD state. Measurement of RAD51 foci formation is one such functional assay. RAD51, a DNA recombinase, is a downstream HR protein that enables high-fidelity dsDNA repair by enabling DNA strand invasion into the sister chromatid, supported by the BRCA1/PALB2/BRCA2 complex. Preclinically, reduced DNA damaged induced nuclear RAD51 foci has been associated with BRCA1/2 mutations in addition to response to PARPi in both ovarian and breast cancer models and in small numbers of patient samples.65,66 Although measuring reduced RAD51 foci as a surrogate for HRD holds promise, translating a real-time RAD51 foci assay into the clinic remains challenging and several hurdles need to be overcome before it is ready for clinical use. This includes determining the optimal source of tissue, mode of evaluation of RAD51 foci and standardisation of definition of RAD51-positive cells.67 Furthermore, this approach will not identify defects in the HR pathway downstream of RAD51 and the RAD51 signal is normally elicited by exogenous DNA damage, further reducing the clinical feasibility of implementing this method as standard of care.11
Currently, there is a lack of evidence to support the clinical validity of functional assays in predicting PARPi response, however these assays hold potential for determining real-time HRD status and should be incorporated into prospective clinical trials as a priority.
HRD Nomenclature
It is noteworthy that the HRD tests that are currently being used in the clinic or that have been evaluated within published randomised clinical trials measure a genotype (deleterious gene mutation, methylation and/or genomic scar assay) that correlates with an HRD phenotype and deficient HRR, but they do not measure HRR directly. For tumors with positive GIS or LOH scores, the term “HRD” is applied universally. However, there is disagreement as to how to label those tumors which test negative for HRD. The terms “HR proficient (HRP)”, “HRD negative” and “HRD test negative” have all been coined.68 Using current assays, we believe the term HRP is best avoided as it implies the tumor can overcome dsDNA breaks, meaning PARPi will not work. It also suggests a categorical HRR deficiency or proficiency, based on a test which we know to be imperfect. There are several examples of observed PARPi response, albeit to a smaller magnitude, in HRD test-negative patient subgroups,4,6,11,12 suggesting that these tests fail to reliable identify all patients with defective HRR. Therefore, we would suggest the preferred term applied to those that test negative for HRD on current assays is either HRD negative or HRD test negative.
HRD as a Predictive Biomarker for PARPi Response
PARP Inhibitor Maintenance in Recurrent Ovarian Cancer
The first randomised Phase 2 trial (Study 19) PARPi maintenance trial, Study 19, randomised 265 patients to receive olaparib or placebo until disease progression or unacceptable toxicity. Patients must have had either a complete response (CR) or partial response (PR) to prior platinum chemotherapy. BRCA1/2 mutation status was not required for trial inclusion but was determined retrospectively.69 Overall, median progression-free survival (PFS) was increased from 4.8 to 8.4 months (HR 0.35 95% CI 0.25–0.49; p<0.001).69 Subsequent analysis by BRCA1/2 status revealed a greater benefit in the BRCA1/2 mutant group (HR 0.18; 95% CI 0.10–0.31), although a significant benefit was still observed in the BRCA1/2 wild-type group (HR 0.54; 95% CI 0.34–0.85).3 SOLO2 the confirmatory randomised phase 3 trial, enrolled patients with germline BRCA1/2 mutant HGOC, following a response to platinum-based chemotherapy to receive olaparib or placebo. There was a significant improvement in PFS with olaparib (19.1 versus 5.5 months, HR: 0.30; 95% CI 0.22–0.41, Table 1).5 A similar benefit for maintenance PARPi in platinum-sensitive, relapsed BRCA1/2 mutated HGOC has been demonstrated with both niraparib (NOVA trial) and rucaparib (ARIEL 3 trial, Table 1).4,6
Both the NOVA and ARIEL3 trials also included germline BRCA1/2 wild-type patients and a benefit was observed for all patients compared to placebo, regardless of BRCA1/2 status (Table 1); NOVA (BRCA1/2 wild-type 9.3 versus 3.9 months HR: 0.45; 95% CI 0.34–0.61) and ARIEL3 (intention to treat population 10.8 versus 5.4 months HR: 0.36 95% CI 0.30–0.45).4,6 Exploratory analyses were performed in patients who were grouped according to HRD status using either the myChoice CDx (NOVA) or Foundation One CDx (ARIEL3) HRD assays.4,6 In both studies, a PFS benefit was observed in all subgroups, regardless of BRCA1/2 or HRD status, but there was an incremental reduction in benefit from BRCA1/2 mutated to HRD/BRCA1/2 wild-type to HRD test negative (Table 1).4,6 However, neither HRD assay was reliably able to recognise a subgroup of patients who had no benefit. The false-negative rate in these trials may be due to the fact that patients were highly selected for platinum sensitivity, which is a strong surrogate for HRD. Based on these data, olaparib, niraparib and rucaparib are now all licensed as maintenance treatment in recurrent HGOC that have responded to platinum-based therapy, regardless of BRCA1/2 status.8,15,70–73 These PARPi have now been adopted as standard of care maintenance therapy in recurrent HGOC. Although a significant improvement in PFS with PARPi maintenance therapy has been demonstrated in patients with recurrent platinum-sensitive disease, this has not consistently translated into an improvement in overall survival (OS). The improvement in OS seen with olaparib in patients with a BRCA1/2 mutant patients within the SOLO2 trial (median OS 51.7 months with olaparib and 38.8 months with placebo (HR 0.74, 95% CI 0.54–1.00) did not achieve statistical significance (p=0.054).74 In the NOVA trial with niraparib there was a numerically superior median OS of 45.9 months niraparib versus 43.2 months with placebo in the gBRCA1/2 cohort, but the HR was 0.93 (95% CI 0.63–1.36). In the non-gBRCA1/2 cohort the median OS was 38.5 months versus 39.1 months with placebo, and the HR was 1.10 (95% CI 0.83–1.46). However, interpretation of the results is difficult as the analysis was confounded by a high rate of crossover and missing follow-up data.75 Cross-over may an important factor to explain why significant benefits in PFS, or PFS2 are not translated into survival benefits, but other factors, such as the development of platinum resistance after PARPi may also be responsible. A worse PFS following platinum rechallenge has been seen patients who received olaparib in the SOLO2 trial.76 There are concerns about drug resistance following PARPi used as monotherapy in place of chemotherapy. In the ARIEL 4 trial, patients with BRCA1/2 mutant relapsed ovarian cancer were randomised to receive rucaparib or standard chemotherapy, with the chemotherapy choice determined by platinum-free interval.77 Overall, there was an increase in PFS with rucaparib (7.4 months in the rucaparib group versus 5.7 months in the chemotherapy group (HR 0.64 [95% CI 0.49–0.84].77 At a planned interim analysis, a detrimental effect in terms of OS was observed for rucaparib compared to the chemotherapy-containing control arm (19.6 months and 27.1 months, respectively, HR 1.550, 95% CI: 1.085,-2.214).78
PARP Inhibitors in First-Line Maintenance Therapy
The majority of patients with recurrent HGOC will ultimately progress and die from their cancer, despite the notable improvements with PARPi maintenance therapy. First-line therapy is the best opportunity for cure and recent studies have investigated whether the early introduction of PARP inhibitors following cytoreductive surgery and platinum-based chemotherapy results in a greater benefit than observed in the recurrent setting. Early efforts focused on BRCA1/2 mutant HGOCs. In the SOLO1 trial, patients with newly diagnosed FIGO stage 3 or 4 HGOC and a BRCA1/2 mutation who had responded to platinum-based chemotherapy were randomised to receive olaparib or placebo.1 Maintenance treatment with olaparib led to an exceptional improvement in outcome with a 70% reduction in the risk of disease progression or death compared to placebo (60 vs 27% HR 0.30, 95% CI 0.23–0.41) with a median PFS of 56.0 months with olaparib versus 13.8 months on placebo (HR 0.33, 95% CI 0.25–0.48), and 48% of olaparib treated patients free from progression at five years, compared to 21% on placebo,1,79 leading to FDA (2018) and EMA (2019) approval in this setting.8,15 The PAOLA-1, PRIMA and VELIA and ATHENA randomised phase 3 trials subsequently investigated the value of maintenance PARPi in BRCA1/2 wild-type patients, in addition to BRCA1/2 mutation populations (Table 1).2,12,13,80 As with SOLO1, the presence of a BRCA1/2 mutation reliably predicted a benefit to PARPi with a similar degree of gain to that seen in the relapsed setting (HR range 0.30–0.44) although the duration of benefit observed was longer.2,12,13 Each of these trials explored the role of HRD as a predictive biomarker to determine PARPi response, with HRD stratification in the PRIMA and ATHENA-MONO trials. A consistent pattern was observed; the largest benefit was seen in the BRCA1/2 cohort, followed by the HRD-positive cohort, with minimal or no benefit observed in the HRD-negative subgroup (Table 1).2,12,13,80 For example, in the PRIMA study which randomised between niraparib with placebo with patients stratified by HRD-score (myChoice CDx) BRCA1/2 wild-type/HRD patients benefited from niraparib with a median PFS of 19.6 months versus 8.2 months (HR; 0.5, 95% CI 0.31–0.83). The trial was not powered to detect benefit in the HRD-negative subgroup although exploratory analyses indicate some benefit, albeit of a lesser magnitude (HR 0.68; 95% CI 0.49–0.94).12 In the PAOLA1 study, olaparib (or placebo) was added to maintenance therapy with the anti-angiogenic drug, bevacizumab. A benefit from olaparib was observed in the BRCA1/2 mutant (PFS 37.2 months versus 21.7 months with placebo, HR 0.31; 95% CI, 0.20–0.47) and BRCA1/2 wild-type/HRD tumors (PFS 28.1 vs 16.6 months, HR 0.43; 95% CI 0.28–0.66) but not in the HRD-negative tumors (16.0 to 16.9 months, HR 0.92; 95% CI 0.72–1.17).13 Based on these results the FDA and EMA approved olaparib and bevacizumab maintenance therapy for HRD HGOC and niraparib maintenance therapy for all HGOCs following first-line platinum-based chemotherapy.15,70,81,82 A similar pattern of benefit was seen in the ATHENA-MONO trial, and in the LOH low group (using FoundationOne CDx assay for HRD) the HR was 0.65 (95% CI 0.45–0.95). However, rucaparib has not yet been licensed for first-line use.80 VELIA was a three-arm trial, the PARPi veliparib was added concurrently with chemotherapy in two arms, continuing as maintenance in one arm, and the third arm received placebo throughout both phases.2 There was no benefit with concurrent veliparib use only and as observed within the other studies, the greatest benefit from maintenance therapy was observed within the BRCA1/2 mutant population. In the BRCA1/2 wild-type tumors a smaller benefit was observed from the addition of veliparib given with chemotherapy and as maintenance therapy in the HRD-positive (HR 0.80; 95% CI 0.64–0.997) and HRD-negative (HR; 0.81; 95% CI 0.6–1.09) groups. Comparisons between trials are difficult, particularly as the VELIA trial used an unvalidated HRD cut-off score compared to the other two first-line studies.2 To date, veliparib has not been submitted for licensing for the treatment of ovarian cancer.
These studies demonstrate that in the first-line setting, maintenance PARPi following chemotherapy leads to an unprecedented increase in PFS, especially within the BRCA1/2 mutant population. Overall survival data are immature, but it is anticipated that the significant PFS gains will translate into an overall survival benefit and even increased rates of cure. This benefit extends to the BRCA1/2 wild-type/HRD patients, although to a lesser degree and highlights the importance of using discriminating HRD assays. The role of PARPi for HRD-negative patients in the first-line setting is less established with differing results obtained between PRIMA and ATHENA-MONO, and PAOLA1/VELIA studies.
HRD as a Prognostic Biomarker
It has long been recognised that BRCA1/2 mutant cancer are associated with better prognosis than BRCA1/2 wild-type cancers.83,84 This extends to HRD, even in the absence of PARPi use.85–87 For example, in the placebo arms of the PRIMA trial, the median PFS was 10.9, 8.2, and 5.4 months, respectively, for the BRCA1/2 mutant, HRD/(BRCA1/2 wild-type) and HRD-negative cohorts.12 In large clinical datasets, using whole-genome data, HRD was significantly associated with longer OS, an effect that persists even after exclusion of BRCA1/2 patients.86
Conclusions
One of the key challenges with HRD testing is to decide on a “gold-standard”. There are advantages and disadvantages with the current genomic, functional, clinical and molecular tests. The genomic scar HRD tests validated to date, have value in predicting the magnitude of benefit from PARPi and can be used to direct treatment selection. However, their use is limited by a failure to consistently predict a group of patients who do not benefit (particularly in the platinum-sensitive recurrent setting), and they fail to address the complex and dynamic nature of the HRD phenotype. As such, there is a need to develop better biomarkers to identify current HR status and this may require composite tests in order to maximise the potential for PARPi in patients with HGOCs.
Disclosure
REM declares consultancies from Merck/Merck Sharp & Dohme, GlaxoSmithKline, AstraZeneca, Ellipses, Shionogi, Clovis Oncology and GI Innovation; Speaker bureau from Roche, GlaxoSmithKline, AstraZeneca and Clovis Oncology; Travel grants from GlaxoSmithKline, AstraZeneca; Clinical trial funding from Merck/Merck Sharp & Dohme.
JAL has received lecture fees from Clovis Oncology, AstraZeneca, GlaxoSmithKline, Eisai, Neopharm; and served on advisory boards for Clovis Oncology, Artios Pharma, AstraZeneca, Merck/Merck Sharp & Dohme, Mersana, Regeneron, VBL Therapeutics, Eisai, Nuvation, Bristol Myers Squibb, and Tesaro/GlaxoSmithKline; personal fees from Immunogen and Pfizer; and received research grants from AstraZeneca and Merck/Merck Sharp & Dohme. The other authors report no conflicts of interest in this work.
References
1. Moore K, Colombo N, Scambia G., et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495–2505. doi:10.1056/NEJMoa1810858
2. Coleman RL, Fleming GF, Brady MF, et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med. 2019;381(25):2403–2415. doi:10.1056/NEJMoa1909707
3. Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–861. doi:10.1016/S1470-2045(14)70228-1
4. Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164. doi:10.1056/NEJMoa1611310
5. Pujade-Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274–1284. doi:10.1016/S1470-2045(17)30469-2
6. Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–1961. doi:10.1016/S0140-6736(17)32440-6
7. EMA. EMA approval of olaparib; 2018. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003726/human_med_001831.jsp&mid=WC0b01ac058001d124.
8. FDA U. FDA approval of olaprib; 2017 [
9. Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. doi:10.1038/nature10166
10. Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–775. doi:10.1158/1078-0432.CCR-13-2287
11. Miller RE, Leary A, Scott CL, et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann Oncol. 2020;31(12):1606–1622. doi:10.1016/j.annonc.2020.08.2102
12. Gonzalez-Martin A, Pothuri B, Vergote I, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391–2402. doi:10.1056/NEJMoa1910962
13. Ray-Coquard I, Pautier P, Pignata S, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381(25):2416–2428. doi:10.1056/NEJMoa1911361
14. FDA U. FDA approves olaparib plus bevacizumab as maintenance treatment for ovarian, fallopian tube, or primary peritoneal cancers; 2020 [
15. EMA. Olaparib EMA approval EMA; 2021. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/lynparza.
16. Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12(9):587–598. doi:10.1038/nrc3342
17. Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7(4):a016600. doi:10.1101/cshperspect.a016600
18. Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355(6330):1152–1158. doi:10.1126/science.aam7344
19. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi:10.1038/nature03443
20. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi:10.1038/nature03445
21. O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60(4):547–560. doi:10.1016/j.molcel.2015.10.040
22. Norquist B, Wurz KA, Pennil CC, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29(22):3008–3015. doi:10.1200/JCO.2010.34.2980
23. Colombo N, Ledermann JA. [email protected] EGCEa. Updated treatment recommendations for newly diagnosed epithelial ovarian carcinoma from the ESMO clinical practice guidelines. Ann Oncol. 2021;32(10):1300–1303. doi:10.1016/j.annonc.2021.07.004
24. Kanchi KL, Johnson KJ, Lu C, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun. 2014;5:3156. doi:10.1038/ncomms4156
25. Dougherty BA, Lai Z, Hodgson DR, et al. Biological and clinical evidence for somatic mutations in BRCA1 and BRCA2 as predictive markers for olaparib response in high-grade serous ovarian cancers in the maintenance setting. Oncotarget. 2017;8(27):43653–43661. doi:10.18632/oncotarget.17613
26. Mirza MR, Avall Lundqvist E, Birrer MJ, et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): a randomised, phase 2, superiority trial. Lancet Oncol. 2019;20(10):1409–1419. doi:10.1016/S1470-2045(19)30515-7
27. Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87. doi:10.1016/S1470-2045(16)30559-9
28. Loveday C, Turnbull C, Ramsay E, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011;43(9):879–882. doi:10.1038/ng.893
29. Meindl A, Hellebrand H, Wiek C, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42(5):410–414. doi:10.1038/ng.569
30. Rafnar T, Gudbjartsson DF, Sulem P, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011;43(11):1104–1107. doi:10.1038/ng.955
31. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–120. doi:10.1038/nrc.2015.21
32. Bajrami I, Frankum JR, Konde A, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74(1):287–297. doi:10.1158/0008-5472.CAN-13-2541
33. McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–8115. doi:10.1158/0008-5472.CAN-06-0140
34. Hodgson DR, Dougherty BA, Lai Z, et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br J Cancer. 2018;119(11):1401–1409. doi:10.1038/s41416-018-0274-8
35. Marshall CH, Sokolova AO, McNatty AL, et al. Differential response to olaparib treatment among men with metastatic castration-resistant prostate cancer harboring BRCA1 or BRCA2 versus ATM mutations. Eur Urol. 2019;76(4):452–458. doi:10.1016/j.eururo.2019.02.002
36. Eccles BK, Copson E, Maishman T, Abraham JE, Eccles DM. Understanding of BRCA VUS genetic results by breast cancer specialists. BMC Cancer. 2015;15:936. doi:10.1186/s12885-015-1934-1
37. Caswell-Jin JL, Gupta T, Hall E, et al. Racial/ethnic differences in multiple-gene sequencing results for hereditary cancer risk. Genet Med. 2018;20(2):234–239. doi:10.1038/gim.2017.96
38. Slavin TP, Van Tongeren LR, Behrendt CE, et al. Prospective study of cancer genetic variants: variation in rate of reclassification by ancestry. J Natl Cancer Inst. 2018;110(10):1059–1066. doi:10.1093/jnci/djy027
39. Bernards SS, Pennington KP, Harrell MI, et al. Clinical characteristics and outcomes of patients with BRCA1 or RAD51C methylated versus mutated ovarian carcinoma. Gynecol Oncol. 2018;148(2):281–285. doi:10.1016/j.ygyno.2017.12.004
40. Cunningham JM, Cicek M, Larson N, et al. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2, and RAD51C status. Sci Rep. 2014;4:4026. doi:10.1038/srep04026
41. Lheureux S, Lai Z, Dougherty BA, et al. Long-term responders on olaparib maintenance in high-grade serous ovarian cancer: clinical and molecular characterization. Clin Cancer Res. 2017;23(15):4086–4094. doi:10.1158/1078-0432.CCR-16-2615
42. Swisher EM, Gonzalez RM, Taniguchi T, et al. Methylation and protein expression of DNA repair genes: association with chemotherapy exposure and survival in sporadic ovarian and peritoneal carcinomas. Mol Cancer. 2009;8:48.
43. Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27(8):1449–1455. doi:10.1093/annonc/mdw142
44. Abkevich V, Timms KM, Hennessy BT, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. 2012;107(10):1776–1782. doi:10.1038/bjc.2012.451
45. Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5(11):1137–1154. doi:10.1158/2159-8290.CD-15-0714
46. Esteller M, Silva JM, Dominguez G, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92(7):564–569. doi:10.1093/jnci/92.7.564
47. Sun T, Ruscito I, Dimitrova D, et al. Genetic versus epigenetic BRCA1 silencing pathways: clinical effects in primary ovarian cancer patients: a study of the tumor bank ovarian cancer consortium. Int J Gynecol Cancer. 2017;27(8):1658–1665. doi:10.1097/IGC.0000000000001071
48. Zhu X, Zhao L, Lang J. The BRCA1 methylation and PD-L1 expression in sporadic ovarian cancer. Int J Gynecol Cancer. 2018;28(8):1514–1519. doi:10.1097/IGC.0000000000001334
49. Kondrashova O, Nguyen M, Shield-Artin K, et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017;7(9):984–998. doi:10.1158/2159-8290.CD-17-0419
50. Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521(7553):489–494. doi:10.1038/nature14410
51. Prieske K, Prieske S, Joosse SA, et al. Loss of BRCA1 promoter hypermethylation in recurrent high-grade ovarian cancer. Oncotarget. 2017;8(47):83063–83074. doi:10.18632/oncotarget.20945
52. Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–1120. doi:10.1038/nature06633
53. Birkbak NJ, Wang ZC, Kim JY, et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012;2(4):366–375. doi:10.1158/2159-8290.CD-11-0206
54. Popova T, Manie E, Rieunier G, et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72(21):5454–5462. doi:10.1158/0008-5472.CAN-12-1470
55. Timms KM, Abkevich V, Hughes E, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014;16(6):475. doi:10.1186/s13058-014-0475-x
56. Timms KM, Mills GB, Perry M, Gutin A, Lanchbury J, Brown R. Comparison of genomic instability test scores used for predicting PARP activity in ovarian cancer. J Clin Oncol. 2020;38(15_suppl):1586. doi:10.1200/JCO.2020.38.15_suppl.1586
57. Loverix L, Busschaert P, Vanderstichele A, et al. Predictive value of the Leuven HRD test compared with Myriad myChoice PLUS 468 ovarian cancer samples from the PAOLA-1/ENGOT-ov25 trial. Gynecol Oncol. 2022;166:S51–S52.
58. Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi:10.1038/nature12477
59. Hillman RT, Chisholm GB, Lu KH, Futreal PA. Genomic rearrangement signatures and clinical outcomes in high-grade serous ovarian cancer. J Natl Cancer Inst. 2018;110(3):265–272. doi:10.1093/jnci/djx176
60. Macintyre G, Goranova TE, De Silva D, et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat Genet. 2018;50(9):1262–1270. doi:10.1038/s41588-018-0179-8
61. Alexandrov LB, Kim J, Haradhvala NJ, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94–101. doi:10.1038/s41586-020-1943-3
62. Gulhan DC, Lee JJ, Melloni GEM, Cortes-Ciriano I, Park PJ. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet. 2019;51(5):912–919. doi:10.1038/s41588-019-0390-2
63. Davies H, Glodzik D, Morganella S, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23(4):517–525. doi:10.1038/nm.4292
64. Telli ML, Timms KM, Reid J, et al. Homologous Recombination Deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22(15):3764–3773. doi:10.1158/1078-0432.CCR-15-2477
65. Mukhopadhyay A, Plummer ER, Elattar A, et al. Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: sensitivity to PARP inhibitors, platinum, and survival. Cancer Res. 2012;72(22):5675–5682. doi:10.1158/0008-5472.CAN-12-0324
66. Hill SJ, Decker B, Roberts EA, et al. Prediction of DNA repair inhibitor response in short-term patient-derived ovarian cancer organoids. Cancer Discov. 2018;8(11):1404–1421. doi:10.1158/2159-8290.CD-18-0474
67. Fuh K, Mullen M, Blachut B, et al. Homologous recombination deficiency real-time clinical assays, ready or not? Gynecol Oncol. 2020;159(3):877–886. doi:10.1016/j.ygyno.2020.08.035
68. Stewart MD, Merino Vega D, Arend RC, et al. Homologous recombination deficiency: concepts, definitions, and assays. Oncologist. 2022;27(3):167–174. doi:10.1093/oncolo/oyab053
69. Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–1392. doi:10.1056/NEJMoa1105535
70. EMA. Niraparib EMA approval EMA; 2021. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/zejula.
71. EMA. Rucaparib EMA approval EMA; 2021. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/rubraca.
72. FDA U. FDA approval of niraparib; 2017. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/niraparib-zejula.
73. FDA U. FA approval of rucaparib as maintenance therapy; 2018. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-rucaparib-maintenance-treatment-recurrent-ovarian-fallopian-tube-or-primary-peritoneal.
74. Poveda A, Floquet A, Ledermann JA, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021;22(5):620–631. doi:10.1016/S1470-2045(21)00073-5
75. Matulonis U, Herrstedt J, Oza A, Mahner S. Long-term safety and secondary efficacy endpoints in the ENGOT-OV16/NOVA Phase III trial of niraparib in recurrent ovarian cancer. Gynecol Oncol. 2021;162:S24–S5. doi:10.1016/S0090-8258(21)00693-4
76. Frenel JS, Kim JW, Aryal N, et al. Efficacy of subsequent chemotherapy for patients with BRCA1/2-mutated recurrent epithelial ovarian cancer progressing on olaparib versus placebo maintenance: post-hoc analyses of the SOLO2/ENGOT Ov-21 trial. Ann Oncol. 2022. doi:10.1016/j.annonc.2022.06.011
77. Kristeleit R, Oza AM. Rucaparib for recurrent ovarian cancer with BRCA1 and BRCA2 mutations - Authors’ reply. Lancet Oncol. 2022;23(7):e315. doi:10.1016/S1470-2045(22)00343-6
78. EMA. Rucaparib (Rubraca®): interim data from Study CO-338-043 (ARIEL4) show a decrease in overall survival compared to standard of care: EMA; 2022 [
79. Banerjee S, Moore KN, Colombo N, et al. Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021;22(12):1721–1731. doi:10.1016/S1470-2045(21)00531-3
80. Monk BJ, Parkinson C, Lim MC, et al. A randomized, Phase III trial to evaluate rucaparib monotherapy as maintenance treatment in patients with newly diagnosed ovarian cancer (ATHENA-MONO/GOG-3020/ENGOT-ov45). J Clin Oncol;2022. JCO2201003. doi:10.1200/JCO.22.01003
81. FDA U. FDA approves niraparib for first-line maintenance of advanced ovarian cancer; 2020 [
82. FDA U. olaparib plus bevacizumab as maintenance treatment for ovarian, fallopian tube, or primary peritoneal cancers; 2021. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-plus-bevacizumab-maintenance-treatment-ovarian-fallopian-tube-or-primary.
83. Ben David Y, Chetrit A, Hirsh-Yechezkel G, et al. Effect of BRCA mutations on the length of survival in epithelial ovarian tumors. J Clin Oncol. 2002;20(2):463–466. doi:10.1200/JCO.2002.20.2.463
84. Westin SN, Coleman RL, Fellman BM, et al. EFFORT: eFFicacy Of adavosertib in parp ResisTance: a randomized two-arm non-comparative Phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer. J Clin Oncol. 2021;39(15_suppl):5505. doi:10.1200/JCO.2021.39.15_suppl.5505
85. Morse CB, Toukatly MN, Kilgore MR, et al. Tumor infiltrating lymphocytes and homologous recombination deficiency are independently associated with improved survival in ovarian carcinoma. Gynecol Oncol. 2019;153(2):217–222. doi:10.1016/j.ygyno.2019.02.011
86. Ewing A, Meynert A, Churchman M, et al. Structural variants at the BRCA1/2 loci are a common source of homologous repair deficiency in high-grade serous ovarian carcinoma. Clin Cancer Res. 2021;27(11):3201–3214. doi:10.1158/1078-0432.CCR-20-4068
87. How JA, Jazaeri AA, Fellman B, et al. Modification of homologous recombination deficiency score threshold and association with long-term survival in epithelial ovarian cancer. Cancers. 2021;13(5). doi:10.3390/cancers13050946
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.