")
Back to Journals » Clinical, Cosmetic and Investigational Dentistry » Volume 10
Osteoimmunology of tumor necrosis factor-alpha, IL-6, and RANTES/CCL5: a review of known and poorly understood inflammatory patterns in osteonecrosis
Authors Lechner J , Rudi T, von Baehr V
Received 19 August 2018
Accepted for publication 3 October 2018
Published 9 November 2018 Volume 2018:10 Pages 251—262
DOI https://doi.org/10.2147/CCIDE.S184498
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Christopher E. Okunseri
Johann Lechner,1 Tatjana Rudi,2 Volker von Baehr3
1Clinic for Integrative Dentistry, Munich 81547, Germany; 2Institute for Epidemiological Studies, Berlin 10709, Germany; 3Department of Immunology and Allergology, Institute for Medical Diagnostics in MVZ GbR, Berlin 12247, Germany
Background: The immune and bone systems are closely linked via cytokine cross-talk. This interdisciplinary field of research is referred to as osteoimmunology and pertains to inflammatory and osteoarticular diseases that feature the primary expression of tumor necrosis factor-alpha (TNF-α) and IL-6.
Objective: Are there bone resorptive processes wherein chronic inflammatory conditions are not linked to TNF-α and IL-6 expression, but rather to the expression of other cytokines?
Materials and methods: A comprehensive literature search was performed in PubMed Central.
Discussion: Although all diseases with cytokines involved in bone resorption (TNF-α and IL-6) are at the forefront of destructive inflammatory processes, there is one exception in the literature: fatty oxide osteoporosis/osteolysis in the jawbone (FDOJ), which is associated with significant bone softening. However, it should be noted that TNF-α and IL-6 fall below the levels found in a healthy jawbone in this condition. Another conspicuous finding is that there is a nearly 35-fold overexpression of the chemokine RANTES/CCL5 (R/C) in all FDOJ cases studied thus far in the literature.
Conclusion: FDOJ appears to represent a unique cytokine and inflammatory pattern from osteolysis in the body. R/C can be defined as the dominant carrier of a “maxillomandibular osteoimmunology”.
Keywords: bone resorption, chemokine RANTES, CCL5, tumor necrosis factor-alpha, maxillomandibular osteoimmunology, maxillomandibular
Background
Recently, researchers have begun to gain a greater understanding of immune cell and bone cell communication, and some have developed a new model of bone remodeling, in which various stages of bone formation and resorption, which coexist in a dynamic interplay, are strictly controlled by the immune system.1 The immune and bone systems seem to be closely linked, as they share common signaling pathways and regulatory mechanisms. The understanding of these mechanisms and pathways has led to new insights and resulted in interdisciplinary research and clinical fields, both of which are related to each other and are now part of a field called osteoimmunology. The complex network of messengers – cytokines, ILs, and chemokines – and their receptors serve as the biochemical and physiological basis for osteoimmunology. To ensure that all physiological processes in the organism interact smoothly (which occurs in ~6×1014 cells in the body), seamless communication must take place. This intricate and complex communication primarily occurs via mediators that have only been discovered in the last 20 years: cytokines and chemokines. Further, since cells in the central nervous system can increase cytokine distribution via neurochemical processes or viral infections,2 the area of “psychoneuroimmunology” was born.3 Under the conditions of cytokine cell communication, which serves the basis of an immunological network, we describe the field of “maxillomandibular osteoimmunology”, which is concerned with inflammation and osteoporosis in the jaw area.
Research question
In this review, we examine scientific reviews from PubMed that have explored the involvement of inflammatory cytokines tumor necrosis factor-alpha (TNF-α) and IL-6 in osteoimmunological conditions that result in destructive reactions in bone metabolism. Specifically, we wish to address the following question: Does any literature exist on the cytokine patterns of chronic, subclinical inflammation in the jaw area and research on a specific “maxillomandibular osteoimmunology?”
Materials and methods
The studies presented herein were performed as a case–control study and were deemed to be retrospective in nature. Approval was granted by IMD-Berlin forensic accredited Institute DIN EN 15189/DIN EN 17025. All patients provided their written consent to participate in the studies, and the samples and data were collected in the course of routine practice, that is, regular oral surgery procedures. For this study, we searched the literature from PubMed Central on destructive inflammatory processes in bone. An additional focus was placed on authors’ publications relating to cytokine profiles in fatty degenerative osteolysis/osteonecrosis in the jawbone (FDOJ). The flow diagram in Figure 1 displays the literature search strategy.
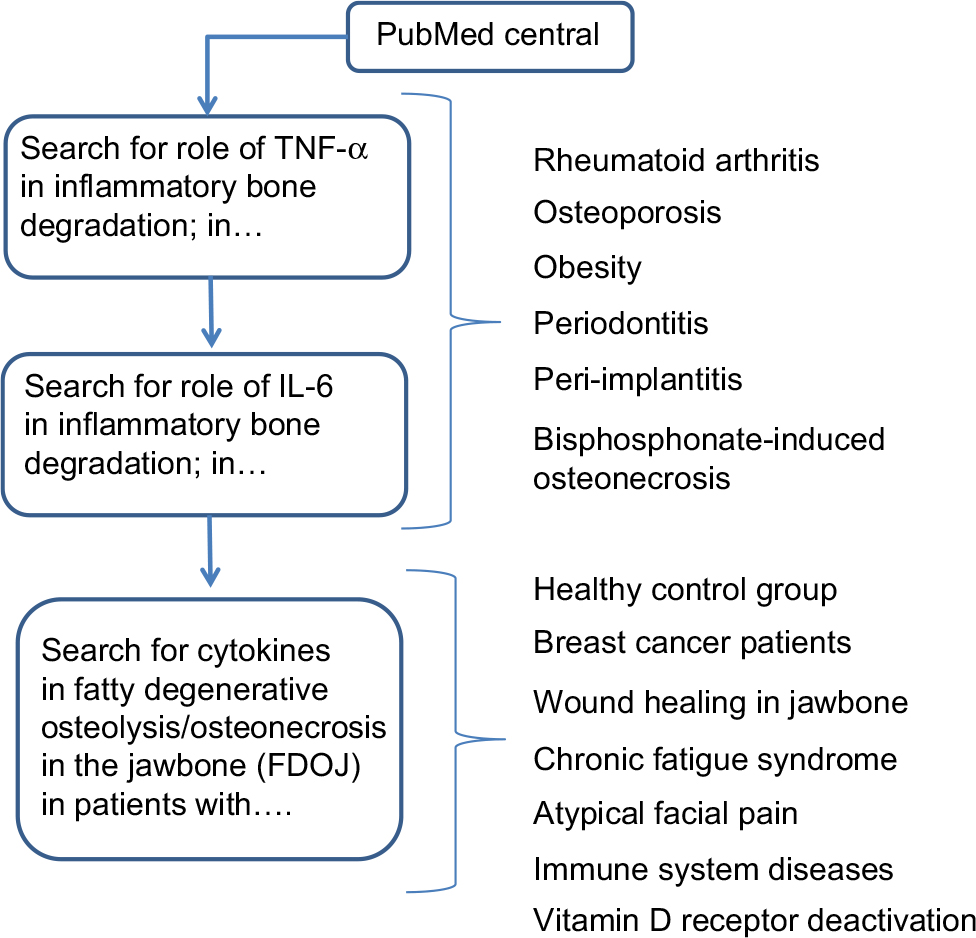 | Figure 1 Diagram flow displays the literature search strategy. Abbreviation: TNF-α, tumor necrosis factor-alpha. |
TNF-α and IL-6 in inflammatory bone degradation
Bone metabolism requires a delicate interplay that depends on the interaction of several factors. Osteoclasts (OCs) and osteoblasts (OBs) determine the structure and strength of the skeletal mass based on their respective roles in bone resorption and regeneration. Bone remodeling is a spatially coordinated, life-long process whereby old bone is removed by OCs and replaced by new bone through bone-forming OBs. Bone resorption depends on receptor activator of nuclear factor kappa-B ligand (RANKL), a cytokine in the TNF family. RANKL is essential for the formation, activity, and survival of OCs in normal and pathological conditions of bone remodeling. RANKL binds to the receptor activator of nuclear factor kappa-B (RANK) on the surface of OC precursor cells. The catabolic effects of RANKL are hindered by osteoprotegerin (OPG), an enzyme and another member of the TNF receptor family. OPG binds to RANKL, thus preventing the activation of the associated receptor, RANK. Thus, OC activity is dependent, at least in part, on the relative balance between RANKL and OPG (Figure 2). Simultaneously, TNF-α and IL-6 play a role in bone resorption and OC genesis in OB inhibition.
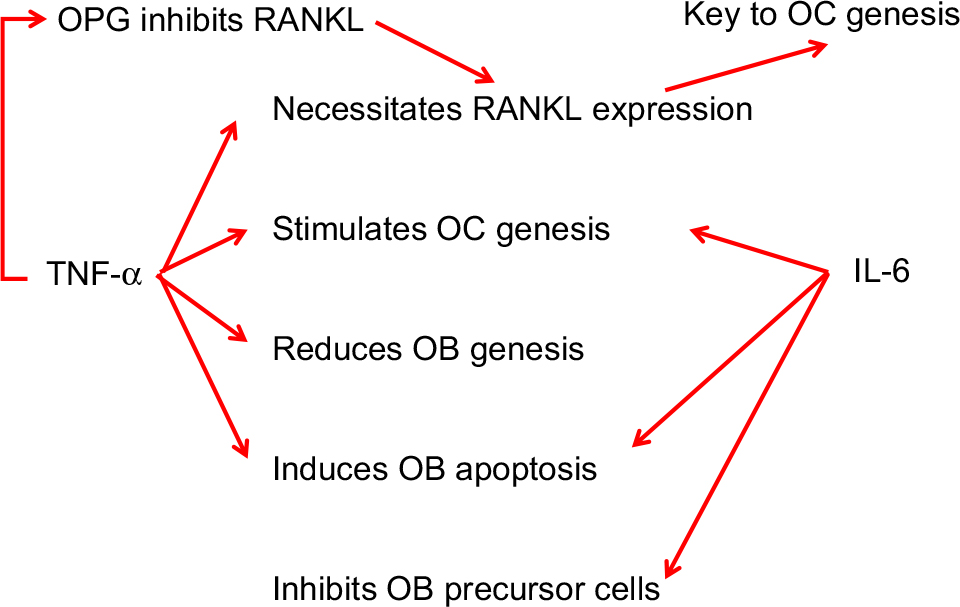 | Figure 2 Role of TNF-α and IL-6 in inflammatory bone degradation. Abbreviations: OB, osteoblast; OC, osteoclast; OPG, osteoprotegerin; TNF-α, tumor necrosis factor-alpha. |
Bone resorption by TNF-α and IL-6 in rheumatoid arthritis
One example of an osteoimmunological connection between inflammatory processes and osteoporosis is rheumatoid arthritis (RA). RA is an autoimmune disease characterized by inflammation of the synovial joint, leading to severe structural damage and bone destruction. IL-6 is one of the most abundant sera and synovial fluids in the inflamed joints of RA patients. Increased bone resorption is the most important pathogenetic mechanism in RA, featuring intra-articular bone erosion and joint damage. Inflammatory OC genesis and OC activation can be observed in cells obtained from the inflamed synovial fluid of patients with RA.4 Reduced bone density in the spine and hip and a higher prevalence of osteoporosis have been reported in RA patients.5 In early untreated RA, bone density is related to the presence and progression of joint damage.6 Autoimmune reactions include RANKL expression and subsequent OC genesis. Activated immune cells at sites of inflammation produce a wide range of proinflammatory and OC gene cytokines, resulting in bone erosion, osteitis, and peri-inflammatory bone loss. Local peri-inflammatory bone loss and osteitis occur early on and predict the erosive bone destruction in RA.7 In addition, peri-inflammatory bone formation is impaired, leading to the nonhealing of erosions, allowing for aggressive, local inflammation between synovitis, osteitis, and local bone loss. RANKL is highly expressed in the synovium of RA, and inflammatory bone damage primarily results from abnormally high RANKL expression. Therefore, OCs play a central role in the pathogenesis of RA.6,7
In addition to activated T lymphocytes and macrophages, RANKL-expressing B cells play a central role in the induction of bone erosion in RA.8 The interaction between immune and mesenchymal cells (MCs) in joints, including synovial fibroblasts that are characterized by their hyperactive and hyperproliferative properties that respond to an inflammatory environment, is paramount in rheumatoid inflammation.9 Synovial cells in RA overproduce both inflammatory cytokines and matrix-degrading enzymes. Synovial membrane hyperplasia is characterized by the hyperproliferation of synovial fibroblasts, as well as by mass infiltration of inflammatory immune cells, including CD4+ T cells and internal immune cells, in the affected joints.
In the development of RA, MCs orchestrate the systemic immune response that occurs in a local joint disorder to foster osteoimmunological connections. MCs contribute to Th17-mediated chronic inflammation (CI) by promoting the migration of Th17 cells to the inflamed joint, as well as the concomitant increase in IL-17 production. Th17-related cytokines enhance OC genesis, mainly via synovial fibroblasts. Thus, the interaction between immune MCs plays a key role in CI and bone destruction in RA. In particular, pathogenic, autoreactive immune cells migrate into joints and activate the resident in the joint MCs, such as synovial fibroblasts. Bone is thus a target of inflammation in RA. Monoclonal antibodies (MAb) to various proinflammatory cytokines and their receptors (such as TNF-α) are useful in preventing/reversing pone erosions and systemic osteoporosis.10 TNF-α, in turn, stimulates OC formation and activity through several mechanisms, including increased RANKL production. In addition, TNF-α upregulates CD40 formation in OBs and stromal cells, suppressing OPG. In summary, the cytokine TNF-α in RA is at the forefront of osteoimmunological bone damage, bone metabolism dysregulation, and a systemic immune response.
Bone resorption by TNF-α and IL-6 in osteoporosis
If bone and the immune system are functionally integrated through complex homeostatic networks, then osteoporosis can also be seen as a chronic, immune-mediated, inflammatory disease based on several osteoimmunological aspects. For instance, clinical and biological features divide osteoporosis into many other inflammatory conditions and other immune-mediated diseases.11 Osteoimmunology also promotes new pathogenetic and clinical interpretations of the osteoporosis, which is a well-known condition.7 For example, it is known that menopause leads to relevant changes in the immune system and skeletal structure due to estrogen breakdown. The estrogen deficiency induces a marked increase of inflammatory mediators, including IFN-γ, M-CSF, TNF-α, IL-1, IL-6, and IL-7, which drive the development of osteoporosis. The increased TNF-α production by activated T cells plays a central role in bone loss. In osteoporosis, cells that express RANKL and secrete OC-gene cytokines, primarily TNF-α, IL-1, IL-6, and IL-17, become active. These promote OC precursor formation, which enhances systemic inflammation and, via other inflammatory cells, leads to impaired OB proliferation and osteoporosis development. That the RANK/RANKL/OPG signaling pathway is central to the pathogenesis of osteoporosis is confirmed by the increased antiresorptive capacity of denosumab, an anti-RANKL MAb used in osteoporosis therapy.10 Inflammation leads to disturbances in the immunoskeletal interface.12 Excessive or abnormal immune activation induces osteoporosis, such as in autoimmune diseases, infections, and postmenopausal osteoporosis. This was eventually discovered to be a pathogenetic factor, even in the face of major osteoporotic risks such as age and estrogen deficiency.13,14
Disrupted OC and OB activity, dysregulated of bone turnover, and consequent increases in bone resorption and osteoporosis characterize disturbed bone metabolism. Activated immune cells at sites of inflammation produce a broad spectrum of proinflammatory and OC-genic cytokines, resulting in bone erosions, osteitis, and peri-inflammatory and systemic bone loss. Osteoporosis has long been regarded as the consequence of the estrogen breakdowns associated with menopause. The fact that immune cells are involved in the bone changes related to menopause has led to a shift in the concept of osteoporosis as a function of osteoimmunology, which is currently regarded as an alternative inflammatory disease.15 Postmenopausal osteoporosis is a clear example of the mutual influence of the immune system, as well as of the bone and endocrine systems. Like many other hormones, estrogen – in addition to specific breast and reproductive systems – also has receptors on immune cells, bones, and bone marrow precursors. These effects lead to the expansion of activated T lymphocytes, resulting in the overproduction of inflammatory cytokines and chronic OC stimulation, which is responsible for bone loss and increased fracture risk.16 Inflammation-induced catabolic signals enhance OB and muscle cell apoptosis, causing both osteoporosis and sarcopenia.16 Since osteoporosis was not considered an inflammatory condition in the past, osteoimmunology represents a new approach that warrants further exploration.17 The stimulation of OC genesis induced by the high concentrations of RANKL and TNF-α leads to bone resorption. In addition, TNF-α reduces bone-tissue formation by increasing OB apoptosis, thereby decreasing OB activity (see Figure 1). Osteoporosis can thus be considered a systemic model of integrated cooperative signaling pathways and cytokines, with primary involvement of TNF-α and IL-6. The proinflammatory cytokines TNF-α and IL-6 are crucial in osteoporosis, as they contribute to acute and CI and are potent triggers of bone resorption.17
The role of TNF-α and IL-6 in obesity
Obesity is a potential risk factor for osteoporosis. In this case, the most important pathogenetic mechanism that leads to alterations in bone tissue also appears to be an inflammation.18 Thus, from this point of view, osteoporosis might be considered an immune-mediated disease in which immune activation by the induction of cytokine production and inflammation results in decreased bone remodeling. Adipose tissue, primarily visceral adipose tissue, may increase bone resorption through the production of IL-6 and TNF-α, which stimulate OC activity by regulating the RANKL/RANK/OPG signaling pathway.19 The inflammatory TNF-α induces OB apoptosis and reduces osteoblastogenesis. The predominant bone marrow T cells are activated by CD8+ lymphocytes that secrete relatively high levels of effector cytokines, mainly TNF-α. Adipokines continue to modulate OB and OC activity. In obese individuals with increased visceral fat, there is a significant increase in several inflammatory markers, such as C-reactive protein, IL-1, IL-6, and TNF-1, which alter the quality of bone and make it more fragile. TNF-α and IL-6 also inhibit the production of adiponectin and adipocytes,20 further inhibiting the stimulation of OB proliferation, maturation, and function. Expansion of RANKL secretion and the inhibition of OPG secretion by OB are additional disruptive factors in bone formation. The stimulation of OC genesis induced by high concentrations of RANKL and TNF-α leads to bone resorption. In addition. TNF-α reduces the formation of bone tissue by enhancing OB apoptosis (Figure 2).
The role of TNF-α and IL-6 in maxillomandibular bone resorption and inflammatory oral medical conditions
Alveolar bone loss in periodontitis and the role of TNF-α and IL-6
The trigger for the inflammatory cascade is the subgingival biofilm in periodontitis, which is separate from material-related inflammation. Bacterial mediators that diffuse through the junctional epithelium (eg, lipopolysaccharides) activate macrophages that reside periodontally to secrete proinflammatory cytokines, particularly IL-1, TNF-α, and IL-6. These cytokines cause local activation of the vascular epithelium, which promotes the infiltration of other immune cells in the area of inflammation (chemotaxis). In periodontitis, the inflammatory processes that are initiated by the aforementioned cytokines result in tissue destruction and are of critical importance. IL-1, TNF-α, and IL-6 activate OCs by increasing RANKL expression, which are associated with alveolar bone resorption. In addition, TNF-α and IL-6 increase the release of tissue-degrading matrix metalloproteinase-8 (MMP-8) by fibroblasts and granulocytes, which is measurable from the tooth pocket and constitutes an important prognostic marker.21 The causal genetic focus of bone destruction and tooth loss in periodontitis is on TNF-α and IL-6. The progressive destructive lesion in periodontitis is characterized by a prominent response of B cells and plasma cells, high IL-1 and IL-6 levels, and periodontal tissue destruction, including alveolar bone loss.22 Osteolytic lesions in periodontitis are triggered by bacterial plaques in the gingival sulcus and on the tooth surface. TNF-α can stimulate OC genesis, while other cytokines stimulate RANKL expression, which leads to the formation of OC and enhances OC activity.23 TNF-α levels are upregulated in the gingival sulcus fluid at sites where bone and attachment loss has recently occurred.24–26 A cause-and-effect relationship between TNF-α and periodontitis-related bone loss was demonstrated. The administration of recombinant TNF-α accelerates periodontal destruction in a rat periodontitis model.27 Specifically, TNF-α and IL-6 have been associated with increased RANKL production, OC genesis, and bone resorption.28 Increased expression of IL-6 is found in the gingival sulcular fluid in periodontitis patients, as compared with healthy controls.29 In addition to TNF-α and IL-6, several chemokines have also been detected in the gingiva or gingival crevicular fluid, including RANTES/CCL5 (R/C). This chemokine is present in greater amounts in active periodontitis than in inactive periodontitis.30,31 That R/C participates in periodontal bone resorption is supported by findings that demonstrate the binding of R/C to the corresponding receptors, CCR1 and CCR5.32 Patients with generalized aggressive periodontitis showed significantly higher R/C levels in the gingival crevicular fluid when compared with healthy individuals. R/C levels were positively correlated with probing depth and clinical attachment loss, suggesting that R/C, associated with TNF-α and IL-6, may activate and recruit inflammatory and immune cells in the aggressive periodontal environment, thus playing a key role in this process.33
Bone degradation in peri-implantitis and the role of TNF-α and IL-6
The pathogenetic mechanisms are the same in periodontitis and peri-implantitis. In addition, bone degradation is aggravated by a possible titanium incompatibility and sensitization. This process is physiological, in so far as macrophages – upon contact with titanium oxide particles – react with the release of proinflammatory cytokines (essentially, TNF-α and IL-1). However, the extent of this immune response is highly individual by TNF-α and IL-6 overexpression. The intensity of cytokine release depends on genetic variants (polymorphisms) of the proinflammatory cytokines (IL-1 and TNF-α) involved, as well as of the anti-inflammatory cytokines (including the IL-1 receptor antagonist [IL-1ra]).34,35
Bone degradation in bisphosphonate-induced osteonecrosis and the role of TNF-α and IL-6
Osteoporosis, with a reduction of mineral content in the jawbone, is counteracted by bisphosphonate doses. Increasingly more problems in the jawbone occur in the form of bisphosphonate-induced pine necrosis (bisphosphonate-induced osteonecrosis of the jawbone [BIOJ]) in the treatment of bone-metastasizing tumors. BIOJ typically develops after the intravenous administration of bisphosphonates. The prognosis of such bone necrosis is poor; surgical interventions are contraindicated, and antibiotics are ineffective due to a lack of blood flow in the dead bone segment. The result is implant loss, tooth loosening, tooth loss, and loss of whole sections of the jawbone. In contrast to the aforementioned diseases, there are only a few studies in the literature that address the levels of TNF-α and IL-6 in BIOJ. Bisphosphonates are potent inhibitors of OC activity in both primary and secondary osteoporosis, as associated with autoimmune diseases.5,36 Autoimmune reactions induce RANKL expression and subsequent OC genesis. The OCs resorb the mineral matrix of the bone and release bone morphogenetic protein and insulin-like growth factors which, in normal cases, induce normal stem cells that promote OB differentiation and aid in the formation of new bone. If the function of OCs is inhibited too much, dying OCs will not be replaced and the capillary network of the bone will not be sustained, leading to bone necrosis.10
Although BIOJ bacterial colonization is frequent, it remains unclear whether infections play a primary causative role in this process or is secondary to the appearance of the lesion, and whether it initially occurs in bone or soft tissue.37 Due to bacterial infections of the necrotic bone components, as well as the accompanying inflammatory dissolution of the oral mucosa, a mediator profile can also be assumed for BIOJ, which is primarily supported by the inflammatory cytokines TNF-α and IL-6. The oral cavity is prone to traumatic injury and bacterial contamination given a thin mucosal layer that exists alongside abundant and diverse microbiological flora.38 A variety of anaerobic bacterial species were detected: Porphyromonas gingivalis was the predominant organism, followed by Fusobacterium nucleatum subspecies polymorphum. All samples additionally contained Actinomyces and Prevotella. Specifically, anaerobes play a fundamental role in the pathophysiology of BIOJ, similar to that found in osteoradionecrosis.39 The effects of bacteria-induced bone destruction and the interactions between the OCs and cytokines involved in this process should be further investigated.40,41 As pointed out by the Task Force Report of the American Society for Bone and Mineral Research,42 the differential diagnosis of BIOJ must specifically exclude other widespread intra-oral diseases, as there is a clear demarcation of various osteolyses in BIOJ, including osteoradionecrosis, neuralgia-inducing cavitational osteonecrosis (NICO), and bone tumors or metastases. A structural or etiological equation of “NICO” and BIOJ are excluded by the authors.42
Bone degeneration in fatty degenerative osteonecrosis of the jawbone
No other organ is subject to more invasive surgical procedures than the tooth–jaw region, such as tooth extractions in mixed dentition, operative wisdom tooth removal, root fillings on inflamed teeth, and dental implantation, all of which involve possible wound-healing disorders. The resulting impaired healing and insufficient new bone formation of dental bone, which take on the form of silent inflammation, are thus inevitable.43 Such jawbone cavitations are medullary areas that are characterized by dying or dead bone marrow and can remain asymptomatic for years as fatty degenerative osteolysis/osteonecrosis in the jawbone (FDOJ). In 1989, Bouquot introduced the term “neuralgia-inducing cavitational osteonecrosis (NICO)” into the literature to describe a chronic, osteolytic, medullary alteration in the jawbone with neurological side effects.44,45 This neglected FDOJ event is characterized by conspicuous R/C overexpression. Proinflammatory triggers lead to the stimulation of various networked signaling pathways that can lead to chronic diseases.46 The mesenchymal stem cells in the bone marrow of the jaw primarily differentiate into OBs and adipocytes; however, if this differentiation is disturbed, it can lead to a reduction in bone mass based on the number of increased adipocytes and decreased OBs.47 Osteolytic and osteoporotic processes, such as those that occur in FDOJ, are the result. These processes lead to the dissolution of the bony trabecular structures within the medullary cancellous bone of the jawbone, which is the essential morphological feature of FDOJ (Figure 3).
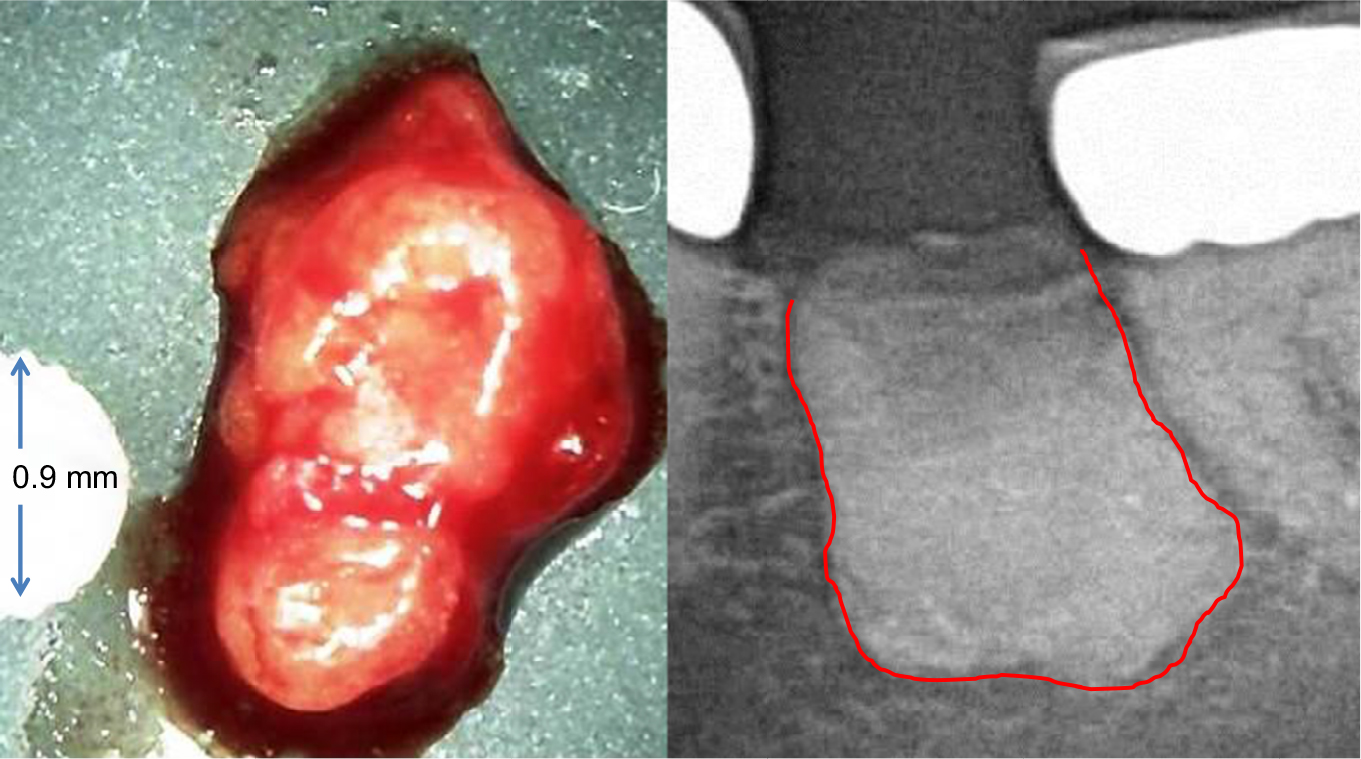 | Figure 3 The left panel shows a lump of fatty-degenerated medullary bone; the right panel shows the extent of osteonecrosis, made visible by contrast medium. |
Cytokine profile of fatty degenerative osteonecrosis/osteolysis in the jawbone
In previous publications, we visualized the cytokine profile of typical FDOJ lesions (n=31) for the first time using multiplex analysis; we then compared these cases with a healthy population (n=3).48 In the healthy control group, IL-6 levels averaged 26.58 pg/mL (±82.48), TNF-α levels averaged 1.21 pg/mL (±4.06), and R/C levels averaged 281.12 pg/mL (±637.24). Comparable values for healthy patients and normal jawbones were not available for comparative purposes in the literature. Comparing both cohorts, the results were quite striking: there were seven cytokines in the FDOJ cohort (n=31), which showed high amounts of IL-1ra, R/C, and FGF-2. R/C showed a median level of 3810.9 pg/mL with a standard deviation of 2566.9 and a 25th percentile of 2085.4. Thus, the mediator expressed in the mediator space of the jawbone was the chemokine R/C alone, while in the 31 examined bone samples, TNF-1 and IL-6 were found in one-third and one-tenth of healthy jawbone (HJB) cases, respectively. The study confirmed that FDOJ was able to produce the overexpression of the inflammatory messenger, R/C.
A small series of tests in a group of 23 breast cancer patients showed consistent findings in comparison to healthy bone (n=19); it was found that TNF-α levels decreased by one-third, while IL-6 decreased by one-tenth. R/C was found to be highly overexpressed in 23 disease cases. No changes in the expression levels of other immune mediators were observed. These data provide compelling evidence to confirm that FDOJ produces high levels of R/C, a cytokine that is implicated in breast cancer and metastasis.49 (Figure 4).
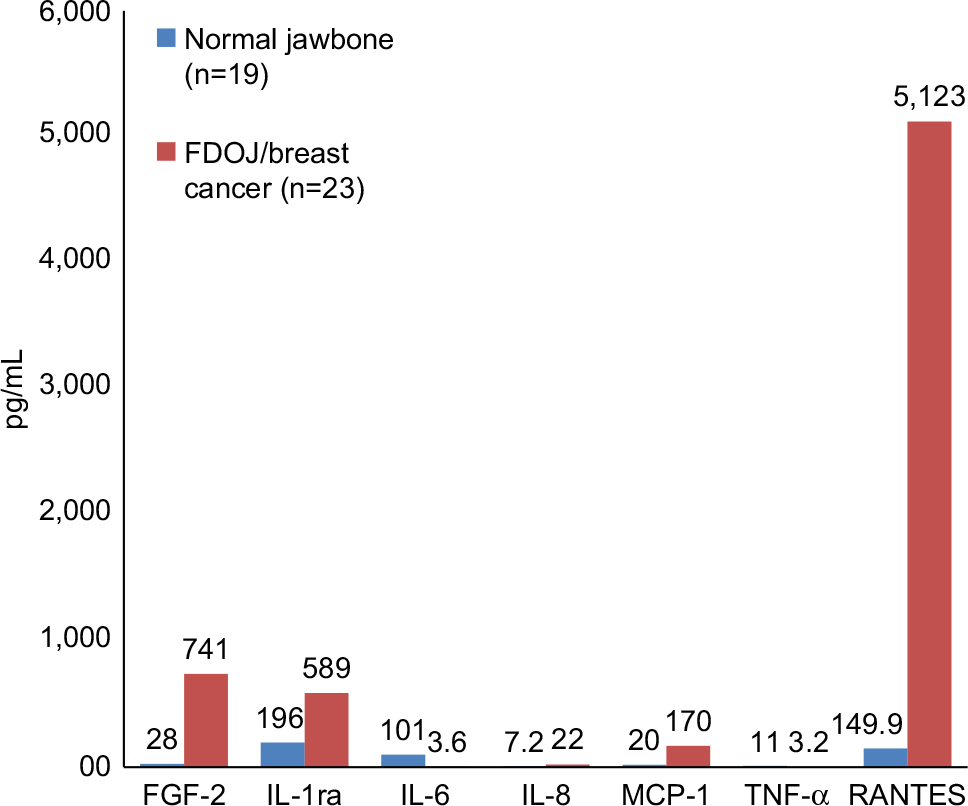 | Figure 4 Seven cytokines in FDOJ (pg/mL) in breast cancer patients (n=23; red columns) when compared with healthy jawbone (controls; n=19; blue columns). Abbreviations: FDOJ, fatty degenerative osteolysis/osteonecrosis in the jawbone; IL-1ra, IL-1 receptor antagonist; TNF-α, tumor necrosis factor-alpha. |
In a further study on wound healing in jawbone,43 we examined samples of jawbone for seven cytokines by multiplex analysis in three groups of jawbone areas. Specimens obtained from 16 patients were analyzed in areas of former surgery in the retromolar wisdom tooth area; other specimens from 16 patients were analyzed in the jawbone, apically of teeth with root fillings; and specimens from 19 patients were from HJB. Each of the retromolar and apical jawbone samples showed clinically fatty-degenerated and osteonecrotic medullary changes. All FDOJ samples showed R/C as the only extremely overexpressed cytokine. FDOJ cohorts showed a 30-fold mean overexpression of R/C when compared with healthy controls. As R/C is discussed in literature as a possible contributor to inflammatory diseases, we hypothesize that FDOJ in areas of improper and incomplete wound healing in the jawbone might act as hyperactivated signaling pathways, while serving as an unknown source of “silent inflammation”.
Another study elucidated the question of whether FDOJ contributes to the development of chronic fatigue syndrome (CFS) by the induction of inflammatory mediators.50 To clarify neurological interrelations, specimens from 21 CFS patients were analyzed in areas of former surgery in the retromolar wisdom tooth area. Each of the 21 retromolar jawbone samples showed clinical FDOJ, and all showed high expression levels of R/C. FDOJ cohorts showed a 30-fold mean overexpression of RANTES when compared with healthy controls. We thus hypothesize that FDOJ in areas of improper and incomplete wound healing in the jawbone may hyperactivate R/C signaling pathways and may represent an unknown cause, thus far, for the development of CFS (Figure 5).
 | Figure 5 In 21 patients with chronic fatigue syndrome, areas of former surgery in the retromolar wisdom tooth area showed clinical FDOJ and a 30-fold mean overexpression of RANTES/CCL5 when compared with healthy controls (n=19). Abbreviations: CFS, chronic fatigue syndrome; FDOJ, fatty degenerative osteolysis/osteonecrosis in the jawbone; IL-1ra, IL-1 receptor antagonist; MV, medium value; TNF-α, tumor necrosis factor-alpha. |
In 15 patients with atypical facial pain (AFP) and trigeminal neuralgia (TRN), we examined FDOJ samples to assess the expression of seven cytokines by multiplex analysis and compared these results with HJBs to elucidate the possible causative role of chronic subclinical inflammation in the jawbone of patients with AFP and TRN in terms of the local overexpression of R/C.51 Proinflammatory chemokines like R/C desensitize µ-opioid receptors in the periphery sensory neurons; it has been suggested that R/C modifies the nociceptive reaction. As a result, each of these FDOJ samples exhibited R/C as the only highly overexpressed cytokine. The FDOJ cohort with AFP/TRN showed a mean 30-fold overexpression of R/C when compared with HJBs. We here hypothesize that the surgical clearing of FDOJ might diminish R/C signaling pathways in neurons and contribute to resolving chronic neurological pain in AFP/TRN patients.
In an additional paper, we examined the relationship between immunology and bone metabolism among 43 FDOJ patients and those with immune system diseases.52 Recent research on vitamin D indicates that autoimmune diseases may be correlated with vitamin D receptor deactivation (VDR-deac), which occurs when the receptor is no longer able to transcribe antimicrobial agents. Excess 1,25-dihydroxyvitamin D (1,25D) is not converted to 25-hydroxyvitamin D (25D); thus, high 1,25D levels may be accompanied by low 25D values. Since 1,25D promotes OC activity and may thereby cause osteoporosis, FDOJ may also be associated with VDR-deac. The clinical data here demonstrate the interaction between VDR-deac and proinflammatory R/C overexpression in FDOJ patients (Figure 6).
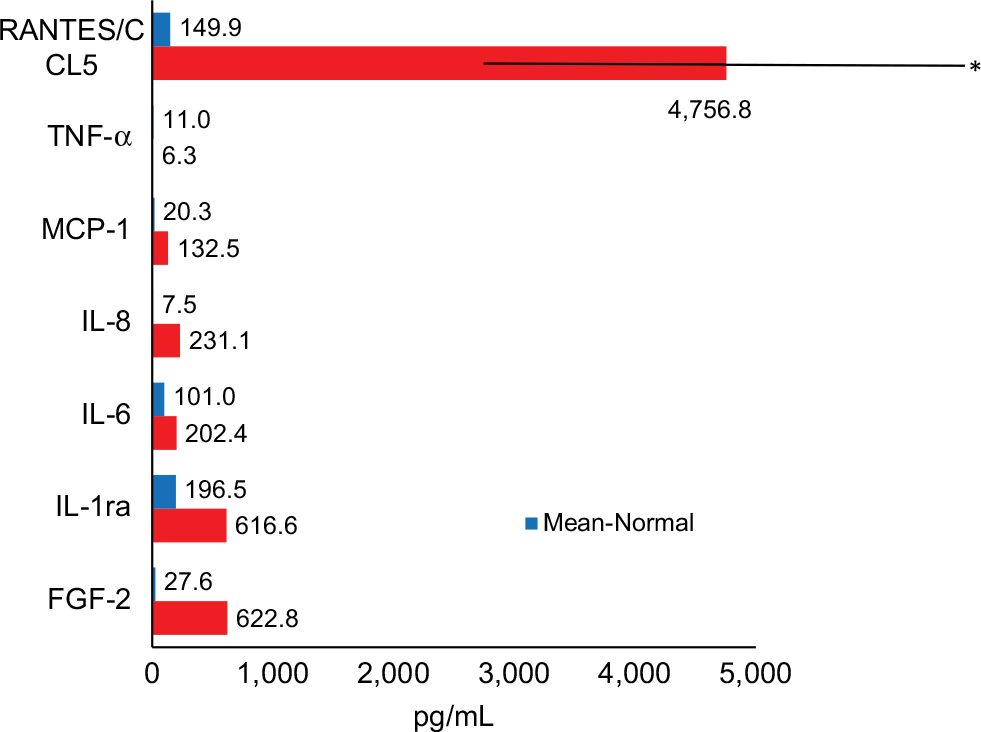 | Figure 6 Forty-three FDOJ patients with vitamin D receptor deactivation showed high RANTES/CCL5 overexpression when compared with healthy jawbone (n=19). Abbreviations: FDOJ, fatty degenerative osteolysis/osteonecrosis in the jawbone; IL-1ra, IL-1 receptor antagonist; TNF-α, tumor necrosis factor-alpha. |
Summary of FDOJ cytokine profiles from the literature
To summarize the expression profiles of the seven publications examined herein, the results are as follows: for the healthy control group (n=19), a mean IL-6 level of 101.0 pg/mL (±82.48) was documented, while that for TNF-α was 11.0 pg/mL (±4.06), and for R/C was 149.9 pg/mL (±637.24); for the immune system group, the mean IL-6 level was 0 pg/mL (±0), while that for TNF-α was 1.2 pg/mL (±4.06), and that for R/C was 2811.25 pg/mL (±6.37); for the breast cancer group, the mean IL-6 level was 0.9 pg/mL (±2.066), that for TNF-α was 0.0 pg/mL (±0), and that for R/C was 5122.73 pg/mL (±2987.85); for the X-ray group, the mean IL-6 level was 0 pg/mL (±0), while that for TNF-α was 0.0 pg/mL (±0), and that for R/C was 6518.67 pg/mL (±2154.80); for the wound-healing group, the mean IL-6 level was 1.3 pg/mL (±4.5), while that for TNF-α was 0.0 pg/mL (±0), and that for R/C was 4,294.81 pg/mL (±2260.88); for the trigeminal group, the mean IL-6 level was 0.23 pg/mL (±0.92), while that for TNF-α was 0.0 pg/mL (±0), and that for R/C was 4399.74 pg/mL (±2,745.54); for the CFS group, the mean IL-6 level was 0 pg/mL (±0), while that for TNF-α was 0.0 pg/mL (±0), and that for R/C was 5315.92 pg/mL (±2.660.72); for the vitamin D group, the mean IL-6 level was 0 pg/mL (±0), that for TNF-α was 0.0 pg/mL (±0), and that for R/C was 3285.54 pg/mL (±1829.28). Differences in the statistical workup of the published data can be explained by the different uses of means and medians. Crucial, however, are the dimensions of the different cytokine expressions. Figure 7 shows the summary of the cytokine profiles and the juxtaposition of TNF-α, IL-6, and R/C expression in FDOJ, as per the publications identified thus far in PubMed. The overexpressed R/C signaling in all seven sets of tests was significant, as were the underexpressed TNF-α and IL-6 levels in the FDOJ samples.
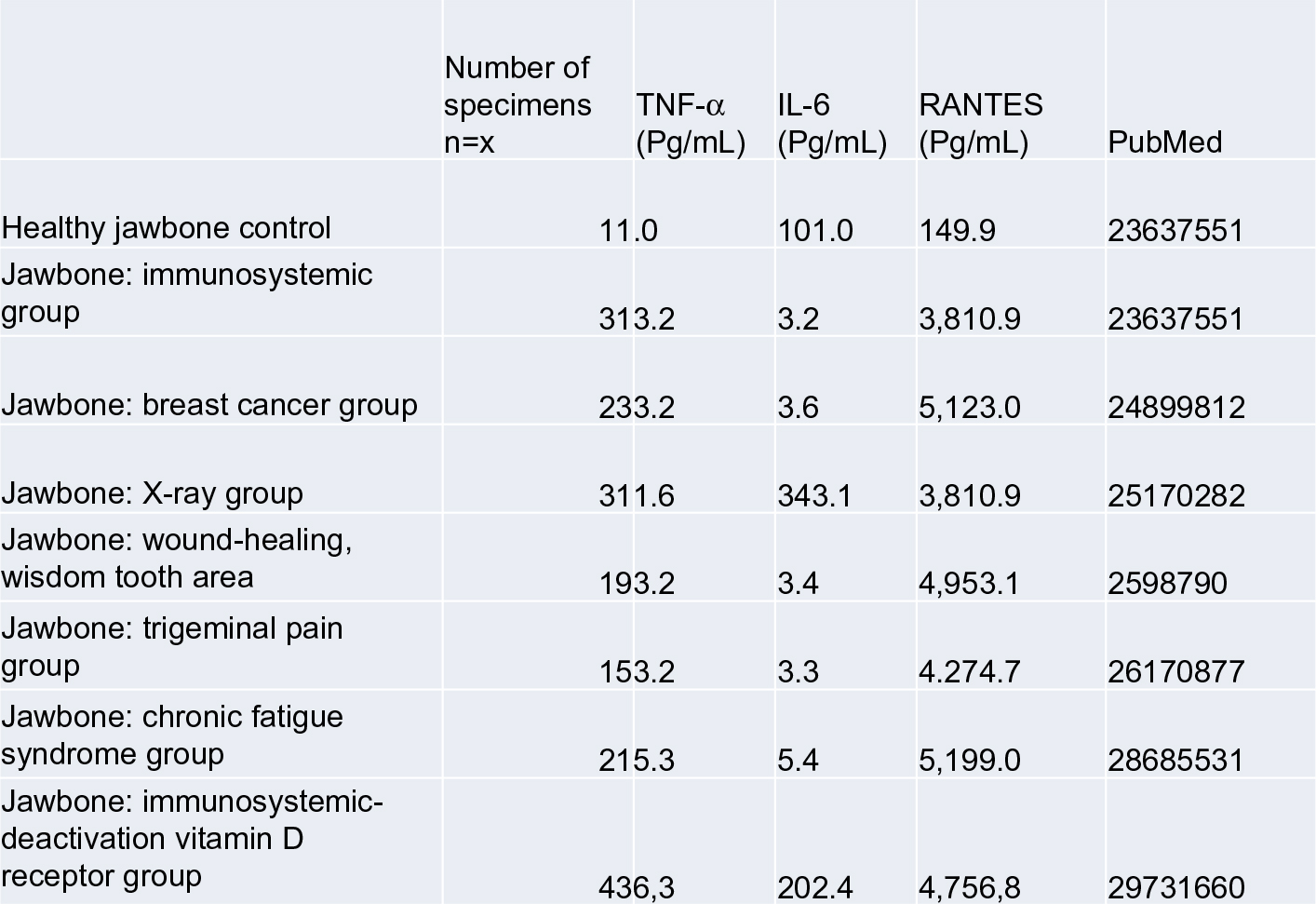 | Figure 7 Cytokine expression in FDOJ areas from seven different patient populations, as cited from PubMed-indexed publications. Abbreviations: FDOJ, fatty degenerative osteolysis/osteonecrosis in the jawbone; TNF-α, tumor necrosis factor-alpha. |
The summary of the cytokine profile and the statistical comparisons of TNF-α, IL-6, and R/C expression from the FDOJ samples obtained from different disease groups (Figure 8) show a significant downregulation of TNF-α and IL-6, and they also highlight the significant upregulation of R/C.
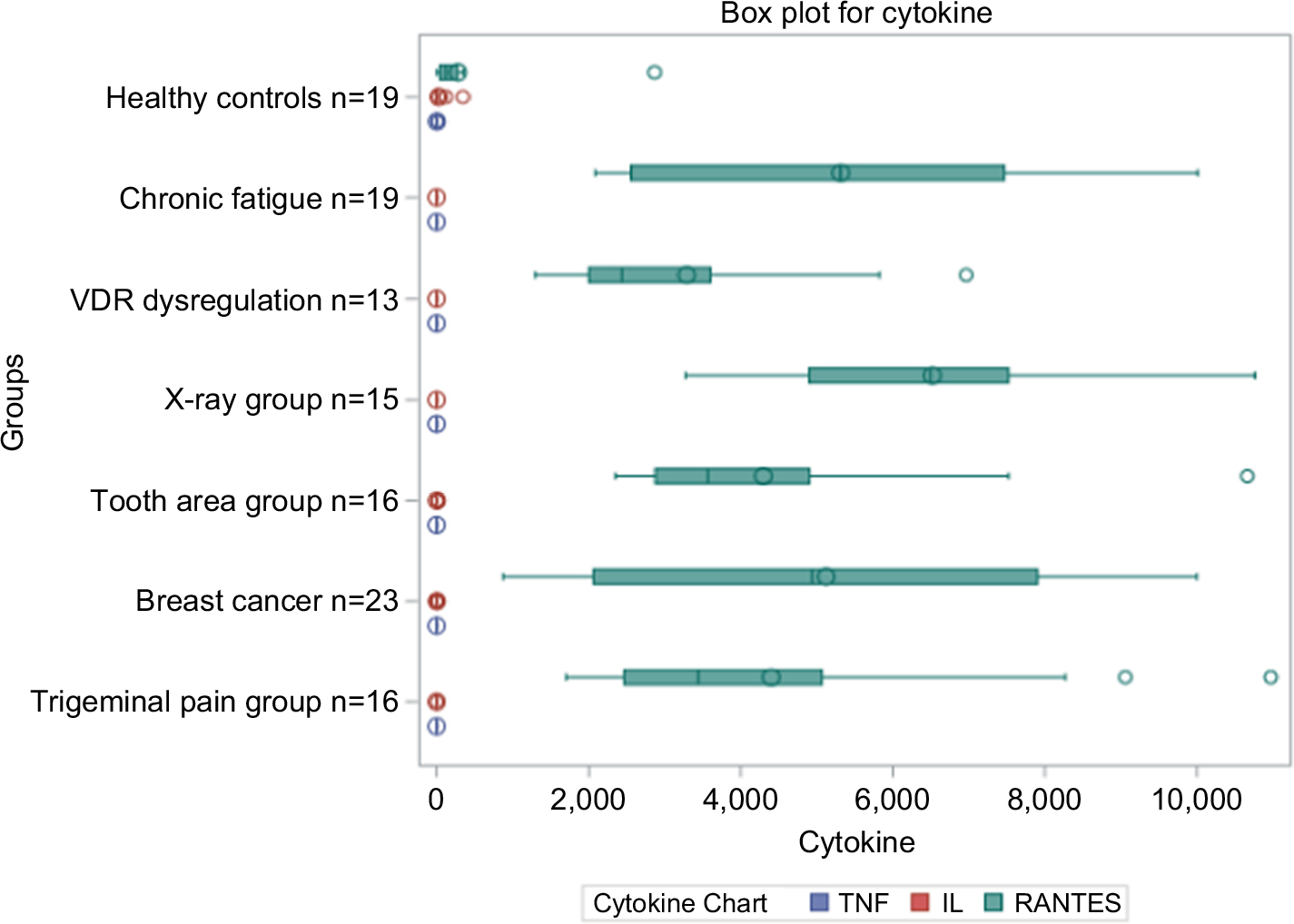 | Figure 8 Summary of the cytokine profiles and statistical comparison of TNF-α, IL-6, and RANTES/CCL5 expression from FDOJ samples into different disease groups; marked downregulation of TNF-α and IL-6 was observed, as was excessive upregulation of RANTES/CCL5. Abbreviations: FDOJ, fatty degenerative osteolysis/osteonecrosis in the jawbone; TNF-α, tumor necrosis factor-alpha; VDR, vitamin D receptor. |
Discussion: in bone resorption, except for FDOJ, TNF-a is the primary inflammatory cytokine
Bone-resorptive disease and the key role played by TNF-α and IL-6
An extensive literature search showed that TNF-α and IL-6 play key inflammatory roles in all disease patterns associated with osteonecrosis. Specifically, TNF-α promotes RANKL expression and thus impacts bone resorption, while TNF-α inhibits the maturation of OB progenitor cells and OB activity. TNF-α also induces OB apoptosis, inhibits the genes involved in bone formation such as the 1,25-(OH)2 D3 receptor, stimulates OC formation and OC activity via increased RANKL production, and suppresses OPG. Endothelial cells that are activated by TNF-α attract circulating OC precursor cells to sites of inflammation, as driven by the expression of cell adhesion molecules, which become active OCs in bone resorption upon stimulation of RANKL53 (see Figure 1).
The proinflammatory cytokine IL-6 is secreted by macrophages, fibroblasts, epithelial cells, and T-cells. IL-6 is involved in the direct activation of OCs in bone resorption. IL-6 further increases MMP-8 release in epithelial cells, granulocytes, fibroblasts, and macrophages. In vitro studies show that IL-6 trans-signaling promotes OC genesis by increasing RANKL expression in OBs and T-cells.54,55 IL-6 has also been shown to inhibit OB precursor cell proliferation while inducing apoptosis at the late stages of the differentiation process.6 Animal models highlight the deleterious effect of IL-6 on bone. Transgenic mice overexpressing IL-6 show increased bone turnover with reduced OB and increased OC numbers, resulting in osteopenia.56 In FDOJ, the low IL-6 level results in less bone-protective influences and to osteolysis, as high IL-6 levels promote the development of OPG and thus would be anti-resorptive.
Bone-resorptive diseases and the role of overexpressed RANTES/CCL5 despite low levels of TNF-α and IL-6
With respect to R/C, the literature shows that it plays a small role in bone resorption. Although the chemokine R/C plays a role as an inflammatory factor in some diseases, it is only directly associated with disturbed bone metabolism in nonpainful RA.8 In contrast, massive bone resorption is found in FDOJ, which presents a unique cytokine profile that is completely different from other bone resorptive conditions. Specifically, TNF-α secretion and IL-6 secretion are both below healthy levels in FDOJ bone, while R/C is overexpressed about 30-fold, as our studies show.48 The unique cytokine profile of R/C overexpression and bone dissolution in FDOJ areas with disrupted bone remodeling by OBs and OCs are explained by the following processes:
- Macrophages and fibroblasts produce R/C, MCP-1/CCL2, and IL-10, which are positively involved in OC resorption.
- Pathological increases of chemokine R/C is activated when OB stimulates chemotactic recruitment and RANKL formation of resorptive OC, exacerbating local osteolysis.
- R/C enhances RANKL-mediated OC formation from primary bone marrow cells.
- Consequently, R/C chemokines (a source of FDOJ) may be involved in inflammatory and cancerous bone loss, as they are elevated in the following skeletal disorders: RA, periodontitis, implant loosening, and osteolytic tumors.57
Osteobiology of R/C overexpression in FDOJ areas
Does R/C overexpression in FDOJ areas make biological sense? R/C overexpression appears to have a repair function in the osteolytic structures of an FDOJ, particularly considering the effect of R/C on OB (Figure 9):
 | Figure 9 Osteobiology of RANTES/CCL5 overexpression. |
Since R/C secretion in OC is physiologically highly regulated by Ca2+ ions, increased Ca2+ released from the bone matrix during bone resorption induces R/C secretion of both OBs and OCs. This upregulation of R/C expression of OB (OB serves as the source of R/C and other chemokines in various pathological conditions) specifically promotes OC migration to the site of resorption as R/C controls the migration of bone cells to the site of osteolytic damage.58 In the form of a mutual amplification loop, the expression of R/C and its receptors in both OB and OC allow for the expressed R/C to function in both autocrine and paracrine mechanisms in bone.59
Summary
The data on cytokine patterns in FDOJ,43,48–53 published by us for the first time, demonstrate extremely low TNF-α values and make “inflammatory erosion” by TNF-α-based osteolytic jawbone formation unlikely in the long term. Bacterial events, which are usually associated with high levels of TNF-α and IL-6 expression, could be ruled out within the FDOJ area.60 Thus, R/C overexpression alone is involved in the characteristic and bone-degrading element of an FDOJ. TNF-α and IL-6 signaling in all other bone resorptive processes are excluded (Figure 10).
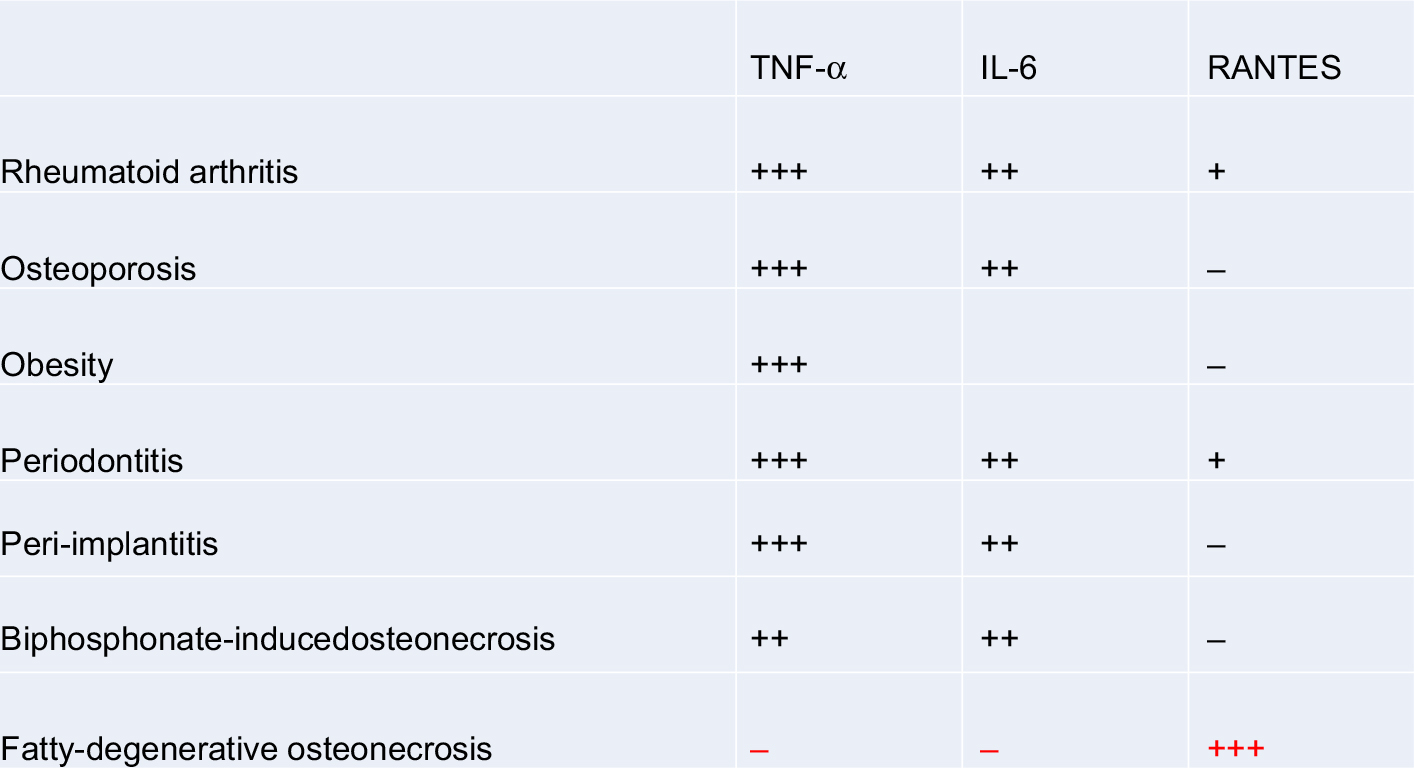 | Figure 10 FDOJ is the only bone resorption process to show R/C overexpression; when compared with other bone resorption processes, FDOJ shows a corresponding reduction in TNF-α and IL-6 expression, while all other bone resorption-related diseases are characterized by TNF-α and IL-6 overexpression. Notes: black + indicates high expression; black +++ indicates extreme high expression; black – indicates no expression; red +++ indicates extraordinary high expression. Abbreviations: FDOJ, fatty degenerative osteolysis/osteonecrosis in the jawbone; R/C, RANTES/CCL5; TNF-α, tumor necrosis factor-alpha. |
Conclusion
Only in FDOJ areas emerge typical patterns, which are different from all other mediator-related signaling patterns found in other bone resorptive processes. This renders FDOJ as a qualitatively unique inflammatory event, as the literature does not describe any other osteolytic disease equivalent mediator-related signaling patterns in the organism. We identified the chemokine R/C as a neglected key factor; however, at the same time, it is the dominant carrier of a chronic, low-grade inflammation within a “maxillomandibular osteoimmunology”. The complete elucidation of these highly complex interactions requires further research. For the benefit of systemically ill patients, medicine and dentistry should thus devote greater attention to “maxillomandibular osteoimmunology” and the associated characteristic signaling patterns.
Acknowledgment
English-language editing of this manuscript was provided by Journal Prep.
Disclosure
The authors report no conflicts of interest in this work.
References
Greenblatt MB, Shim JH. Osteoimmunology: a brief introduction. Immune Netw. 2013;13(4):111–115. | ||
Anisman H. Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. J Psychiatry Neurosci. 2009;34(1):4–20. | ||
Geiss A, Varadi E, Steinbach K, Bauer HW, Anton F. Psychoneuroimmunological correlates of persisting sciatic pain in patients who underwent discectomy. Neurosci Lett. 1997;237(2–3):65–68. | ||
Greisen SR, Einarsson HB, Hvid M, Hauge EM, Deleuran B, Kragstrup TW. Spontaneous generation of functional osteoclasts from synovial fluid mononuclear cells as a model of inflammatory osteoclastogenesis. APMIS. 2015;123(9):779–786. | ||
Jones D, Glimcher LH, Aliprantis AO. Osteoimmunology at the nexus of arthritis, osteoporosis, cancer, and infection. J Clin Invest. 2011;121(7):2534–2542. | ||
Deal C. Bone loss in rheumatoid arthritis: systemic, periarticular, and focal. Curr Rheumatol Rep. 2012;14(3):231–237. | ||
Geusens P, Lems WF. Osteoimmunology and osteoporosis. Arthritis Res Ther. 2011;13(5):242. | ||
Karmakar S, Kay J, Gravallese EM. Bone damage in rheumatoid arthritis: mechanistic insights and approaches to prevention. Rheum Dis Clin North Am. 2010;36(2):385–404. | ||
Hot A, Miossec P. Effects of interleukin (IL)-17A and IL-17F in human rheumatoid arthritis synoviocytes. Ann Rheum Dis. 2011;70(5):727–732. | ||
Brown JP, Roux C, Ho PR, et al. Denosumab significantly increases bone mineral density and reduces bone turnover compared with monthly oral ibandronate and risedronate in postmenopausal women who remained at higher risk for fracture despite previous suboptimal treatment with an oral bisphosphonate. Osteoporos Int. 2014;25(7):1953–1961. | ||
Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7(4):292–304. | ||
D’Amelio P, Sassi F. Osteoimmunology: from mice to humans. Bonekey Rep. 2016;5:802. | ||
De Martinis M, Di Benedetto MC, Mengoli LP, Ginaldi L. Senile osteoporosis: is it an immune-mediated disease? Inflamm Res. 2006;55(10):399–404. | ||
Rauner M, Sipos W, Thiele S, Pietschmann P. Advances in osteoimmunology: pathophysiologic concepts and treatment opportunities. Int Arch Allergy Immunol. 2013;160(2):114–125. | ||
Weitzmann MN. The role of inflammatory cytokines, the RANKL/OPG axis, and the immunoskeletal interface in physiological bone turnover and osteoporosis. Scientifica (Cairo). 2013;2013:125705. | ||
Li H, Lu Y, Qian J, et al. Human osteoclasts are inducible immunosuppressive cells in response to T cell-derived IFN-γ and CD40 ligand in vitro. J Bone Miner Res. 2014;29(12):2666–2675. | ||
Ginaldi L, Mengoli LP, De Martinis M. Osteoporosis, inflammation and ageing. In: Fu-lop T, Franceschi C, Hirokawa K, Pawelec G, editors. Handbook on Immunosenescence: Basic Understanding and Clinical Applications. New York: Springer-Verlag Press; 2009:1329–1352. | ||
Zupan J, Jeras M, Marc J. Osteoimmunology and the influence of pro-inflammatory cytokines on osteoclasts. Biochem Med (Zagreb). 2013;23(1):43–63. | ||
Sigl V, Penninger JM. RANKL/RANK – from bone physiology to breast cancer. Cytokine Growth Factor Rev. 2014;25(2):205–214. | ||
Dispirito JR, Mathis D. Immunological contributions to adipose tissue homeostasis. Semin Immunol. 2015;27(5):315–321. | ||
Chen HY, Cox SW, Eley BM, Mäntylä P, Rönkä H, Sorsa T. Matrix metalloproteinase-8 levels and elastase activities in gingival crevicular fluid from chronic adult periodontitis patients. J Clin Periodontol. 2000;27(5):366–369. | ||
Ohlrich EJ, Cullinan MP, Seymour GJ. The immunopathogenesis of periodontal disease. Aust Dent J. 2009;54(Suppl 1):S2–S10. | ||
Graves DT, Oates T, Garlet GP. Review of osteoimmunology and the host response in endodontic and periodontal lesions. J Oral Microbiol. 2011;3:5304. | ||
Salvi GE, Brown CE, Fujihashi K, et al. Inflammatory mediators of the terminal dentition in adult and early onset periodontitis. J Periodontal Res. 1998;33(4):212–225. | ||
Kurtiş B, Tüter G, Serdar M, et al. Gingival crevicular fluid levels of monocyte chemoattractant protein-1 and tumor necrosis factor-alpha in patients with chronic and aggressive periodontitis. J Periodontol. 2005;76(11):1849–1855. | ||
Boström L, Linder LE, Bergström J. Clinical expression of TNF-alpha in smoking-associated periodontal disease. J Clin Periodontol. 1998;25(10):767–773. | ||
Gaspersic R, Stiblar-Martincic D, Osredkar J, Skaleric U. Influence of subcutaneous administration of recombinant TNF-alpha on ligature-induced periodontitis in rats. J Periodontal Res. 2003;38(2):198–203. | ||
Bar-Shavit Z. Taking a toll on the bones: regulation of bone metabolism by innate immune regulators. Autoimmunity. 2008;41(3):195–203. | ||
Takeichi O, Haber J, Kawai T, Smith DJ, Moro I, Taubman MA. Cytokine profiles of T-lymphocytes from gingival tissues with pathological pocketing. J Dent Res. 2000;79(8):1548–1555. | ||
Gemmell E, Carter CL, Seymour GJ. Chemokines in human periodontal disease tissues. Clin Exp Immunol. 2001;125(1):134–141. | ||
Gamonal J, Acevedo A, Bascones A, Jorge O, Silva A. Characterization of cellular infiltrate, detection of chemokine receptor CCR5 and interleukin-8 and RANTES chemokines in adult periodontitis. J Periodontal Res. 2001;36(3):194–203. | ||
Garlet GP, Martins W, Ferreira BR, Milanezi CM, Silva JS. Patterns of chemokines and chemokine receptors expression in different forms of human periodontal disease. J Periodontal Res. 2003;38(2):210–217. | ||
Emingil G, Atilla G, Hüseyinov A. Gingival crevicular fluid monocyte chemoattractant protein-1 and RANTES levels in patients with generalized aggressive periodontitis. J Clin Periodontol. 2004;31(10):829–834. | ||
Olmedo D, Fernández MM, Guglielmotti MB, Cabrini RL. Macrophages related to dental implant failure. Implant Dent. 2003;12(1):75–80. | ||
Jacobi-Gresser E, Huesker K, Schütt S. Genetic and immunological markers predict titanium implant failure: a retrospective study. Int J Oral Maxillofac Surg. 2013;42(4):537–543. | ||
Schwarz P, Jørgensen NR, Abrahamsen B. Status of drug development for the prevention and treatment of osteoporosis. Expert Opin Drug Discov. 2014;9(3):245–253. | ||
Hansen T, Kunkel M, Weber A, James Kirkpatrick C. Osteonecrosis of the jaws in patients treated with bisphosphonates – histomorphologic analysis in comparison with infected osteoradionecrosis. J Oral Pathol Med. 2006;35(3):155–160. | ||
Cunha BA. Osteomyelitis in elderly patients. Clin Infect Dis. 2002;35(3):287–293. | ||
Støre G, Eribe ER, Olsen I. DNA-DNA hybridization demonstrates multiple bacteria in osteoradionecrosis. Int J Oral Maxillofac Surg. 2005;34(2):193–196. | ||
Russo TA. Agents of actinomycosis. In: Mandell GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. 4th ed. New York: Churchill Livingstone; 1995:2280–2288. | ||
Nair SP, Meghji S, Wilson M, Reddi K, White P, Henderson B. Bacterially induced bone destruction: mechanisms and misconceptions. Infect Immun. 1996;64(7):2371–2380. | ||
Khosla S, Burr D, Cauley J, et al; American Society for Bone and Mineral Research. Bisphosphonate-associated osteonecrosis of the jaw: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2007;22(10):1479–1491. | ||
Lechner J, Baehr von V. Chemokine RANTES/CCL5 as an unknown link between wound healing in the jawbone and systemic disease: is prediction and tailored treatments in the horizon? EPMA J. 2015:6(1):10. | ||
Bouquot JE, McMahon RE, Glueck CJ. Bone marrow edema: mild or nascent variant of ischemic bone disease. J Oral Maxillofac Surg. 2008;66(1):205–208. | ||
Neville B, Damm DD, Allen C, Bouquot J. Oral and Maxillofacial Pathology. 1st–3rd ed. St. Louis, MO: Saunders; 1995–2008. | ||
Townsend MJ, Mckenzie AN. Unravelling the net? Cytokines and diseases. J Cell Sci. 2000;113:3549–3550. | ||
Wang C, Meng H, Wang X, Zhao C, Peng J, Wang Y. Differentiation of bone marrow mesenchymal stem cells in osteoblasts and adipocytes and its role in treatment of osteoporosis. Med Sci Monit. 2016;22:226–233. | ||
Lechner J, von Baehr V. RANTES and fibroblast growth factor 2 in jawbone cavitations: triggers for systemic disease? Int J Gen Med. 2013;6:277–290. | ||
Lechner J, von Baehr V. Hyperactivated signaling pathways of chemokine RANTES/CCL5 in osteopathies of jawbone in breast cancer patients – case report and research. Breast Cancer (Auckl). 2014;8:89–96. | ||
Lechner J, Huesker K, Von Baehr V. Impact of RANTES from jawbone on chronic fatigue syndrome. J Biol Regul Homeost Agents. 2017;31(2):321–327. | ||
Lechner J, von Baehr V. Peripheral neuropathic facial/trigeminal pain and RANTES/CCL5 in jawbone cavitation. Evid Based Complement Alternat Med. 2015;2015:582520. | ||
Lechner J, Aschoff J, Rudi T. The vitamin D receptor and the etiology of RANTES CCL expressive fatty degenerative osteolysis of the jawbone an interface between osteoimmunology and bone metabolism. Int J Gen Med. 2018;11:155–166. | ||
Sućur A, Katavić V, Kelava T, Jajić Z, Kovačić N, Grčević D. Induction of osteoclast progenitors in inflammatory conditions: key to bone destruction in arthritis. Int Orthop. 2014;38(9):1893–1903. | ||
Palmqvist P, Persson E, Conaway HH, Lerner UH. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J Immunol. 2002;169(6):3353–3362. | ||
Wong PK, Quinn JM, Sims NA, van Nieuwenhuijze A, Campbell IK, Wicks IP. Interleukin-6 modulates production of T lymphocyte-derived cytokines in antigen-induced arthritis and drives inflammation-induced osteoclastogenesis. Arthritis Rheum. 2006;54(1):158–168. | ||
De Benedetti F, Rucci N, Del Fattore A, et al. Impaired skeletal development in interleukin-6-transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum. 2006;54(11):3551–3563. | ||
Yu X, Huang Y, Collin-Osdoby P, Osdoby P. CCR1 chemokines promote the chemotactic recruitment, RANKL development, and motility of osteoclasts and are induced by inflammatory cytokines in osteoblasts. J Bone Miner Res. 2004;19(12):2065–2077. | ||
Yano S, Mentaverri R, Kanuparthi D, et al. Functional expression of beta-chemokine receptors in osteoblasts: role of regulated upon activation, normal T cell expressed and secreted (RANTES) in osteoblasts and regulation of its secretion by osteoblasts and osteoclasts. Endocrinology. 2005;146(5):2324–2335. | ||
Bonecchi R, Galliera E, Borroni EM, Corsi MM, Locati M, Mantovani A. Chemokines and chemokine receptors: an overview. Front Biosci (Landmark Ed). 2009;14:540–551. | ||
Lechner J, Schuett S, von Baehr V. Aseptic-avascular osteonecrosis: local “silent inflammation” in the jawbone and RANTES/CCL5 overexpression. Clin Cosmet Investig Dent. 2017;9:99–109. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.