")
Back to Journals » Orthopedic Research and Reviews » Volume 15
Osteoporosis, Fractures, and Blindness Due to a Missense Mutation in the LRP5 Receptor
Authors Littman J , Phornphutkul C, Saade C, Katarincic J, Aaron R
Received 3 December 2022
Accepted for publication 16 March 2023
Published 22 March 2023 Volume 2023:15 Pages 39—45
DOI https://doi.org/10.2147/ORR.S400111
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Clark Hung
Jake Littman,1 Chanika Phornphutkul,2 Celine Saade,3 Julia Katarincic,1 Roy Aaron1
1Department of Orthopedic Surgery, Warren Alpert Medical School of Brown University, Providence, RI, USA; 2Division of Human Genetics, Department of Pediatrics, Hasbro Children’s Hospital, Warren Alpert Medical School of Brown University, Providence, RI, USA; 3Division of Ophthalmology, Lifespan Physician Group, Warren Alpert Medical School of Brown University, Providence, RI, USA
Correspondence: Roy Aaron, Department of Orthopedic Surgery, Warren Alpert Medical School of Brown University, 2 Dudley Street. Suite 200, Providence, RI, 02905, USA, Tel +1 401-274-9660, Fax +1 401-270-1560, Email [email protected]
Abstract: Familial exudative vitreoretinopathy (FEVR) is a genetic disorder whose presentation can include osteoporosis, multiple fractures, and incomplete retinal angiogenesis leading to retinal detachment and blindness if left untreated. Discussed herein are the cases of two pediatric siblings who presented to the orthopedic service with multiple fractures and, through interdisciplinary management, were diagnosed with FEVR and treated appropriately before permanent visual impairment. The skeletal manifestations of FEVR, which have not been explored in depth in prior literature, are described. One sibling presented to orthopedic services for evaluation of a closed distal radius fracture sustained while playing sports. A comprehensive history revealed he had suffered at least four appendicular fractures in his lifetime, and dual-energy x-ray absorptiometry (DEXA) scan revealed his bone density to be in the first percentile for his age. Concurrent evaluation of his younger sibling revealed a similar history of multiple fractures and low bone density. Referral to genetic services and ensuing whole-exome sequencing revealed a likely pathogenic variant in both siblings’ LRP5 gene, the only known causative mutation for FEVR that leads to skeletal manifestations. While FEVR is well known in genetic and ophthalmologic settings, greater awareness of FEVR and other genetic disorders that predispose to fractures in pediatric populations is warranted in orthopedic settings. This will lead to reduced sequelae in pediatric patients with genetic disorders and improved interdisciplinary expertise. The story of these siblings illustrates that a high index of suspicion for genetic diseases is essential when treating children with osteoporosis and growth delays.
Keywords: familial exudative vitreoretinopathy, FEVR, low-density lipoprotein receptor-related protein 5, LRP5, gracile bones, skeletal dysmorphogenesis
Introduction
The diagnosis of children with multiple sequential fractures and osteoporosis is especially challenging. One comes into contact with genetic conditions that are rare and unfamiliar to the orthopedic surgeon but can cause multisystem functional disabilities if not diagnosed accurately. The orthopedic surgeon often sees these patients first and is therefore challenged with the sensitivity to recognize the presence of genetic abnormalities. One such genetic condition is familial exudative vitreoretinopathy (FEVR). FEVR is a hereditary disorder whose phenotype can include osteoporosis, multiple fractures, and incomplete retinal angiogenesis leading to retinal detachment and permanent visual impairment or blindness if left untreated. Other genetic diseases can present similarly and also lead to blindness or other functional disabilities.
Described here are the cases of two pediatric siblings who presented to the orthopedic service with multiple sequential fractures and growth delays and, through interdisciplinary management, were diagnosed with FEVR and treated appropriately before permanent visual and skeletal impairment. The skeletal characteristics and the ocular and genetic abnormalities are presented. These cases illustrate that a high index of suspicion for genetic diseases is essential when treating children with multiple fractures, especially when they are accompanied by growth disturbances. Remaining vigilant for the potential for underlying genetic pathology can reduce the frequency and consequences of fractures and even prevent blindness and other functional disabilities for pediatric patients.
Case Report
Two siblings with sequential fractures and delayed growth were seen in the Osteoporosis and Bone Health Clinic. A 13-year-old boy presented with a closed distal radius fracture sustained while playing sports. A comprehensive history revealed he had suffered at least three other appendicular fractures, including of both radial growth plates at different ages and a metacarpal fracture, all secondary to trauma. Physical examination revealed his height to be 60.3’ (8th percentile) and his weight to be 84 lbs (4th percentile). He appeared well-nourished with a normal stance and gait, a normal spine without compression fracture, and normal range of motion in the lumbar spine. His elbows were hyperextensible but his knees were not. Sclerae were normal. His hearing was normal, and no frontal bossing was present. A radiographic skeletal survey revealed widened medullary canals with thin cortices typical of osteoporosis, gracile bones, and valgus proximal femoral neck-shaft angles (Figure 1). Dual-energy X-ray absorptiometry (DEXA) scan showed his bone density to be two standard deviations below the mean for his age (Figure 2).
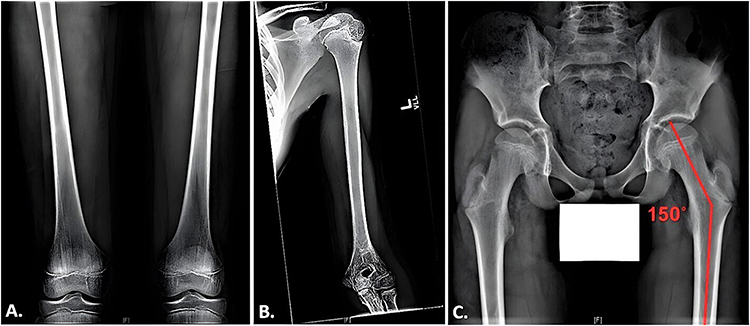 |
Figure 1 Radiographic findings in a 13-year-old boy with FEVR. (A) Anteroposterior views of the femora demonstrating thin, gracile bones. (B) Anterior view of the left humerus showing a widened medullary canal and thin cortices. (C) Anteroposterior view of the pelvis exhibiting coxa valga with a neck-shaft angle approximating 150 degrees. |
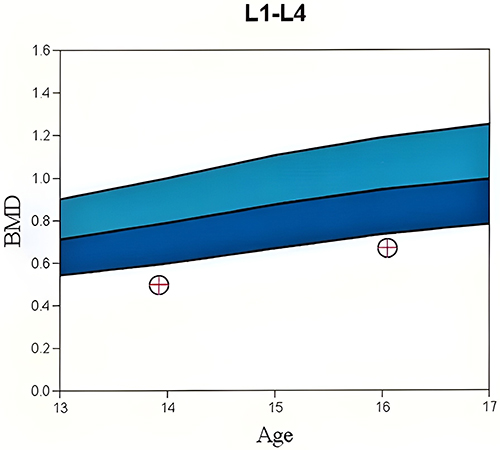 |
Figure 2 DEXA Z-scores in a 13-year-old boy with FEVR. Bone mineral density (BMD) at initial presentation (leftmost circle with cross) showed a Z-score of −3.0 (1st percentile). A Z-score represents a patient’s BMD as the number of standard deviations away from the mean compared to an age- and sex-matched cohort and is calculated using the raw BMD score obtained by the DEXA scan (y-axis). Lighter and darker blue areas on the graph represent up to two standard deviations above average BMD for age/sex (Z-scores of 0 to 2 or 50th to 95th percentile) and up to two standard deviations below average BMD for age/sex (Z-scores of −2 to 0 or 5th to 50th percentile), respectively. Two years after initial presentation, this patient had kept pace with the expected bone density increase rate for his age (rightmost circle with cross) but was still osteoporotic with a Z-score of −2.6 (1st percentile). |
Concurrent evaluation of his 10-year-old sister revealed a similar history of multiple sequential fractures, including a distal phalanx fracture, a fracture of both bones of the forearm requiring surgical stabilization, and an impacted 2-part fracture of the proximal humerus. Physical examination showed her height to be 58.3’ (68th percentile) and her weight to be 97 lbs (42nd percentile). Like her brother, she appeared well-nourished with a normal stance and gait and a normal spine without compression fracture. Her elbows were hyperextensible but her knees were not. Sclerae were normal. She had no frontal bossing. A radiographic skeletal survey revealed gracile bones and widened medullary canals and thin cortices typical of osteoporosis (Figure 3). A dental Panorex film demonstrated impaired dentinogenesis of 5 adult teeth (Figure 4). DEXA scan revealed her bone density to be at the 1st percentile for her age.
 |
Figure 3 Radiographic findings in a 10-year-old girl with FEVR. (A) Anteroposterior view of the left humerus exhibiting gracile bones and thin cortices. (B) Anteroposterior film of the right forearm showing gracile bones. (C) Anteroposterior view of tibias revealing endosteal reabsorption with widened medullary canals and thin cortices characteristic of osteoporosis. |
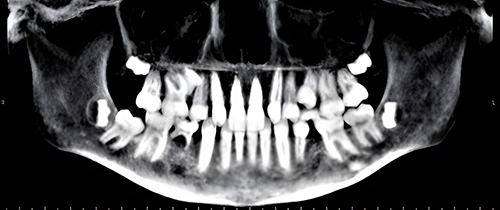 |
Figure 4 Panorex film of a 10-year-old girl with FEVR. Impaired dentinogenesis is manifested by the absence of the maxillary right and left lateral incisors, maxillary right and left 2nd pre-molars, and right mandibular 1st pre-molar. |
Referral to the genetics service was made. Based on clinical features, the differential diagnosis was broad but included disorders resulting in low bone density. Whole-exome sequencing identified a likely pathogenic variant in both siblings resulting in a guanine to adenine substitution at nucleotide 1481 in the cDNA sequence for the low-density lipoprotein receptor-related protein 5 (LRP5) gene (c.1481G>A). This results in an arginine to glutamine substitution at amino acid 494 in the LRP5 protein sequence (p.R494Q). Mutations in the LRP5 gene have been described to cause both skeletal and ocular dysmorphogenesis,1 and an ophthalmologic consult was obtained. Eye examination, including fluorescein angiography, revealed a temporal retinal capillary dropout and a retinal hole in the older sibling’s right eye, possibly representing an atrophic hole due to surrounding peripheral ischemia, and he was diagnosed with FEVR (Figure 5). Fluorescein angiography also revealed a temporal retinal capillary dropout in the younger sibling’s left eye, and she was diagnosed with FEVR as well.
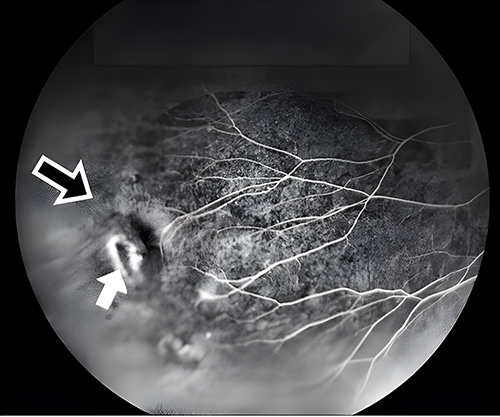 |
Figure 5 Mid-phase fluorescein angiography of the retina of a 13-year-old boy with FEVR. Capillary dropout, defined as areas of hypofluorescence and indicating reduced perfusion (open arrow), and a retinal hole (solid arrow) can be seen. |
The older sibling was treated with a laser retinopexy in the right eye as prophylaxis for retinal detachment (Figure 6). Both have since been followed by orthopedic and ophthalmologic services. At 2 years follow-up, vision has been preserved in both children. Both have ceased playing contact sports, and no new fractures have occurred. DEXA scans demonstrate progressive slow increase of bone density for their ages, though they remain below normal range (Figure 2). Indications for pharmacotherapy for bone density were discussed and reviewed with the patient’s parents. Currently, there is no FDA-approved medication with which to treat children with osteoporosis. Given that both children’s fractures were trauma related, no vertebral compression fractures were apparent, and both children were entering puberty at the time of evaluation, pharmacological treatment for low bone density is being omitted until clinical progression via additional fractures or lack of bone mass accumulation. However, the importance of adequate calcium and vitamin D intake was emphasized.2
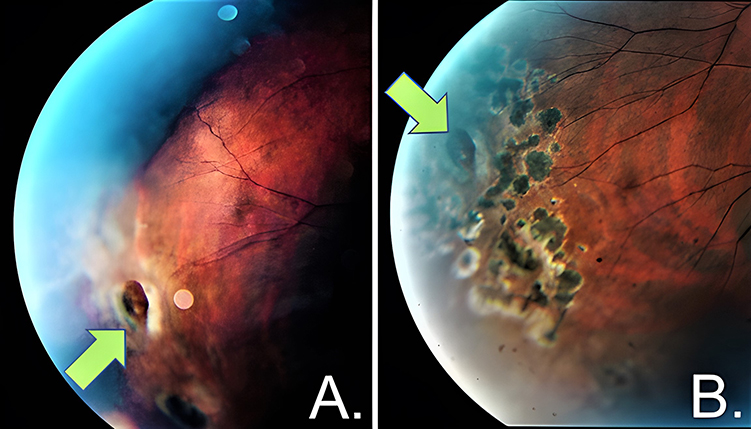 |
Figure 6 Fundus photographs exhibiting the temporal area of the right eye of a 13-year-old boy with FEVR before and after laser retinopexy treatment. (A) Atrophic hole in temporal periphery before treatment (green arrow). (B) Atrophic hole in temporal periphery after treatment (green arrow) surrounded by 360° laser scar extended to area of capillary dropout to prevent neovascularization or further atrophic hole in that area. |
Discussion
Several genetic mutations affect both the skeleton and the eye. The patients presented here displayed heterozygous mutations in the LRP5 gene. Total loss-of-function of this gene results in the osteoporosis-pseudoglioma syndrome (OPPG), whose phenotype is characterized by congenital or infancy-onset blindness and severe osteoporosis.1,3 Most forms of FEVR affect only the eye and may be caused by mutations in LRP5 or in about 14 other genes including FZD4, TSPAN12, NDP, and KIF11.4 Incomplete loss-of-function mutations in LRP5 can result in a distinct form of FEVR whose phenotype includes osteoporosis and predisposition to fracture in addition to the incomplete or anomalous vascularization of the peripheral retina for which the condition is known.4–6 Because OPPG and LRP5-mutation-induced FEVR arise secondary to similar genetic mechanisms, it has been proposed that the conditions are in fact a continuum and should not be considered separate entities.5,7,8
While the skeletal characteristics of adolescents with OPPG have been described,9,10 the skeletal phenotypes of patients with LRP5-mutation-induced FEVR have not been explored in depth. The cases presented here indicate that prominent skeletal characteristics of the c.1481G>A mutation in LRP5 include growth delay, osteoporosis, fractures, thin gracile bones, coxa valga, and impaired dentinogenesis. Observation of children with these skeletal features should alert the clinician to the possibility of underlying genetic abnormalities and stimulate referral for genetic characterization.
The LRP5 gene encodes for the LRP5 transmembrane receptor, which is a major contributor to bone health and is a key receptor for the canonical Wnt signaling pathway. The specific mutation identified in the cases presented here was a guanine to adenine substitution at gene locus 1481 (c.1481G>A). This results in an arginine to glutamine substitution (p.R494Q) in the LRP5 transmembrane receptor, impairing its function and reducing Wnt signaling. The Wnt pathway is highly conserved and highly complex, with Wnt proteins regulating a variety of cellular processes including cell fate determination, motility, polarity, primary axis formation, organogenesis, and stem cell renewal.11 The Wnt pathway’s role in the pathogenesis of FEVR’s ocular manifestations is not entirely understood, though it has been hypothesized that the canonical Wnt pathway functions to upregulate endothelial cell growth during retinal angiogenesis, presenting a plausible mechanism for the dysvascular retina observed in FEVR.6
The phenotype of the skeletal manifestations of FEVR is related to mutations in the LRP5 gene. The role of Wnt and the LRP5 transmembrane receptor in bone formation and remodeling is not entirely understood and is an area of active investigation.12,13 Proposed mechanisms by which inhibited LRP5 transmembrane receptor functioning and canonical Wnt pathway signaling affect the skeleton include 1) direct downregulation of osteoblast proliferation, function, or differentiation;1,14 2) direct effects on osteocyte function,15,16 possibly by way of interference with osteocyte-mediated skeletal mechanotransduction;17 3) LRP5-mediated osteoclast differentiations that increase resorptive capacity;18 and, perhaps most controversially, 4) a Wnt pathway-independent, gut-derived serotonin-mediated endocrine axis involved in osteoblast proliferation.19
FEVR can present as a consequence of several genetic mutations and can be acquired through autosomal dominant, autosomal recessive, or X-linked patterns of inheritance.20 The prevalence of FEVR in the general population is unknown, but one multicenter study performing screening ocular exams in about 200,000 newborns found the incidence in that population to be 0.11%.21 A 2022 systematic review and meta-analysis including over 3200 FEVR patients found the frequency of the LRP5 mutation to be 13.6%, the highest of any known mutation linked to FEVR.22 Out of the known causative mutations for FEVR, only those of the LRP5 locus have been linked to reduced bone mass and predisposition to fracture.6 In the patients reported here, heterozygous c.1481G>A (p.R494Q) mutations in LRP5 were discovered and determined to be the likely cause of their disorder, consistent with the association of this mutation with FEVR and OPPG.23
Conclusion
FEVR is one of the several genetic diseases that can lead to osteoporosis, fractures, and vision loss. The evaluation of pediatric patients with these conditions is difficult, in part, because rare genetic conditions presenting with reduced bone mass and fractures often resemble osteogenesis imperfecta. A high index of suspicion by the orthopedic surgeon for genetic diseases is essential in children with fractures and decreased bone density. Not to do so can be tragic for the young patients, but careful and sensitive analysis of children with low bone density can reveal conditions for which interventions may yield great benefits to the patients and considerable gratification for the surgeon. Precise diagnosis is often challenging and usually requires a multidisciplinary approach. The key point for the orthopedic surgeon is that suspicion for these developmental mutations should be a part of the evaluation of children with multiple fractures and slow physical development, and the threshold should be low for obtaining measurements of bone density and genetic consultations. In these cases, blindness can be avoided and the frequency and consequences of fractures mitigated.
Abbreviations
FEVR, familial exudative vitreoretinopathy; DEXA, dual-energy X-ray absorptiometry; LRP5, low-density lipoprotein receptor-related protein 5; BMD, bone mineral density; OPPG, osteoporosis-pseudoglioma.
Consent
Written informed consent was obtained by the patients and their parent to have the case details and accompanying images published.
Institutional Review Board
The IRB was contacted and the authors were assured that, as this is a case report and no research was being conducted, this report was not under their jurisdiction.
Acknowledgments
The authors acknowledge and appreciate the patients herein described and their family for their cooperation and enthusiasm for reporting.
Funding
Funding was provided by The Miriam Hospital.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gong Y, Slee RB, Fuca N, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107(4):513–523. doi:10.1016/s0092-8674(01)00571-2
2. Bachrach LK, Gordon CM, Sills IN; Section on Endocrinology. Bone densitometry in children and adolescents. Pediatrics. 2016;138(4):e20162398. doi:10.1542/peds.2016-2398
3. Levasseur R, Lacombe D, de Vernejoul MC. LRP5 mutations in osteoporosis-pseudoglioma syndrome and high-bone-mass disorders. Joint Bone Spine. 2005;72(3):207–214. doi:10.1016/j.jbspin.2004.10.008
4. Mao J, Chen Y, Fang Y, et al. Clinical characteristics and mutation spectrum in 33 Chinese families with familial exudative vitreoretinopathy. Ann Med. 2022;54(1):3286–3298. doi:10.1080/07853890.2022.2146744
5. Kheir V, Munier FL, Aubry-Rozier B, Schorderet DF. Potential blindness in children of patients with hereditary bone disease. Osteoporos Int. 2016;27(2):841–844. doi:10.1007/s00198-015-3245-4
6. Tauqeer Z, Yonekawa Y. Familial exudative vitreoretinopathy: pathophysiology, diagnosis, and management. Asia Pac J Ophthalmol. 2018;7(3):176–182. doi:10.22608/APO.201855
7. Gilmour DF. Familial exudative vitreoretinopathy and related retinopathies. Eye. 2015;29(1):1–14. doi:10.1038/eye.2014.70
8. Wang Z, Liu CH, Huang S, Chen J. Wnt signaling in vascular eye diseases. Prog Retin Eye Res. 2019;70:110–133. doi:10.1016/j.preteyeres.2018.11.008
9. Homaei A, Chegini V, Saffari F. Clinical response to treatment with teriparatide in an adolescent with osteoporosis-pseudoglioma syndrome (OPPG): a case report. Int J Endocrinol Metab. 2022;20(2):e121031. doi:10.5812/ijem-121031
10. Abdel-Hamid MS, Elhossini RM, Otaify GA, Abdel-Ghafar SF, Aglan MS. Osteoporosis-pseudoglioma syndrome in four new patients: identification of two novel LRP5 variants and insights on patients’ management using bisphosphonates therapy. Osteoporos Int. 2022;33(7):1501–1510. doi:10.1007/s00198-022-06313-1
11. Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4(2):68–75. doi:10.4161/org.4.2.5851
12. Johnson ML. LRP5 and bone mass regulation: where are we now? Bonekey Rep. 2012;1:1. doi:10.1038/bonekey.2012.1
13. Goltzman D. LRP5, serotonin, and bone: complexity, contradictions, and conundrums. J Bone Miner Res. 2011;26(9):1997–2001. doi:10.1002/jbmr.462
14. Kato M, Patel MS, Levasseur R, et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157(2):303–314. doi:10.1083/jcb.200201089
15. Cui Y, Niziolek PJ, MacDonald BT, et al. Lrp5 functions in bone to regulate bone mass. Nat Med. 2011;17(6):684–691. doi:10.1038/nm.2388
16. Cui Y, Niziolek PJ, MacDonald BT, et al. Reply to Lrp5 regulation of bone mass and gut serotonin synthesis. Nat Med. 2014;20(11):1229–1230. doi:10.1038/nm.3697
17. Bullock WA, Pavalko FM, Robling AG. Osteocytes and mechanical loading: the Wnt connection. Orthod Craniofac Res. 2019;22(Suppl1):175–179. doi:10.1111/ocr.12282
18. Khrystoforova I, Shochat-Carvalho C, Harari R, et al. Zebrafish mutants reveal unexpected role of Lrp5 in osteoclast regulation. Front Endocrinol. 2022;13:985304. doi:10.3389/fendo.2022.985304
19. Yadav VK, Ryu JH, Suda N, et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135(5):825–837. doi:10.1016/j.cell.2008.09.059
20. Qu N, Li W, Han DM, et al. Mutation spectrum in a cohort with familial exudative vitreoretinopathy. Mol Genet Genomic Med. 2022;10(9):e2021. doi:10.1002/mgg3.2021
21. Tang H, Li N, Li Z, et al. Fundus examination of 199 851 newborns by digital imaging in China: a multicentre cross-sectional study. Br J Ophthalmol. 2018;102(12):1742–1746. doi:10.1136/bjophthalmol-2018-312366
22. Wang X, Chen J, Xiong H, Yu X. Genotype-phenotype associations in familial exudative vitreoretinopathy: a systematic review and meta-analysis on more than 3200 individuals. PLoS One. 2022;17(7):e0271326. doi:10.1371/journal.pone.0271326
23. Xiao H, Tong Y, Zhu Y, Peng M. Familial exudative vitreoretinopathy-related disease-causing genes and Norrin/β-catenin signal pathway: structure, function, and mutation spectrums. J Ophthalmol. 2019;2019:5782536. doi:10.1155/2019/5782536
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.