")
Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 5
Orphan drugs for sickle vaso-occlusion: dawn of a new era of targeted treatment
Authors Dampier C
Received 4 May 2015
Accepted for publication 14 August 2015
Published 2 November 2015 Volume 2015:5 Pages 99—112
DOI https://doi.org/10.2147/ODRR.S46305
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Lise Aagaard
Carlton Dampier1,2
1Emory University School of Medicine, Emory University, 2AFLAC Cancer and Blood Disorders Center, Children's Healthcare of Atlanta, Atlanta, GA, USA
Abstract: While an orphan disease in the USA, sickle cell disease (SCD), a group of genetic disorders of hemoglobin structure and function, is a major public health problem in much of the rest of the world, particularly sub-Saharan Africa. The pathophysiology of SCD stems from the formation of sickle hemoglobin polymers that deform the erythrocyte into a characteristic sickle shape, the rapidity of which is regulated by its intracellular hemoglobin concentration. Subsequent vaso-occlusion is dependent on adhesion of sickled erythrocytes, and perhaps other cellular elements, including leucocytes and platelets, to abnormal vascular endothelium using a number of receptor–ligand pairs. This propensity for vaso-occlusion may be enhanced by altered vascular tone from excessive amounts of vaso-constrictive factors or diminished amounts of vasodilatory factors. Acute pain is the hallmark symptom caused by sickle polymer formation and subsequent vaso-occlusion, and is represented in the endpoints of most previous and current clinical trial designs. Numerous failures of prior investigational agents have frustrated clinicians and patients alike. Hydroxyurea is currently the only US Food and Drug Administration-approved drug for SCD and reduces the frequency of vaso-occlusive complications in many individuals. A considerable therapeutic need remains as hydroxyurea usage is currently not approved for all types of SCD, is not always clinically effective, and requires frequent monitoring. Recent improvements in our understanding of SCD pathophysiology have generated many new therapeutic targets and associated investigational agents. For example, a number of more specific fetal hemoglobin inducers and several therapies to reduce sickle polymer formation are being tested in preclinical and early phase clinical trials. Several agents that target receptor–ligand interactions which mediate cellular adhesion to vascular endothelium have shown considerable promise and are entering Phase III trials, but present some continuing challenges in clinical trial design and conduct. Gene therapy trials are poised to start and offer the potential for curative therapy.
Keywords: sickle cell disease, pain, clinical trials, investigational agents
Introduction
The replacement of glutamine with valine at the sixth amino acid position in the β-globin subunit is the mutation diagnostic for sickle hemoglobin. In sufficient concentrations, molecules of this abnormal hemoglobin polymerize upon deoxygenation into long polymers that physically deform the erythrocyte into the characteristic “sickle” shape and ultimately obstruct blood flow leading to subsequent local tissue ischemia.1 Such polymer formation can also occur to varying degrees by the copolymerization of sickle hemoglobin with other structural hemoglobin variants, such as hemoglobin C, D, E, or Oarab. When inherited as a compound heterozygote with sickle hemoglobin, mutations in the β-globin gene that cause premature termination of normal β-globin protein production, or that produce structural hemoglobin variants that are ineffectively synthesized or are markedly unstable (β-thalassemias), also result in a sickling disorder.2,3
Early clinical trials of potential treatments to prevent sickle hemoglobin polymerization and subsequent vaso-occlusion were hampered by a limited understanding of the complex pathophysiology of sickle cell disease (SCD). The many basic science advances in the last 30 years have provided a number of different drug targets that have been explored in clinical trials (Figure 1). However, an effective pharmacologic strategy remains elusive as most of these agents studied to date have failed in late stage trials. The increasing focus on translational medicine and advances in drug development have brought a number of new highly targeted agents to mid and late stage SCD clinical trials that are the focus of this review, and show promise of finally providing effective therapies for many of the acute complications of SCD in children and adults.
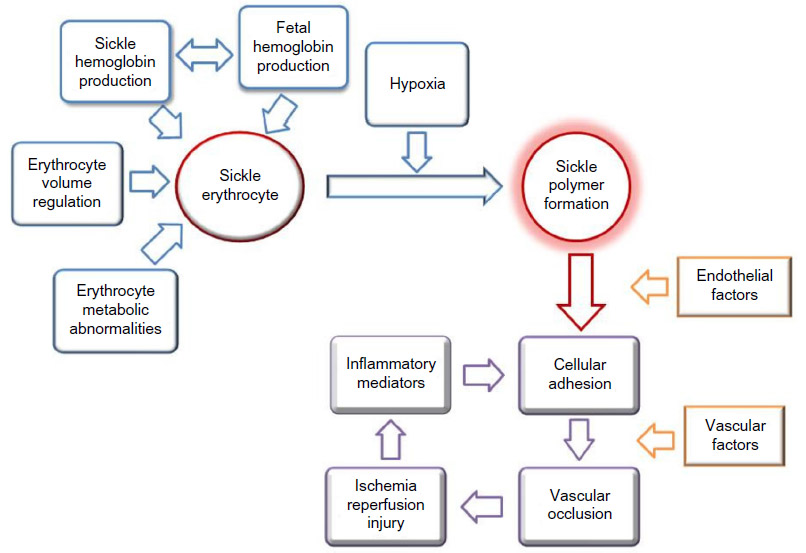 | Figure 1 Pathophysiology of sickle vaso-occlusion. |
First a brief history lesson: SCD as the first molecular disease
James Herrick provided the very first clinical description of SCD in the USA over 100 years ago, when he reported a 20-year old male of West Indian origin with fever, pain, anemia, pallor, jaundice, leg ulcers, and sickle-shaped red blood cells.4 Subsequent case series over the next 40 years provided more in-depth clinical descriptions, and key observations on the role of spleen,5 central nervous system lesions,6 and infectious complications.7 The hypothesis for the role of vaso-occlusion is attributed to Diggs and Ching in 1934.8 In 1940, the key effect of deoxygenation on sickle cells was noted by Sherman9 and the “vicious” cycle of sickling was proposed by Ham and Castle.10 The likely ameliorating role of high fetal hemoglobin in newborns with SCD was described by Watson in 1948,11 while in 1956, Ingram demonstrated that a glutamic acid to valine substitution at position 6 of the β-globin chain was the causative mutation in sickle cell anemia (SCA, homozygous SS).12
Pauling et al’s 1949 characterization of SCA as a molecular disease13 engendered the expectation that a quick cure would be developed now that the genetic abnormality had been identified; yet this dream remains largely unfulfilled. While many individuals are less symptomatic with the widespread introduction of hydroxyurea (HU) therapy (see “HU – A ‘leadoff home run’ for fetal hemoglobin inducers”), and a few are cured by allogeneic hematopoietic stem-cell transplantation,14 treatment for most SCD complications relies on supportive care that has changed little from the 1950s.
SCD – USA orphan disease but a global health problem
The sickle mutation is believed to have arisen independently many thousands of years ago in at least three areas of Africa, and in the Middle East or Indian subcontinent.15,16 The prevalence of the sickle gene was maintained and expanded in these areas by reproductive advantage as the heterozygous condition, sickle trait, provided some degree of protection from severe malaria.17 The sickle gene was introduced into the Western hemisphere primarily by Africans forcibly taken to the Americas through the transatlantic slave trade, with largest numbers being distributed to the USA, Brazil, and the Caribbean.
Based on estimates of sickle gene frequencies, it is expected that over 300,000 newborns with SCA were born globally in 2010; an estimated 80% of newborns with SCA occurred in sub-Saharan Africa alone, with three countries (Nigeria, India, and the Democratic Republic of the Congo) representing 57% of the annual number of newborns with SCA globally.18 Estimates suggest that over 6,000 annual births and 100,000–150,000 Latin Americans are affected by SCD.19 By comparison, estimates of annual births with SCA in the USA or in the UK are in the range of 1,000–2,000, with estimates of over 100,000 individuals living with SCD in the USA,20 consistent with orphan disease status. SCA represents a large proportion of SCD in the Americas, UK, and certain regions of Africa, while higher proportions of hemoglobin SC are observed in Burkina Faso and hemoglobin Sβ-thalassemias in Greece and India.21 Continued immigration from countries with a high sickle hemoglobin prevalence to high-income countries, such as the USA and UK, will contribute to the increasing health burden of these hemoglobinopathies in the future.22
While it represents some significant logistical challenges, the availability of this large non-USA sickle cell patient population is a significant advantage for enrolling in large clinical trials and has allowed several current Phase III SCD clinical trials to meet enrollment milestones in a much more timely fashion then with USA sites alone. In contrast, once marketing approval has been obtained, the likely high cost of most new drugs may be a substantial barrier to their use in these low-income countries with the highest prevalence of affected individuals. Lessons learned from the introduction and use of antiretroviral therapies in these low- and middle-income countries may be relevant to future use of anti-sickling therapies.23
Pathophysiology – a multiplicity of potential targets
The pathophysiology of vaso-occlusion in SCD is quite complex and involves both erythrocyte sickling and subsequent vascular occlusion24 (Figure 1). The former is related to the rapidity and extent of sickle polymer formation, which is regulated by cell volume and hemoglobin concentrations,25 while the latter is dependent on adhesion of sickled erythrocytes, and perhaps other cellular elements, including leucocytes and platelets, to abnormal vascular endothelium using a number of receptor–ligand pairs.26,27 This propensity for vaso-occlusion may be enhanced by altered vascular tone from excessive amounts of vaso-constrictive factors, such as ET-1,28 or diminished amounts of vaso-dilatory factors, particularly nitric oxide (NO).29
As part of the “vicious” cycle of vaso-occlusion, the subsequent ischemic tissue injury results in the accumulation of endogenous factors released from cells that reside or infiltrate into the injured area. These factors represent a wide array of signaling molecules, including cytokines, and chemokines, extracellular proteases, peptides, eicosanoids and related lipids, neurotrophins, as well as protons, which damage local nociceptive nerves leading to increased spontaneous firing that is perceived as acute pain,30 the hallmark complication of this disorder. Many factors contribute to a substantial inflammatory response, which may be a key feature of SCD pathobiology that is likely enhanced by the consequences of reperfusion injury.31
While the focus for much of the treatment of SCD has been on these acute vaso-occlusive events (Figure 2), much of the morbidity and mortality encountered in children, and particularly in adults, is related to chronic organ damage from vasculopathy in the brain, lungs, kidney, and eyes.32 This injury to vascular endothelium also is a complex process,33 likely fueled by the inflammation resulting from recurrent sickling/vaso-occlusion, oxidative stress,34 and hypercoagulability.35 The central pathophysiology that relates these features of a “vascular sub-phenotype” of SCD to vaso-occlusion appears to be the downstream effects of sickling-related intravascular hemolysis and its impact on NO biology (Figure 3).36,37
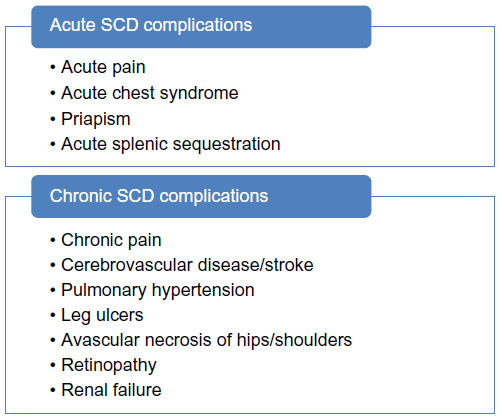 | Figure 2 Acute and chronic complications of sickle cell disease (SCD). |
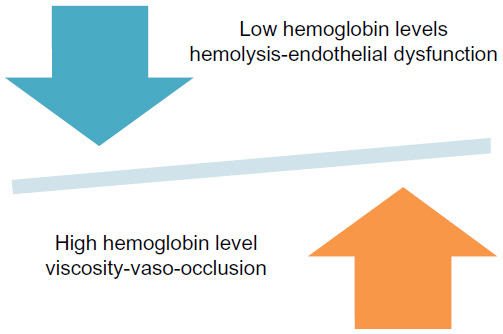 | Figure 3 Competing effects of hemoglobin in sickle cell disease pathophysiology. |
Protean clinical manifestations provide many potential trial endpoints
The many complications of SCD provide a number of clinically relevant events to consider as potential endpoints for interventional clinical trials (Figure 2). Pain in SCD can begin early in the first year of life as levels of fetal hemoglobin decline.38 Most infants with pain in the first year of life have pain locations (hands/feet) and signs or symptoms (swelling or tenderness) consistent with dactylitis, or “hand-foot syndrome”, which becomes less common in older children, and rare after 5–7 years of age.39,40 Children with SCD often experience increasingly frequent acute pain through childhood into adolescence or young adulthood.41 While frequent hospitalizations are the major source of health care utilization, the large majority of these painful episodes are managed at home, particularly in children.42 Most episodes are relatively brief, typically 2–3 days, with pain of moderate intensity, usually in lower legs, back, or chest wall.43 A small number of children, often in their early adolescent years, change from a pattern of episodic acute pain to frequently recurrent acute pain or chronic pain.44,45 SCD pain is an even more complex experience in adults, with features of both acute recurrent pain and chronic pain,46,47 and is more often managed in an acute care setting compared to children.48 In a large national history study, more frequent episodes of acute pain were associated with higher baseline hematocrit and lower levels of fetal hemoglobin.44 A pattern of frequent pain, defined as three or more yearly serious pain episodes, conferred an increased risk for mortality.44 Comorbidities such as mental health issues often contribute to more frequent health care utilization in SCD adults with chronic pain.49,50
These episodes of more intense pain require initial evaluation and treatment in acute care settings, usually Emergency Departments, which can be challenging in busy urban hospitals.51 Some larger SCD programs have developed dedicated acute treatment settings, “day hospitals” with some success,52 but despite aggressive analgesic therapy, hospitalization for continued management of these episodes remain frequent.53 Inpatient management usually consists of supportive care with intravenous (IV) fluids and analgesics until the pain subsides.54
Change in pain intensity, rather than change in pain frequency, is another obvious pain-related endpoint, but measurement may require different assessment measures if both pediatric and adult participants are included. A significant amount of pain may remain at the end of hospitalization in those patients with coexistent chronic pain, making it difficult to ascertain when acute vaso-occlusive-related pain has resolved. Reduction in standard of care analgesic usage is a frequent pain-related endpoint in analgesic trials, but is complex in acute vaso-occlusive pain as many patients may have substantial degrees of opioid tolerance from prior chronic oral opioid usage, and analgesic therapy often needs to be individualized. Most such acute pain studies ultimately utilize a composite of these measures as a primary endpoint, which continues to be refined based on feasibility and performance in subsequent studies.
Approximately 10%–20% of these pain episodes requiring hospitalization are further complicated by the development of new pulmonary infiltrates associated with fever and hypoxia, often within the first 48–72 hours of admission, referred to as an acute chest syndrome.55,56 Some of these pulmonary episodes represent an infectious process, particularly in young children. However, in the setting of continued vaso-occlusion, most infiltrates, particularly in the lung bases,57 are caused by pulmonary parenchymal damage from vaso-occlusion occurring in the distal pulmonary circulation. These events typically prolong the length of hospitalization, and are treated with antibiotics, bronchodilators, and blood transfusions. Some patients with unusually severe disease will require mechanical ventilation and other respiratory support, many of whom will also develop neurologic events and acute kidney injury.58
Vaso-occlusion also occurs less commonly in other vascular beds, including the spleen, where life-threatening trapping of sickled erythrocytes and platelets can cause acute splenic sequestration with acute left upper quadrant visceral pain from rapid enlargement of the splenic capsule.59 Similarly, vaso-occlusion in the penile circulation causes acute episodes of painful priapism that is typically treated with aspiration and alpha-adrenergic agents.60
Several other complications likely represent a combination of progressive vascular changes and vaso-occlusion. Progressive endothelial damage occurring in the large vessels of the cerebral vascular circulation, likely related to increased blood flow from chronic anemia, leads to overt strokes in children61 and adults.62 More prevalent neurologically silent infarction from vascular occlusion in smaller vessels also occurs, but is of less certain etiology.63 Routine screening of young SCD children with transcranial Doppler ultrasound has dramatically decreased the incidence of large vessel strokes.64 While most children identified with abnormal transcranial ultrasound studies require lifelong transfusion therapy,65 some may benefit from daily use of HU instead.66
Vascular changes related to hemolysis-related damage also accounts for the development of pulmonary hypertension in adolescents and young adults,67 usually diagnosed by an elevated tricuspid regurgitant jet velocity on cardiac echocardiography and confirmed by cardiac catheterization.68 An increased risk for mortality has been associated with a tricuspid regurgitant jet velocity ≥2.5 m/s, an N-terminal pro-brain natriuretic peptide (NT-pro-BNP) level ≥160 pg/mL, or right heart catheterization-confirmed pulmonary hypertension.69 Prostacyclin agonist or endothelin receptor antagonist therapies have been successful in selected patients.
While uncommon in the pediatric age group, leg ulcers, typically over the medial malleolus of either or both ankles, can be a source of considerable pain and disability and are difficult to manage.70 The role of venous stasis or other vascular factors remains controversial.71 Avascular necrosis in the vertebral column, shoulder, or hips, from vascular compromise of the cortical bone and/or bone marrow circulations can be a source of acute pain as well as chronic pain in adolescent and young adult patients. Some patients progress to joint collapse, particularly at the head of the femur, which ultimately requires joint replacement for relief of chronic pain or to improve physical functioning.72
Severe anemia, characteristic of SCA and Sß0 thalassemia, also is a potential therapeutic target. Lower hemoglobin levels are associated with a higher frequency of stroke, renal disease, leg ulcers, and pulmonary hypertension secondary to hemolysis-related endothelial dysfunction,37 but a lower frequency of vaso-occlusive pain,73 likely as a result of lower blood viscosity. This typifies the delicate balance in treating sickle cell complications with investigational agents where a therapeutic improvement in one feature may conversely have a negative impact on the frequency of other complications (Figure 3).
Hemostatic abnormalities related to sickling and hemolysis-related changes are also common.74 Changes include thrombin activation, altered levels of anticoagulant proteins, impaired fibrinolysis, and platelet activation. Platelet activation may be particularly problematic as it may contribute to cell–cell interactions that may impact sickle erythrocyte-endothelial cell adhesion and subsequent vaso-occlusion. Antithrombotic therapies have been clinically useful to treat thrombotic complications of SCD, and have shown some preliminary benefit as a potential vaso-occlusive therapy.
Therapeutic trials
Design considerations for vaso-occlusion therapy trials
The design of new therapeutics for sickle vaso-occlusion presents many challenges. Numerous agents have been developed to intervene in the many facets of SCD pathophysiology (Figure 4). There remains limited understanding of the potential efficacy that could be obtained from intervening at any one particular point in the process leading to vaso-occlusion and subsequent tissue injury (Figure 1), but the most successful therapies to date have directly impacted sickle polymer formation rather than any of its “downstream” consequences.
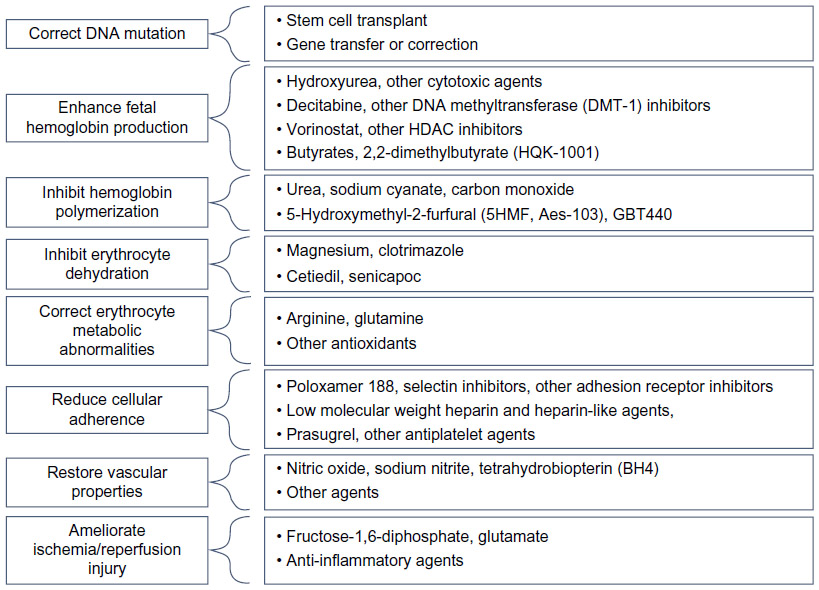 | Figure 4 Therapeutic targets and corresponding investigation agents in sickle cell disease. |
Comparison of test article to placebo in the context of best standard care is the most ethically appropriate design, particularly for children, but the clinical practice for analgesics, fluids, oxygen, and transfusions all vary considerably across clinical sites.54 This site-to-site variability may need to be considered in statistical analysis strategies. In the absence of an effective surrogate marker or ability to measure vaso-occlusion in humans, studies of therapies for vaso-occlusion have had to rely on clinical outcomes of “crisis resolution” as endpoints. These too have been challenging as the most obvious clinical endpoint, duration of hospitalization, can be impacted by social considerations, transportation issues, and treatment of other comorbidities such as fever or acute chest syndrome.
Most agents, including HU, have been tested only in individuals with the more severe sickling genotypes of SS and Sβ0-thalassemia, so their effectiveness in other sickling genotypes is poorly understood. While this knowledge and treatment gap may be less of an issue in the pediatric population where these other sickling phenotypes are often less symptomatic, it significantly impacts the availability of therapies for the adult sickle cell population where most individuals are symptomatic regardless of sickle genotype. Inclusion of all sickling disorders in late stage trials would address this critical problem,75 but requires careful attention to inclusion/exclusion criteria and analysis of safety assessments as baseline hematologic and metabolic parameters will vary by sickle genotype. If endpoints are symptom-based, the enrollment may need to be stratified by sickle genotype to balance across study arms, which may impact sample size considerations.
Participant age is another important design consideration. The frequency of vaso-occlusive pain and other SCD complications are age-dependent,44 so designs that require a minimum pain frequency may have difficulty recruiting adequate numbers of young children unless a large number of clinical sites are utilized. Similarly, some complications such as leg ulcers, priapism, or hip avascular necrosis are uncommon in children. Age-related changes in drug distribution, metabolism, and clearance are well-understood pharmacologic issues in children,76 but SCD children also have substantially increased renal blood flow and resultant increases in glomerular filtration rates.77 These changes may impact the therapeutic levels of agents or their metabolites that have substantial degrees of renal clearance, which may require further pharmacokinetic testing and alternative dosing strategies when expanding clinical trials into younger SCD populations.
“Painful lessons” – past treatment failures
In contrast to the success of a number of multisite clinical trials exploring the role of transfusion therapy or HU for central nervous system complications of SCD,65,78,79 the therapeutic arena in sickle cell vaso-occlusion therapy has been dominated by a number of multisite studies with negative results. Early studies by the first SCD clinical trial group, the Cooperative Urea Trials Group, evaluated the relative effectiveness of anti-sickling therapy with alkali or urea, compared to invert sugar, administered intravenously in the treatment of the painful episodes of SCA over a 48-hour period using a double-blind protocol in the early 1970s.80 While the clinical trial design was remarkably good by today’s standards, neither urea nor alkali administration was found to be superior to invert sugar alone in shortening the duration of crisis episodes.
The pioneering studies by Hebbel et al81 and Kaul et al,82 demonstrating the tendency for sickle erythrocytes to adhere to vascular endothelium, in vitro and in vivo, suggested anti-adhesion therapy could stop and/or reverse sickle vaso-occlusion. In an initial pilot study, a nonionic block copolymer surfactant with hemorheologic and antithrombotic properties, poloxamer 188, reduced total analgesic use and pain intensity, and showed trends for shorter duration of painful episodes and total days of hospitalization.83 However, a subsequent larger randomized, placebo-controlled Phase III trial showed a statistically significant but clinically disappointing small (9-hour) decrease in the duration of painful episodes compared with the patients receiving placebo.84 In subgroup analysis, children and patients who were receiving concomitant HU had a more significant reduction in crisis duration (16–21 hours). Whether these findings represented more efficacy of this agent in these subgroups, or a more effective statistical analysis strategy, was uncertain. After a hiatus of almost 15 years, this agent is being studied again in an ongoing pediatric and young adult Phase III trial (MAST Therapeutics, NCT01737814) using a different endpoint, analgesic usage, and a different statistical analysis plan.
The strong dependence of the rate of sickle polymerization on the intracellular hemoglobin concentration suggested that modulation of erythrocyte volume might be an effective therapeutic strategy. An early study of induced hyponatremia with fluids and desmopressin proved too difficult to control,85 but encouraging pilot studies with magnesium86 and with clotrimazole87 suggested that inhibition of the calcium activated potassium channel (Gardos channel) could be effective in increasing erythrocyte volume and reducing sickling. Senicapoc, a novel Gardos channel inhibitor, increased hemoglobin concentration with a concomitant decrease in total reticulocyte number and various markers of erythrocyte destruction in a Phase II study,88 but a subsequent Phase III study was stopped early by its data safety monitoring board (DSMB) due to a lack of efficacy when it determined that despite improvements in anemia and hemolysis, there was no significant improvement in the rate of sickle cell painful episodes.89 Many have speculated that the increase in hematocrit and resulting blood viscosity offset the beneficial anti-sickling effect of an increased erythrocyte volume reflecting the complex nature of sickle polymer formation and vaso-occlusion.
Similarly, IV magnesium also has been studied in two randomized, double-blind, placebo-controlled trials in children requiring admission to hospital for treatment of severe pain with IV analgesics.90,91 Magnesium was well tolerated, but had no effect on hospital duration, pain scores, or cumulative analgesia use in either study. However, a number of important future feasibility issues were successfully addressed in these studies, including recruitment and initial administration of study drug in the Emergency Department setting.
The compelling role for NO in the pathobiology of vaso-occlusion prompted a number of attempts to manipulate this pathway as a therapeutic target. A large randomized placebo-controlled Phase III study of NO inhalation therapy was subsequently conducted, using a primary endpoint, which has been adapted for many subsequent clinical trials, of time to resolution of painful crisis, defined by: 1) freedom from parenteral opioid use for 5 hours; 2) pain relief as assessed by visual analog pain scale scores of 6 cm or lower (on 0–10 cm scale); 3) ability to walk; and 4) patient’s and family’s decision, with physician consensus, that the remaining pain could be managed at home. Unfortunately, there was no significant change in the primary endpoint between the inhaled NO and placebo groups, nor were there significant differences in secondary outcome measures, including length of hospitalization, visual analog pain scale scores, or cumulative opioid usage.92
A number of other clinical trials have explored alternative targets to normalized NO biology. The use of sodium nitrite as alternative source of NO has also been explored, largely by investigators at the National Institutes of Health Clinical Center.93 The proposed mechanism involves a novel physiological function of human hemoglobin as an oxygen- and pH-dependent nitrite reductase potentially converting nitrite to NO along the physiological oxygen gradient in arterial blood, accentuating vasodilation in hypoxic tissue. Sodium nitrite infusions were well tolerated without hypotension, clinically significant methemoglobinemia or other untoward events in Phase I/II clinical trials,94 and demonstrated dose-dependent increases in forearm blood flow. Subsequent pilot studies of sodium nitrite during vaso-occlusive crises have not been promising, but the drug is being explored as a topical agent for treatment of sickle leg ulcers (NCT01316796).95
NO production could also be restored and endothelial dysfunction reduced or reversed by restoring production of NO. Since tetrahydrobiopterin (BH4) is an essential cofactor required for the activity of eNOS, supplementation with exogenous BH4 or therapeutic approaches that increase endogenous amounts of BH4 could be possible therapeutic strategies. To explore this concept, BioMarin conducted a Phase IIa dose-escalation trial in SCD of oral sapropterin, a synthetic BH4 approved for the treatment of phenylketonuria.96 While well tolerated, the effects on physiological and biochemical markers of endothelial function in SCD were unimpressive and further studies have not been pursued.
Ischemia-related tissue damage, with its own complex pathophysiology,31 is the likely downstream consequence of sickle vaso-occlusion. Numerous drug development programs have attempted to identify cytoprotective agents for tissues ranging from the heart to the brain, including the development of fructose-1,6-diphosphate by Cypros Pharmaceuticals, which was intended to restore adenosine triphosphate (ATP) levels in ischemic tissues.97 An initial dose-escalation Phase IIa trial of 47 SCD patients evaluated a single dose at three dose levels of Fructose di-phosphate (FDP) (50, 100 and 250 mg/kg). The primary endpoint of the study was the level of pain intensity reported during the first 24 hours following drug or placebo administration. All of the drug-treated patients demonstrated reductions in pain levels, which reached statistical significance at the two lower dose groups. Unfortunately, a larger Phase III study in hospitalized vaso-occlusive pain was stopped early for futility, perhaps reflecting a lack of efficacy in more established pain, which may be a potential issue for similar Phase III trial designs of agents intended to reverse rather than prevent sickle vaso-occlusion.
Newer agents – the next generation of targeted therapies
Enhancement of fetal hemoglobin production
Fetal hemoglobin, as its name implies, is the dominant hemoglobin produced later in human fetal development. It begins to be replaced by adult hemoglobin shortly after birth in a process called “hemoglobin switching”, the biochemical and genetic basis of which has been extensively studied.98 Recovery from a period of anemia or hypoxia in response to increased erythropoietin production, so-called “stress erythropoiesis”,99 is another clinical situation that reverses the fetal-to-adult hemoglobin switch. This process was studied in animals with anemia induced with a myelosuppressive chemotherapy agent, 5-azacytidine.100 Subsequent studies in humans, including those with SCD, documented similar increases in fetal hemoglobin, but this agent was thought to be too toxic for long-term use. HU was subsequently studied as a fetal hemoglobin inducer in SCD as it was a less toxic myelosuppressive agent with some long-term experience in the treatment of polycythemia vera.101
HU – A “leadoff home run” for fetal hemoglobin inducers
HU remains the only US Food and Drug Administration-approved disease-modifying therapy for SCD, although repeated scheduled transfusions of red blood cells can also decrease disease severity.102 The approval was based on a single randomized clinical trial, the MSH (Multicenter Study of Hydroxyurea for Sickle Cell Anemia), that enrolled 299 adults (mean age of 30.5 years), and almost all had SCA.103 The primary endpoint was a reduction in the frequency of painful crises over a 2-year period, but the trial was closed earlier than expected, after demonstration that the median pain rate was reduced by almost 50% (2.5 vs 4.5 events per year) in patients assigned to HU therapy. Hospitalizations, episodes of chest syndrome, and numbers of transfusions were also lower in patients treated with HU. A subsequent follow-up study of these trial participants demonstrated a continued clinical improvement in morbidity and mortality.104 A similar Phase III trial of HU in SCA infants and young children did not meet its primary endpoint of preventing a decline in organ function, but showed similar reductions in the frequency of acute pain, chest syndromes, and transfusions.105,106
The large Phase III MSH trial was preceded by a number of pilot studies,107 and was supported by previous studies of cytotoxic chemotherapeutic agents in animals108 and humans,109 and a wealth of clinical and laboratory data on the role of fetal hemoglobin in ameliorating sickle polymer formation. The clinical benefit of higher levels of fetal hemoglobin was also supported by the relatively mild clinical manifestations among Bedouin Arabs from the Saudi Arabian peninsula and certain tribes from central India with individuals who had homozygous sickle hemoglobin and relatively high amounts of fetal hemoglobin,110 and the beneficial effect of high fetal hemoglobin in newborns and young infants with SCD.38 This “perfect storm” of substantial clinical benefit, impressive supportive preclinical and laboratory data, and strong correlative clinical models, allowed regulatory approval with a single pivotal Phase III trial, but has set a high standard for comparison in future regulatory deliberations for new therapies.
The efficacy of HU for the treatment of vaso-occlusion in SCD is generally attributed to its ability to increase fetal hemoglobin. However, the cellular mechanisms by which this occurs are not completely understood, and other mechanisms may account for the clinical benefit of this agent. For example, HU reduces the expression of adhesion molecules on sickle erythrocytes,111 and cytoreduction of neutrophils, monocytes, and reticulocytes may also have a beneficial effect on blood viscosity or cell–cell interactions.112
Other fetal hemoglobin inducers
A number of additional fetal hemoglobin inducers have been developed based on the evolving research on the importance of epigenetic changes, such as DNA methylation and histone modification, in regulating fetal hemoglobin production.113 These agents will likely be initially tested as alternatives to HU, recognizing that some individuals taking this drug may not achieve a clinically effective response, particularly in those with low initial fetal hemoglobin levels.114,115 Other patients may benefit from a drug that requires less frequent monitoring, or that has less reproductive risk. Trial designs that test fetal hemoglobin inducers with different mechanisms of action separately and in combination may ultimately be necessary to achieve clinically useful increases in broad populations of SCD adults and children.
DNA hypomethylation agents that act by inhibiting DNA methyltransferase initially built on the success of 5-azacytidine using a derivative, 5-aza-2′-deoxycytidine (decitabine). Substantial elevations of fetal hemoglobin have been demonstrated in a number of Phase I and II trials of decitabine in SCD adults,116 but further development has been hampered by the absence of an oral dosing form, and occurrence of dose-related neutropenia and thrombocytosis.
HDAC inhibitors have been another promising class of agents to induce fetal hemoglobin production in SCD.117 Short chain fatty acids such as butyrate cause significant increases in fetal hemoglobin levels, but the practical use of IV butyrate analogs has been complicated by their relatively low potency and short half-life. Similarly, a Phase II clinical trial of a recently developed oral dosing form, 2,2-dimethylbutyrate (HQK-1001),118 was recently terminated early for lack of effects. With their clinical use in a variety of hematologic malignancies and solid tumor malignancies, newer HDAC inhibitors have been explored in Phase I and II trials in SCD, including vorinostat and panobinostat.117 Unfortunately, vorinostat’s Phase II clinical trial was recently closed due to the lack of measurable effects. A number of newer more selective HDAC inhibitors are in preclinical development. Thalidomide, a drug used for multiple myeloma treatment with immunomodulatory and anti-angiogenic properties, upregulates γ-globin gene expression in vitro.113 Pomalidomide, a thalidomide derivative, has been used in a phase I study in SCD adults (NCT01522547).
Polymerization inhibitors
While initially envisioned once the mechanism of sickle polymerization was characterized, this therapeutic area has made little headway until recently. A naturally occurring aromatic aldehyde, 5-hydroxymethyl-2-furfural (5-HMF), binds to the valine residue of hemoglobin producing a left shift in the oxygen equilibrium curve, which reduces erythrocyte sickling in animal models.119 Reduction in Psickle-mediated Ca2+ by 5-HMF following erythrocyte sickling has also been observed suggesting that this agent may also have an additional anti-sickling effect by inhibiting the deleterious sequelae of Gardos channel activation.120 Single doses of 5-HMF were well tolerated in a Phase I study, and showed changes in blood oxygen levels during a hypoxia challenge test consistent with the effects of a left-shifting allosteric hemoglobin modifier.121 This agent is completing Phase II trials in SCD sponsored by AesRx, LLC, and is expected to enter Phase III trials late in 2015 with sponsorship by Baxter.
Another novel orally bioavailable small molecule, GTx011 (GBT440) (Global Blood Therapeutics, South San Francisco, CA, USA), that binds to the N-terminal α-globin chain of hemoglobin via a reversible Schiff base has been evaluated in preclinical studies. Inhibition of sickle hemoglobin polymerization in vitro improved erythrocyte deformability and blood viscosity,122 decreased the p50 value of human blood, and in a murine model of SCD (Townes SS mice), GTx011 prevented ex vivo sickling of RBCs and prolonged erythrocyte half-life.123 This agent has entered Phase I testing in SCD patients (NCT 02285088).
Anti-adhesion therapies/anti-hemostatic agents
Much of the current enthusiasm and activity in SCD clinical trials rests in this therapeutic area, especially for agents targeting selectins,124 although agents targeting integrin-mediated interactions are also being developed or repurposed for SCD. For example, a humanized anti-P-selectin monoclonal antibody (SelG1) is being developed by Selexys Pharmaceuticals as a treatment for SCD. A Phase I clinical study has evaluated the safety, pharmacokinetics, pharmacodynamics, and immunogenicity of SelG1 in a single-center, double-blind, placebo-controlled, ascending single dose and multiple dose study of IV-administered SelG,125 and a Phase II study in adults with SCD is underway.
An alternative approach, a small molecule pan-selectin inhibitor (GMI-1070, rivipansel) is being developed by GlycoMimetics, Inc., and now Pfizer, to inhibit blood cell adhesion in sickle vaso-occlusive episodes. After a successful initial Phase I/II study in SCD,126 a subsequent Phase II study of GMI-1070 was conducted in a prospective multicenter, randomized, placebo-controlled, double-blind, study of 76 SCD children and adults hospitalized for vaso-occlusive pain.127 Comparing active treatment group to placebo, clinically meaningful reductions in mean and median times to crisis resolution of 41 and 63 hours were demonstrated. Also in secondary analyses, mean cumulative IV opioid analgesic use was reduced by 83% with GMI-1070 vs placebo (P=0.010). A randomized placebo-controlled Phase III trial sponsored by Pfizer using crisis resolution as its primary endpoint has recently been opened. This study is currently unique in its inclusion of all sickle genotypes, in both children and adults.
Eli Lilly is conducting a therapeutic development program in SCD for prasugrel, a thienopyridine P2Y12 adenosine diphosphate receptor antagonist that inhibits adenosine diphosphate-mediated platelet activation and aggregation. Drug exposure in Phase I studies was well tolerated, with no bleeding-related events in patients with SCD.128 Mean drug concentration–time profiles and platelet reactivity were comparable between healthy subjects and patients with SCD. Subsequent Phase II studies in SCD adults observed a modest decrease in mean pain rate and intensity in the prasugrel arm compared with placebo.129 Platelet surface P-selectin and plasma soluble P-selectin, biomarkers of in vivo platelet activation, were significantly reduced in SCD patients receiving prasugrel compared with placebo suggesting potential effects on sickle erythrocyte adhesion. A Phase III randomized placebo-controlled pediatric trial is ongoing using reduction in pain frequency as the primary study endpoint (NCT01794000). Additional novel features of this trial design include the counting of home-managed pain using electronic diaries, and the extensive use of non-US clinical sites.
Vascular agents
Small pilot studies to enhance NO levels by providing increased amounts of its substrate, arginine, have had some success and are advancing to Phase II studies.130,131 A number of preclinical studies have evaluated the utility of carbon monoxide form of polyethylene glycol (PEG)ylated hemoglobin (SANGUINATE™) in ischemia/reperfusion injury settings, including myocardial infarction,132 and is now in development for SCD by Prolong Pharmaceuticals.
Cytoprotective agents
Therapies explored to date have been disappointing. However, recent progress has been made with the development of oral glutamine to provide increased defense against oxidant stress in sickle erythrocytes. Glutamine supplementation was expected to increase NAD and NADH levels by increased transport and utilization of glutamine in sickle erythrocytes, which was confirmed in an initial pilot study.133 A subsequent Phase II randomized, double-blind, placebo-controlled, parallel-group, multicenter study evaluated the safety and efficacy of l-glutamine therapy. Eighty-one patients aged 5 years or older diagnosed with SCA or Sβ0-thalassemia were randomized to oral l-glutamine at 0.3 g/kg or placebo twice daily for 48 weeks. At 6 months of therapy, l-glutamine treatment reduced the frequency of hospitalization (nearly 40% reduction) and there was a major trend for the decrease in frequency of painful crises (over 50% reduction) favoring the l-glutamine treatment arm.134 These results have been replicated in a larger Phase III clinical trial, but the results have not yet been published in a peer-reviewed publication, and further studies will likely be needed for regulatory approval.
Advent of curative therapies
Stem cell transplant
HLA-identical sibling bone marrow transplantation in children with severe SCD currently provides a curative outcome in more than 90% of the recipients.14 Health-related quality of life largely returns to that of the normal population by 1 year post-transplant, and acute and some chronic sickle complications also resolve in this time period.135 While some progress is being made in using unrelated and alternative donor sources in the absence of suitable HLA-matched siblings,136 this remains a fundamental barrier to more widespread use of this therapeutic strategy. Similarly, the current risk of loss of fertility, and the risk of post-transplant complications remain unacceptable to many potentially eligible families.137
Gene therapy
Correction of the genetic defect in the patient’s own hematopoietic stem cells would obviate the need for alternative sources of bone marrow to produce normal red blood cells, prevent therapy-related immunological side effects (graft vs host disease/graft rejection), and obviate the need for prophylactic therapies such as HU.138 Gene insertion therapies have been developed to insert γ-globin–based or modified β-globin–based vectors for SCD gene therapy, largely using lentiviruses. Both approaches have shown proof of concept in mouse models of SCD.139,140 Bluebird Bio, Inc. has initiated a Phase I trial in adults with severe SCD to evaluate the safety of transplantation of autologous CD34+ stem cells transduced ex vivo with the LentiGlobin BB305 lentiviral vector inserting a bioengineered recombinant β-globin that prevents axial and lateral contacts in the sickle polymer originally developed by Levasseur et al.141 A similar Phase I study using autologous CD34+ stem cells transduced by a vector which substitutes the exons of γ-globin gene for β-globin in the β-globin vector has been opened at Cincinnati Children’s Hospital. Myeloablative chemotherapy is used to provide space for the transduced stem cell to grow, so this procedure will share some of the risks of bone marrow transplantation, as well as yet to be established risk of the gene therapy. However, the survival advantage of erythrocytes protected from sickle hemoglobin polymerization by introduction of novel genes provides an opportunity for phenotypic correction of SCD even with modest levels of transduced stem cells similar to that observed with donor chimerism of 10%–30% in patients after traditional bone marrow transplant.142
Final thoughts
A number of clinical trial design-related issues remain to be resolved. Since patients typically manage the initial portion of their vaso-occlusive pain at home, studies of those agents intended to accelerate crisis resolution will invariably contain individuals with a heterogeneous distribution of crisis duration before being exposed to the test article. Whether the duration of prior vaso-occlusion impacts the potential efficacy of these agents is largely unknown. Given the complexity of sickle vaso-occlusion pathophysiology, it is likely that combination therapy may provide optimum inhibition of sickle polymerization and its consequences to maximize clinical benefit.143 However, few studies have attempted such multiple combination comparisons so feasible and regulatory acceptable study designs remain largely unexplored. The regulatory considerations for investigational agents that might provide only modest incremental benefit in ameliorating sickle cell-related complications have yet to be clarified. Finally if effective, short-term intervention studies for acute complications, such as pain, will need to be followed by much larger and more expensive studies to examine their impact on less frequent vaso-occlusion complications, such as acute chest syndrome, or priapism. Longer term multi-year studies will also be needed to examine their impact on preventing chronic organ damage in spleen, kidneys, and the lung.
Disclosure
The author reports previous consulting relationships with Baxter, Biogen Idec, GlycoMimetics, Inc., and Lilly; current consulting relationships with Pfizer Inc., and grant support from Lilly and Pfizer, Inc. The author reports no other conflicts of interest in this work.
References
Barabino GA, Platt MO, Kaul DK. Sickle cell biomechanics. Annu Rev Biomed Eng. 2010;12:345–367. | |
Steinberg MH. Sickle cell anemia, the first molecular disease: overview of molecular etiology, pathophysiology, and therapeutic approaches. ScientificWorldJournal. 2008;8:1295–1324. | |
Thein SL. Genetic insights into the clinical diversity of beta thalassaemia. Br J Haematol. 2004;124(3):264–274. | |
Prabhakar H, Haywood C, Molokie R. Sickle cell disease in the United States: looking back and forward at 100 years of progress in management and survival. Am J Hematol. 2010;85(5):346–353. | |
Diggs LW. Siderofibrosis of the spleen in sickle cell anemia. Journal of the American Medical Association. 1935;104(7):538–541. | |
Hughes JG, Diggs L, Gillespie CE. The involvement of the nervous system in sickle-cell anemia. The Journal of Pediatrics. 1940;17(2):166–184. | |
Barrett-Connor E. Bacterial infection and sickle cell anemia. An analysis of 250 infections in 166 patients and a review of the literature. Medicine (Baltimore). 1971;50(2):97–112. | |
Diggs LW, Ching RE. Pathology of sickle cell anemia. Southern Medical Journal. 1934;27(10):839–845. | |
Sherman IJ. The sickling phenomenon, with special reference to the differentiation of sickle cell anemia from the sickle cell trait. Bulletin of the Johns Hopkins Hospital. 1940;67:309–324. | |
Ham TH, Castle WB. Relation of increased hypotonic fragility and of erythrostasis to the mechanism of hemolysis in certain anemias. Trans Assoc Am Physicians. 1940;55:127–132. | |
Watson J. Sickling in Negro newborns – its possible relationship to fetal hemoglobin. American Journal of Medicine. 1948;5(1):159–160. | |
Ingram VM. Specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature. 1956; 178(4537):792–794. | |
Pauling L, Itano HA, Singer SJ, Wells IC. Sickle cell anemia, a molecular disease. Science. 1949;110(2865):543–548. | |
Walters MC. Update of hematopoietic cell transplantation for sickle cell disease. Curr Opin Hematol. 2015;22(3):277–233. | |
Serjeant GR. One hundred years of sickle cell disease. Br J Haematol. 2010;151(5):425–429. | |
Williams TN, Weatherall DJ. World distribution, population genetics, and health burden of the hemoglobinopathies. Cold Spring Harb Perspect Med. 2012;2(9):a011692. | |
Piel FB, Patil AP, Howes RE, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. 2010;1:104. | |
Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10(7):e1001484. | |
Huttle A, Maestre GE, Lantigua R, Green NS. Sickle cell in sickle cell disease in Latin America and the United States. Pediatr Blood Cancer. 2015;62(7):1131–1136. | |
Hassell KL. Population estimates of sickle cell disease in the US. Am J Prev Med. 2010;38(4 Suppl):S512–S521. | |
Saraf SL, Molokie RE, Nouraie M, et al. Differences in the clinical and genotypic presentation of sickle cell disease around the world. Paediatr Respir Rev. 2014;15(1):4–12. | |
Piel FB, Tatem AJ, Huang Z, Gupta S, Williams TN, Weatherall DJ. Global migration and the changing distribution of sickle haemoglobin: a quantitative study of temporal trends between 1960 and 2000. Lancet Glob Health. 2014;2(2):e80–e89. | |
Vella S, Schwartländer B, Sow SP, Eholie SP, Murphy RL. The history of antiretroviral therapy and of its implementation in resource-limited areas of the world. Aids. 2012;26(10):1231–1241. | |
Ferrone FA. Polymerization and sickle cell disease: a molecular view. Microcirculation. 2004;11(2):115–128. | |
Vekilov PG. Sickle-cell haemoglobin polymerization: is it the primary pathogenic event of sickle-cell anaemia? Brit J Haematol. 2007;139(2):173–184. | |
Telen MJ. Role of adhesion molecules and vascular endothelium in the pathogenesis of sickle cell disease. Hematology Am Soc Hematol Educ Program. 2007:84–90. | |
Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A. 2002;99(5):3047–3051. | |
Ergul S, Brunson CY, Hutchinson J, et al. Vasoactive factors in sickle cell disease: In vitro evidence for endothelin-1-mediated vasoconstriction. Am J Hematol. 2004;76(3):245–251. | |
Wood KC, Hsu LL, Gladwin MT. Sickle cell disease vasculopathy: a state of nitric oxide resistance. Free Radic Biol Med. 2008;44(8):1506–1528. | |
Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139(2):267–284. | |
Eltzschig HK, Eckle T. Ischemia and reperfusion – from mechanism to translation. Nat Med. 2011;17(11):1391–1401. | |
Kassim AA, Debaun MR. Sickle Cell Disease, Vasculopathy, and Therapeutics. Annu Rev Med. 2013;64:451–466. | |
Kato GJ, Hebbel RP, Steinberg MH, Gladwin MT. Vasculopathy in sickle cell disease: Biology, pathophysiology, genetics, translational medicine, and new research directions. Am J Hematol. 2009;84(9):618–625. | |
Heistad DD. Oxidative stress and vascular disease 2005 Duff lecture. Arterioscler Thromb Vasc Biol. 2006;26(4):689–695. | |
Sparkenbaugh E, Pawlinski R. Interplay between coagulation and vascular inflammation in sickle cell disease. Brit J Haematol. 2013; 162(1):3–14. | |
Kato GJ, Taylor JG 6th. Pleiotropic effects of intravascular haemolysis on vascular homeostasis. Br J Haematol. 2010;148(5):690–701. | |
Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21(1):37–47. | |
Bailey K, Morris JS, Thomas P, Serjeant GR. Fetal haemoglobin and early manifestations of homozygous sickle cell disease. Arch Dis Child. 1992;67(4):517–520. | |
Stevens MC, Padwick M, Serjeant GR. Observations on the natural history of dactylitis in homozygous sickle cell disease. Clin Pediatr (Phila). 1981;20(5):311–317. | |
Dampier C, Ely B, Brodecki D, et al. Pain characteristics and age-related pain trajectories in infants and young children with sickle cell disease. Pediatr Blood Cancer. 2014;61(2):291–296. | |
Smith WR, Scherer M. Sickle-cell pain: advances in epidemiology and etiology. Hematology Am Soc Hematol Educ Program. 2010;2010:409–415. | |
Dampier C, Ely E, Brodecki D, O’Neal P. Home management of pain in sickle cell disease: a daily diary study in children and adolescents. J Pediatr Hematol Oncol. 2002;24(8):643–647. | |
Dampier C, Setty BN, Eggleston B, Brodecki D, O’Neal P, Stuart M. Vaso-occlusion in children with sickle cell disease: clinical characteristics and biologic correlates. J Pediatr Hematol Oncol. 2004;26(12):785–790. | |
Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991;325(1):11–16. | |
Raphael JL, Mei M, Mueller BU, Giordano T. High resource hospitalizations among children with vaso-occlusive crises in sickle cell disease. Pediatr Blood Cancer. 2012;58(4):584–590. | |
Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med. 2008;148(2):94–101. | |
Taylor LE, Stotts NA, Humphreys J, Treadwell MJ, Miaskowski C. A review of the literature on the multiple dimensions of chronic pain in adults with sickle cell disease. J Pain Symptom Manage. 2010;40(3):416–435. | |
Hemker BG, Brousseau DC, Yan K, Hoffmann RG, Panepinto JA. When children with sickle-cell disease become adults: Lack of outpatient care leads to increased use of the emergency department. Am J Hematol. 2011;86(10):863–865. | |
Carroll CP, Haywood C Jr, Lanzkron S. Prediction of onset and course of high hospital utilization in sickle cell disease. J Hosp Med. 2011; 6(5):248–255. | |
Carroll PC, Haywood C, Hoot MR, Lanzkron S. A Preliminary Study of Psychiatric, Familial, and Medical Characteristics of High-utilizing Sickle Cell Disease Patients. Clin J Pain. 2013;29(4):317–323. | |
Tanabe P, Hafner JW, Martinovich Z, Artz N. Adult emergency department patients with sickle cell pain crisis: results from a quality improvement learning collaborative model to improve analgesic management. Acad Emerg Med. 2012;19(4):430–438. | |
Benjamin LJ, Swinson GI, Nagel RL. Sickle cell anemia day hospital: an approach for the management of uncomplicated painful crises. Blood. 2000;95(4):1130–1136. | |
Zempsky WT. Evaluation and Treatment of Sickle Cell Pain in the Emergency Department: Paths to a Better Future. Clin Pediatr Emerg Med. 2010;11(4):265–273. | |
Miller ST, Kim HY, Weiner D, et al. Inpatient management of sickle cell pain: A ‘snapshot’ of current practice. Am J Hematol. 2012;87(3):333–336. | |
Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood. 1994;84(2):643–649. | |
Vichinsky EP, Styles LA, Colangelo LH, Wright EC, Castro O, Nickerson B. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood. 1997;89(5):1787–1792. | |
Dessap AM, Deux JF, Habibi A, et al. Lung imaging during acute chest syndrome in sickle cell disease: computed tomography patterns and diagnostic accuracy of bedside chest radiograph. Thorax. 2014;69(2):144–151. | |
Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. 2000;342(25):1855–1865. | |
Brousse V, Elie C, Benkerrou M, et al. Acute splenic sequestration crisis in sickle cell disease: cohort study of 190 paediatric patients. Br J Haematol. 2012;156(5):643–648. | |
Crane GM, Bennett NE Jr. Priapism in sickle cell anemia: emerging mechanistic understanding and better preventative strategies. Anemia. 2010;2011:297364. | |
Switzer JA, Hess DC, Nichols FT, Adams RJ. Pathophysiology and treatment of stroke in sickle-cell disease: present and future. Lancet Neurol. 2006;5(6):501–512. | |
Strouse JJ, Jordan LC, Lanzkron S, Casella JF. The excess burden of stroke in hospitalized adults with sickle cell disease. Am J Hematol. 2009;84(9):548–552. | |
DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood. 2012;119(20):4587–4596. | |
Adams R, McKie V, Nichols F, et al. The use of transcranial ultrasonography to predict stroke in sickle cell disease. New Engl J Med. 1992;326(9):605–610. | |
Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. New Engl J Med. 1998;339(1):5–11. | |
Ware RE, Zimmerman SA, Sylvestre PB, et al. Prevention of secondary stroke and resolution of transfusional iron overload in children with sickle cell anemia using hydroxyurea and phlebotomy. J Pediatr. 2004;145(3):346–352. | |
Potoka KP, Gladwin MT. Vasculopathy and Pulmonary Hypertension in Sickle Cell Disease. Am J Physiol Lung Cell Mol Physiol. 2015;308(4):L314–L324. | |
Klings ES, Machado RF, Barst RJ, et al. An Official American Thoracic Society Clinical Practice Guideline: Diagnosis, Risk Stratification, and Management of Pulmonary Hypertension of Sickle Cell Disease. Am J Resp Crit Care. 2014;189(6):727–740. | |
Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350(9):886–895. | |
Delaney KM, Axelrod KC, Buscetta A, et al. Leg ulcers in sickle cell disease: current patterns and practices. Hemoglobin. 2013;37(4):325–332. | |
Minniti CP, Delaney KM, Gorbach AM, et al. Vasculopathy, inflammation, and blood flow in leg ulcers of patients with sickle cell anemia. Am J Hematol. 2014;89(1):1–6. | |
Aguilar C, Vichinsky E, Neumayr L. Bone and joint disease in sickle cell disease. Hematol Oncol Clin North Am. 2005;19(5):929–941, viii. | |
Taylor JG 6th, Nolan VG, Mendelsohn L, Kato GJ, Gladwin MT, Steinberg MH. Chronic hyper-hemolysis in sickle cell anemia: association of vascular complications and mortality with less frequent vasoocclusive pain. PloS One. 2008;3(5):e2095. | |
Pakbaz Z, Wun T. Role of the hemostatic system on sickle cell disease pathophysiology and potential therapeutics. Hematol Oncol Clin North Am. 2014;28(2):355–374. | |
Debaun MR, Field JJ. Limitations of clinical trials in sickle cell disease: a case study of the Multi-center Study of Hydroxyurea (MSH) trial and the Stroke Prevention (STOP) trial. Hematology Am Soc Hematol Educ Program. 2007:482–488. | |
Wood AJ, Kearns GL, Abdel-Rahman SM, et al. Developmental pharmacology – drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157–1167. | |
Etteldorf JN, Tuttle A, Clayton GW. Renal function studies in pediatrics: I. Renal hemodynamics in children with sickle cell anemia. AMA Am J Dis Child. 1952;83(2):185–191. | |
Lee MT, Piomelli S, Granger S, et al. Stroke Prevention Trial in Sickle Cell Anemia (STOP): extended follow-up and final results. Blood. 2006;108(3):847–852. | |
DeBaun MR, Gordon M, McKinstry RC, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N Engl J Med. 2014;371(8):699–710. | |
Felts M. Clinical trials of therapy for sickle cell vaso-occlusive crises. Cooperative urea trials group. JAMA. 1974;228:1120–1124. | |
Hebbel RP, Eaton JW, Steinberg MH, White JG. Erythrocyte/endothelial interactions and the vasocclusive severity of sickle cell disease. Prog Clin Biol Res. 1981;55:145–162. | |
Kaul DK, Fabry ME, Nagel RL. Microvascular sites and characteristics of sickle cell adhesion to vascular endothelium in shear flow conditions: pathophysiological implications. Proc Natl Acad Sci U S A. 1989;86(9):3356–3360. | |
Adams-Graves P, Kedar A, Koshy M, et al. RheothRx (poloxamer 188) injection for the acute painful episode of sickle cell disease: a pilot study. Blood. 1997;90(5):2041–2046. | |
Orringer EP, Casella JF, Ataga KI, et al. Purified poloxamer 188 for treatment of acute vaso-occlusive crisis of sickle cell disease: A randomized controlled trial. JAMA. 2001;286(17):2099–2106. | |
Charache S, Moyer MA, Walker WG. Treatment of acute sickle cell crises with a vasopressin analogue. Am J Hematol. 1983;15(4):315–319. | |
De Franceschi L, Bachir D, Galacteros F, et al. Oral magnesium supplements reduce erythrocyte dehydration in patients with sickle cell disease. J Clin Invest. 1997;100(7):1847–1852. | |
Brugnara C, Gee B, Armsby CC, et al. Therapy with oral clotrimazole induces inhibition of the Gardos channel and reduction of erythrocyte dehydration in patients with sickle cell disease. J Clin Invest. 1996; 97(5):1227–1234. | |
Ataga KI, Smith WR, De Castro LM, et al. Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood. 2008;111(8):3991–3997. | |
Ataga KI, Reid M, Ballas SK, et al. Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: a phase III randomized, placebo-controlled, double-blind study of the gardos channel blocker senicapoc (ICA-17043). Br J Haematol. 2011;153(1):92–104. | |
Badaki-Makun O, Scott JP, Panepinto JA, et al. Intravenous magnesium for pediatric sickle cell vaso-occlusive crisis: methodological issues of a randomized controlled trial. Pediatr Blood Cancer. 2014;61(6):1049–1054. | |
Goldman RD, Mounstephen W, Kirby-Allen M, Friedman JN. Intravenous magnesium sulfate for vaso-occlusive episodes in sickle cell disease. Pediatrics. 2013;132(6):e1634–e1641. | |
Gladwin MT, Kato GJ, Weiner D, et al. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. JAMA. 2011;305(9):893–902. | |
Kato GJ, Gladwin MT. Evolution of novel small-molecule therapeutics targeting sickle cell vasculopathy. JAMA. 2008;300(22):2638–2646. | |
Mack AK, McGowan Ii VR, Tremonti CK, et al. Sodium nitrite promotes regional blood flow in patients with sickle cell disease: a phase I/II study. Br J Haematol. 2008;142(6):971–978. | |
Minniti CP, Gorbach AM, Xu D, et al. Topical sodium nitrite for chronic leg ulcers in patients with sickle cell anaemia: a phase 1 dose-finding safety and tolerability trial. Lancet Haematol. 2014;1(3):e95–e103. | |
Katusic ZS, d’Uscio L, Nath KA. Vascular protection by tetrahydrobiopterin: progress and therapeutic prospects. Trends Pharmacol Sci. 2009;30(1):48–54. | |
Marangos PJ, Fox AW, Riedel BJ, Royston D, Dziewanowska ZE. Potential therapeutic applications of fructose-1,6-diphosphate. Expert Opin Investig Drugs. 1998;7(4):615–623. | |
Sankaran VG, Xu J, Orkin SH. Advances in the understanding of haemoglobin switching. Br J Haematol. 2010;149(2):181–194. | |
Socolovsky M. Molecular insights into stress erythropoiesis. Curr Opin Hematol. 2007;14(3):215–224. | |
Fathallah H, Atweh GF. Induction of fetal hemoglobin in the treatment of sickle cell disease. Hematology Am Soc Hematol Educ Program. 2006:58–62. | |
Dover GJ, Charache S. Chemotherapy and hemoglobin F synthesis in sickle cell disease. Ann N Y Acad Sci. 1989;565:222–227. | |
Miller ST, Wright E, Abboud M, et al. Impact of chronic transfusion on incidence of pain and acute chest syndrome during the Stroke Prevention Trial (STOP) in sickle-cell anemia. J Pediatr. 2001;139(6):785–789. | |
Charache S, Barton FB, Moore RD, et al. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine (Baltimore). 1996;75(6):300–326. | |
Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289(13):1645–1651. | |
Thornburg CD, Files BA, Luo Z, et al. Impact of hydroxyurea on clinical events in the BABY HUG trial. Blood. 2012;120(22):4304–4310. | |
Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377(9778):1663–1672. | |
Dover GJ, Charache S. Increasing fetal hemoglobin production in sickle cell disease: results of clinical trials. Prog Clin Biol Res. 1987;251:455–466. | |
Letvin NL, Linch DC, Beardsley GP, McIntyre KW, Nathan DG. Augmentation of fetal-hemoglobin production in anemic monkeys by hydroxyurea. N Engl J Med. 1984;310(14):869–873. | |
Dover GJ, Charache S, Boyer SH, Vogelsang G, Moyer M. 5-Azacytidine increases HbF production and reduces anemia in sickle cell disease: dose-response analysis of subcutaneous and oral dosage regimens. Blood. 1985;66(3):527–532. | |
Pembrey ME, Wood WG, Weatherall DJ, Perrine RP. Fetal hemoglobin production and sickle gene in oases of Eastern Saudi-Arabia. Br J Haematol. 1978;40(3):415–429. | |
Bridges K, Barabino G, Brugnara C, et al. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood. 1996;88(12):4701–4710. | |
Okpala I. The intriguing contribution of white blood cells to sickle cell disease–a red cell disorder. Blood Rev. 2004;18(1):65–73. | |
Costa D, Capuano M, Sommese L, Napoli C. Impact of epigenetic mechanisms on therapeutic approaches of hemoglobinopathies. Blood Cells Mol Dis. 2015;55(2):95–100. | |
Charache S, Dover GJ, Moore RD, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood. 1992;79(10):2555–2565. | |
Steinberg MH, Chui DH, Dover GJ, Sebastiani P, Alsultan A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014;123(4):481–485. | |
Saunthararajah Y, Molokie R, Saraf S, et al. Clinical effectiveness of decitabine in severe sickle cell disease. Br J Haematol. 2008;141(1):126–129. | |
Okam MM, Ebert BL. Novel approaches to the treatment of sickle cell disease: the potential of histone deacetylase inhibitors. Expert Rev Hematol. 2012;5(3):303–311. | |
Reid ME, El Beshlawy A, Inati A, et al. A double-blind, placebo-controlled phase II study of the efficacy and safety of 2,2-dimethylbutyrate (HQK-1001), an oral fetal globin inducer, in sickle cell disease. Am J Hematol. 2014;89(7):709–713. | |
Abdulmalik O, Safo MK, Chen Q, et al. 5-hydroxymethyl-2-furfural modifies intracellular sickle haemoglobin and inhibits sickling of red blood cells. Br J Haematol. 2005;128(4):552–561. | |
Hannemann A, Cytlak UM, Rees DC, Tewari S, Gibson JS. Effects of 5-hydroxymethyl-2-furfural on the volume and membrane permeability of red blood cells from patients with sickle cell disease. J Physiol. 2014;592(18):4039–4049. | |
American Society of Hematology [homepage on the Internet]. Stern W, Matthews D, McKew JC, Shen J, Kato GJ. A phase 1, first-in-man, dose–response study of Aes-103 (5-HMF), an anti-sickling, allosteric modifier of hemoglobin oxygen affinity in healthy normal volunteers. American Society of Hematology; 2012. Available from: https://ash.confex.com/ash/2012/webprogram/Paper46463.html. Accessed August 19, 2015. | |
Patel M, Cabrales P, Dufu K, Metcalf B, Sinha U. GTx011, an Anti-Sickling Compound, Improves SS Blood Rheology By Reduction of HbS polymerization Via Allosteric Modulation of O2 Affinity. Blood. 2014;124(21):1370. | |
Dufu K, Oksenberg D, Zhou C, Hutchaleelaha A, Archer DR. GTx011, a Potent Allosteric Modifier of Hemoglobin Oxygen Affinity, Prevents RBC Sickling in Whole Blood and Prolongs RBC Half-Life in Vivo in a Murine Model of Sickle Cell Disease. Blood. 2014; 124(21):217. | |
Okpala I. Investigational selectin-targeted therapy of sickle cell disease. Expert Opin Investig Drugs. 2015;24(2):229–238. | |
Mandarino D, Kawar Z, Alvarez R, Falconer D, Rollins SA, Rother RP. Placebo-Controlled, Double-Blind, First-In-Human, Ascending Single Dose and Multiple Dose, Healthy Subject Study Of Intravenous-Administered SelG1, a Humanized Anti-P-Selectin Antibody In Development For Sickle Cell Disease. Blood. 2013;122(21):970–970. | |
Styles L, Wun T, De Castro LM, et al. GMI-1070, a pan-selectin inhibitor: safety and PK in a phase 1/2 study in adults with sickle cell disease. Blood. 2010;116(21):685–686. | |
Telen MJ, Wun T, McCavit TL, et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood. 2015;125(17):2656–2664. | |
Jakubowski JA, Zhou C, Small DS, et al. A phase 1 study of prasugrel in patients with sickle cell disease: pharmacokinetics and effects on ex vivo platelet reactivity. Br J Clin Pharmacol. 2013;75(6):1433–1444. | |
Wun T, Soulieres D, Frelinger AL, et al. A double-blind, randomized, multicenter phase 2 study of prasugrel versus placebo in adult patients with sickle cell disease. J Hematol Oncol. 2013;6:17. | |
Morris CR, Kuypers FA, Larkin S, et al. Arginine therapy: a novel strategy to induce nitric oxide production in sickle cell disease. Br J Haematol. 2000;111(2):498–500. | |
Morris CR, Kuypers FA, Lavrisha L, et al. A randomized, placebo-controlled trial of arginine therapy for the treatment of children with sickle cell disease hospitalized with vaso-occlusive pain episodes. Haematologica. 2013;98(9):1375–1382. | |
Ananthakrishnan R, Li Q, O’Shea KM, et al. Carbon monoxide form of PEGylated hemoglobin protects myocardium against ischemia/reperfusion injury in diabetic and normal mice. Artif Cells Nanomed Biotechnol. 2013;41(6):428–436. | |
Niihara Y, Zerez CR, Akiyama DS, Tanaka KR. Oral L-glutamine therapy for sickle cell anemia: I. subjective clinical improvement and favorable change in red cell NAD redox potential. Am J Hematol. 1998;58(2):117–121. | |
Niihara Y, Macan H, Eckman J, Koh H, Cooper M. L-Glutamine Therapy Reduces Hospitalization for Sickle Cell Anemia and Sickle β-Thalassemia Patients at Six Months–A Phase II Randomized Trial. Clin Pharmacol Biopharm. 2014;3(116):2. | |
Bhatia M, Kolva E, Cimini L, et al. Health-related quality of life after allogeneic hematopoietic stem cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2015;21(4):666–672. | |
Shenoy S, Boelens JJ. Advances in unrelated and alternative donor hematopoietic cell transplantation for nonmalignant disorders. Curr Opin Pediatr. 2015;27(1):9–17. | |
Meier ER, Dioguardi JV, Kamani N. Current attitudes of parents and patients toward hematopoietic stem cell transplantation for sickle cell anemia. Pediatr Blood Cancer. 2015;62(7):1277–1284. | |
Chandrakasan S, Malik P. Gene therapy for hemoglobinopathies: the state of the field and the future. Hematol Oncol Clin North Am. 2014;28(2):199–216. | |
Perumbeti A, Higashimoto T, Urbinati F, et al. A novel human gamma-globin gene vector for genetic correction of sickle cell anemia in a humanized sickle mouse model: critical determinants for successful correction. Blood. 2009;114(6):1174–1185. | |
Romero Z, Urbinati F, Geiger S, et al. β-globin gene transfer to human bone marrow for sickle cell disease. J Clin Invest. 2013;123(8):3317–3330. | |
Levasseur DN, Ryan TM, Reilly MP, McCune SL, Asakura T, Townes TM. A recombinant human hemoglobin with anti-sickling properties greater than fetal hemoglobin. J Biol Chem. 2004;279(26):27518–27524. | |
Krishnamurti L, Kharbanda S, Biernacki MA, et al. Stable long-term donor engraftment following reduced-intensity hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2008;14(11):1270–1278. | |
Steinberg MH. Clinical trials in sickle cell disease: adopting the combination chemotherapy paradigm. Am J Hematol. 2008;83(1):1–3. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.