")
Back to Journals » OncoTargets and Therapy » Volume 9
Orchestrating epigenetic roles targeting ocular tumors
Authors Wen X, Lu L, Zhang H, Fan X
Received 7 August 2015
Accepted for publication 7 November 2015
Published 29 February 2016 Volume 2016:9 Pages 1001—1009
DOI https://doi.org/10.2147/OTT.S93995
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jianmin Xu
Xuyang Wen*, Linna Lu*, He Zhang, Xianqun Fan
Department of Ophthalmology, Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: Epigenetics is currently one of the most promising areas of study in the field of biomedical research. Scientists have dedicated their efforts to studying epigenetic mechanisms in cancer for centuries. Additionally, the field has expanded from simply studying DNA methylation to other areas, such as histone modification, non-coding RNA, histone variation, nucleosome location, and chromosome remodeling. In ocular tumors, a large amount of epigenetic exploration has expanded from single genes to the genome-wide level. Most importantly, because epigenetic changes are reversible, several epigenetic drugs have been developed for the treatment of cancer. Herein, we review the current understanding of epigenetic mechanisms in ocular tumors, including but not limited to retinoblastoma and uveal melanoma. Furthermore, the development of new pharmacological strategies is summarized.
Keywords: ocular tumors, epigenetics, retinoblastoma, uveal melanoma, epigenetic drugs
Introduction
Since Gregor Johann Mendel formulated the basic concepts of heredity, giving rise to the basic understanding of genetics, the mystery of genetics has been constantly explored for over a century. From the DNA double helix to base sequence, scientists opened a door into the micro world. A multitude of virulent genes were identified, and an increasing number of drugs have been developed to cure genetic diseases. The molecular underpinnings of terminal diseases, such as cancer, have been unveiled, and gene therapy has gradually become a reality.1–3
However, with the broadening of genetic research, scientists found that phenotypic variations could not be identified for nucleotide sequence variations in every case, and it was noted that some genes could interact with their environment to yield a phenotype. The word “epigenetics” was subsequently coined by Conrad Waddington in 1940. With the development of this field of study, the word epigenetics now refers specifically to the study of meiotically heritable changes in gene expression that occur without changes in the DNA sequence.4
Epigenetic regulation includes several phenomena, such as DNA methylation, histone modifications, alterations in non-coding RNA, histone variation, nucleosome location, and chromosome remodeling. These changes can work together or individually to affect gene expression which is involved in a wide variety of pathologies, including cancer.5
Among these phenomena, DNA methylation, histone modifications, and alterations in non-coding RNA are the regulations on linear DNA level. Quantity of research has been carried out to explore the relationship between ocular tumors and these variations.6–8 These findings are explained in further detail below.
However, the genome does not exist as a linear entity within actual cells. Inside the nucleus, the genome is organized in three-dimensional space9 and many regulations are carried out in this pattern, such as histone variation, nucleosome location, and chromosome remodeling. As the basic components of a nucleosome, histones are essential to chromatin structure and function.10 In order to adapt to various states of chromatin, several kinds of histone variants are incorporated in a nucleosome.11–13 These variants can finally affect the gene expression including transcriptional activation and deactivation.14 Nucleosome location is another critical factor for transcriptional regulation and DNA repair. The acquisition of transcription information depends on the location of the nucleosome in DNA, and the stability or removal of nucleosome positioning may be important factors influencing gene transcription regulation.15–17 Furthermore, since Dekker et al discovered the method of chromosome conformation capture (3C),18 C technology has enabled and significantly accelerated the exploration of chromosome remodeling.19–22 The long-range interaction between promoters and enhancers/repressors significantly influences gene transcription, and spatial interactions between these elements are integral to their function.23,24 However, these regulatory models have not been reported in ocular tumors to date, and this area may be of great interest for future studies. These phenomena are not explained in further detail below.
Ocular tumors are a malignant disease that can seriously affect an individual’s health and quality of life. The prognosis typically ranges from facial deformity and vision loss to death. Similar to other types of tumors, the occurrence and development of ocular tumors has been primarily attributed to direct damage to specific genes.25,26 However, with the discovery of disease-related epigenetic mechanisms, epigenetic disruptions have been increasingly found to affect tumorigenesis.
DNA methylation in ocular tumors
DNA methylation represents one of the earliest identified epigenetic modification pathways. DNA methylation typically occurs at the 5′ end of the cytosine within CpG dinucleotides and leads to gene silencing. CpG sites are methylated by DNMTs, a family of enzymes with three main members: DNMT1, DNMT3A, and DNMT3B.6 DNMT1 is a persistent DNA methyltransferase that maintains existing methylation patterns following DNA replication. DNMT3A and DNMT3B primarily target previously unmethylated CpGs and participate in the regulation of cell growth and differentiation.27 Furthermore, except for these DNMTs, some other chromatin binding proteins such as HP1 also play an important role in DNA methylation.28 DNMT3A and DNMT3B can interact with mHP1α and direct DNA methylation.29 In normal cells, CpG island methylation typically occurs during developmental phenomena, such as X-chromosome inactivation.30 Recent findings suggest that many tumor suppressor genes are methylated, thus leading to tumorigenesis.31–33
RB1 inactivation is the primary cause of retinoblastoma (RB); its inactivation is typically caused by loss-of-function mutations and the most recent study showed that RB function might be sexually dimorphic.34,35 In addition, RB1 has been reported to be involved in many other tumors.36 However, many differential gene expression profiles of RB tumors in comparison with normal retinas have recently been characterized, and many of these differences are caused by epigenetic changes.37–39 A previous study reported five unilateral RB patients with no mutation in the RB1 gene. The 5′ end of the RB1 gene, including its promoter region and exon 1, exhibits hypermethylation in these patients.40 Another study also reported nine unilateral, sporadic RBs with hypermethylation in the 5′ region of the RB1 gene.41 Numerous other hypermethylated genes were recently found to be involved in the pathogenesis of RB. Hypermethylation of the promoter region of the RASSF1A gene has been detected in 82% to 89% of RB cases,42–44 and promoter hypermethylation of the MGMT gene has been observed in lower stage RB patients.43,45 Another study investigated the methylation status of 25 tumor suppressor genes in 12 RB tumors compared with corresponding normal retinas. Hypermethylation of several cancer-related genes was detected: MGMT (58%), NEUROG1 (52%), MSH6 (50%), CD44 (42%), PAX5 (42%), and GATA5 (25%).42,46 Interestingly, deletions of some of these tumor suppressor genes may drive RB.46 Moreover, high-density methylation of numerous other genes, such as TFF3,47 the apoptotic effector CASP8,48,49 the DNA repair gene MLH1,50 APC-2,31 and the RB2/p130 gene, is involved in RB51,52 (Table 1).
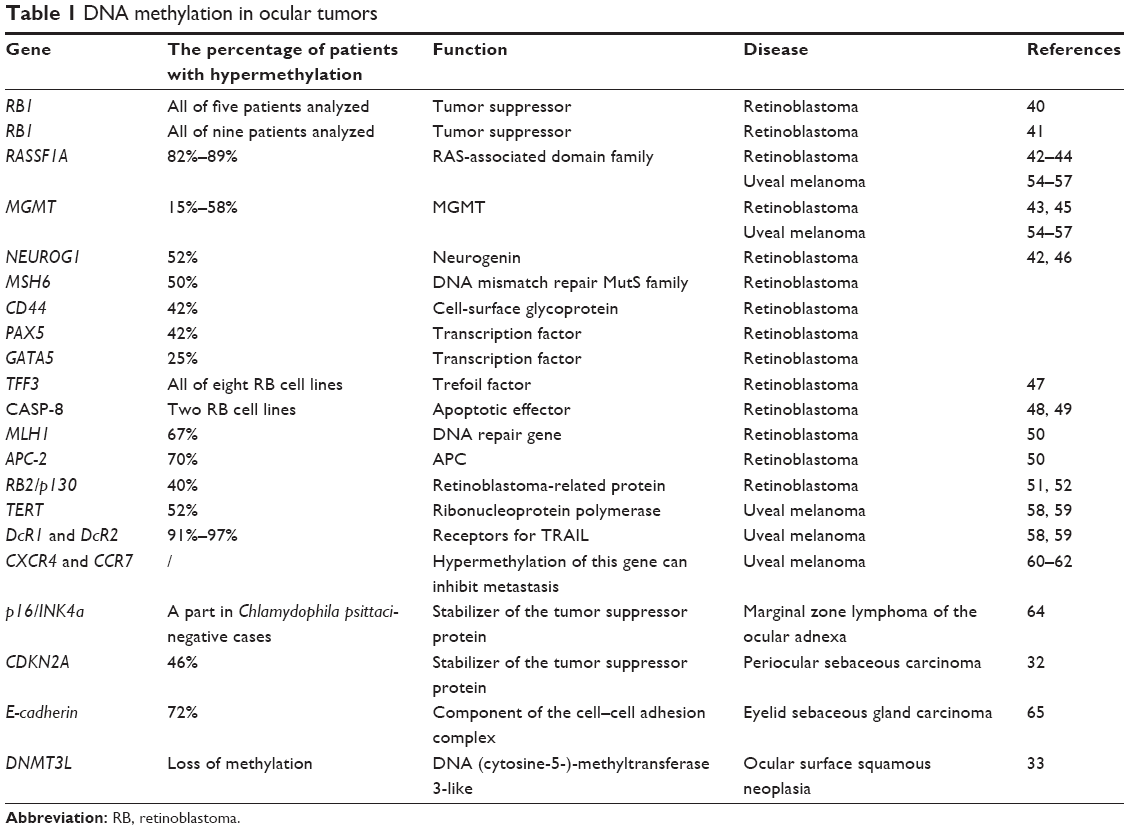 | Table 1 DNA methylation in ocular tumors |
The pathogenesis of uveal melanoma (UM) varies among patients.53 RASSF1A and MGMT appear to have similar effects in UM and RB. A positive correlation was noted between RASSF1A or MGMT promoter methylation and the development of UM.54–57 Furthermore, hypermethylation of the hTERT promoter and the TRAIL receptors DcR1 and DcR2 was detected at a relatively high frequency in cases of UM.58,59 Another study demonstrated that CXCR4 and CCR7 expression in UM enabled directional migration of these tumor cells to the liver, and that the demethylating agent 5-aza-2′-deoxycytidine (5-Aza) upregulates the repressed CXCR4 gene via demethylation.60–62 Another recent study reported that 5-Aza causes significant decreases in growth, invasion, and clonogenicity in UM. In addition, 5-Aza decreased the number of metastases from the eye to the lung in a murine xenograft model.63
In addition to these two main malignant ocular tumors, DNA methylation has been reported in other ocular tumors. Methylation of the p16/INK4a gene promoter was noted in marginal zone lymphoma of the ocular adnexa,64 whereas hypermethylation of the CDKN2A gene promoter was demonstrated to have an effect on periocular sebaceous carcinoma and was associated with younger patient age.32 Methylation of the E-cadherin promoter region was detected in 72% of eyelid sebaceous gland carcinoma, and this effect could contribute to the reduced disease-free survival of patients.65 Furthermore, a study in Drosophila suggested that the downregulation of Rbf due to DNA hypermethylation was associated with eye cancer,66 and a loss of methylation at the DNMT3L promoter was detected in ocular surface squamous neoplasia33 (Table 1).
Histone modifications in ocular tumors
Multiple post-translational modifications have been noted on histones. The enzymes involved in such modifications include HAT, HDAC, HMT, HDM, ubiquitin ligase, deubiquitinase, kinase, and phosphatase.7,67 Genome-wide studies have demonstrated that these modifications in specific regions can lead to the activation or repression of gene expression. For example, the post-translational modifications of histones, including monomethylation of H4K20 and H2BK5; trimethylation of H3K4, H3K36 and H3K79; and acetylation of H3K9 and H3K14, activate gene expression. In contrast, dimethylation of H3K9 and trimethylation of H3K9 and H3K27, lead to the repression of gene expression.68–70 Similar to DNA methylation, many chromatin binding proteins can also affect histone modifications. HP1 can specifically recognize and bind to methylated histone H3K9 which leads to epigenetic silencing.71,72 In addition, HP1 proteins have been demonstrated to harbor a wide variety of modifications such as phosphorylation, acetylation, ubiquitination, and so on.73,74
A study performed in 25 human cancer cell lines revealed that the global loss of monoacetylation and trimethylation of histone H4 is a common hallmark of human tumor cells.75 With regard to eye cancer, by studying tumorigenesis in the Drosophila eye, it was noted that deacetylated H3K9 and methylated H3K27 of Pipsqueak and Lola contributed to the tumor phenotype.66 Another study reported that the HMT EZH2 could act on MHC2TA promoter IV (CIITA-PIV) chromatin, resulting in high levels of trimethylated histone H3K27me3. This modification leads to the silencing of MHC2TA in UM cells and ultimately causes tumorigenesis.76,77 Moreover, HDAC inhibitors, such as valproic acid, reverse the effect of BAP1 loss in UM and inhibit tumor metastasis78,79 (Table 2).
 | Table 2 Histone modifications in ocular tumors |
Non-coding RNA in ocular tumors
Non-coding RNA refers to RNA that does not code for proteins. These RNAs are transcribed from the genome and directly exercise their biological function at the RNA level. According to the length of non-coding RNA, these RNAs are divided into three types: RNA length less than 50 bp, such as microRNA (miRNA) and small interfering RNA; RNA with lengths of greater than 200 bp, such as long non-coding RNA (lncRNA); and other RNAs with a length between 50 bp and 500 bp, such as ribosomal RNA and transfer RNA. Previous studies of non-coding RNA have primarily focused on miRNAs, which can result in gene silencing via translational repression or target mRNA degradation. These studies reported that miRNAs are associated with several human diseases, including ocular tumors.80–84 Recently, lncRNA has been receiving increasing amounts of attention. Mounting evidence suggests that lncRNA plays an important role in epigenetic regulation and affects cell proliferation and differentiation.85–88
Reduced expression levels of let-7, which can dramatically repress oncogenes, such as HMGA2, c-Myc, and members of the Ras family, have been observed in RB cases.89–92 Other studies have reported that miR-34a is a tumor suppressor miRNA in RB.83,93 Furthermore, miR-24, 125b, 191, 181a, and 423 are also decreased in RB.94 Regarding oncogenic miRNA, the miR-17~92 cluster is over-expressed in primary RB tumors and cell lines.80,95 miR-17~92 is a target of E2F, and loss of RB1 may lead to increased expression of miR-17~92 through depressed E2F activity.96,97 Moreover, a recent report has suggested that hypoxia-induced miR-181b enhances angiogenesis of RB cells by targeting PDCD10 and GATA698,99 (Table 3).
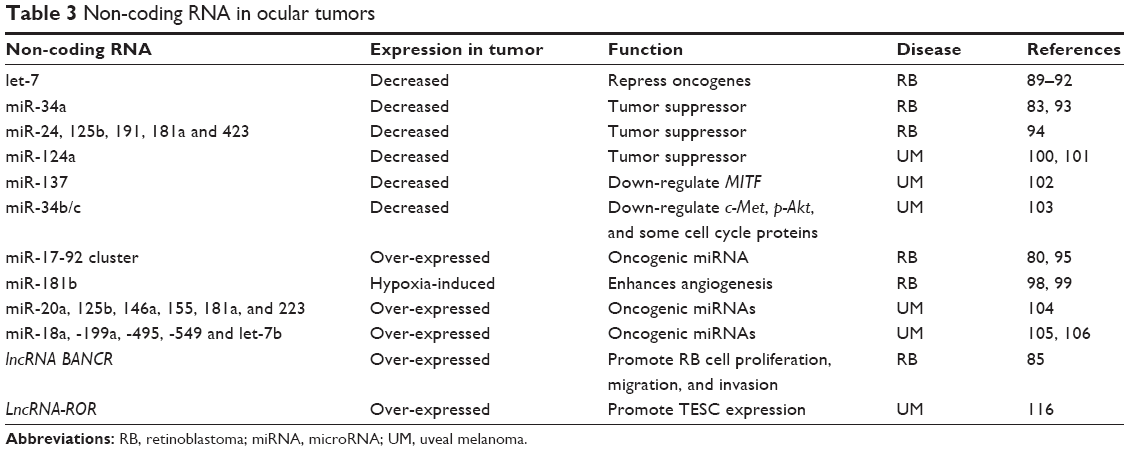 | Table 3 Non-coding RNA in ocular tumors |
Several studies of miRNAs have also been conducted in UM. As for tumor suppressor miRNAs, miR-124a is significantly downregulated, and its re-expression in UM cell lines dramatically decreases cell proliferation, migration, and invasion.100,101 The same phenomenon has been observed for miR-137102 and miR-34b/c.103 Furthermore, the oncogene MITF is downregulated by overexpression of miR-137 in UM,102 whereas c-Met, p-Akt, and some cell cycle proteins are downregulated by miR-34b/c.103 Regarding oncogenic miRNAs, five miRNAs, including miR-20a and -125b, affect tumor metastasis in UM patients.104 Moreover, miR-18a, -199a, -495, -549, and let-7b are also overexpressed in UM105,106 (Table 3).
In recent years, lncRNA has been confirmed to facilitate common cancer hallmarks, such as replicative immortality, resistance to cell death, and evasion of growth suppression.88 Various lncRNAs, including H19, PANDA, and lncRNA-ROR, are associated with tumorigenesis to varying degrees both in vitro and in vivo.36,107–115 However, to our knowledge, only a few studies have been conducted on ocular tumors to date. One such study suggested that lncRNA BANCR was over-expressed in RB tissues and cell lines. Patients with increased lncRNA BANCR expression exhibited poorer survival, and knocking down lncRNA BANCR expression significantly suppressed RB cell proliferation, migration, and invasion in vitro.85 Another study of UM was performed by our group. We recently found that lncRNA-ROR played an important role in ocular melanoma. LncRNA-ROR occupied and activated the TESC promoter by repelling the histone G9A methyltransferase and promoting the release of histone H3K9 methylation. Suppression of ROR in tumors resulted in silencing of TESC expression followed by restoration of G9A-mediated histone H3K9 methylation in the TESC promoter, which significantly reduced tumor growth and metastasis116 (Table 3).
Epigenetic drugs for ocular tumor treatment
One of the most important reasons for the focus on epigenetics in recent years is that epimutations are reversible in contrast with genetic mutations, which are difficult to completely restore. Epigenetic drugs can restore the normal epigenetic landscape in cancer cells by several modes, such as inhibiting enzymes of the epigenetic machinery. Currently, several epigenetic drugs have been approved by the US Food and Drug Administration (FDA) for the treatment of cancer. The DNMT inhibitors Vidaza (5-aza-cytidine) and decitabine (5-Aza) are approved for use in patients. The former is effective for the treatment of myelodysplastic syndrome and acute non-lymphocytic leukemia, whereas the latter is the improved version that exhibits enhanced efficacy in a variety of malignancies.117 The HDAC inhibitors vorinostat (suberoylanilide hydroxamic acid) and romidepsin (FK228, depsipeptide) are effective for cutaneous T cell lymphoma.118,119 In addition to these epigenetic drugs approved by the FDA, even more drugs are currently undergoing clinical trials or are at the stage of laboratory research.120–125
In ocular tumors, DNMT and HDAC inhibitors also play an anticancer role. In addition, 5-Aza decreased the number of metastases from the eye to the lung in a murine xenograft model;63 and HDAC inhibitors, such as valproic acid, may have therapeutic potential for inducing differentiation and prolonged dormancy of micrometastatic disease in UM.78,79 Regarding RB, the proto-oncogene SYK is upregulated in cases of RB and is required for tumor cell survival. Targeting SYK with the small-molecule inhibitor BAY61-3606 or R406 could remarkably induce RB tumor cell death in vitro and in vivo.126,127
Future prospects
Due to technological developments and high-throughput technologies, the study of epigenetic processes is currently possible at a much broader level than only single genes.128,129 Differences in epigenetic marks, such as lncRNA and histone modifications, between cancer cells and normal cells can be easily explored through microarray analysis, chromatin immunoprecipitation sequencing, and RNA sequencing. With the popularization of techniques, such as C technology, the study of chromosome remodeling will continue to attract the attention of researchers. Although lncRNA and chromosome remodeling have already become an interesting area of study for mechanisms of tumorigenesis, research in this area in ocular tumors remains minimal. Additional studies should be conducted to enable a better understanding of such epigenetic mechanisms in ocular tumors.
Finally, in view of the use of epigenetic drugs for cancer treatment, the identification of optimal doses for single and combined therapies requires careful analysis. Furthermore, because most of the epigenetic drugs are nonspecific, they may be a “double-edged sword” and cause unpredictable side effects. Thus, it is necessary for doctors to design personalized treatment programs. Hopefully, more accurate personalized treatments that are free of side effects can be identified to cure malignant tumors in the future.
Acknowledgments
This work was supported by the Scientific Research Program of The National Health and Family Planning Commission of China (201402014), The National Natural Science Foundation of China (grant 31470757), The Program for Professor of Special Appointment (Eastern Scholar) at the Shanghai Institutions of Higher Learning (1410000159), The Shanghai PuJiang Program (13PJ1405700), SMC-ChenXing Yong Scholar Program (2014, Class B), and The Science and Technology Commission of Shanghai (grants 14JC1404100, 14JC1404200, and 14430723100). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors have no conflicts of interest to disclose.
References
Jia RB, Zhang P, Zhou YX, et al. VEGF-targeted RNA interference suppresses angiogenesis and tumor growth of retinoblastoma. Ophthalmic Res. 2007;39(2):108–115. | ||
Pan H, Jia R, Zhang L, et al. BAP1 regulates cell cycle progression through E2F1 target genes and mediates transcriptional silencing via H2A monoubiquitination in uveal melanoma cells. Int J Biochem Cell Biol. 2015;60:176–184. | ||
Zhang H, Wang H, Zhang J, et al. Enhanced therapeutic efficacy by simultaneously targeting two genetic defects in tumors. Mol Ther. 2009;17(1):57–64. | ||
Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17(3):330–339. | ||
Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. | ||
Aoki A, Suetake I, Miyagawa J, et al. Enzymatic properties of de novo-type mouse DNA (cytosine-5) methyltransferases. Nucleic Acids Res. 2001;29(17):3506–3512. | ||
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. | ||
Liz J, Esteller M. IncRNAs and microRNAs with a role in cancer development. Biochim Biophys Acta. Epub 2015 Jul 4. | ||
Gorkin DU, Leung D, Ren B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell. 2014;14(6):762–775. | ||
Bernstein E, Hake SB. The nucleosome: a little variation goes a long way. Biochem Cell Biol. 2006;84(4):505–517. | ||
Sirotkin AM, Edelmann W, Cheng G, Klein-Szanto A, Kucherlapati R, Skoultchi AI. Mice develop normally without the H1(0) linker histone. Proc Natl Acad Sci U S A. 1995;92(14):6434–6438. | ||
Ausio J, Abbott DW. The many tales of a tail: carboxyl-terminal tail heterogeneity specializes histone H2A variants for defined chromatin function. Biochemistry. 2002;41(19):5945–5949. | ||
Wouters-Tyrou D, Martinage A, Chevaillier P, Sautiere P. Nuclear basic proteins in spermiogenesis. Biochimie. 1998;80(2):117–128. | ||
Costa S, Shaw P. Chromatin organization and cell fate switch respond to positional information in Arabidopsis. Nature. 2006;439(7075):493–496. | ||
McGhee JD, Felsenfeld G, Eisenberg H. Nucleosome structure and conformational changes. Biophys J. 1980;32(1):261–270. | ||
Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98(3):285–294. | ||
Schalch T, Duda S, Sargent DF, Richmond TJ. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436(7047):138–141. | ||
Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295(5558):1306–1311. | ||
Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. | ||
Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467(7314):430–435. | ||
Zhang H, Jiao W, Sun L, et al. Intrachromosomal looping is required for activation of endogenous pluripotency genes during reprogramming. Cell Stem Cell. 2013;13(1):30–35. | ||
Zhang H, Zeitz MJ, Wang H, et al. Long noncoding RNA-mediated intrachromosomal interactions promote imprinting at the Kcnq1 locus. J Cell Biol. 2014;204(1):61–75. | ||
Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature. 2009;461(7261):199–205. | ||
de Laat W, Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature. 2013;502(7472):499–506. | ||
Price EA, Price K, Kolkiewicz K, et al. Spectrum of RB1 mutations identified in 403 retinoblastoma patients. J Med Genet. 2014;51(3):208–214. | ||
Dommering CJ, van der Hout AH, Meijers-Heijboer H, Marees T, Moll AC. IVF and retinoblastoma revisited. Fertil Steril. 2012;97(1):79–81. | ||
Wang Y, Leung FC. An evaluation of new criteria for CpG islands in the human genome as gene markers. Bioinformatics. 2004;20(7):1170–1177. | ||
Kwon SH, Workman JL. The changing faces of HP1: From heterochromatin formation and gene silencing to euchromatic gene expression: HP1 acts as a positive regulator of transcription. Bioessays. 2011;33(4):280–289. | ||
Bachman KE, Rountree MR, Baylin SB. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J Biol Chem. 2001;276(34):32282–32287. | ||
Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. | ||
Beta M, Chitipothu S, Khetan V, Biswas J, Krishnakumar S. Hypermethylation of adenomatosis polyposis coli-2 and its tumor suppressor role in retinoblastoma. Curr Eye Res. 2015;40(7):719–728. | ||
Liau JY, Liao SL, Hsiao CH, Lin MC, Chang HC, Kuo KT. Hypermethylation of the CDKN2A gene promoter is a frequent epigenetic change in periocular sebaceous carcinoma and is associated with younger patient age. Hum Pathol. 2014;45(3):533–539. | ||
Manderwad GP, Gokul G, Kannabiran C, Honavar SG, Khosla S, Vemuganti GK. Hypomethylation of the DNMT3L promoter in ocular surface squamous neoplasia. Arch Pathol Lab Med. 2010;134(8):1193–1196. | ||
Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820–823. | ||
Sun T, Warrington NM, Luo J, et al. Sexually dimorphic RB inactivation underlies mesenchymal glioblastoma prevalence in males. J Clin Invest. 2014;124(9):4123–4133. | ||
Tsang WP, Ng EK, Ng SS, et al. Oncofetal H19-derived miR-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis. 2010;31(3):350–358. | ||
Saxena P, Kaur J. Differential expression of genes in retinoblastoma. Clin Chim Acta. 2011;412(23–24):2015–2021. | ||
Ganguly A, Shields CL. Differential gene expression profile of retinoblastoma compared to normal retina. Mol Vis. 2010;16:1292–1303. | ||
Benavente CA, Dyer MA. Genetics and epigenetics of human retinoblastoma. Annu Rev Pathol. 2015;10:547–562. | ||
Sakai T, Toguchida J, Ohtani N, Yandell DW, Rapaport JM, Dryja TP. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am J Hum Genet. 1991;48(5):880–888. | ||
Ohtani-Fujita N, Dryja TP, Rapaport JM, et al. Hypermethylation in the retinoblastoma gene is associated with unilateral, sporadic retinoblastoma. Cancer Genet Cytogenet. 1997;98(1):43–49. | ||
Cohen Y, Merhavi-Shoham E, Avraham RB, Frenkel S, Pe’er J, Goldenberg-Cohen N. Hypermethylation of CpG island loci of multiple tumor suppressor genes in retinoblastoma. Exp Eye Res. 2008;86(2):201–206. | ||
Choy KW, Lee TC, Cheung KF, et al. Clinical implications of promoter hypermethylation in RASSF1A and MGMT in retinoblastoma. Neoplasia. 2005;7(3):200–206. | ||
Harada K, Toyooka S, Maitra A, et al. Aberrant promoter methylation and silencing of the RASSF1A gene in pediatric tumors and cell lines. Oncogene. 2002;21(27):4345–4349. | ||
Choy KW, Pang CP, To KF, Yu CB, Ng JS, Lam DS. Impaired expression and promotor hypermethylation of O6-methylguanine-DNA methyltransferase in retinoblastoma tissues. Invest Ophthalmol Vis Sci. 2002;43(5):1344–1349. | ||
Livide G, Epistolato MC, Amenduni M, et al. Epigenetic and copy number variation analysis in retinoblastoma by MS-MLPA. Pathol Oncol Res. 2012;18(3):703–712. | ||
Philippeit C, Busch M, Dunker N. Epigenetic control of trefoil factor family (TFF) peptide expression in human retinoblastoma cell lines. Cell Physiol Biochem. 2014;34(3):1001–1014. | ||
Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer. 2007;46(7):617–634. | ||
Poulaki V, Mitsiades CS, McMullan C, et al. Human retinoblastoma cells are resistant to apoptosis induced by death receptors: role of caspase-8 gene silencing. Invest Ophthalmol Vis Sci. 2005;46(1):358–366. | ||
Choy KW, Pang CP, Fan DS, et al. Microsatellite instability and MLH1 promoter methylation in human retinoblastoma. Invest Ophthalmol Vis Sci. 2004;45(10):3404–3409. | ||
Tosi GM, Trimarchi C, Macaluso M, et al. Genetic and epigenetic alterations of RB2/p130 tumor suppressor gene in human sporadic retinoblastoma: implications for pathogenesis and therapeutic approach. Oncogene. 2005;24(38):5827–5836. | ||
De Falco G, Giordano A. pRb2/p130: a new candidate for retinoblastoma tumor formation. Oncogene. 2006;25(38):5333–5340. | ||
Coupland SE, Lake SL, Zeschnigk M, Damato BE. Molecular pathology of uveal melanoma. Eye (Lond). 2013;27(2):230–242. | ||
Maat W, van der Velden PA, Out-Luiting C, et al. Epigenetic inactivation of RASSF1a in uveal melanoma. Invest Ophthalmol Vis Sci. 2007;48(2):486–490. | ||
Maat W, Beiboer SH, Jager MJ, Luyten GP, Gruis NA, van der Velden PA. Epigenetic regulation identifies RASEF as a tumor-suppressor gene in uveal melanoma. Invest Ophthalmol Vis Sci. 2008;49(4):1291–1298. | ||
Merhavi E, Cohen Y, Avraham BC, et al. Promoter methylation status of multiple genes in uveal melanoma. Invest Opthalmol Vis Sci. 2007;48(10):4403–4406. | ||
Voelter V, Diserens AC, Moulin A, et al. Infrequent promoter methylation of the MGMT gene in liver metastases from uveal melanoma. Int J Cancer. 2008;123(5):1215–1218. | ||
Moulin AP, Clement G, Bosman FT, Zografos L, Benhattar J. Methylation of CpG island promoters in uveal melanoma. Br J Ophthalmol. 2008;92(2):281–285. | ||
Venza M, Visalli M, Catalano T, et al. Impact of DNA methyltransferases on the epigenetic regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expression in malignant melanoma. Biochem Biophys Res Commun. 2013;441(4):743–750. | ||
Li H, Niederkorn JY, Sadegh L, Mellon J, Chen PW. Epigenetic regulation of CXCR4 expression by the ocular microenvironment. Invest Ophthalmol Vis Sci. 2013;54(1):234–243. | ||
Li H, Alizadeh H, Niederkorn JY. Differential expression of chemokine receptors on uveal melanoma cells and their metastases. Invest Ophthalmol Vis Sci. 2008;49(2):636–643. | ||
Li H, Yang W, Chen PW, Alizadeh H, Niederkorn JY. Inhibition of chemokine receptor expression on uveal melanomas by CXCR4 siRNA and its effect on uveal melanoma liver metastases. Invest Ophthalmol Vis Sci. 2009;50(12):5522–5528. | ||
Rajaii F, Asnaghi L, Enke R, Merbs SL, Handa JT, Eberhart CG. The demethylating agent 5-Aza reduces the growth, invasiveness, and clonogenicity of uveal and cutaneous melanoma. Invest Ophthalmol Vis Sci. 2014;55(10):6178–6186. | ||
Carugi A, Onnis A, Antonicelli G, et al. Geographic variation and environmental conditions as cofactors in Chlamydia psittaci association with ocular adnexal lymphomas: a comparison between Italian and African samples. Hematol Oncol. 2010;28(1):20–26. | ||
Jayaraj P, Sen S, Sharma A, et al. Epigenetic inactivation of the E-cadherin gene in eyelid sebaceous gland carcinoma. Br J Dermatol. 2012;167(3):583–590. | ||
Ferres-Marco D, Gutierrez-Garcia I, Vallejo DM, Bolivar J, Gutierrez-Avino FJ, Dominguez M. Epigenetic silencers and Notch collaborate to promote malignant tumours by Rb silencing. Nature. 2006;439(7075):430–436. | ||
Allis CD, Berger SL, Cote J, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131(4):633–636. | ||
Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128(4):707–719. | ||
Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. | ||
Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, Zhang MQ. Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genomics. 2009;10:143. | ||
Bannister AJ, Zegerman P, Partridge JF, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410(6824):120–124. | ||
Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410(6824):116–120. | ||
Lomberk G, Bensi D, Fernandez-Zapico ME, Urrutia R. Evidence for the existence of an HP1-mediated subcode within the histone code. Nat Cell Biol. 2006;8(4):407–415. | ||
LeRoy G, Weston JT, Zee BM, et al. Heterochromatin protein 1 is extensively decorated with histone code-like post-translational modifications. Mol Cell Proteomics. 2009;8(11):2432–2442. | ||
Fraga MF, Ballestar E, Villar-Garea A, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37(4):391–400. | ||
Holling TM, Bergevoet MW, Wilson L, et al. A role for EZH2 in silencing of IFN-gamma inducible MHC2TA transcription in uveal melanoma. J Immunol. 2007;179(8):5317–5325. | ||
Radosevich M, Song Z, Gorga JC, Ksander B, Ono SJ. Epigenetic silencing of the CIITA gene and posttranscriptional regulation of class II MHC genes in ocular melanoma cells. Invest Ophthalmol Vis Sci. 2004;45(9):3185–3195. | ||
Landreville S, Agapova OA, Matatall KA, et al. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin Cancer Res. 2012;18(2):408–416. | ||
Field MG, Harbour JW. Recent developments in prognostic and predictive testing in uveal melanoma. Curr Opin Ophthalmol. 2014;25(3):234–239. | ||
Conkrite K, Sundby M, Mukai S, et al. miR-17~92 cooperates with RB pathway mutations to promote retinoblastoma. Genes Dev. 2011;25(16):1734–1745. | ||
Li BQ, Zhang J, Huang T, Zhang L, Cai YD. Identification of retinoblastoma related genes with shortest path in a protein-protein interaction network. Biochimie. 2012;94(9):1910–1917. | ||
Jo DH, Kim JH, Park WY, Kim KW, Yu YS. Differential profiles of microRNAs in retinoblastoma cell lines of different proliferation and adherence patterns. J Pediatr Hematol Oncol. 2011;33(7):529–533. | ||
Dalgard CL, Gonzalez M, deNiro JE, O’Brien JM. Differential microRNA-34a expression and tumor suppressor function in retinoblastoma cells. Invest Ophthalmol Vis Sci. 2009;50(10):4542–4551. | ||
Reis AHO, Vargas FR, Lemos B. More epigenetic hits than meets the eye: microRNAs and genes associated with the tumorigenesis of retinoblastoma. Front Genet. 2012;3:284. | ||
Su S, Gao J, Wang T, Wang J, Li H, Wang Z. Long non-coding RNA BANCR regulates growth and metastasis and is associated with poor prognosis in retinoblastoma. Tumour Biol. 2015;36(9):7205–7211. | ||
Oliveira JR, Bertolin TC, Andrade D, et al. Specificity studies on Kallikrein-related peptidase 7 (KLK7) and effects of osmolytes and glycosaminoglycans on its peptidase activity. Biochim Biophys Acta. 2015;1854(1):73–83. | ||
Audas TE, Lee S. Stressing out over long noncoding RNA. Biochim Biophys Acta. Epub 2015 Jul 2. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120(5):635–647. | ||
Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315(5818):1576–1579. | ||
Sampson VB, Rong NH, Han J, et al. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007;67(20):9762–9770. | ||
Mu G, Liu H, Zhou F, et al. Correlation of overexpression of HMGA1 and HMGA2 with poor tumor differentiation, invasion, and proliferation associated with let-7 down-regulation in retinoblastomas. Hum Pathol. 2010;41(4):493–502. | ||
Martin J, Bryar P, Mets M, et al. Differentially expressed miRNAs in retinoblastoma. Gene. 2013;512(2):294–299. | ||
Huang JC, Babak T, Corson TW, et al. Using expression profiling data to identify human microRNA targets. Nat Methods. 2007;4(12):1045–1049. | ||
Nittner D, Lambertz I, Clermont F, et al. Synthetic lethality between Rb, p53 and Dicer or miR-17-92 in retinal progenitors suppresses retinoblastoma formation. Nat Cell Biol. 2012;14(9):958–965. | ||
Sylvestre Y, De Guire V, Querido E, et al. An E2F/miR-20a autoregulatory feedback loop. J Biol Chem. 2007;282(4):2135–2143. | ||
Woods K, Thomson JM, Hammond SM. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J Biol Chem. 2007;282(4):2130–2134. | ||
Xu X, Ge S, Jia R, et al. Hypoxia-induced miR-181b enhances angiogenesis of retinoblastoma cells by targeting PDCD10 and GATA6. Oncol Rep. 2015;33(6):2789–2796. | ||
Xu X, Jia R, Zhou Y, et al. Microarray-based analysis: identification of hypoxia-regulated microRNAs in retinoblastoma cells. Int J Oncol. 2011;38(5):1385–1393. | ||
Jazirehi AR, Torres-Collado AX, Nazarian R. Epigenetic regulation of melanoma tumor suppressor miRNA-124a. Epigenomics. 2013;5(3):251–252. | ||
Chen X, He D, Dong XD, et al. MicroRNA-124a is epigenetically regulated and acts as a tumor suppressor by controlling multiple targets in uveal melanoma. Invest Ophthalmol Vis Sci. 2013;54(3):2248–2256. | ||
Chen X, Wang J, Shen H, et al. Epigenetics, microRNAs, and carcinogenesis: functional role of microRNA-137 in uveal melanoma. Invest Ophthalmol Vis Sci. 2011;52(3):1193–1199. | ||
Dong F, Lou D. MicroRNA-34b/c suppresses uveal melanoma cell proliferation and migration through multiple targets. Mol Vis. 2012;18:537–546. | ||
Achberger S, Aldrich W, Tubbs R, Crabb JW, Singh AD, Triozzi PL. Circulating immune cell and microRNA in patients with uveal melanoma developing metastatic disease. Mol Immunol. 2014;58(2):182–186. | ||
Worley LA, Long MD, Onken MD, Harbour JW. Micro-RNAs associated with metastasis in uveal melanoma identified by multiplexed microarray profiling. Melanoma Res. 2008;18(3):184–190. | ||
Radhakrishnan A, Badhrinarayanan N, Biswas J, Krishnakumar S. Analysis of chromosomal aberration (1, 3, and 8) and association of microRNAs in uveal melanoma. Mol Vis. 2009;15:2146–2154. | ||
Yang F, Huo XS, Yuan SX, et al. Repression of the long noncoding RNA-LET by histone deacetylase 3 contributes to hypoxia-mediated metastasis. Mol Cell. 2013;49(6):1083–1096. | ||
Takahashi K, Yan IK, Haga H, Patel T. Modulation of hypoxia-signaling pathways by extracellular linc-RoR. J Cell Sci. 2014;127(Pt 7):1585–1594. | ||
Choudhry H, Albukhari A, Morotti M, et al. Tumor hypoxia induces nuclear paraspeckle formation through HIF-2alpha dependent transcriptional activation of NEAT1 leading to cancer cell survival. Oncogene. 2015;34(34):4546. | ||
Gomez-Maldonado L, Tiana M, Roche O, et al. EFNA3 long noncoding RNAs induced by hypoxia promote metastatic dissemination. Oncogene. 2015;34(20):2609–2620. | ||
Yang F, Zhang H, Mei Y, Wu M. Reciprocal regulation of HIF-1alpha and lincRNA-p21 modulates the Warburg effect. Mol Cell. 2014;53(1):88–100. | ||
Hung T, Wang Y, Lin MF, et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011;43(7):621–629. | ||
Negishi M, Wongpalee SP, Sarkar S, et al. A new lncRNA, APTR, associates with and represses the CDKN1A/p21 promoter by recruiting polycomb proteins. PloS One. 2014;9(4):e95216. | ||
Marin-Bejar O, Marchese FP, Athie A, et al. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome Biol. 2013;14(9):R104. | ||
Xu Y, Wang J, Qiu M, et al. Upregulation of the long noncoding RNA TUG1 promotes proliferation and migration of esophageal squamous cell carcinoma. Tumour Biol. 2015;36(3):1643–1651. | ||
Fan J, Xing Y, Wen X, et al. Long non-coding RNA ROR decoys gene-specific histone methylation to promote tumorigenesis. Genome Biol. 2015;16:139. | ||
Garcia JS, Jain N, Godley LA. An update on the safety and efficacy of decitabine in the treatment of myelodysplastic syndromes. Onco Targets Ther. 2010;3:1–13. | ||
O’Connor OA, Heaney ML, Schwartz L, et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol. 2006;24(1):166–173. | ||
Piekarz RL, Frye R, Turner M, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27(32):5410–5417. | ||
Beumer JH, Parise RA, Newman EM, et al. Concentrations of the DNA methyltransferase inhibitor 5-fluoro-2′-deoxycytidine (FdCyd) and its cytotoxic metabolites in plasma of patients treated with FdCyd and tetrahydrouridine (THU). Cancer Chemother Pharmacol. 2008;62(2):363–368. | ||
Flotho C, Claus R, Batz C, et al. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia. 2009;23(6):1019–1028. | ||
Keppler BR, Archer TK. Chromatin-modifying enzymes as therapeutic targets – Part 1. Expert Opin Ther Targets. 2008;12(10):1301–1312. | ||
Lara E, Mai A, Calvanese V, et al. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene. 2009;28(6):781–791. | ||
Mai A, Cheng D, Bedford MT, et al. Epigenetic multiple ligands: mixed histone/protein methyltransferase, acetyltransferase, and class III deacetylase (sirtuin) inhibitors. J Med Chem. 2008;51(7):2279–2290. | ||
Gustafson CB, Yang C, Dickson KM, et al. Epigenetic reprogramming of melanoma cells by vitamin C treatment. Clin Epigenetics. 2015;7(1):51. | ||
Zhang J, Benavente CA, McEvoy J, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481(7381):329–334. | ||
Benavente CA, McEvoy JD, Finkelstein D, et al. Cross-species genomic and epigenomic landscape of retinoblastoma. Oncotarget. 2013;4(6):844–859. | ||
Jin F, Li Y, Dixon JR, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503(7475):290–294. | ||
Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–322. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.