")
Back to Journals » Cancer Management and Research » Volume 12
Osimertinib for Front-Line Treatment of Locally Advanced or Metastatic EGFR-Mutant NSCLC Patients: Efficacy, Acquired Resistance and Perspectives for Subsequent Treatments
Authors Denis MG, Bennouna J
Received 16 October 2020
Accepted for publication 28 November 2020
Published 8 December 2020 Volume 2020:12 Pages 12593—12602
DOI https://doi.org/10.2147/CMAR.S218751
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmet Emre Eşkazan
Marc G Denis,1,2 Jaafar Bennouna2,3
1Department of Biochemistry and Molecular Biology, Nantes University Hospital, Nantes, France; 2INSERM U1232, CRCINA, Nantes, France; 3Thoracic Oncology Unit, Nantes University Hospital, Nantes, France
Correspondence: Marc G Denis
Department of Biochemistry and Molecular Biology, Nantes University Hospital, 9 Quai Moncousu, Nantes 44093, France
Tel +33 240 08 40 01
Email [email protected]
Abstract: Non-small cell lung cancer (NSCLC) is one of the most efficient models for precision medicine in oncology. The most appropriate therapeutic for the patient is chosen according to the molecular characteristics of the tumor, schematically distributed between immunogenicity and oncogenic addiction. For this last concept, advanced NSCLC with epidermal growth factor receptor (EGFR) mutation is one of the most illustrative models. EGFR-tyrosine kinase inhibitors (TKIs) are the therapeutic backbone for this type of tumor. The recent development of a third-generation TKI, osimertinib, has been a new step forward in the treatment of NSCLC patients. In this article, we first review the clinical development of osimertinib and highlight its efficacy results. We then present the most frequent tumor escape mechanisms when osimertinib is prescribed in first line: off-target (MET amplification, HER2 amplification, BRAF mutation, gene fusions, histologic transformation) and on-target mechanisms (EGFR mutation). Finally, we discuss subsequent biomarker-driven treatment strategies.
Keywords: lung cancer, EGFR mutation, targeted therapy, acquired resistance
Introduction
Over the past 20 years, therapeutic innovation in oncology, i.e. targeted therapies or immune checkpoint inhibitors, has literally exploded. Overall, the discovery of new anticancer drugs was prompted in part by the identification of predictive biomarkers, but also fostered by innovative designs for early-phase clinical trials. Consequently, international patient management guidelines evolve regularly to adapt to the use of new targets and new drugs.1–3 In particular, the development of EGFR inhibitors has played a major role in the evolution of targeted therapeutic approaches.4 Most EGFR-mutated NSCLC patients respond to first- and second-generation EGFR TKIs, but they develop resistance after an average of 1 year.5 The “gatekeeper” EGFR T790M mutation is the most commonly acquired resistance mechanism in these tumors and is described in 50–60% of patients at progression.6 Third-generation TKIs have been developed to target mutant EGFR harboring this T790M mutation. Osimertinib (AZD9291) is one of these. It is an oral, irreversible EGFR tyrosine kinase inhibitor that is selective for EGFR tyrosine kinase inhibitor–sensitizing mutations and the T790M resistance mutation.7,8 Its clinical development was initially performed in T790M patients treated in a second-line setting in the AURA clinical trials, subsequently being developed in a front-line setting.
The AURA Program
The AURA program, encompassing large Phase I and II trials, fulfilled the criteria for a faster drug development process in oncology.9 The study design was qualified as adaptive and sequential. Patients were eligible if they had EGFR-mutated NSCLC, progressing after an EGFR-TKI and expressing the T790M mutation in most cases. Initially, patients were enrolled in a typical phase I schedule with starting doses at 20 mg, doubling in increments to 240 mg. Overall, 31 patients were treated, 6 patients at each dose level of 20 mg, 40 mg, 80 mg, 160 mg, and 7 patients at 240 mg. The protocol anticipated expansion cohorts in each subgroup in case of objective response. Consequently, the 5 initial cohorts were expanded for a total of 222 patients. The EGFR T790M mutation was detected by tumor tissue biopsy in 62% (138/222) of those patients. It was not detected, or unknown, in 62 patients (28%) and 22 patients (10%), respectively. The ORR was 61% for patients with EGFR T790M with a very high rate of disease control (95%) and a median PFS of 9.6 months (Table 1). The absence of the T790M mutation led to a low ORR, 21% with a Disease Control Rate (DCR) of 61% and a median PFS of 2.8 months. Based on these data, the Phase II extension of the AURA study screened 401 patients with EGFR-mutated NSCLC progressing after first- or second-generation EGFR-TKIs.10 From 327 tissue samples, 207 were positive for the T790M mutation and finally 201 patients received osimertinib at the recommended dose of 80 mg. Efficacy results were closed to the phase I data with an ORR of 62%, with no difference between second and third-line or more (62% and 61%), and a DCR of 90%. The median PFS was 12.3 months. Though ORRs were closed between patients with exon 19 deletion and L858R (64% and 57%), the median PFS appeared to be a little better in the first subgroup, 12.5 versus 9.6 months. The AURA 2 is a multicenter Phase 2 study involving the same eligibility criteria as the former study: 472 patients were screened, and 210 received osimertinib.11 The ORR was 70% for the population evaluable for response (n=199). Patients with tumors co-occurring with EGFR exon 19 deletion have a higher objective response rate than those with EGFR L858R mutation, 77% versus 59%. The median PFS was 9.9 months and the median duration of response was 11.4 months. Prescription of osimertinib, in second and later-line settings did not impact the ORR (73% and 69%). A long-term follow-up was reported for both studies giving a median OS of 24.3 months for the AURA extension and 28.3 months for AURA 2.12 In the pooled analysis of the two studies, the median OS was clearly higher in patients with exon 19 deletion compared to L858R mutation, 29.1 and 21.4 months, respectively.12 The same pooled data also confirmed the strong and sustained efficacy of osimertinib in brain metastases.13 A total of 128 patients were included with Central Nervous Lesions (CNS), 78 non measurable and 50 measurable. The CNS ORR was 54% (27/50) with a DCR of 92%. Osimertinib efficacy was also observed in the subgroup of patients with leptomeningeal metastases as demonstrated in the BLOOM study and in a pooled analysis of the AURA studies. The AURA program was finalized by a randomized Phase 3 study, AURA3, comparing osimertinib to platinum + pemetrexed in 419 patients with EGFR-mutated NSCLC, progressing after EGFR-TKI and expressing the T790M mutation.14 Efficacy parameters were in favor of the oral drug for ORR (71 versus 31%), and median PFS [10.1 vs. 4.4 months (HR 0.30; 95% CI, 0.23 to 0.41)], except for median OS [26.8 versus 22.5 months (HR 0.87; 95% CI: 0.67–1.12)] (Table 2).
 |
Table 1 The AURA Program: Efficacy Results of Osimertinib in EGFR-Mutated NSCLC |
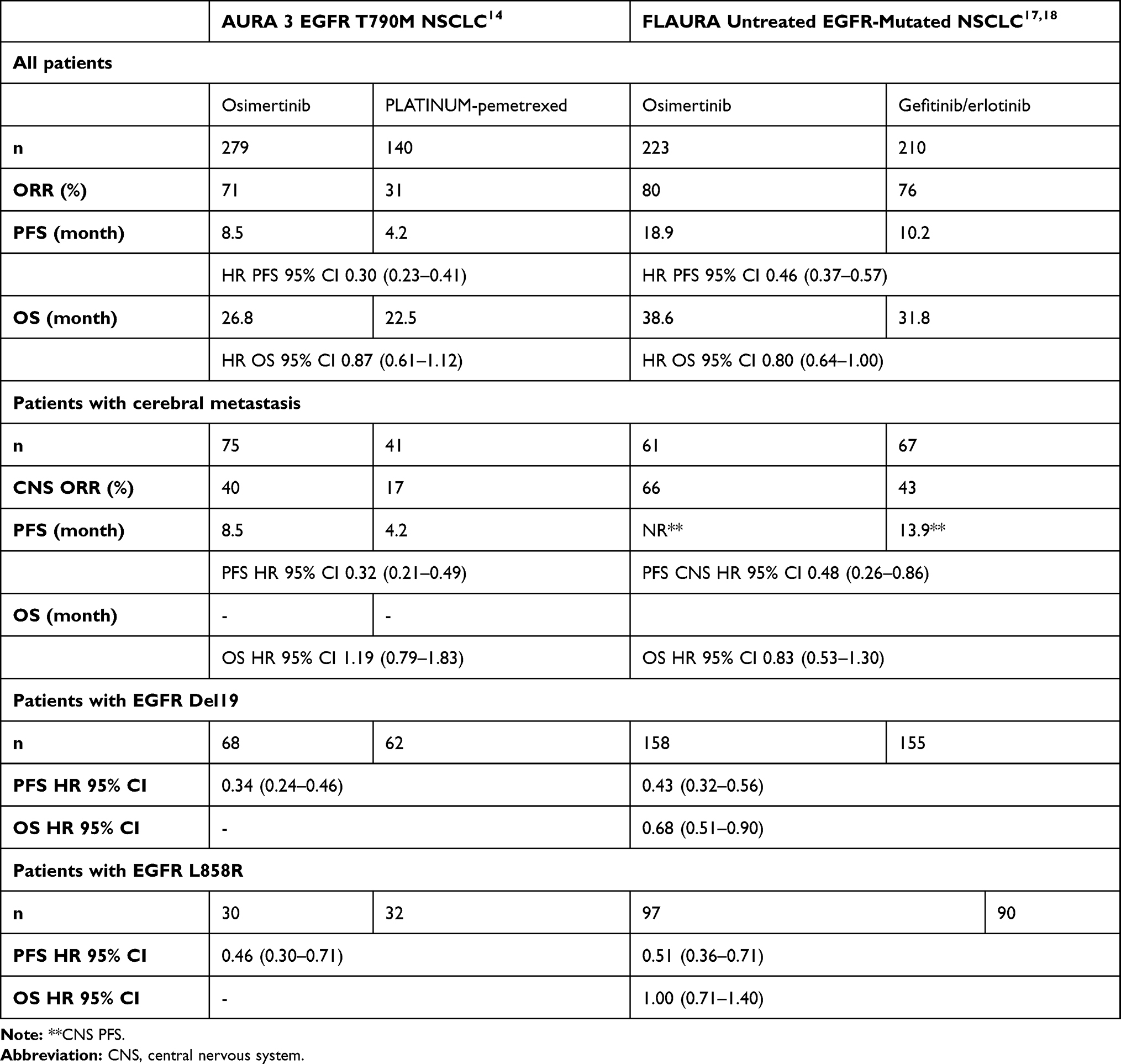 |
Table 2 Randomized Phase 3 Studies with Osimertinib in EGFR-Mutated NSCLC |
The FLAURA Study
Although it has been demonstrated that it could be acquired through deamination of 5-methylcytosine induced by TKIs,15 the EGFR T790M resistance mutation is thought to be originally under-represented as a minority clone and grows in 60% of cases under first- or second-generation EGFR-TKI.16 Based on this rationale of a pre-existing T790M clone in non-TKI-exposed tumors, osimertinib was compared to gefitinib or erlotinib in non-pretreated EGFR-mutated advanced NSCLC.17,18 The FLAURA study is an international randomized phase 3 study enrolling 62% of patients from Asia and 38% from the rest of world. The median PFS was 18.9 months in the osimertinib arm and 10.2 months in the standard arm with gefitinib or erlotinib [HR95% CI 0.46 (0.37–0.57)] (Table 2). The 8-month PFS advantage in favor of osimertinib could be translated for OS, 38.6 months versus 31.8 months with first-generation EGFR TKI [HR95% CI 0.799 (0.641–0.997)]. No difference was seen for the Asian population [HR95% CI 0.995 (0.752–0.1.319)], contrary to the non-Asian population [HR95% CI 0.542 (0.378–0.772)]. In the osimertinib arm, 47% of patients received first subsequent (second line) anticancer treatment. In the first-generation EGFR TKI arm, 65% of patients were treated at progression with a subsequent therapy, of which 47% with osimertinib (corresponding to 31% of all patients randomized in this standard arm).
Overall, osimertinib could be considered as the best choice in light of all these relevant parameters according to FLAURA. This is reinforced by the drug’s very good safety profile, comparable to first-generation TKI: grade 3–4 rash, 1% versus 7%; grade 3–4 diarrhea 2% versus 2%; paronychia 1% versus 1%. The presence of brain metastasis (BM) at diagnosis is another point to promote the use of osimertinib first. FLAURA enrolled 61 (22%) patients with BM in the osimertinib arm.19 There were 67 (24%) in the gefitinib or erlotinib arm (Table 2). For this population, not stratified at the inclusion, the median intracranial PFS was not reached with osimertinib and limited to 13.9 months with first-generation TKI (HR 0.48, 95% CI: 0.26–0.86). Moreover, the risk of an exclusive CNS progression at 12 months was 8% with osimertinib and 24% with gefitinib or erlotinib. In all patients included in the FLAURA study, osimertinib markedly reduced CNS progression (6% with osimertinib and 15% with first-generation TKI). Overall, the median PFS of patients with BM were 15.2 and 9.6 months (HR 0.47, 95% CI: 0.30–0.74) in the osimertinib and first-generation TKI arms, respectively. The HR for OS in this small subgroup of 116 patients with BM was 0.83 (0.53–1.30).
The results of the FLAURA study could be also explored according to the type of EGFR mutation, L858R and Del19. For the L858R mutation, the magnitude of the benefit seemed to be less pronounced. The median PFS was 21.4 months (osimertinib) versus 11.0 months (gefitinib or erlotinib) in patients with exon 19 deletion (HR 0.43, 95CI: 0.32–0.56) and 14.5 months versus 9.5 months in patients with exon 21 L858R mutation (HR 0.51, 95CI: 0.36–0.71) (Table 3).17 More striking was the median OS, largely superior with osimertinib for exon 19 deletion (HR 0.68, 95% CI: 0.51–0.90) in contrast to L858R mutation (HR 1.00, 95% CI: 0.71–1.40).18 This subgroup analysis leaves unresolved the issue of the multiline strategy for L858R EGFR-mutated advanced NSCLC.
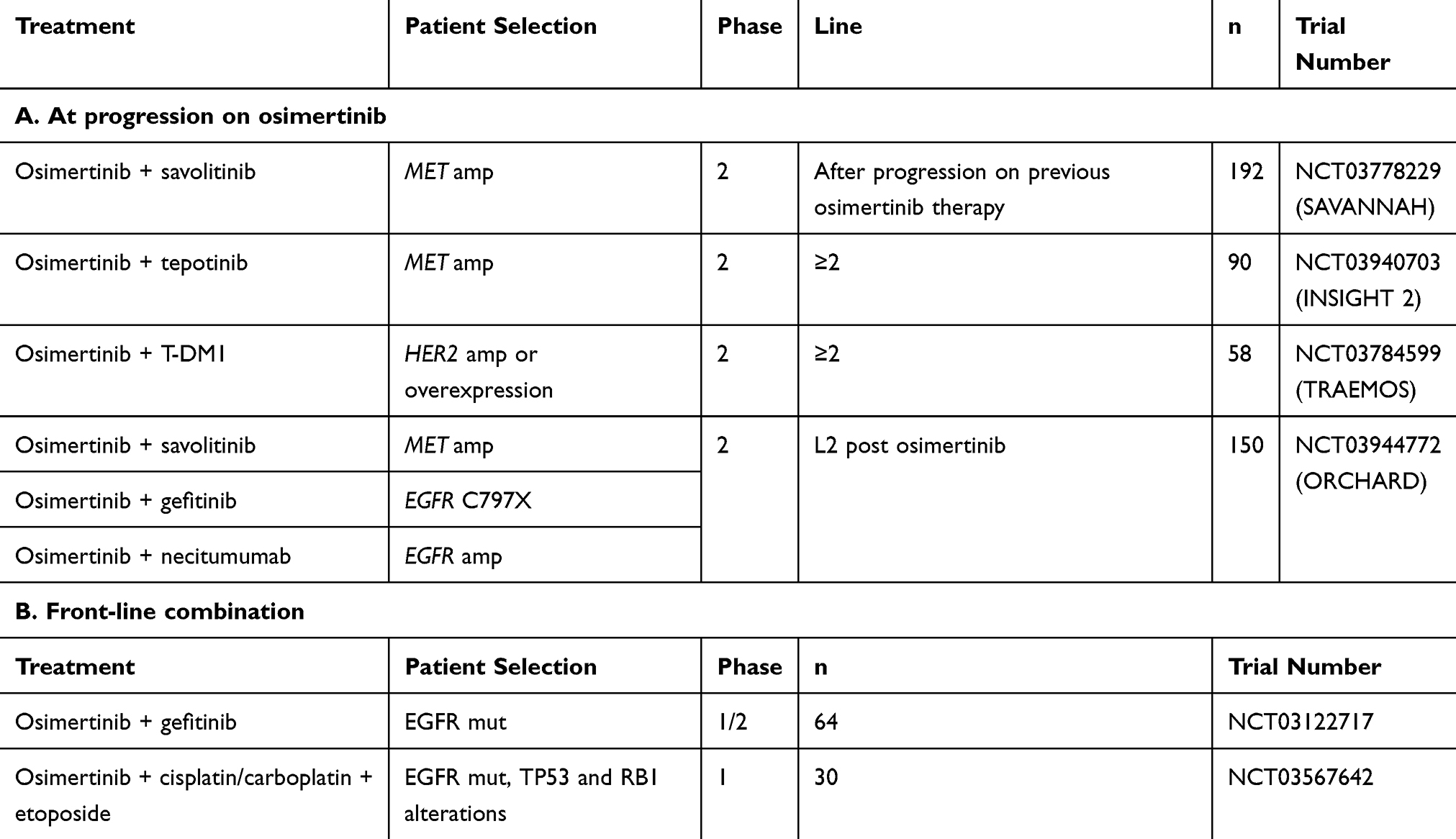 |
Table 3 Clinical Trials with Osimertinib |
The question of the osimertinib effect was raised in patients with uncommon EGFR mutations. A multicenter phase 2 study was conducted with osimertinib in 36 patients with uncommon EGFR mutations.20 The median PFS was 9.5 months with a PR in seven patients (77.8%) with L861Q, 10 (52.6%) with G719A/C/D/S/X mutation and three (37.5%) with S768I mutation. Responses to osimertinib in patients with EGFR exon 20 insertion were only mentioned in clinical case reports.21,22
Resistance Mechanisms
Despite the clear clinical benefit of osimertinib, most patients finally escape. Many studies and clinical cases have reported mechanisms of resistance to osimertinib in EGFR-mutated patients previously treated with first- or second-generation TKIs and harboring the resistance EGFR T790M mutation. A variety of molecular alterations explaining the relapse of these patients have been described in this setting.23,24 Many on-target EGFR mutations have been described. The most common being EGFR C797S,25,26 though many other EGFR mutations have been reported.27–30 Off-target mechanisms of resistance have also been described, including mutation,31 gene amplification10,32 and gene fusion.33,34 Finally, histologic and phenotypic transformation from NSCLC to Small Cell Lung Cancer (SCLC) has also been described.
These different alterations have been described in research articles and collected in several recent reviews.35,36 Some have also been identified when osimertinib is administered as a first-line treatment, and they will be described in greater detail below (Figure 1).
Resistance to Front-Line Osimertinib
With practice changing and approval of osimertinib for front-line treatment, the identification of mechanisms of resistance in this setting is important, both to select the best therapeutic option in subsequent lines of treatment, or in combination with osimertinib, to prevent the appearance of these resistance mechanisms.
MET Amplification
The first indication of MET amplification came from the analysis of circulating tumor DNA (ctDNA) in patients enrolled in the FLAURA trial. Of the 279 patients in the osimertinib arm, plasma was collected at progression in 91 patients who tested positive in plasma at baseline for the activating EGFR mutation.37 ctDNA was analyzed by next-generation sequencing (NGS, Guardant Health; Guardant360 73 gene panel or Omni 500 gene panel). The most frequent alteration was amplification of the MET (hepatocyte growth factor (HGF) receptor) gene (15% of cases). Since plasma-based techniques are assumed to have a lower sensitivity than tissue-based techniques to assess copy number changes, this alteration might be even more frequent. Indeed, in a very recent report, MET amplification was found to occur in 66% (n = 6/9) of first-line osimertinib-treated patients.38 Analyses of larger series of patients are required to achieve a more precise indication of the frequency of this alteration. This is the primary endpoint of the ongoing MELROSE trial.39
MET is expressed on the epithelial cells of numerous organs where it is involved in regulation of angiogenesis, cell survival, invasion, and proliferation. Binding of the ligand HGF to its receptor activates the MET tyrosine kinase, leading to the initiation of a cascade of downstream signals including activation of the Ras/Raf/MEK/ERK and PI3K/Akt pathways.40 Activating MET mutations have been reported in a variety of tumors.41–43 MET amplification has been described at baseline in patients with NSCLC, and also proposed as one of the off-target mechanisms of resistance to first- and second-generation EGFR TKIs.6,44,45
MET is an interesting target as several inhibitors have been described and are being studied in clinical trials (Table 3). The SAVANNAH trial is currently enrolling patients that have been treated with osimertinib and presenting a MET amplification (as determined by FISH and/or NGS) or a MET overexpression (IHC). MET-amplified patients can also be enrolled in one cohort of the ORCHARD trial. In both cases, patients are treated with a combination of osimertinib and savolitinib (AZD6094, HMPL-504, volitinib), a potent and highly selective small-molecule inhibitor of MET tyrosine kinase.46,47 An interesting positive signal recently came from the expansion cohorts of the Phase 1b TATTON study.48 Adult patients with locally advanced or metastatic, MET-amplified, EGFR mutation-positive NSCLC who had progressed on EGFR TKIs were enrolled. There were 138 patients in expansion cohort B (osimertinib 80 mg and savolitinib 600 or 300 mg daily). Objective partial responses were observed in 66 (48%; 95% CI 39–56) patients. In cohort B1 (patients previously treated with a third-generation EGFR TKI) the response rate was 30% (20–43; 21/69 patients).48
Tepotinib, another highly selective, type Ib and orally administered MET inhibitor49 is also being investigated in combination with osimertinib in EGFR-mutated, MET-amplified, locally advanced or metastatic NSCLC that has acquired resistance to prior EGFR TKI in the INSIGHT 2 study (NCT03940703) (Table 3).
EGFR Mutations
Acquired EGFR mutations have also been described in the FLAURA trial. In the osimertinib arm, there was no evidence of acquired EGFR T790M, but the EGFR C797S mutation was detected in the plasma of 6 patients (6/91; 7%).37 In their analysis of resistance mechanisms to osimertinib in a French multicentric retrospective study, Mehlman et al described the existence of a C797S mutation in 2 out of the 3 patients treated in first line.50
The C797S mutation was initially described in EGFR T790M patients progressing on osimertinib.25 The substitution of the cysteine on which osimertinib covalently binds to the ATP-binding site of mutant EGFR7 explains its loss of efficacy in this case. When used in EGFR T790M patients, the efficacy of 1st or 2nd generation EGFR TKI is very limited, due to this resistance mutation.
When osimertinib is used front-line, the C797S mutation appears in the absence of T790M. Cell lines expressing exon 19 deletion and a C797S mutation have been developed.26 These cells were resistant to WZ4002, another third-generation EGFR TKI, but sensitive to gefitinib and, to a lesser extent, to afatinib. These in vitro data support the notion that patients with a C797S mutation acquired during front-line osimertinib treatment may respond to first-generation TKIs. The combination of osimertinib and gefitinib in such patients is being investigated in one cohort of the ORCHARD trial (Table 3). Similar results were described with erlotinib and afatinib in mice models.51 Many other EGFR mutations have been described in patients progressing on osimertinib. Similarly to what has been described for the C797S mutation, some of these mutations have been found to respond to EGFR TKI in cellular models.52
In vitro, combination of first-line osimertinib and erlotinib prevented the emergence of secondary EGFR mutations.51 The objective response rate of osimertinib in combination with gefitinib in EGFR inhibitor-naïve advanced EGFR mutant lung cancer is being investigated in the NCT03122717 phase 1/2 trial (Table 3).
Besides these two most common alterations, a number of other acquired modifications have been detected in the FLAURA trial.
HER2 Amplification
Alterations of the human epidermal growth factor receptor 2 (HER2) gene, a member of the ERBB family of tyrosine kinase receptors, have been described in small subsets of NSCLC patients. These alterations were initially identified as oncogenic drivers. HER2 gene amplification accounts for 1–3% of untreated NSCLC tumors,53,54 and mutations (mainly located on exon 20), represent 2–4% of cases.54–56
Both HER2 mutation and amplification have now been described as resistance mechanisms to first-generation EGFR TKIs. Amplification was detected in 12% of EGFR TKI-resistant cell lines,57 and in 13% of NSCLC patients with acquired resistance to EGFR TKI.6 HER2 mutations have been detected in 2% of patients treated with osimertinib in the FLAURA trial.37
Trastuzumab, a humanized monoclonal antibody directed against HER2, is approved for the treatment of breast and gastric cancer patients. A single-arm phase II study evaluated the safety and efficacy of trastuzumab and paclitaxel in NSCLC patients expressing HER2 after progression on EGFR TKI. An objective response rate of 46% was observed, with a 100% response observed in the 4 patients with an HER2 amplification (more than 10 copies/nucleus).58 Another phase 2 study evaluated trastuzumab-emtansine (T-DM1) treatment in NSCLC patients overexpressing HER2. Three of 4 responders had HER2 gene amplification.59 Furthermore, in vitro experiments suggested that T-DM1 can overcome HER2-mediated resistance in patients with EGFR-mutated NSCLC.60 The TRAEMOS trial, a single-arm open-label multi-center phase II study, is investigating disease control rate after 3 months of treatment with trastuzumab-emtansine/osimertinib combination therapy in patients with advanced EGFR mutation-positive NSCLC with HER2 bypass track resistance (Table 3).
BRAF Mutation
Acquired BRAF V600E mutations have been described as resistance mechanism of T790M patients progressing on osimertinib.61,62 A few cases of BRAF V600E mutation acquired upon front-line osimertinib have also been described.37,63 Combined treatment using dabrafenib/trametinib/osimertinib showed modest efficacy in EGFR T790M patients with osimertinib-induced BRAF V600E.62 Interestingly, however, cell lines that present a BRAF V600E as a resistance mechanism to osimertinib showed sensitivity to osimertinib and encorafenib, a BRAF inhibitor already approved for the treatment, in combination with binemitinib (MEK inhibitor), of advanced and metastatic BRAF V600 melanoma.61 To our knowledge, there is currently no clinical trial underway with the objective of evaluating the combination of osimertinib + BRAF inhibitor + MEK inhibitor in these patients.
Gene Fusions
Besides gene mutation and amplification, gene rearrangements have been described in EGFR TKI-resistant NSCLC patients.
In the FLAURA trial, one patient was found to present an SPTBN1-ALK gene fusion at progression on osimertinib.37 Offin et al reported two cases of NSCLC patients developing EML4-ALK rearrangement at progression on osimertinib after erlotinib-induced T790M mutation. Both patients received clinical benefit from a combination of osimertinib and an ALK inhibitor (crizotinib or alectinib).34 Similarly, Schoenfeld et al reported durable responses in two patients with tumors that acquired ALK fusion that who were treated with osimertinib and ALK inhibitors (crizotinib, alectinib and lorlatinib).64 The combination of an ALK inhibitor with osimertinib in such patients warrants further investigation.
Acquired BRAF rearrangements have also been described as mechanisms of resistance to osimertinib.64 Induction of BRAF gene fusion in cell lines conferred resistance to growth inhibition by osimertinib.65 Interestingly, a pan-RAF inhibitor was demonstrated to block growth of all produced cell lines with mutant EGFR and BRAF fusion.65
RET rearrangements have been described in 1–3% of NSCLC.66,67 KIF5B-RET and CCDC6-RET fusions have been found in cases of EGFR-mutated NSCLC that had progressed on first- or second-generation EGFR TKI.68 CCDC6-RET fusions have also been detected in 2 out of 41 patients with osimertinib resistance biopsies.33 The authors treated two patients with EGFR-mutant NSCLC and RET-mediated resistance with osimertinib and BLU-667 (pralsetinib). The combination was well-tolerated and led to rapid radiographic response in both patients. In vitro experiments demonstrated that this osimertinib/BLU-667 combination was effective both in the presence and in the absence of T790M.
Histologic Transformation
Finally, besides altered drug target and bypass track activation, morphological alterations have been described at progression in EGFR TKI treatment. Histologic transformation of NSCLC into SCLC has been reported many times, both in the case of treatment with a first-generation inhibitor6 and with osimertinib.64,69,70 In this case, the therapeutic strategy consists in using the reference treatments for SCLC. This transformation appears to be more frequent in NSCLC presenting, at diagnosis, an alteration of the TP53 and RB1 genes. It is therefore tempting to prevent transformation to SCLC by a combination of osimertinib, platinum (cisplatin or carboplatin) and etoposide in patients with EGFR/TP53/RB1 alterations. The NCT03567642 phase 1 trial is intended to determine the safety and toxicity profile of this treatment combination in metastatic EGFR mutant lung cancers with concurrent RB1 and TP53 alterations. In a systematic analysis of resistance mechanisms to first-line osimertinib in EGFR mutant lung cancer patients, Schoenfeld et al identified histologic transformation in 15% of cases (9/62 patients), both in the first-line setting and in the later-line setting.64 They described three cases of small cell transformation and one case of pleiomorphic transformation. But the most frequent transformation they reported was squamous cell transformation (five cases).64 Varying treatment strategies were used and outcomes were mixed with limited follow-up.
Conclusions
Other molecular alterations have been described in small subsets of patients progressing on osimertinib.28,37,64 Systematic analysis of tissue biopsies taken at progression will likely lead to the identification of additional mechanisms of resistance.39 The use of tissue biopsies is essential in this setting since plasma genotyping cannot identify histological transformation and gene amplification is more difficult to demonstrate due to the background of normal genomic DNA. Recent work has shown the benefit of combining a first-generation EGFR inhibitor with platinum-based chemotherapy71,72 or with an angiogenesis inhibitor.73 Similar approaches combining osimertinib with chemotherapy (FLAURA2; NCT04035486) or bevacizumab (ECOG5182; NCT04181060) are being evaluated in Phase III randomized clinical trials. The analysis of resistance mechanisms in these settings will be of great interest. Optimization of the first line of treatment and identification of resistance mechanisms to guide subsequent treatments will ultimately improve patient survival.
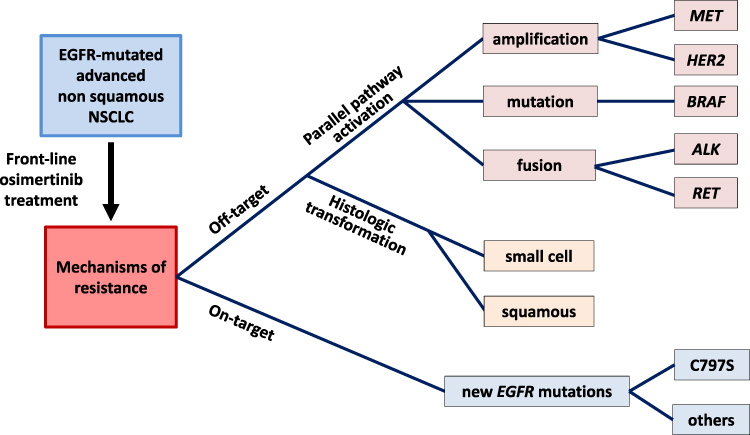 |
Figure 1 The tree structure of main resistance mechanisms to first-line osimertinib in advanced EGFR-mutated NSCLC. |
Disclosure
Professor Marc G Denis reports grants and personal fees from Boehringer Ingelheim and personal fees from Astra Zeneca, during the conduct of the study; and grants and personal fees from Takeda, and personal fees from Roche Diagnostics and BMS, outside the submitted work. Professor Jaafar Bennouna reports grants and personal fees from Astra Zeneca, during the conduct of the study; and personal fees from BMS, Roche, MSD, and Servier, outside the submitted work. The authors report no other potential conflicts of interest for this work.
References
1. Wu YL, Planchard D, Lu S, et al. Pan-Asian adapted clinical practice guidelines for the management of patients with metastatic non-small-cell lung cancer: a CSCO-ESMO initiative endorsed by JSMO, KSMO, MOS, SSO and TOS. Ann Oncol. 2019;30(2):171–210. doi:10.1093/annonc/mdy554
2. Planchard D, Popat S, Kerr K, et al. Correction to: metastatic non-small cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2019;30(5):863–870. doi:10.1093/annonc/mdy474
3. Planchard D, Popat S, Kerr K, et al. Metastatic non-small cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):iv192–iv237. doi:10.1093/annonc/mdy275
4. Hulo P, Coupez D, Denis MG, Bennouna J. EGFR mutation-positive NSCLC: factors to consider when deciding first-line therapy. Expert Rev Anticancer Ther. 2020;20(5):365–372. doi:10.1080/14737140.2020.1757439
5. Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013;31(8):1070–1080. doi:10.1200/JCO.2012.43.3912
6. Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. doi:10.1158/1078-0432.CCR-12-2246
7. Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–1061. doi:10.1158/2159-8290.CD-14-0337
8. Finlay MR, Anderton M, Ashton S, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem. 2014;57(20):8249–8267. doi:10.1021/jm500973a
9. Janne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–1699. doi:10.1056/NEJMoa1411817
10. Yang JC, Ahn MJ, Kim DW, et al. Osimertinib in pretreated T790M-positive advanced non-small-cell lung cancer: AURA study phase II extension component. J Clin Oncol. 2017;35(12):1288–1296. doi:10.1200/JCO.2016.70.3223
11. Goss G, Tsai CM, Shepherd FA, et al. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2016;17(12):1643–1652. doi:10.1016/S1470-2045(16)30508-3
12. Ahn MJ, Tsai CM, Shepherd FA, et al. Osimertinib in patients with T790M mutation-positive, advanced non-small cell lung cancer: long-term follow-up from a pooled analysis of 2 phase 2 studies. Cancer. 2019;125(6):892–901. doi:10.1002/cncr.31891
13. Goss G, Tsai CM, Shepherd FA, et al. CNS response to osimertinib in patients with T790M-positive advanced NSCLC: pooled data from two phase II trials. Ann Oncol. 2018;29(3):687–693. doi:10.1093/annonc/mdx820
14. Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. 2017;376(7):629–640. doi:10.1056/NEJMoa1612674
15. El Kadi N, Wang L, Davis A, et al. The EGFR T790M mutation is acquired through AICDA-mediated deamination of 5-methylcytosine following TKI treatment in lung cancer. Cancer Res. 2018;78(24):6728–6735. doi:10.1158/0008-5472.CAN-17-3370
16. Denis MG, Vallee A, Theoleyre S. EGFR T790M resistance mutation in non small-cell lung carcinoma. Clin Chim Acta. 2015;444:81–85. doi:10.1016/j.cca.2015.01.039
17. Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378(2):113–125. doi:10.1056/NEJMoa1713137
18. Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382(1):41–50. doi:10.1056/NEJMoa1913662
19. Reungwetwattana T, Nakagawa K, Cho BC, et al. CNS response to osimertinib versus standard epidermal growth factor receptor tyrosine kinase inhibitors in patients with untreated EGFR-mutated advanced non-small-cell lung cancer. J Clin Oncol. 2018;36(33):
20. Cho JH, Lim SH, An HJ, et al. Osimertinib for patients with non-small-cell lung cancer harboring uncommon EGFR mutations: a multicenter, open-label, phase II trial (KCSG-LU15-09). J Clin Oncol. 2020;38(5):488–495. doi:10.1200/JCO.19.00931
21. Piotrowska Z, Fintelmann FJ, Sequist LV, Jahagirdar B. Response to osimertinib in an EGFR exon 20 insertion-positive lung adenocarcinoma. J Thorac Oncol. 2018;13(10):e204–e206. doi:10.1016/j.jtho.2018.05.017
22. Fang W, Huang Y, Hong S, et al. EGFR exon 20 insertion mutations and response to osimertinib in non-small-cell lung cancer. BMC Cancer. 2019;19(1):595. doi:10.1186/s12885-019-5820-0
23. Wang YN, Lee HH, Chou CK, et al. Angiogenin/ribonuclease 5 is an EGFR ligand and a serum biomarker for erlotinib sensitivity in pancreatic cancer. Cancer Cell. 2018;33(4):752–769 e758. doi:10.1016/j.ccell.2018.02.012
24. Oxnard GR, Hu Y, Mileham KF, et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 2018;4(11):1527–1534. doi:10.1001/jamaoncol.2018.2969
25. Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21(6):560–562. doi:10.1038/nm.3854
26. Niederst MJ, Hu H, Mulvey HE, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. 2015;21(17):3924–3933. doi:10.1158/1078-0432.CCR-15-0560
27. Oztan A, Fischer S, Schrock AB, et al. Emergence of EGFR G724S mutation in EGFR-mutant lung adenocarcinoma post progression on osimertinib. Lung Cancer. 2017;111:84–87. doi:10.1016/j.lungcan.2017.07.002
28. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. 2019;121(9):725–737.
29. Zhang Q, Zhang XC, Yang JJ, et al. EGFR L792H and G796R: two novel mutations mediating resistance to the third-generation EGFR tyrosine kinase inhibitor osimertinib. J Thorac Oncol. 2018;13(9):1415–1421. doi:10.1016/j.jtho.2018.05.024
30. Song Y, Jia Z, Wang Y, et al. Potential treatment strategy for the rare osimertinib resistant mutation EGFR L718Q. J Thorac Dis. 2020;12(5):2771–2780. doi:10.21037/jtd.2020.03.29
31. Chabon JJ, Simmons AD, Lovejoy AF, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi:10.1038/ncomms11815
32. Wang Y, Li L, Han R, Jiao L, Zheng J, He Y. Clinical analysis by next-generation sequencing for NSCLC patients with MET amplification resistant to osimertinib. Lung Cancer. 2018;118:105–110. doi:10.1016/j.lungcan.2018.02.007
33. Piotrowska Z, Isozaki H, Lennerz JK, et al. Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. 2018;8(12):1529–1539. doi:10.1158/2159-8290.CD-18-1022
34. Offin M, Somwar R, Rekhtman N, et al. Acquired ALK and RET gene fusions as mechanisms of resistance to osimertinib in EGFR-mutant lung cancers. JCO Precis Oncol. 2018;2.
35. Yang Z, Yang J, Chen Y, Shao YW, Wang X. Acquired EGFR L718V mutation as the mechanism for osimertinib resistance in a T790M-negative non-small-cell lung cancer patient. Target Oncol. 2019;14(4):369–374. doi:10.1007/s11523-019-00652-6
36. Yang Z, Yang N, Ou Q, et al. Investigating novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non-small cell lung cancer patients. Clin Cancer Res. 2018;24(13):3097–3107. doi:10.1158/1078-0432.CCR-17-2310
37. Ramalingam SS. LBA50 - mechanisms of acquired resistance to first-line osimertinib: preliminary data from the phase III FLAURA study. Ann Oncol. 2018;29:mdy424–063.
38. Roper N, Brown AL, Wei JS, et al. Clonal evolution and heterogeneity of osimertinib acquired resistance mechanisms in EGFR mutant lung cancer. Cell Rep Med. 2020;1:1.
39. Bennouna J, Girard N, Audigier-Valette C, et al. Phase II study evaluating the mechanisms of resistance on tumor tissue and liquid biopsy in patients with EGFR-mutated non-pretreated advanced lung cancer receiving osimertinib until and beyond radiologic progression: the MELROSE trial. Clin Lung Cancer. 2020;21(1):e10–e14. doi:10.1016/j.cllc.2019.09.007
40. Eder JP, Vande Woude GF, Boerner SA, LoRusso PM. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009;15(7):2207–2214. doi:10.1158/1078-0432.CCR-08-1306
41. Kwak Y, Kim SI, Park CK, Paek SH, Lee ST, Park SH. C-MET overexpression and amplification in gliomas. Int J Clin Exp Pathol. 2015;8(11):14932–14938.
42. Lee JH, Han SU, Cho H, et al. A novel germ line juxtamembrane met mutation in human gastric cancer. Oncogene. 2000;19(43):4947–4953. doi:10.1038/sj.onc.1203874
43. Jardim DL, de Melo Gagliato D, Falchook G, et al. MET abnormalities in patients with genitourinary malignancies and outcomes with c-MET inhibitors. Clin Genitourin Cancer. 2015;13(1):e19–26. doi:10.1016/j.clgc.2014.06.017
44. Ou SI, Agarwal N, Ali SM. High MET amplification level as a resistance mechanism to osimertinib (AZD9291) in a patient that symptomatically responded to crizotinib treatment post-osimertinib progression. Lung Cancer. 2016;98:59–61. doi:10.1016/j.lungcan.2016.05.015
45. Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14(10):2895–2899. doi:10.1158/1078-0432.CCR-07-2248
46. Jia H, Dai G, Weng J, et al. Discovery of (S)-1-(1-(Imidazo[1,2-a]pyridin-6-yl)ethyl)-6-(1-methyl-1H-pyrazol-4-yl)-1H-[1,2, 3]triazolo[4,5-b]pyrazine (volitinib) as a highly potent and selective mesenchymal-epithelial transition factor (c-Met) inhibitor in clinical development for treatment of cancer. J Med Chem. 2014;57(18):7577–7589.
47. Gan HK, Millward M, Hua Y, et al. First-in-human Phase I Study of the selective MET inhibitor, savolitinib, in patients with advanced solid tumors: safety, pharmacokinetics, and antitumor activity. Clin Cancer Res. 2019;25(16):4924–4932. doi:10.1158/1078-0432.CCR-18-1189
48. Sequist LV, Han JY, Ahn MJ, et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020;21(3):373–386. doi:10.1016/S1470-2045(19)30785-5
49. Falchook GS, Kurzrock R, Amin HM, et al. First-in-man Phase I trial of the selective MET inhibitor tepotinib in patients with advanced solid tumors. Clin Cancer Res. 2020;26(6):1237–1246. doi:10.1158/1078-0432.CCR-19-2860
50. Mehlman C, Cadranel J, Rousseau-Bussac G, et al. Resistance mechanisms to osimertinib in EGFR-mutated advanced non-small-cell lung cancer: a multicentric retrospective French study. Lung Cancer. 2019;137:149–156. doi:10.1016/j.lungcan.2019.09.019
51. Starrett JH, Guernet AA, Cuomo ME, et al. Drug sensitivity and allele specificity of first-line osimertinib resistance EGFR mutations. Cancer Res. 2020;80(10):2017–2030. doi:10.1158/0008-5472.CAN-19-3819
52. Nishino M, Suda K, Kobayashi Y, et al. Effects of secondary EGFR mutations on resistance against upfront osimertinib in cells with EGFR-activating mutations in vitro. Lung Cancer. 2018;126:149–155. doi:10.1016/j.lungcan.2018.10.026
53. Li BT, Ross DS, Aisner DL, et al. HER2 amplification and HER2 mutation are distinct molecular targets in lung cancers. J Thorac Oncol. 2016;11(3):414–419. doi:10.1016/j.jtho.2015.10.025
54. Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14(8):535–546.
55. Barlesi F, Mazieres J, Merlio JP, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet. 2016;387(10026):1415–1426. doi:10.1016/S0140-6736(16)00004-0
56. Mazieres J, Peters S, Lepage B, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013;31(16):1997–2003. doi:10.1200/JCO.2012.45.6095
57. Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2(10):922–933. doi:10.1158/2159-8290.CD-12-0108
58. de Langen AJ, Jebbink M, Hashemi SMS, et al. Trastuzumab and paclitaxel in patients with EGFR mutated NSCLC that express HER2 after progression on EGFR TKI treatment. Br J Cancer. 2018;119(5):558–564. doi:10.1038/s41416-018-0194-7
59. Peters S, Stahel R, Bubendorf L, et al. Trastuzumab emtansine (T-DM1) in patients with previously treated HER2-overexpressing metastatic non-small cell lung cancer: efficacy, safety, and biomarkers. Clin Cancer Res. 2019;25(1):64–72. doi:10.1158/1078-0432.CCR-18-1590
60. Cretella D, Saccani F, Quaini F, et al. Trastuzumab emtansine is active on HER-2 overexpressing NSCLC cell lines and overcomes gefitinib resistance. Mol Cancer. 2014;13:143. doi:10.1186/1476-4598-13-143
61. Ho -C-C, Liao W-Y, Lin C-A, Shih J-Y, Yu C-J, Yang JC. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J Thorac Oncol. 2017;12(3):567–572. doi:10.1016/j.jtho.2016.11.2231
62. Meng P, Koopman B, Kok K, et al. Combined osimertinib, dabrafenib and trametinib treatment for advanced non-small-cell lung cancer patients with an osimertinib-induced BRAF V600E mutation. Lung Cancer. 2020;146:358–361. doi:10.1016/j.lungcan.2020.05.036
63. Minari R, Bordi P, La Monica S, et al. Concurrent acquired BRAF V600E mutation and MET amplification as resistance mechanism of first-line osimertinib treatment in a patient with EGFR-mutated NSCLC. J Thorac Oncol. 2018;13(6):e89–e91. Doi:10.1016/j.jtho.2018.03.013
64. Schoenfeld AJ, Chan JM, Kubota D, et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin Cancer Res. 2020;26(11):2654–2663. doi:10.1158/1078-0432.CCR-19-3563
65. Vojnic M, Kubota D, Kurzatkowski C, et al. Acquired BRAF rearrangements induce secondary resistance to EGFR therapy in EGFR-mutated lung cancers. J Thorac Oncol. 2019;14(5):802–815. doi:10.1016/j.jtho.2018.12.038
66. Kohno T, Ichikawa H, Totoki Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18(3):375–377. doi:10.1038/nm.2644
67. Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18(3):378–381. doi:10.1038/nm.2658
68. Klempner SJ, Bazhenova LA, Braiteh FS, et al. Emergence of RET rearrangement co-existing with activated EGFR mutation in EGFR-mutated NSCLC patients who had progressed on first- or second-generation EGFR TKI. Lung Cancer. 2015;89(3):357–359. doi:10.1016/j.lungcan.2015.06.021
69. Li L, Wang H, Li C, Wang Z, Zhang P, Yan X. Transformation to small-cell carcinoma as an acquired resistance mechanism to AZD9291: a case report. Oncotarget. 2017;8(11):18609–18614. doi:10.18632/oncotarget.14506
70. Ham JS, Kim S, Kim HK, et al. Two cases of small cell lung cancer transformation from EGFR mutant adenocarcinoma during AZD9291 treatment. J Thorac Oncol. 2016;11(1):e1–e4. doi:10.1016/j.jtho.2015.09.013
71. Noronha V, Patil VM, Joshi A, et al. Gefitinib versus gefitinib plus pemetrexed and carboplatin chemotherapy in EGFR-mutated lung cancer. J Clin Oncol. 2020;38(2):124–136. doi:10.1200/JCO.19.01154
72. Hosomi Y, Morita S, Sugawara S, et al. Gefitinib alone versus gefitinib plus chemotherapy for non-small-cell lung cancer with mutated epidermal growth factor receptor: NEJ009 Study. J Clin Oncol. 2020;38(2):115–123. doi:10.1200/JCO.19.01488
73. Nakagawa K, Garon EB, Seto T, et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(12):1655–1669. doi:10.1016/S1470-2045(19)30634-5
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.