")
Back to Journals » Drug Design, Development and Therapy » Volume 15
OSU-03012 Disrupts Akt Signaling and Prevents Endometrial Carcinoma Progression in vitro and in vivo
Authors Ding L, Ren C, Yang L, Wu Z, Li F, Jiang D, Zhu Y, Lu J
Received 3 February 2021
Accepted for publication 2 April 2021
Published 30 April 2021 Volume 2021:15 Pages 1797—1810
DOI https://doi.org/10.2147/DDDT.S304128
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Leilei Ding, Chenchen Ren, Li Yang, Zimeng Wu, Feiyan Li, Dongyuan Jiang, Yuanhang Zhu, Jie Lu
Department of Obstetrics and Gynecology, The Third Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, People’s Republic of China
Correspondence: Chenchen Ren; Li Yang
The Third Affiliated Hospital of Zhengzhou University, No. 7, Front Kangfu Street, Zhengzhou, Henan Province, People’s Republic of China
Tel +8613939057999
; +8618697336662
Email [email protected]; [email protected]
Purpose: OSU-03012 is a celecoxib derivative lacking cyclooxygenase-2 inhibitory activity and a potent PDK1 inhibitor which has been shown to inhibit tumor growth in various ways. However, the role of OSU-03012 in endometrial carcinoma (EC) in which the PI3K/Akt signaling pathway highly activated has not been studied. Here, we determined the potency of OSU-03012 in suppressing EC progression in vitro and in vivo, and studied the underlined mechanisms.
Methods: The human EC Ishikawa and HEC-1A cells were used as the in vitro models. CCK8 assay and flow cytometry were conducted to evaluate cell proliferation, cell cycle progression, and apoptosis. The metastatic ability was evaluated using the transwell migration assay. The Ishikawa xenograft tumor model was used to study the inhibitory effects of OSU-03012 on EC growth in vivo. Western blot analysis was performed to evaluate expressions of the cell cycle and apoptosis associated proteins.
Results: OSU-03012 could inhibit the progression of EC both in vitro and in vivo by disrupting Akt signaling. It reduced the metastatic ability of EC, led to G2/M cell cycle arrest and induced apoptosis via the mitochondrial apoptosis pathway.
Conclusion: Our data indicated that OSU-03012 could inhibit the progression of EC in vitro and in vivo. It can potentially be used as the targeted drug for the treatment of EC by inhibiting Akt signaling.
Keywords: OSU-03012, endometrial carcinoma, Akt signaling, the mitochondrial apoptosis pathway
Introduction
Endometrial carcinoma (EC) is an epithelial malignancy that occurs in the endometrium and most commonly occurs in perimenopausal and postmenopausal women. It is the most common gynecologic malignancy in the US, with its incidence and disease-related mortality continuing to increase.1 Although early EC has a positive 5-year relative survival rate (96%), the survival rate for patients with distant metastasis is only 18%.2 Recent progress in chemotherapy and radiation therapy has modestly improved patient prognosis. However, treatment options are limited for metastatic, recurrent patients with poor prognostic factors. Although platinum-based regimens are often used for adjunctive therapy, patients do not respond well to them and have a median survival of less than 1 year and a median progression-free survival (PFS) of 4 months.3 Therefore, innovative and alternative drugs or combination therapy, involving new targeted drugs and standard regimens need to be developed.
Activation of the PI3K/Akt signaling pathway occurs in various malignant tumors, such as breast cancer,4 esophageal cancer,5 endometrial cancer,6 ovarian cancer,7 and colorectal cancer,8 etc. Activated Akt subsequently activates downstream molecules, including mTOR and GSK3β, which mediate important biological processes such as cell proliferation, cell cycle, energy metabolism, migration, angiogenesis, and drug-induced apoptosis.9,10 In solid tumors, EC has the most frequent PI3K/Akt/mTOR pathway mutation rate, with specific changes in 92% of type I and 60% of type II ECs.11 In cancer genome screening, the PI3K/Akt pathway plays a crucial role in advanced EC.12 The abnormal activation of PI3K/Akt pathway is more frequent in type I, mainly with PTEN or PI3KCA related gene mutations.13 The roles of the PI3K/Akt pathway in the pathogenesis of EC is revealed in the proteomic analysis of human endometrial tissues.14 Most studies have shown that hyperactivation of Akt indicates highly aggressive behavior and poor prognosis of EC.15 The value of the biological treatment in advanced EC (type I, II carcinoma) targeting Akt deserves further discussion. These data indicate that the PI3K/Akt signaling pathway plays an essential role in the genesis and development of EC. The study of the regulatory mechanism underlying Akt activation may provide a novel strategy for EC therapy. This approach has led to the flurry of research on specific small-molecule drugs targeting the key components of the PI3K/Akt pathway.6,16
The promising agent OSU-03012 is the third generation celecoxib derivative that lacks COX-2 inhibitory activity; without the associated risks of potential gastrointestinal damage; it is shown to be a potent inhibitor of PDK1 and Akt phosphorylation.17 OSU-03012 inhibits growth of multiple types of cancer cells, including primary chronic lymphocytic leukemia cells,18 pancreatic carcinoma cells,19 esophageal carcinoma cells20 and breast cancer cells.21 Also, OSU-03012 can induce autophagy and activation of reactive oxygen species (ROS).22 The mechanism of action may occur via the inhibition of the PDK1/Akt signaling pathway. As part of the RAPID program, OSU-03012 was administered orally at 200 mg/kg for one month, with no obvious toxicity (Chen et al unpublished data).23 The safety and tolerability of OSU-03012 are under study in Phase I clinical trials involving adults with recurrent or advanced solid tumors or lymphomas.24
As a novel small-molecule inhibitor of the PI3K/Akt signaling pathway, the role of OSU-03012 in EC in which the PI3K/Akt signaling pathway is highly activated has not been studied. Thus, we further investigated the potential of OSU-03012 for the treatment of EC. The compound was evaluated in two EC cell lines, Ishikawa and Hec-1A cell lines. Both cell lines were well characterized for their considerably high levels of activated Akt. In this study, OSU-03012 inhibited the phosphorylation of Akt and its downstream substrate, GSK. OSU-03012 impaired EC cell viability and initiated the apoptotic pathway, inducing nearly 90% cell apoptosis within 48h. In addition, OSU-03012 inhibited tumor growth in the Ishikawa xenograft study.
Materials and Methods
Cell Culture
The human EC cell lines Hec-1A (NTCC, Beijing, China), KLE (Procell, Wuhan, China) were obtained commercially, and Ishikawa was provided by the research group of Professor Ya Xie from the First Affiliated Hospital of Zhengzhou University. The use of the Ishikawa cell line had been approved by the Ethics Committee of the Third Affiliated Hospital of Zhengzhou University (Protocol # 2021-WZ-002). They were cultured in McCoy’5A medium (Procell, Wuhan, China) and DMEM medium (Solarbio, Beijing, China), respectively, which contained 10% serum, 100U/mL penicillin, and 100mg/mL streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
Animal Model and Drug Administration
All studies involving mice were approved by the Ethics Committee of Lab Animal Center of Zhengzhou University (Protocol # ZZU-LAC20201013[06]) and followed the 3R principle for the welfare and treatment of the laboratory animals. Sexually mature female BALB/c-nu mice (aged 35 days), (Spelfer Biotechnology Co. LTD, Beijing) were put in the Lab Animal Center of Zhengzhou University and maintained in a specific sterile environment (12-hour light–dark cycle, constant ambient temperature (21°C±1°C) and humidity (50%±7%)).
Animals were divided into two groups and fed with sterile bedding, water, and food (n =6 mice; before surgery). After one week of adaptation, 10 million Ishikawa cells suspended in 100μL sterile DMEM were injected subcutaneously to form xenografts. Tumor was detected by palpation and tumor volume and weight were measured by a caliper three times per week. Their health was monitored daily. When the tumor volume reached an average of 80 mm3, the mice were selected and randomly divided into two groups with five mice in each group. In the animal studies, OSU-03012 was dissolved in-vehicle solution (0.5% methylcellulose/0.1% Tween 80) and administered orally to EC bearing mice. Two treatment groups were randomly assigned to receive OSU-03012 (100 mg/kg) or vehicle solution (0.5% methylcellulose/0.1% twin-80 in sterile water) gavage daily for three weeks. We used the lowest dosage of OSU-03012 previously reported to successfully inhibit cell proliferation in vivo. Then, executed mice and collected tumors at autopsy. Part of each animal’s tumor was immobilized in paraformaldehyde, while the other part was snap-frozen in liquid nitrogen.
IC50 and Viability Assay Determination by CCK8 Assay
Ishikawa and HEC-1A cells at logarithmic growth stage were selected. The target cells were divided into DMSO group and drug group according to different treatment measures. The concentration of OSU-03012 (Selleck Chemicals, Houston, Texas, USA) was 0μL, 2.5μL, 5μL, 7.5μL, 10μL and 15μL, respectively. It was dissolved in dimethyl sulfoxide (DMSO, Solarbio, Beijing, China), and dilutions were made in McCoy’5A or DMEM to the desired in vitro concentrations. The maximum concentration of DMSO does not exceed 0.1% and will not influence cell growth. The single cell suspension was placed into 96-well plates and incubated in the incubator for 12 hours, 24 hours, 36 hours and 48 hours respectively. In the dark state, 10μL CCK-8 solution (dojindo, Shanghai, China) was added to each well, and then the plates were placed in the incubator at 37°C for 1 hour. The absorbance of each cell at 450nm was measured with a microplate reader (Thermo Fisher Scientific Inc.). The IC50 value and proliferation rate of cells were calculated by SPSS software.
Apoptosis Detection by Flow Cytometry
In order to investigate whether the inhibitory effect of OSU-03012 on cell proliferation is mediated by apoptosis, apoptosis was detected by The YF488A-AnnexinV and PI Cell Apoptosis Detection kit (US EVERBRIGHT INC, Suzhou, China) and analyzed by flow cytometry (BD FACS Canto II). Hec-1A and Ishikawa cells were seeded in 6-well plates and harvested after treatment with OSU-03012 at different concentrations for 48 hours. Then, the cells were labelled with Annexin V-PI stain, and 10,000 randomly selected cells were analyzed by flow cytometry.
Cell Cycle Detection by Flow Cytometry
Hec-1A and Ishikawa cells were seeded in 6-well plates and treated with different concentrations of OSU-03012. After 48 h, cells were collected and fixed in ice-cold 70% ethanol overnight at 4°C. Then, cells were washed with PBS and resuspend in loading buffer containing 50μL PI, 450μL RNase A (KeyGEN BioTECH, Nanjing, China) and stained for 30 min in the dark, at room temperature. We used a flow cytometer (BD FACS Canto II) and the ModFIT software to analyze the cells and calculate the results.
Transwell Assay
Cells were seeded in transwell inserts (Corning Life Sciences) which contained 100μL serum-free medium at a concentration of 5 × 10^4 cells per insert. The transwell inserts were put in a 24-well plate containing 20% serum complete medium. After 24 hours’ incubation, removed the non-migrated cells, fixed the migrated cells with 4% paraformaldehyde and stained them with crystal violet (0.1%). The cells migrated through the chamber were observed by microscope (×200 magnification).
Protein Extraction and Western Blot Analysis
To detect the release of cytochrome C, we separated the mitochondria and cytoplasm as described previously. Hec-1A and Ishikawa cell lysates and mouse EC tissue lysates (specimens stored at −80°C) were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to PVDF membrane, and blocked with 5% BSA prior to incubation with rabbit anti-human cytochrome-C,GSK3β,p-GSK3β,Akt,Bax,Bcl-2,caspase-3,P21 (1:5000 dilution; Proteintech, Wuhan,China) and cleaved caspase-3 (1:1000 dilution; Cell Signaling Technology,Danvers, MA, USA), rabbit anti-human caspase-9 (1:1000 dilution; Wanleibio,Shenyang,China), mouse anti-human p-Akt (1: 5000 dilution; Proteintech,Wuhan,China).The HRP-conjugated Affinipure Goat Anti-Rabbit IgG (1:5000 dilution; Proteintech) was used as secondary antibody.β-Actin (1:5000 dilution; Proteintech,Wuhan,China) and COX IV (1: 500 dilution; Cell Signaling Technology,Danvers, MA, USA) were used as reference proteins. Protein bands were detected using an enhanced chemiluminescence reagent (NCM Biotech, Suzhou, China).
Immunohistochemistry (IHC)
Immunohistochemical staining of p-Akt (1:1000,Proteintech,Wuhan,China) was performed on paraffin-embedded mice EC tissue sections. Briefly, serial 5µm thick tissue sections were subjected to standard immunohistochemical procedures, including dewaxing, hydration, antigen repair, and blocking endogenous peroxidase. After incubated with primary antibodies over night at 4°C, the slides were treated with rabbit anti-mouse IgG. Finally, the immunohistochemical sections were examined microscopically and interpreted by a qualified pathologist.
Data Analysis and Statistics
We repeated all the experiments three times unless otherwise stated. GraphPad Prism 7 software (GraphPad Software Inc., La Jolla, CA, USA) was used for statistical analyses. The results were expressed as mean ± standard deviation. We used One-way ANOVA to compare the mean among three or more groups and t-test to compare the mean between the two groups. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Results
Inhibitors of PDK1 Disrupts Akt Signaling in EC Cells
The molecular structure of OSU-03012 is presented in Figure 1A. The phosphorylated Akt expression levels of these cell lines were then measured. Proteins were extracted from EC cells, and the phosphorylated Akt levels were assessed by Western blot (Figure 1B). Except for the KLE cell line, both Ishikawa and Hec-1A cell lines expressed high levels of p-Akt. No difference in total Akt levels between the two cells was indicated and thus could be used to evaluate the effect of the PDK1 inhibitor OSU-03012 on the Akt pathway.
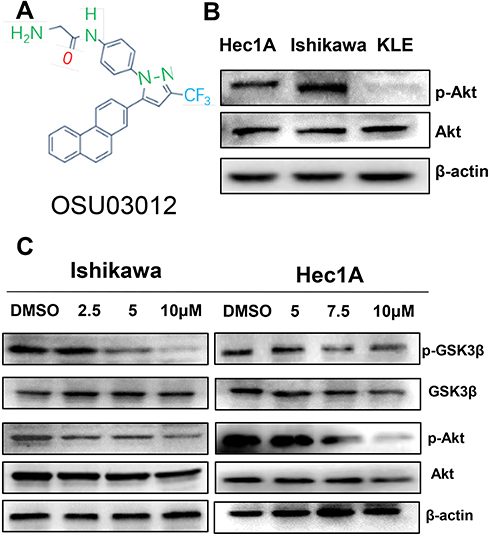 |
Figure 1 OSU-03012 disrupts Akt signaling. (A) The chemical structure of OSU-03012. (B) Examination of phosphorylated Akt (p-Akt) in EC cell lines. (C) Western blot analysis of proteins associated with Akt signaling expression in EC cells treated with OSU-03012 for 48 h. β-actin was used as an internal reference. |
To detect the potential inhibitory effects of the PDK1 inhibitor OSU-03012 on Akt pathway, Hec-1A and Ishikawa cells were treated with either DMSO or OSU-03012 for 48h. Treatment with OSU-03012 induced a decrease in the Akt phosphorylation in both cell lines (Figure 1C and Supplementary Figure S1), and the downstream target GSK3β was subsequently reduced (Figure 1C). These results indicated that Akt signaling could be disrupted by OSU-03012.
OSU-03012 Impairs EC Cells Viability in vitro
OSU-03012 could inhibit the proliferation of EC cells in the time and dose dependent manner (Figure 2E and F). That is, the inhibitory effect of OSU-03012 was gradually enhanced with increases in time and drug concentration. The potency (IC50 value) of the treatment for 48h was used as an indicator to evaluate the OSU-03012 sensitivities of two cell lines. The Ishikawa cell line, with an OSU-03012 potency of 5μm was more sensitive than the Hec-1A cell line with a potency of 7.5μm. The mean IC50 of OSU-03012 on the viability of the Hec-1A cell line at 12h time point was 13.91mmol/L, which changed to 13.25, 8.996 and 7.351 mmol/L at the 24, 36 and 48h time point, respectively. The mean IC50 of OSU-03012 on Ishikawa cell viability at the 12h time point was 7.677mmol/L, which changed to 8.527, 8.484 and 4.938 mmol/L at the 24, 36 and 48h time point, respectively (Figure 2A–D). We conclude that Ishikawa is more sensitive to OSU-03012, compared with Hec-1A.
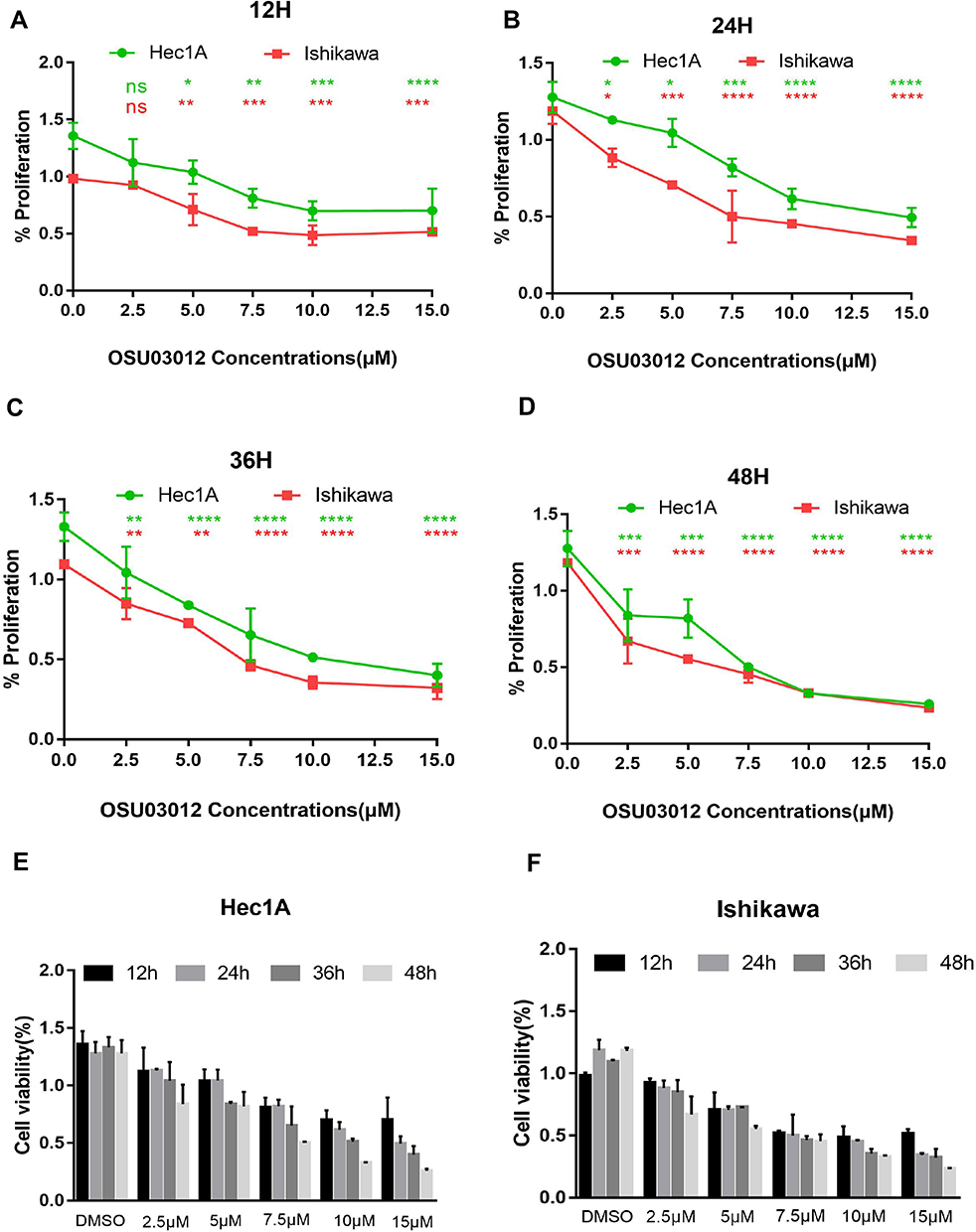 |
Figure 2 The impacts of OSU-03012 on EC cells proliferation. (A–D) CCK8 assay for cell viability of EC cells treated with OSU-03012 at the specified concentrations for 12 h (A), 24h (B), 36h (C), and 72 h (D). (E and F) OSU-03012 impairs EC cells viability in a time and dose-dependent manner. Data was expressed as mean ± SD. t-test was used for the statistical analysis versus the control group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
OSU-03012 Induces Apoptosis in Human EC Cells
We then evaluated whether OSU-03012 induces the apoptosis of EC cells. At the cellular level, flow cytometric analysis of Annexin V/propidium iodide (PI) stained cells demonstrated that OSU-03012 could induce the apoptosis of EC cells (Figure 3A and B). Treatment with OSU-03012 at different concentrations for 48h caused the apoptotic index of the Hec-1A cells to increase from 5.9% to 96.1% and that of the Ishikawa cells to increase from 6.8% to 99.8% (Figure 3C and D). On the basis of the apoptosis data, the IC50s of the Ishikawa and Hec-1A cell lines at the 48h time point was verified to be about 5 and 7.5μM, respectively. Morphological changes in apoptotic characteristics, such as contraction, roundness and floating, were also induced, which became more noticeable over time (Figure 3E and F).
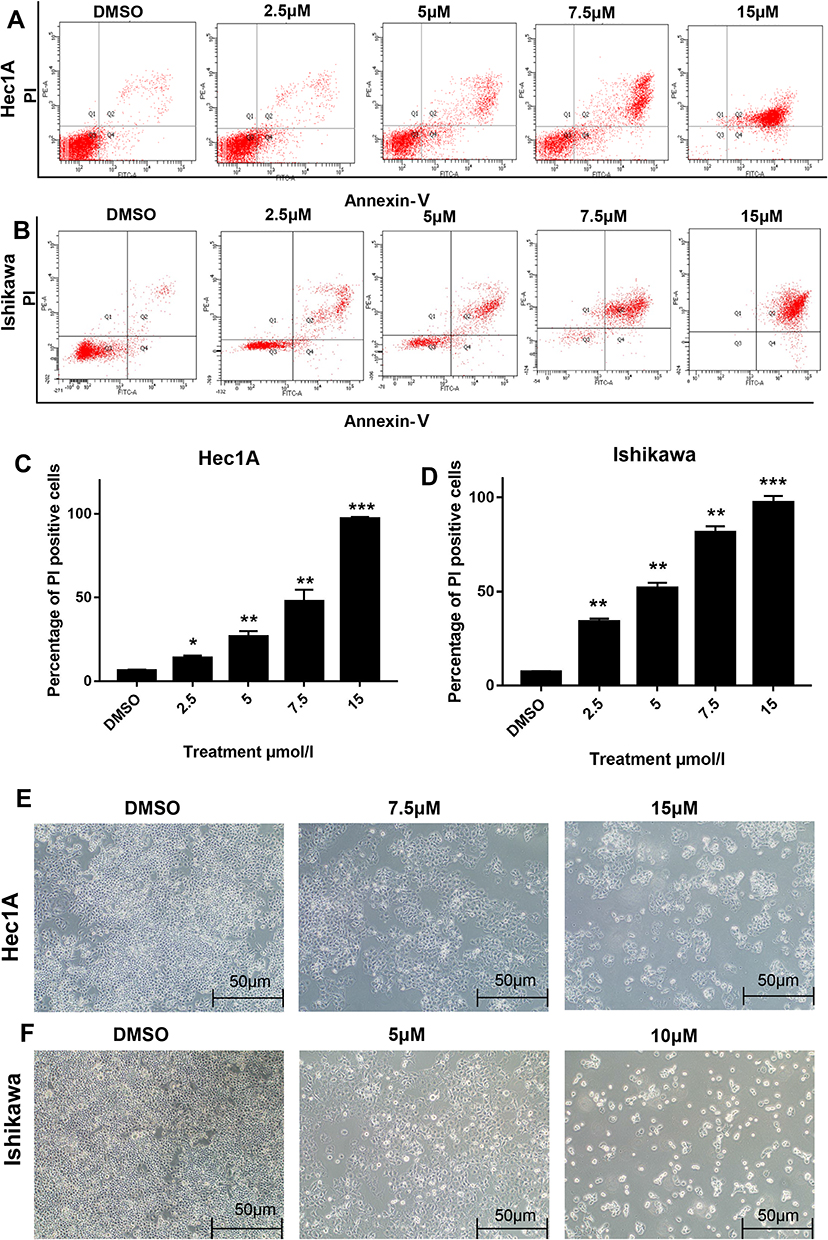 |
Figure 3 OSU-03012 induces apoptosis in human EC cells. (A–D) Flow cytometry analysis of the cell apoptosis in EC cells treated with OSU-03012 for 48h.(A and B) Representative results of apoptosis of EC cells.(C and D) Percentage of PI positive EC cells induced by OSU-03012. (E and F) The cell morphology of apoptosis induced by OSU-03012 in EC cells was observed by the microscopy (100×). Data was expressed as mean ± SD. t-test was used for the statistical analysis versus the control group. *p < 0.05, **p < 0.01, ***p < 0.001. |
OSU-03012 Induces the G2/M Cell Cycle Arrest and Inhibits Human EC Cells Migration
To explore the mechanism underlying the anti-proliferative activity of OSU-03012 in EC cells, we detected the distribution of cell cycle by flow cytometry. The results showed that OSU-03012 resulted in cell cycle arrest at the G2/M phase in both cell lines (Figure 4A–D). The expression of cell cycle-related protein was analyzed by Western blot, which exerted OSU-03012 effects that are in agreement with the inhibition of proliferation. As shown in Figure 4E, OSU-03012 upregulated p21, which is a widely known cyclin-dependent kinase (CDK) inhibitor that leads to cell cycle arrest at G2/M phase by suppressing the activities of various cyclin-CDK complexes required to enter the M phase.25
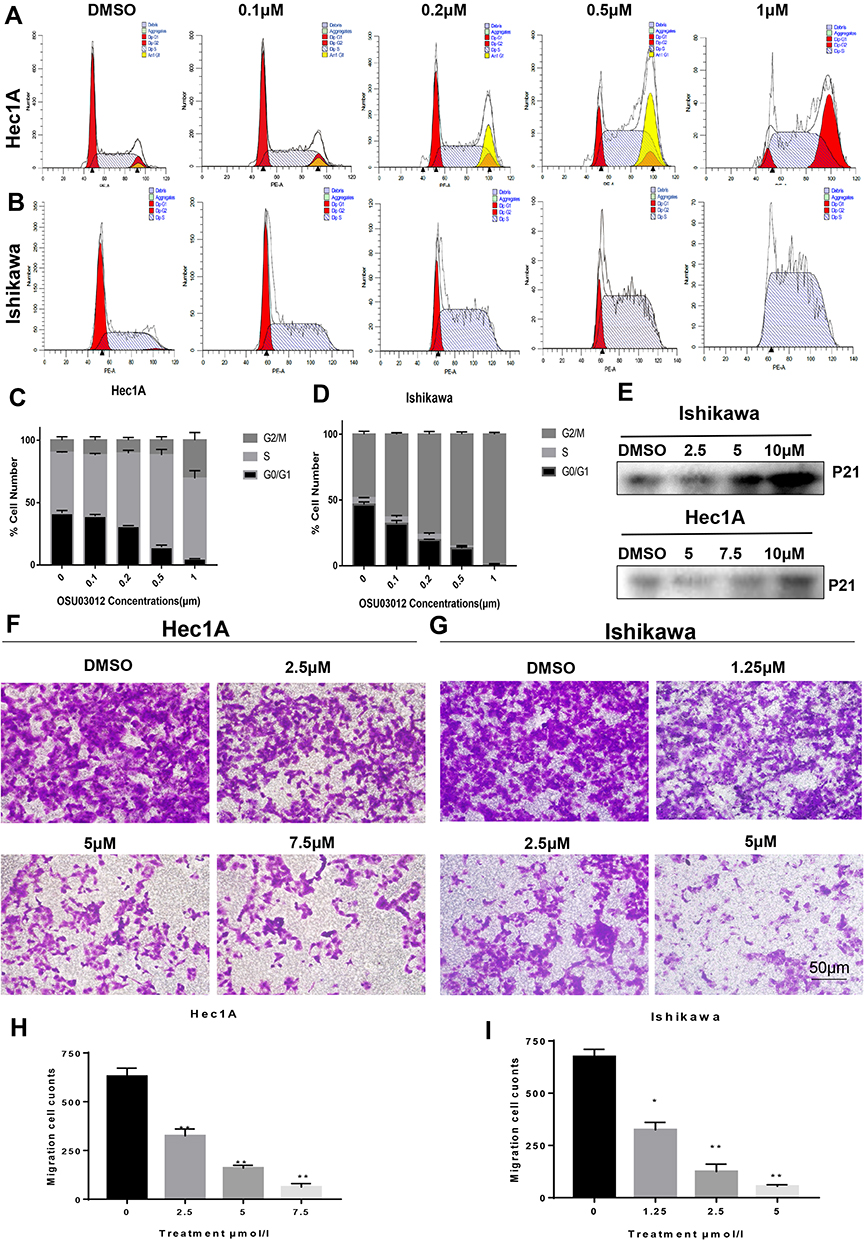 |
Figure 4 OSU-03012 can induce G2/M cell cycle arrest and inhibit the migration ability in EC cells. (A–D) Flow cytometry analysis of the cell cycle distribution in EC cells treated with OSU-03012 for 48h. (A and B) Representative images of flow cytometry result. (C and D) The proportion of cells at different cell cycle phases are showed. (E) Western blot analysis of proteins associated with cell cycle expression in EC cells treated with OSU-03012 for 48 h. The internal reference is the same as Figure 1C. (F and G) Representative images of migrated EC cells treated with OSU-03012 (0–7.5μM) in transwell assay. (200×). (H and I) The histogram of the metastasis rate of EC cells. Data was expressed as mean ± SD. t-test was used for the statistical analysis versus the control group. *p < 0.05, **p < 0.01. |
Metastasis is a hallmark of tumors and is an important standard for evaluating new anticancer drugs. The transwell assay was used to evaluate the effect of OSU-03012 on the metastasis of human EC cell lines. The results showed that OSU-03012 inhibited the migration of the Ishikawa and Hec-1A cells at significant concentrations of 5 and 7.5μM, respectively (Figure 4F–I).
Mitochondrial Apoptosis Mediates the EC Cell-Death Induced by OSU-03012
The mitochondrial apoptotic pathway was closely related to the regulation of Bcl-2 family proteins, mainly composed of anti-apoptotic proteins (eg, Bcl-2) and pro-apoptotic proteins (eg, Bax). Bcl-2 proteins could increase mitochondrial permeability, induce the release of mitochondrial cytochrome C into the cell cytoplasm, and thus activate downstream effector protein caspases to induce cell apoptosis.26 A change in Bax/Bcl-2 ratio triggers the release of cytochrome C from mitochondria to cytoplasm.27 Figure 5A and C showed the downregulation of Bcl-2, upregulation of Bax and the increase of the Bax/Bcl-2 ratio due to the drug treatment. Then, the subcellular fractionation assays showed that treatment of both Hec-1A and Ishikawa cells with OSU-03012 resulted in the mitochondrial release of cytochrome C into the cytosol (Figure 5B). The release of cytochrome c was determined as the mechanism underlying mitochondria-mediated apoptosis.28 The Western blot analysis showed a remarkable dose-dependent caspase-3 activation and caspase-9 cleavage at the molecular level (Figure 5A and Supplementary Figure S1). In summary, these results demonstrate that the cell death induced by OSU-03012 is mediated by a mitochondrial pathway involving caspase-9 activation.
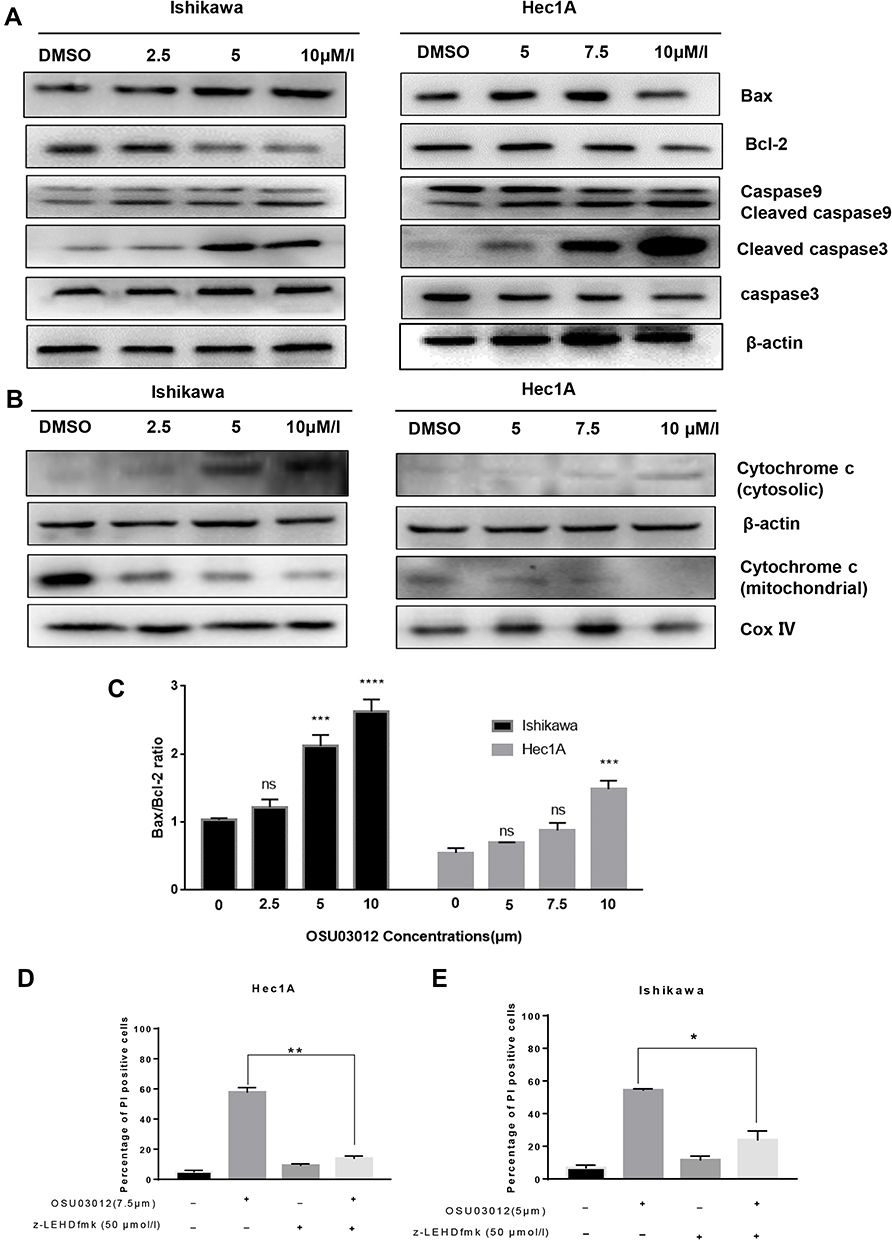 |
Figure 5 Mitochondrial apoptosis mediates the apoptosis of EC cells induced by OSU-03012. (A) Western blot analysis of apoptosis-related proteins expression in EC cells treated with OSU-03012 for 48 h. (B) Western blot analysis of cytochrome C expression in the cytoplasm of EC cells treated with OSU-03012 for 48 h. (C) The ratio of Bax/Bcl-2 in EC cells after treated with OSU-03012 for 48h. (D and E) Inhibition of caspase-9 activity reduced the induction of death of EC cells by OSU-03012. Data was expressed as mean ± SD. t-test was used for the statistical analysis versus the control group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns (Non-significant difference, p > 0.05). |
To test whether caspase-9 is required for anticancer activity of 0SU-03012, Hec-1A and Ishikawa cells were pretreated with 50μM of Z-LEHD-FMK (caspase-9 inhibitor) for 6h before being treated with OSU-03012 (7.5 and 5μM for the Hec1A and Ishikawa cell lines, respectively) for 48h, and the apoptotic rates of the cells were measured by flow cytometry. The killing effect of OSU-03012 on the cells pretreated with Z-LEHD-FMK was significantly weaker than that of the untreated group in both EC cells (Figure 5D and E). The results suggested that caspase-9 activity is required for mediating the effects of OSU-03012.
OSU-03012 Inhibits Tumor Progression in the Ishikawa Xenograft Study
To study the effects of OSU-03012 on tumor progression in vivo, we treated the Ishikawa xenograft with OSU-03012. We used the lowest dosage of OSU-03012 previously reported to successfully inhibit tumor growth in vivo in an Ishikawa xenograft tumor model.22,29,30 OSU-03012 (100 mg/kg daily) and 0.5% methylcellulose/0.1% Tween 80 as the control were orally administered to EC-bearing mice (N = 5 per group) for 3 weeks.
First of all, we can clearly see that compared with the control group, the tumor volume of mice in the OSU-03012 treatment group was significantly reduced (Figure 6A and B).The results showed that the mass and volume of the tumor were significantly reduced with OSU-03012 treatment (0.222 ± 0.07235g, 339.6 ± 103.2 mm3, n=5) relative to the vehicle controls (0.624 ± 0.02694 g, 1170 ± 84.51 mm3, n=5, Figure 6C and E); meanwhile, the body weights of the mice remained consistent throughout the duration of the experiment, exhibiting no serious toxicity at specified dose regimens (Figure 6D). These results demonstrate that oral administration of OSU-03012 can prevent EC progression.
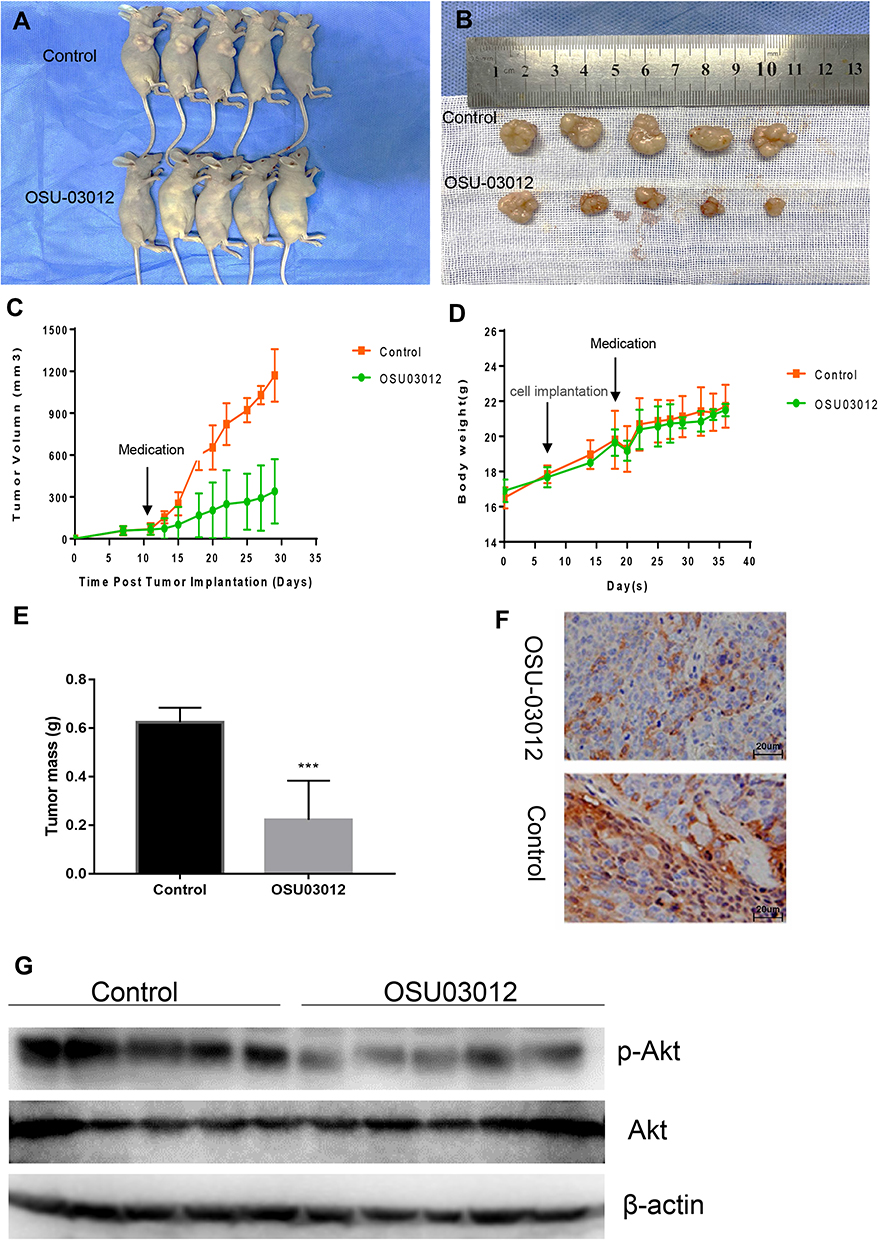 |
Figure 6 OSU-03012 Inhibits Tumor Growth in Ishikawa Xenograft Study. (A)The whole picture of the mice. (B) Dissected tumors from control and OSU-03012 groups of mice. (C) Tumor volumes and (D) body weight of mice was measured every 3 days using a caliper. The data were presented as mean ± SD of five mice. (E) Tumor mass of control and OSU-03012 groups were presented as mean ± SD of five mice. (F) A marked decrease in the percentage of p-Akt positive EC cells was demonstrated in the tissues exposed to OSU-03012 versus the untreated group (400×). (G) Western blot analysis of Akt, p-Akt expression in the dissected tumor tissues of control and OSU-03012 groups. Data was expressed as mean ± SD. t-test was used for the statistical analysis versus the control group. ***p < 0.001. |
Western blot analysis was conducted to detect the effect of OSU-03012 on Akt signaling. The results revealed that p-Akt levels were decreased in tumor tissues exposed to OSU-03012 (Figure 6G). To further verify the disrupted of Akt signaling as a mechanism of tumor suppression at the organizational level, we performed immunohistochemical (IHC) staining on polyformaldehyde-fixed xenografts to determine the expression of p-Akt in all treated tumors. The percentage of p-Akt positive endometrial epithelial cells was significantly decreased in OSU-03012-treated mice relative to the control specimens (Figure 6F). These data suggest that OSU-03012 can disrupt Akt signaling and suppress EC progression in vivo.
Discussion
Advanced EC is generally treated with surgery, radiotherapy, and the standard first-line therapy consisting of carboplatin plus paclitaxel. Recent advances in radiation therapy and chemotherapy have improved the patient’s outcomes; however, the effect is limited on the patients with recurrent EC. Therefore, more effective biomarker-oriented treatment strategies are needed for advanced and recurrent EC patients.31 The use of PI3K/Akt inhibitors depends on the evidence indicating that EC showed the highest change rate in the PI3K/AKT pathway, and mutations in the PI3K/Akt pathway confer poor prognosis in EC.32 Nevertheless, the clinical application of PI3K/Akt inhibitors remains limited because of toxicity and drug resistance.33 Numerous studies have recently shown that OSU-03012 has no impact on COX-2 activity but triggers apoptosis via the inhibition of Akt signaling. The safety and tolerability of OSU-03012 are under study in phase I clinical trials.
In the current study, we identify OSU-03012 as a powerful inhibitor of the PI3K/Akt signaling pathway in EC and report the potent killing efficacy of OSU-03012 against EC in vitro and in vivo. OSU-03012 decreased the phosphorylation of PI3K targeted proteins, such as Akt and GSK3β. It significantly inhibited proliferation and triggered apoptosis in EC cells. Treatment with OSU-03012 induced remarkable inhibition of tumor growth in the xenograft model of EC. At the cellular level, OSU-03012 impaired the viability of EC cell lines at micromolar concentrations. Such inhibition involved G2/M cell cycle arrest and apoptosis via the mitochondrial apoptosis pathway. The cell cycle arrest at the G2/M phase is a DNA damage checkpoint controlled by p21/cdc2/cyclin B1.34 In EC cell lines, OSU-03012 induced P21 upregulation, which is consistent with the inhibition of the p21/cdc2/cyclin B1 complex responsible for G2 to M cell cycle regulation.
During apoptosis, an increase in Bax expression can lead to apoptosis by inhibiting Bcl-2 activity,35 whereas during drug-induced apoptosis,36 the Bcl-2/Bax ratio is more important than solely Bcl-2 for the survival of cells.37 The study demonstrated that Bax expression increased, whereas Bcl-2 expression decreased, suggesting that the increasing ratio of Bax/Bcl-2 can promote cell apoptosis. Impaired mitochondrial function can lead to apoptosis.38 In mitochondria-dependent apoptotic pathways, cytochrome C participates in the activation of caspase-9, which then interacts with procaspase-3 to activate caspase-3. As an apolipoprotein, cytochrome C can hardly detect in the cytoplasm of normal cells.39 However, the release of cytochrome C from mitochondria into cytoplasm can be triggered during apoptosis involving mitochondrial pathways. So, we measured the cytochrome C levels in the cytoplasm and mitochondria. The results showed that cytochrome C was significantly upregulated in the cytoplasm but was reduced in mitochondria after treatment with OSU-03012, indicating that the permeability of the mitochondrial outer membrane of EC cells was enhanced after OSU-03012 treatment. Bax promotes apoptosis, enhances the permeability of mitochondrial outer membrane40 and prompts the release of cytochrome C.41 Caspases are aspartic acid-specific cysteine proteases that play a key role in cell apoptosis.42 Caspase activation causes the lysis and inactivation of key cell proteins, leading to apoptosis. Caspase-9 is the initiator of apoptosis and caspase-3 which is an important apoptotic protein that is initially activated in both Fas/FasL and mitochondrial pathways executes apoptosis. In the current study, the 17kDa cleavage fragment of caspase-3 significantly increased, suggesting that caspase-3 is activated during apoptosis. The data gathered in the present study suggested that OSU-03012 mediated cell apoptosis by targeting Akt on Bax/cytochrome C/caspase-9/caspase-3 dependent pathways (Figure 7). In summary, our results were in agreement with the anticancer effect and mechanisms of OSU-03012 in other cancers.19–21
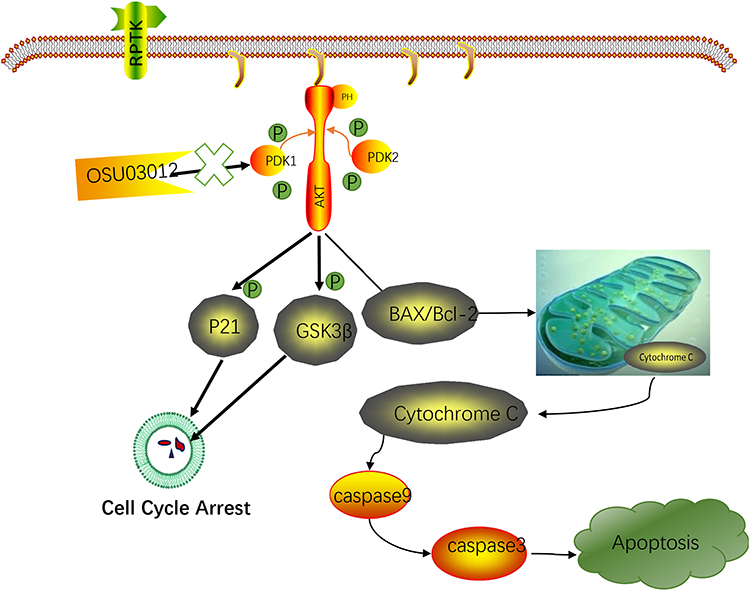 |
Figure 7 Schematic model for OSU-03012 mediated EC tumor inhibition. |
We can also verify the effective growth inhibition of OSU-03012 on the in vivo model at the animal level. Oral administration of OSU-03012 at low doses (100 mg/kg) significantly impeded the surgically induced growth of subcutaneous endometrium carcinomatous lesions. The significant growth inhibition of the Ishikawa xenograft by OSU-03012 provided an in vivo prove of our in vitro results.
Abnormalities in the Akt signaling pathway are frequently reported in various cancers.43 Akt signaling promotes cell survival which is mediated by the activation of the cellular growth factor and the inactivation of pro-apoptotic proteins.44 On the basis of the role of PI3K/AKT/mTOR in cancer progression, clinical trials are currently evaluating the role of PI3K/AKT/mTOR inhibitors in the treatment of metastatic, recurrent, and persistent EC patients.6 Small molecule inhibitors of PI3K/Akt signaling could induce cancer cell apoptosis by reducing the mitochondrial membrane potential. Consequently, GRP78/BIP expression was inhibited,45 caspase-3 was activated, BAX was upregulated, and Bcl-2 was downregulated.46 P16, P21, and P27 have been reported to be upregulated in tumors to induce cell senescence and inhibit cell proliferation.47 These results were consistent with the interruption of PI3K/Akt signaling pathway induced by OSU-03012, resulting in the apoptosis and cell cycle arrest in EC cells. In summary, we confirmed that OSU-03012 prevented the proliferation of EC cells and inhibited the growth of EC lesions in mouse models by disrupting Akt signaling.
On the basis of the aforementioned findings and data from ongoing studies, we anticipate that OSU-03012 can be potentially applied to treat EC in the future. However, further studies need to be conducted to thoroughly evaluate the effectiveness of the compound and to determine whether it has serious side effects. With its clinical application for the treatment of EC considered, it is likely that the efficacy of OSU-03012 will first be tested in conjunction with current standard treatment regimens. Studies combining OSU-03012 with other drugs for EC, such as doxorubicin or carboplatin, are needed. Combining small-molecule compounds with chemotherapy and other anticancer drugs shows strong potential.
Conclusions
Our data indicated that OSU-03012 can inhibit the progression of EC in vitro and in vivo. OSU-03012 mediated the disruption of PI3K/Akt signaling, resulting in the apoptosis of EC cells and cell cycle arrest. It may be used as the targeted drug for the treatment of EC by inhibiting Akt signaling.
Acknowledgments
This research was funded by the Key Project of Science and Technology of Henan, grant number 144/22170001.We thank the research group of Professor Ya Xie from the First Affiliated Hospital of Zhengzhou University for great support in Ishikawa cell line. Thanks for the support and guidance provided by Henan Key Laboratory of Digestive Organ Transplantation.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that there are no conflicts of interests in this work.
References
1. Rahib L, Smith B, Aizenberg R, Rosenzweig A, Fleshman J, Matrisian L. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–2921. doi:10.1158/0008-5472.CAN-14-0155
2. Makker V, Taylor M, Aghajanian C, et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer. J Clin Oncol. 2020;38(26):2981–2992. doi:10.1200/JCO.19.02627
3. Fleming G. Systemic chemotherapy for uterine carcinoma: metastatic and adjuvant. J Clin Oncol. 2007;25(20):2983–2990. doi:10.1200/JCO.2007.10.8431
4. Basho R, Gilcrease M, Murthy R, et al. Targeting the PI3K/AKT/mTOR pathway for the treatment of mesenchymal triple-negative breast cancer: evidence from a phase 1 trial of mTOR inhibition in combination with liposomal doxorubicin and bevacizumab. JAMA Oncol. 2017;3(4):509–515. doi:10.1001/jamaoncol.2016.5281
5. Shi N, Yu H, Chen T. Inhibition of esophageal cancer growth through the suppression of PI3K/AKT/mTOR signaling pathway. Onco Targets Ther. 2019;12:7637–7647. doi:10.2147/OTT.S205457
6. Roncolato F, Lindemann K, Willson M, Martyn J, Mileshkin L. PI3K/AKT/mTOR inhibitors for advanced or recurrent endometrial cancer. Cochrane Database Syst Rev. 2019;10. doi:10.1002/14651858.CD012160.pub2
7. Ediriweera M, Tennekoon K, Samarakoon S. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: biological and therapeutic significance. Semin Cancer Biol. 2019;59:147–160. doi:10.1016/j.semcancer.2019.05.012
8. Qian D, Xiao X, Byun J, et al. PI3K/Akt/mTOR signaling and plasma membrane proteins are implicated in responsiveness to adjuvant dendritic cell vaccination for metastatic colorectal cancer. Clin Cancer Res. 2017;23(2):399–406. doi:10.1158/1078-0432.CCR-16-0623
9. Johnsen J, Segerström L, Orrego A, et al. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene. 2008;27(20):2910–2922. doi:10.1038/sj.onc.1210938
10. Opel D, Poremba C, Simon T, Debatin K, Fulda S. Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res. 2007;67(2):735–745. doi:10.1158/0008-5472.CAN-06-2201
11. Kandoth C, Schultz N, Cherniack A, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. doi:10.1038/nature12113
12. Salvesen H, Carter S, Mannelqvist M, et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc Natl Acad Sci U S A. 2009;106(12):4834–4839. doi:10.1073/pnas.0806514106
13. Bansal N, Yendluri V, Wenham R. The molecular biology of endometrial cancers and the implications for pathogenesis, classification, and targeted therapies. Cancer Control. 2009;16(1):8–13. doi:10.1177/107327480901600102
14. Liu Z, Hong Z, Qu P. Proteomic analysis of human endometrial tissues reveals the roles of PI3K/AKT/mTOR pathway and tumor angiogenesis molecules in the pathogenesis of endometrial cancer. Biomed Res Int. 2020;2020:5273969.
15. Uegaki K, Kanamori Y, Kigawa J, et al. PTEN-positive and phosphorylated-Akt-negative expression is a predictor of survival for patients with advanced endometrial carcinoma. Oncol Rep. 2005;14(2):389–392.
16. Megino-Luque C, Moiola C, Molins-Escuder C, et al. Small-molecule inhibitors (SMIs) as an effective therapeutic strategy for endometrial cancer. Cancers. 2020;12(10):2751. doi:10.3390/cancers12102751
17. Zhu J, Huang J, Tseng P, et al. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res. 2004;64(12):4309–4318. doi:10.1158/0008-5472.CAN-03-4063
18. Johnson A, Smith L, Zhu J, et al. A novel celecoxib derivative, OSU03012, induces cytotoxicity in primary CLL cells and transformed B-cell lymphoma cell line via a caspase- and Bcl-2-independent mechanism. Blood. 2005;105(6):2504–2509. doi:10.1182/blood-2004-05-1957
19. Murata M, Narahara S, Kawano T, et al. Design and function of engineered protein nanocages as a drug delivery system for targeting pancreatic cancer cells via neuropilin-1. Mol Pharm. 2015;12(5):1422–1430. doi:10.1021/mp5007129
20. Liu J, Qin C, Lv W, Zhao Q, Qin C. OSU-03012, a non-Cox inhibiting celecoxib derivative, induces apoptosis of human esophageal carcinoma cells through a p53/Bax/cytochrome c/caspase-9-dependent pathway. Anticancer Drugs. 2013;24(7):690–698. doi:10.1097/CAD.0b013e328362469f
21. West N, Garcia-Vargas A, Chalfant C, Park M. OSU-03012 sensitizes breast cancers to lapatinib-induced cell killing: a role for Nck1 but not Nck2. BMC Cancer. 2013;13:256. doi:10.1186/1471-2407-13-256
22. Gao M, Yeh P, Lu Y, et al. OSU-03012, a novel celecoxib derivative, induces reactive oxygen species-related autophagy in hepatocellular carcinoma. Cancer Res. 2008;68(22):9348–9357. doi:10.1158/0008-5472.CAN-08-1642
23. Kucab J, Lee C, Chen C, et al. Celecoxib analogues disrupt Akt signaling, which is commonly activated in primary breast tumours. Breast Cancer Res. 2005;7(5):R796–807. doi:10.1186/bcr1294
24. Mateo J, De Bono JS, Ramanathan RK, et al. A first-in-human phase I trial of AR-12, a PDK-1 inhibitor, in patients with advanced solid tumors. 2013.
25. Muñoz-Alonso M, Acosta J, Richard C, Delgado M, Sedivy J, León J. p21Cip1 and p27Kip1 induce distinct cell cycle effects and differentiation programs in myeloid leukemia cells. J Biol Chem. 2005;280(18):18120–18129. doi:10.1074/jbc.M500758200
26. Kalkavan H, Green D. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018;25(1):46–55. doi:10.1038/cdd.2017.179
27. Nagaraj N, Anilakumar K, Singh O. Diallyl disulfide causes caspase-dependent apoptosis in human cancer cells through a Bax-triggered mitochondrial pathway. J Nutr Biochem. 2010;21(5):405–412. doi:10.1016/j.jnutbio.2009.01.015
28. Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi:10.1146/annurev.biochem.73.011303.073706
29. Wang Y, Kulp S, Wang D, et al. Targeting endoplasmic reticulum stress and Akt with OSU-03012 and gefitinib or erlotinib to overcome resistance to epidermal growth factor receptor inhibitors. Cancer Res. 2008;68(8):2820–2830. doi:10.1158/0008-5472.CAN-07-1336
30. Weng S, Kashida Y, Kulp S, et al. Sensitizing estrogen receptor-negative breast cancer cells to tamoxifen with OSU-03012, a novel celecoxib-derived phosphoinositide-dependent protein kinase-1/Akt signaling inhibitor. Mol Cancer Ther. 2008;7(4):800–808. doi:10.1158/1535-7163.MCT-07-0434
31. Salvesen H, Haldorsen I, Trovik J. Markers for individualised therapy in endometrial carcinoma. Lancet Oncol. 2012;13(8):e353–e361. doi:10.1016/S1470-2045(12)70213-9
32. Catasus L, Gallardo A, Cuatrecasas M, Prat J. Concomitant PI3K-AKT and p53 alterations in endometrial carcinomas are associated with poor prognosis. Mod Pathol. 2009;22(4):522–529. doi:10.1038/modpathol.2009.5
33. Dedes K, Wetterskog D, Ashworth A, Kaye S, Reis-Filho J. Emerging therapeutic targets in endometrial cancer. Nat Rev Clin Oncol. 2011;8(5):261–271. doi:10.1038/nrclinonc.2010.216
34. Pore S, Hahm E, Kim S, et al. A novel sulforaphane-regulated gene network in suppression of breast cancer-induced osteolytic bone resorption. Mol Cancer Ther. 2020;19(2):420–431. doi:10.1158/1535-7163.MCT-19-0611
35. Noguchi K, Kitanaka C, Yamana H, Kokubu A, Mochizuki T, Kuchino Y. Regulation of c-Myc through phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J Biol Chem. 1999;274(46):32580–32587. doi:10.1074/jbc.274.46.32580
36. Salomons G, Brady H, Verwijs-Janssen M, et al. The Bax alpha:Bcl-2 ratio modulates the response to dexamethasone in leukaemic cells and is highly variable in childhood acute leukaemia. Int J Cancer. 1997;71(6):959–965. doi:10.1002/(SICI)1097-0215(19970611)71:6<959::AID-IJC9>3.0.CO;2-X
37. Susin S, Lorenzo H, Zamzami N, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397(6718):441–446. doi:10.1038/17135
38. Khosravi-Far R, Esposti M. Death receptor signals to mitochondria. Cancer Biol Ther. 2004;3(11):1051–1057. doi:10.4161/cbt.3.11.1173
39. Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274(17):11549–11556. doi:10.1074/jbc.274.17.11549
40. Kale J, Osterlund E, Andrews D. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25(1):65–80. doi:10.1038/cdd.2017.186
41. Rossé T, Olivier R, Monney L, et al. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998;391(6666):496–499. doi:10.1038/35160
42. Thornberry N, Lazebnik Y. Caspases: enemies within. Science. 1998;281(5381):1312–1316. doi:10.1126/science.281.5381.1312
43. Romano G. The role of the dysfunctional Akt-related pathway in cancer: establishment and maintenance of a malignant cell phenotype, resistance to therapy, and future strategies for drug development. Scientifica. 2013;2013:317186. doi:10.1155/2013/317186
44. Nitulescu G, Van De Venter M, Nitulescu G, et al. The Akt pathway in oncology therapy and beyond (Review). Int J Oncol. 2018;53(6):2319–2331. doi:10.3892/ijo.2018.4597
45. Ha D, Lee A. Insulin-like growth factor 1-receptor signaling stimulates GRP78 expression through the PI3K/AKT/mTOR/ATF4 axis. Cell Signal. 2020;75:109736. doi:10.1016/j.cellsig.2020.109736
46. Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. 2019;18(1):26.
47. Rodriguez-Cupello C, Dam M, Serini L, et al. The STRIPAK complex regulates response to chemotherapy through p21 and p27. Front Cell Dev Biol. 2020;8:146. doi:10.3389/fcell.2020.00146
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.