")
Back to Journals » International Journal of Nanomedicine » Volume 12
Oral immunization of mice with Omp31-loaded N-trimethyl chitosan nanoparticles induces high protection against Brucella melitensis infection
Authors Abkar M, Fasihi-Ramandi M, Kooshki H, Sahebghadam Lotfi A
Received 23 August 2017
Accepted for publication 11 October 2017
Published 13 December 2017 Volume 2017:12 Pages 8769—8778
DOI https://doi.org/10.2147/IJN.S149774
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster

Morteza Abkar,1 Mahdi Fasihi-Ramandi,2 Hamid Kooshki,3 Abbas Sahebghadam Lotfi4
1Nanomedicine and Nanobiology Research Center, Shiraz University of Medical Sciences, Shiraz, 2Molecular Biology Research Center, 3Nanobiotechnology Research Center, Baqiyatallah University of Medical Sciences, 4Department of Clinical Biochemistry, Faculty of Medicine, Tarbiat Modares University, Tehran, Iran
Abstract: Brucellosis is a group of closely associated zoonotic bacterial illnesses caused by members of the genus Brucella. B. melitensis Omp31 is a promising candidate for a subunit vaccine against brucellosis. This study surveyed the immunogenicity of Omp31 alone and with incomplete Freund’s adjuvant (Omp31-IFA) and N-trimethyl chitosan (TMC/Omp31) nanoparticles (NPs), as well as the effect of Omp31 immunization route on immunological responses and protection. After expression and purification, the recombinant Omp31 (rOmp31) was loaded onto TMC NPs by ionic gelation. The particle size, loading efficiency and in vitro release of the NPs were examined. Omp31-IFA was administered intraperitoneally, while TMC/Omp31 NPs were administered orally and intraperitoneally. According to the antibody subclasses and cytokine profile, intraperitoneal immunization by Omp31-IFA and TMC/Omp31 NPs induced T helper 1 (Th1) and Th1–Th2 immune responses, respectively. On the other hand, oral immunization with TMC/Omp31 NPs elicited a mixed Th1–Th17 immune response. Data obtained from the cell proliferation assay showed that vaccination with Omp31 stimulated a vigorous antigen-specific cell proliferative response, which could be further increased after oral immunization with TMC/Omp31 NPs. Vaccinated groups of mice when challenged with B. melitensis 16M were found to be significantly protected in the orally administered group in comparison with the intraperitoneally immunized mice. Results of this study indicated that the reason for high protection after oral vaccination can be via elicited Th17 response.
Keywords: brucellosis, Th17, trimethyl chitosan, vaccine, nanoparticle
Introduction
Brucellosis is a group of closely associated zoonotic bacterial illnesses caused by members of the genus Brucella, a group of facultative intracellular Gram-negative, nonmotile and nonspore-forming bacteria.1 There are different Brucella species that infect a wide range of mammals. The disease is mostly acquired by ingestion, inhalation, or direct contact such as conjunctiva or skin lesions contaminated with animal products.2,3
Owing to Brucella intracellular lifestyle, a few antibiotics are useful against these pathogens upon entering their intracellular niche. Antibiotics are applied to treat brucellosis in human beings including rifampicin, tetracycline, trimethoprim–sulfamethoxazole, aminoglycosides, quinolones, and chloramphenicol. Since there is a high possibility for relapse in single-agent therapy, the antibiotics are mostly administered in combination.4,5 Hence, there is a huge demand for efficient vaccines or treatments against human brucellosis. At present, there is no accessible safe vaccine against brucellosis in human beings, and all commercially accessible animal vaccines are based on live attenuated strains of Brucella such as B. melitensis Rev.1 and Brucella abortus S19 and RB51.6 Despite their effective role in controlling brucellosis in animals, these vaccines have some drawbacks such as being infectious in human beings, interfering with diagnosis, causing abortion when administered to pregnant domestic animals, and allowing the regional transmission of vaccine strain.6,7
Owing to disadvantages of live attenuated vaccines, replacing these vaccines by subunit ones would be a great improvement for safety reasons, which would make them suitable for vaccination.8 Several cell surface and intracellular Brucella spp. components were designed and examined as subunit vaccine against brucellosis.9–13 Among these antigens, 31 kDa outer membrane protein (Omp31) conferred protection against B. melitensis and Brucella ovis in BALB/c mice.14 The main subunit vaccine’s drawback is poor immunogenicity. To improve their immunogenicity, these types of vaccines can be formulated into particulate vaccine delivery systems.15,16
Owing to their simplicity and convenience, mucosal immunizations (particularly oral immunization) are a subject of great interest. Overall, advantages of oral immunization include infection control at pathogen entry site, induction of mucosal and systemic immune responses, and no requirement for needle.17,18 However, oral immunization is difficult due to extremely low bioavailability. Development of oral vaccine formulations requires overcoming various obstacles such as the low permeability of large molecules, lack of drug lipophilicity, and fast enzymatic degradation or inactivation in the gastrointestinal tract.19,20 To overcome these problems, different types of polymeric nanoparticles (NPs) have been investigated as delivery systems to the intestine, which can protect their cargo from adverse situations that could affect vaccine bioactivity.21 Particles composed of bioadhesive materials such as N-trimethyl chitosan (TMC), known by its stable positive charges regardless of pH, can increase the vaccine residence time in the intestine and enhance their permeation and immunogenicity. Furthermore, TMC NPs can stimulate maturation of dendritic cells (DCs); hence, they have intrinsic adjuvant properties. These properties make TMC NPs a promising delivery system for immunization.22–24
The aim of this study was to examine the immunogenicity property of Omp31 alone and with TMC NPs and Freund’s adjuvant and to investigate the administration route influence on the type of immune response.
Materials and methods
Animals and ethics statement
Female BALB/c mice (4–6 weeks old; obtained from Pasteur Institute of Iran) were acclimated and randomly divided into six experimental groups. Mice were kept in conventional animal facilities and received water and food ad libitum. All experiments were approved by the ethical committee of Razi Vaccine and Serum Research Institute (No 515.92 GD, 26.1.2010) and performed following the guidelines of ethical committee of Razi Vaccine and Serum Research Institute.
Bacterial strains and plasmid
B. melitensis 16M and B. melitensis Rev.1 were obtained from Razi Vaccine and Serum Research Institute (Karaj, Iran) and were grown on Brucella broth (Difco Laboratories, Detroit, MI, USA) under aerobic conditions at 37°C in a humidified chamber. Escherichia coli DH5α strain (Thermo Fisher Scientific, Waltham, MA, USA) was applied for gene cloning. E. coli BL21 (DE3) and pET28a vector (Novagen, Madison, WI, USA) were used to express recombinant Omp31 (rOmp31).
Antigen production
The selected open reading frame (ORF) was amplified by polymerase chain reaction (PCR) from a synthetic gene (GenBank Accession Number: JQ965699) with forward primer (5′-ACTAGAATTCGCCACCATG GTTGTGGTCAGC-3′) and reverse primer (5′-GGTACTCGAGATTAGTGATGGTGATGGTGATG-3′). The underlined regions of the primer sequences represent the restriction sites for EcoRI and XhoI, respectively. The amplified DNA fragment was cloned into the pET28a vector. The recombinant protein was expressed in E. coli. The recombinant protein expression was validated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The rOmp31 was then purified using affinity chromatography on Ni2+-conjugated chelating sepharose. Protein expressed as inclusion bodies was solubilized with 8 M urea and refolded by serial dialysis against 4, 2, 1 and 0 M of urea in phosphate buffered saline (PBS). The purified protein was finally solubilized in PBS-1% glycine (0.01 M, pH 8.0). The rOmp31 purity was confirmed by Western blotting. Briefly, protein was separated on a 12% SDS-PAGE gel and transferred into polyvinylidene fluoride membrane. The recombinant protein was determined by probing the membrane with mouse anti-His antibodies (1:5,000; Sigma-Aldrich Co., St Louis, MO, USA). The concentration of purified protein was determined by the Bradford method.
Preparation of TMC formulations
TMC was provided by Dr Abbas Sahebghadam Lotfi (Department of Clinical Biochemistry, Faculty of Medicine, Tarbiat Modares University, Tehran, Iran). TMC NPs were prepared by ionic complexation with pentasodium tripolyphosphate (TPP) (Merck, Darmstadt, Germany) as a cross-linking agent. Omp31 and TMC were dissolved in a 5 mM HEPES (Sigma-Aldrich Co.) buffer (pH 7.4) to a final concentration of 0.1 and 1 mg/mL, respectively. TPP was added under continuous stirring to an Omp31:TPP:TMC weight ratio of 1:3.6:10 until the solution became slightly opalescent. The NPs were harvested by centrifugation (30 minutes, 14,000× g) on a glycerol bed to avoid aggregation. The supernatant was removed, and the NPs were resuspended in PBS.
Characterization of antigen-loaded NPs
The size of the NPs was determined by using a NanoSizer ZS (Malvern Instruments, Malvern, UK) in PBS at 25°C. NPs’ morphology was assessed using field emission scanning electron microscopy (FESEM, 7500F; JEOL, Tokyo, Japan). The samples were coated with gold prior to examination by FESEM.
The amount of encapsulated Omp31 in the NPs was determined by measuring the amount of protein remaining in the supernatant by the Bradford method after centrifugation (30 minutes, 14,000× g). The loading efficiency (LE) was computed by the following equation:
|
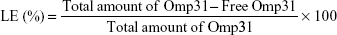
Protein integrity
Omp31-loaded TMC NPs were destabilized by adding 10% (w/v) NaCl to the NPs suspension, resulting in a solution with a protein concentration of 0.41 mg/mL. The protein was electrophoresed at 115 V under reducing conditions in a 12% SDS-PAGE gel.
In vitro release study
Omp31-loaded TMC NPs were separated by centrifugation at 12,000× g and 4°C for 20 minutes. The supernatant was decanted, and the NPs were resuspended in 10 mL of 0.1 M PBS (pH 7.4) and then kept at 37°C under magnetic stirring (150 rpm). At various time intervals, 0.5 mL of the suspension was removed and centrifuged (16,000× g, 20 minutes). The Omp31 concentration in the supernatant was determined by the Bradford method. The same amount of fresh PBS was added to the release medium to reach the primary volume. A sample consisting of only nonloaded TMC NPs was resuspended in PBS to be used as a negative control.
Vaccination
Mice were vaccinated by the oral and intraperitoneal (ip) routes. Six groups of mice either receiving vaccine or as negative control groups are shown in Table 1. The positive control group was administered intraperitoneally on the 15th day with 1×105 CFU of B. melitensis Rev.1. TMC NPs and PBS were used as negative control groups.
 | Table 1 The groups of immunized mice |
Antibody responses
After harvesting of the whole blood, the blood was allowed to clot by leaving it undisturbed at room temperature. This usually took ~30 minutes. The clot was removed by centrifuging at 1,800× g for 15 minutes at 4°C. The resulting supernatant was designated serum. After centrifugation, it was important to immediately transfer the serum into a clean polypropylene tube using a Pasteur pipette. The samples were maintained on ice while handling. Antibody titer and isotypes of IgG, namely, IgG1 and IgG2a, in vaccinated mice sera were determined by the enzyme-linked immunosorbent assay (ELISA) as described previously.25 The threshold value for titer determination was taken as the absorbance plus three times the standard deviation obtained at 1:250 dilution of preimmune sera. Isotypes of IgG were analyzed using anti-mouse IgG1–HRP- and anti-mouse IgG2a–HRP-conjugated antibodies (Santa Cruz Biotechnology Inc., Dallas, TX, USA). Dilution of anti-mouse IgG–HRP, IgG1–HRP and IgG2a–HRP used was 1:8,000 (50 ng/mL).
Anti-Omp31 IgA was determined in fecal extracts by indirect ELISA via a goat anti-mouse IgA-specific HRP conjugate (Santa Cruz Biotechnology Inc.). Fecal extracts were prepared by suspending five fecal pellets in 0.5 mL of extraction buffer (100 mg/mL soybean trypsin inhibitor [Sigma-Aldrich Co.], 10 mg/mL bovine serum albumin [Sigma-Aldrich Co.] and 30 mM disodium EDTA in PBS, pH =7.6). After homogenization and centrifugation at 4°C, the supernatants of the fecal extracts (dilution 1:2) were applied for IgA analysis in feces. Dilution of anti-mouse IgA–HRP used was 1:400 (1 μg/mL). All antibody assays were performed in triplicate.
Cytokine responses
Four weeks after the final immunization, five mice from each group were sacrificed and their spleens were aseptically removed. Spleens from mice were removed and teased apart between two ground glass slides. Cells were washed three times in Hanks’ balanced salt solution (BioWhittaker, Walkersville, MD, USA) and resuspended in Roswell Park Memorial Institute medium containing 10% fetal bovine serum, 5×10−5 M 2-mercaptoethanol, 1% sodium pyruvate, 1% nonessential amino acids, 2 mM l-glutamine, and 10 μg of gentamicin per milliliter. Splenocytes (4×106/well) were seeded in 96-well cell culture plates in the complete medium. Cells were stimulated with 10 μg/mL purified rOmp31 and incubated at 37°C in 5% CO2. Supernatants were harvested from cultures after 48 hours of incubation. Cytokine responses were analyzed by mouse ELISA kits according to the manufacturer’s instructions: IFN-γ, interleukin (IL)-12, IL-4, and IL-17 (R&D Systems, Inc., Minneapolis, MN, USA). All assays were performed in triplicate.
Protection experiments
Vaccinated mice were challenged by ip injection with 2×107 CFUs of B. melitensis 16 M 1 month after the last immunization. One month after being challenged, mice were sacrificed by cervical dislocation and their spleens were removed aseptically. Each spleen was homogenized in a stomacher bag, serially diluted and plated on Brucella agar, and CFUs were counted after 48–72 hours of incubation at 37°C. The results are shown as the mean of the standard deviation of the common logarithm of CFU ± per group. Units of protection were obtained by calculating the differences between the common logarithm of CFU obtained from control mice and the common logarithm of CFU obtained from the corresponding experimental mice groups.
Lymphocyte proliferation assay
Mice were sacrificed 1 month after the last immunization. Splenocytes from vaccinated mice were adjusted to 2×105 cells/well in complete DMEM, and then stimulated with rOmp31 (0.1 μg/mL). The plates were then incubated at 37°C, 5% CO2 and 95% humidity for 72 hours. Lymphocyte proliferation was assessed by MTT assay. In all, 1 mL of 5 μg/mL MTT in incomplete media was prepared and 10 μL was added to each well and incubated in dark at 37°C, 5% CO2 and 95% humidity for 30 minutes. After removal of supernatant from each well, the formazan crystals were solubilized using 75 μL of 5% dimethyl sulfoxide (DMSO). Finally, color density absorbance was measured at 540 nm.
Statistical analysis
Comparative analyses of lymphocyte proliferation assay, antibody responses, and cytokine responses were performed by applying the Student’s t-test with values of p calculated accordingly. The CFU data were normalized by log transformation and evaluated by analysis of variance, followed by Dunnett’s post hoc test.
Results
Antigen production
Omp31 gene amplification produced a 708 bp DNA fragment (Figure 1A). The PCR product was ligated in pET28a vector at EcoRI and XhoI restriction sites in frame with 6× His tag at N and C terminals. The Omp31 gene was expressed in E. coli BL21 (DE3). Recombinant protein expression was analyzed by SDS-PAGE and then confirmed by Western blot using anti-His antibody (Figure 1B and C). Protein concentration was estimated by the Bradford method and then used for vaccination and other experiments.
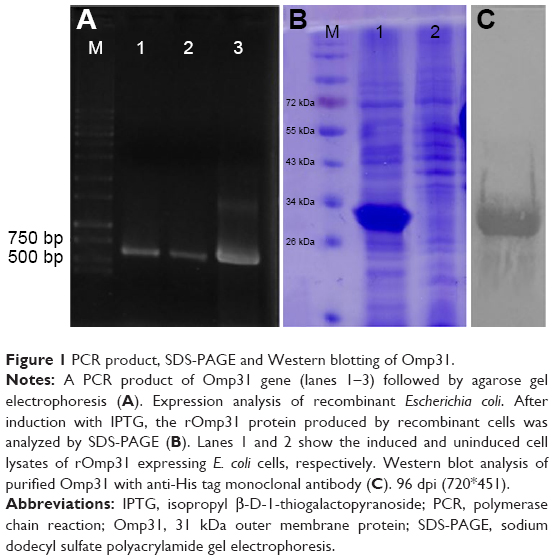 | Figure 1 PCR product, SDS-PAGE and Western blotting of Omp31. |
Characterization of antigen-loaded NPs
Dynamic light scattering (DLS) showed that most TMC/Omp31 NPs had a mean size distribution of 300–400 nm (data not shown). Owing to sample dehydration, SEM images displayed the size of particles to be smaller than measured with DLS (200–300 nm). Furthermore, TMC/Omp31 NPs had a spherical appearance and a smooth surface (Figure 2). The LE of Omp31 was 70.8%±6.3%.
 | Figure 2 Appearance and size of the NPs were characterized by scanning electron micrograph of TMC/Omp31 nanoparticles. |
Protein integrity
The protein integrity of Omp31 after encapsulation was examined by SDS-PAGE analysis. The Omp31 integrity within NPs was found to be intact after protein loading. SDS-PAGE gel analysis results validated the structural integrity of the recombinant protein in the NPs, which shows that the Omp31 integrity has been maintained during the entrapment procedure (data not shown).
In vitro release study
TMC NPs showed ~29% release within the first day, followed by no release over the next 9 days (Figure 3).
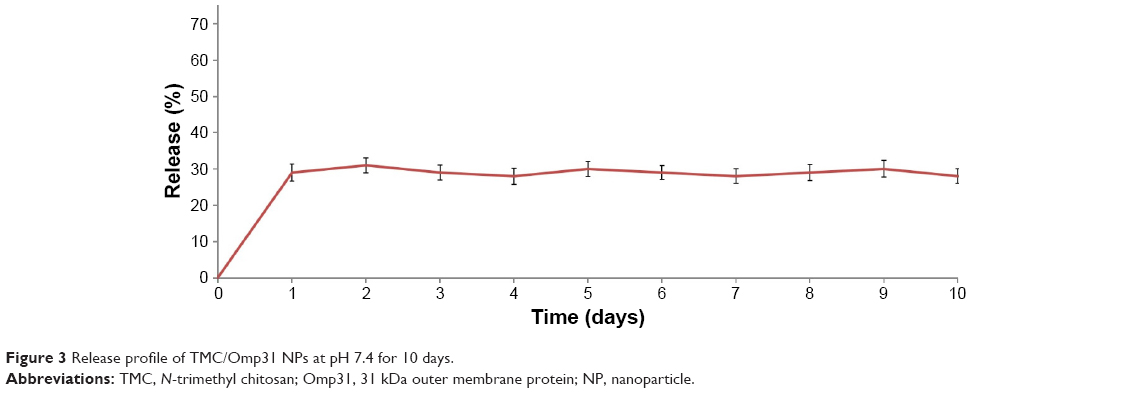 | Figure 3 Release profile of TMC/Omp31 NPs at pH 7.4 for 10 days. |
Antibody responses
Ip immunization with Omp31-IFA, TMC/Omp31, and oral immunization induced specific IgG production. Then, Omp31-specific IgG isotypes (IgG1 and IgG2a) were determined in sera from the immunized animals. The obtained results indicated that the main subtype produced after ip immunization with Omp31-IFA and oral immunization with TMC/Omp31 NPs was IgG2a, whereas both IgG1 and IgG2a titers were induced after ip immunization with TMC/Omp31 NPs (Figure 4A–C).
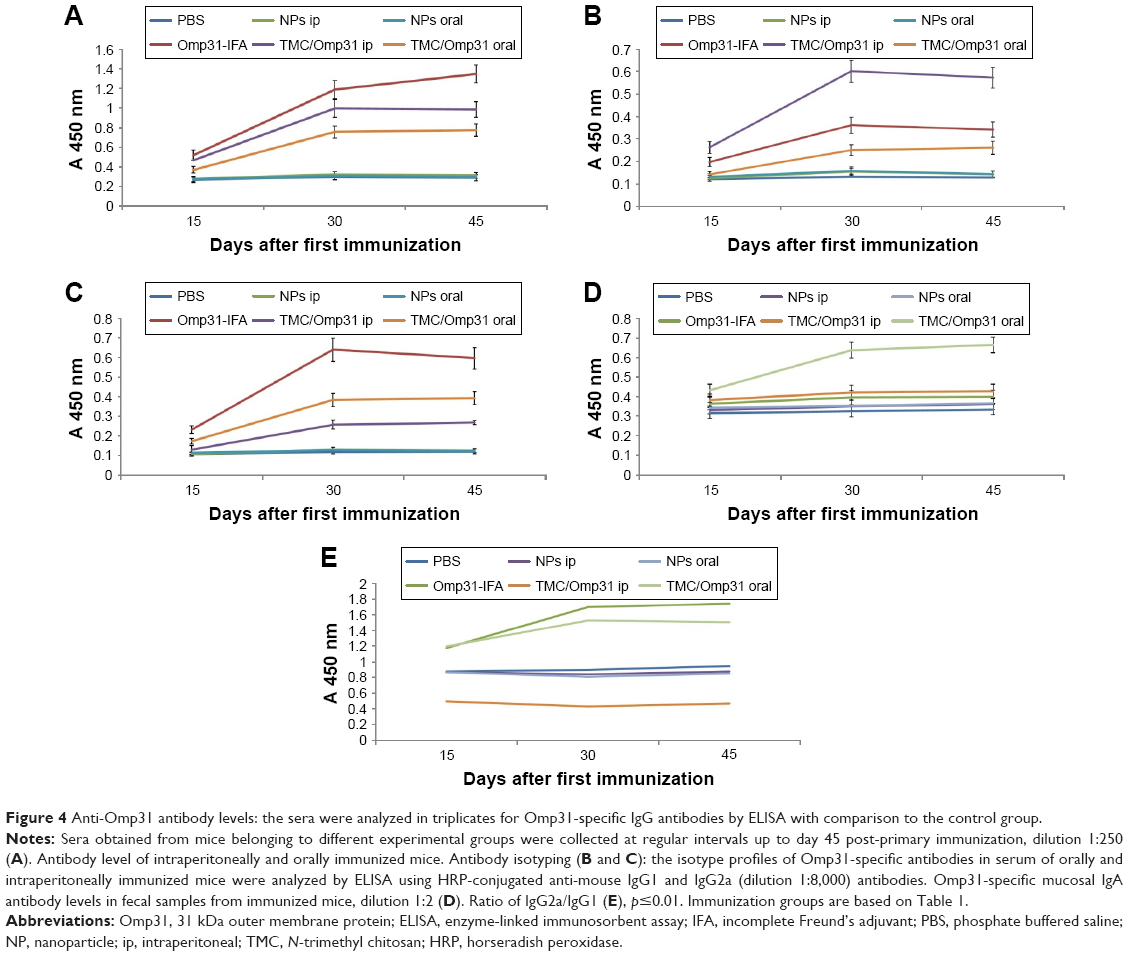 | Figure 4 Anti-Omp31 antibody levels: the sera were analyzed in triplicates for Omp31-specific IgG antibodies by ELISA with comparison to the control group. |
Secretary IgA is a major antibody in mucosal immunity. Ip immunization did not induce any detectable IgA levels in the fecal extracts, whereas oral immunization with TMC/Omp31 NPs showed increased IgA levels (Figure 4D). Additionally, ratio of IgG2a/IgG1 is shown in Figure 4E.
Cytokine responses
According to the cytokine profile, splenocyte culture supernatants from all vaccinated mice contained high levels of IFN-γ and IL-12 compared to the negative control groups. IL-4 production was significantly higher in mice immunized with TMC/Omp31 in ip route but not in the other groups (Figure 5A–C). Additionally, a significantly high IL-17 production was observed in orally vaccinated mice (Figure 5D).
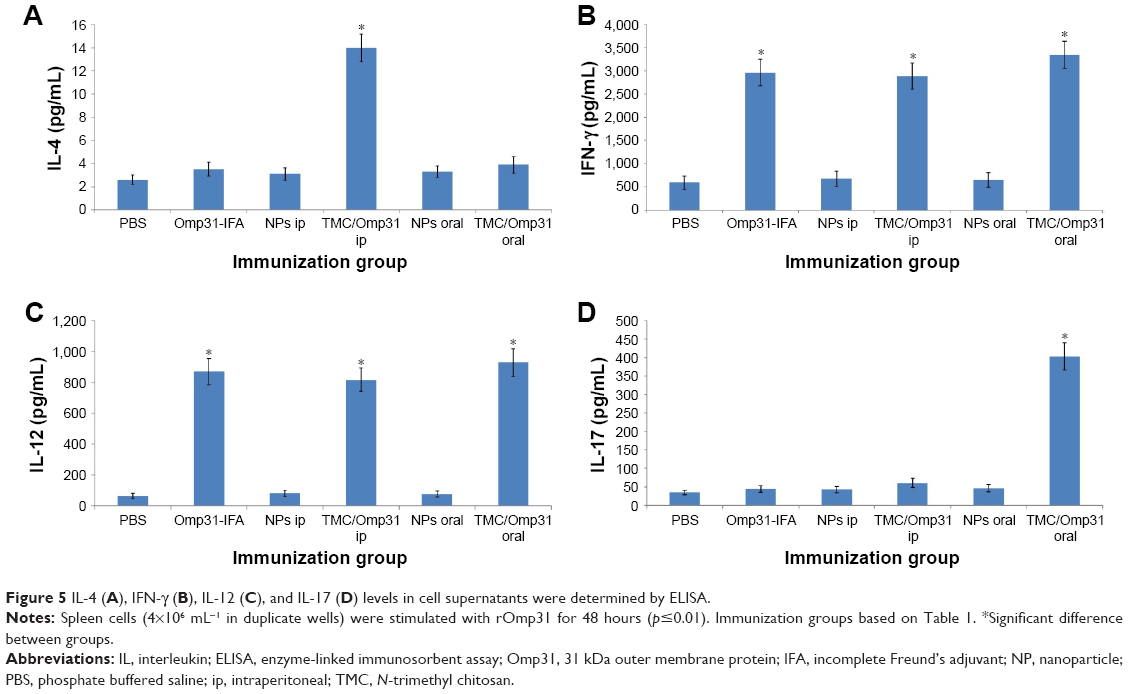 | Figure 5 IL-4 (A), IFN-γ (B), IL-12 (C), and IL-17 (D) levels in cell supernatants were determined by ELISA. |
Protection experiments
The Omp31 ability to protect against virulent B. melitensis 16M challenges was determined in BALB/c mice vaccinated with Omp31. The infection level was computed by determining the CFU numbers in spleens 4 weeks after challenge. As expected, the Rev.1 vaccine displayed high protection with 1.91 log units of protection. However, ip vaccination with TMC/Omp31 NPs and Omp31-IFA produced 1.17 and 1.32 log protection units against B. melitensis, respectively (p≤0.01; Table 2). In comparison with the control group, mice that were immunized with Omp31 orally showed a higher level of protection when challenged with B. melitensis, the log units of protection obtained being 1.77.
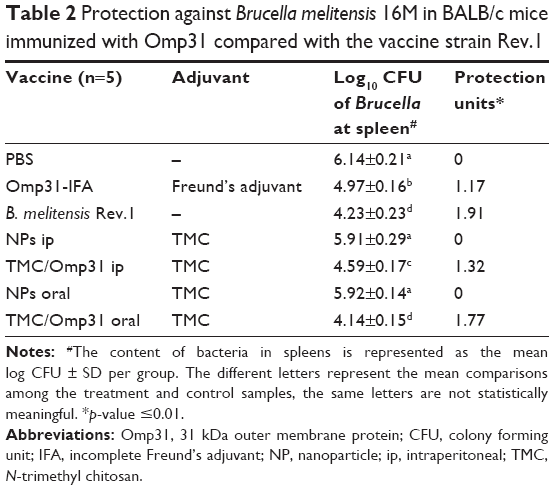 | Table 2 Protection against Brucella melitensis 16M in BALB/c mice immunized with Omp31 compared with the vaccine strain Rev.1 |
Lymphocyte proliferation assay
The results of MTT proliferation assay were displayed as stimulation index (SI). The SI corresponds to the count per minute of induced spleen cells divided by the count per minute of unstimulated spleen cells. As shown in Figure 6, the SI values for intraperitoneally vaccinated mice with Omp31-IFA and TMC/Omp31 were determined to be 1.31 and 1.19, respectively, whereas in orally vaccinated mice, it was found to be 1.84 when stimulated with rOmp31 (p≤0.01; Figure 6). Thus, this high SI value suggests that cell stimulatory activity of rOmp31 may be one of the reasons behind a robust immune response.
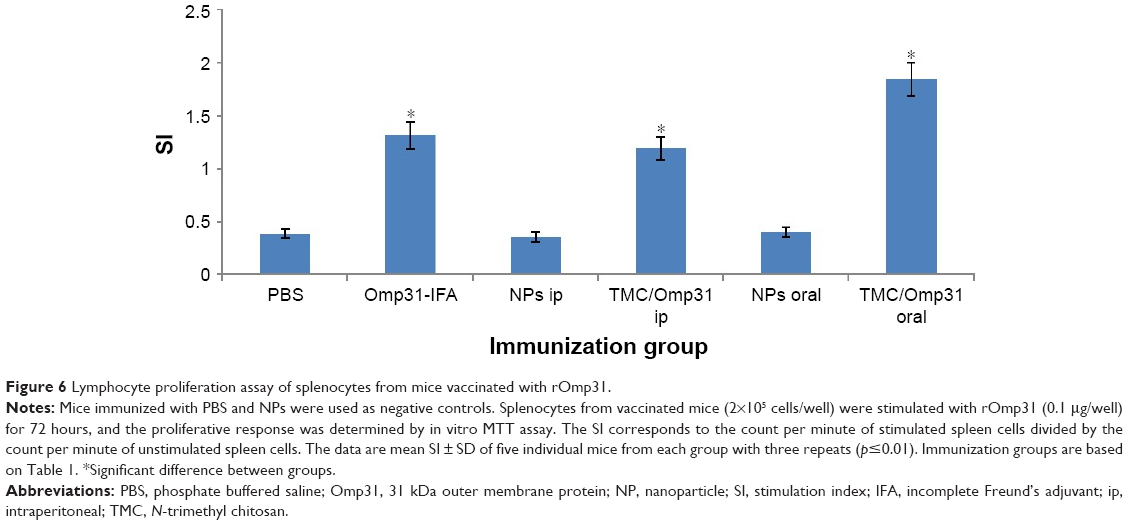 | Figure 6 Lymphocyte proliferation assay of splenocytes from mice vaccinated with rOmp31. |
Discussion
Polymeric NPs in which the antigen is encapsulated were applied for oral vaccination.26–28 Previous studies have shown that chitosan (CS) and TMC NPs are efficient vaccine delivery systems for systemic vaccination.29–32 In another study, Slutter et al compared CS and TMC NPs’ function in oral immunization. They had indicated that application of TMC NPs is a promising strategy for oral immunization.24 Additionally, TMC NPs, but not CS NPs, displayed intrinsic adjuvant effect on DCs. These NPs have successfully been applied for immunization via different administration routes.33–35 In this study, we assessed the TMC NPs’ ability as a delivery system via the oral and ip routes.
The Omp31 release profile from the TMC NPs showed a primary burst release (Figure 3). After the primary surge, equilibrium was reached, indicating no further release during the next 9 days. The data obtained from release study are in accordance with data obtained by Amidi et al32 and Bal et al34 who attributed the burst release to a protein that was weakly bound to the NP surface. This means that most of the Omp31 is encapsulated in the TMC NPs.
The subunit vaccine success is related to its composition and immunization route. Currently, licensed human or animal vaccines are generally administered by the parenteral route, but then again, oral vaccines have many advantages compared to systemic injections.36 As the Brucella bacterium often enters the body via contaminated food and/or water, mucosal immunity can act as a first barrier against the infection before it reaches the bloodstream.37 Hence, one of the goals of this study was the elicitation of anti-Brucella IgA in mucosal sites. There is not defined role for IgA in protection against Brucella infection; however, IgA can reflect upon the induction of common mucosal immune response. Our data indicated that when TMC/Omp31 NPs were orally administered, the specific anti-Omp31 IgA was detected in feces of the immunized mice (Figure 4D). Ip vaccination with both Omp31-IFA and TMC/Omp31 NPs showed high IgG titer, whereas oral vaccination with TMC/Omp31 showed a lower antibody titer. Our results are in line with the observations by Chen et al indicating that subcutaneous immunization with another antigen (urease/TMC NPs) into mice generated high levels of IgG titers but low levels of IgA titers. In contrast, orally administered urease/TMC NPs elicited high titers of both IgA and IgG antibodies.26 However, our results are in contrast with those of the study of Boontha et al that had suggested that oral immunization with TMC/ovalbumin elicits low IgA titers.38 Since the antigen type affected the immune response type, this might be the reason behind the difference.
With respect to the IgG subclass detected via pattern of cytokines produced by CD4+ helper T cells, Omp31-specific IgG1 and the IgG2a antibodies titers were analyzed by ELISA. Significant levels of IgG1 and IgG2a were detected from the mice sera that were intraperitoneally immunized with TMC/Omp31, whereas a high amount of IgG2a was determined in the mice sera that were intraperitoneally and orally immunized with Omp31-IFA and TMC/Omp31, respectively. The IgG2a isotype plays a key role in anti-Brucella immunity, since the binding of its Fc portion to Fc receptors on the surface of phagocytes activates a wide range of antimicrobial responses.39 In a study conducted by Mohanan et al, immunogenicity of protein ovalbumine (OVA)-containing dimethyldioctadecylammonium (DDA) liposomes, PLGA microspheres or TMC NPs or adjuvanted OVA-containing DDA/TDB liposomes, or TMC: LPS NPs or PLGA: CpG microspheres was assessed by various immunization routes. They indicated that IgG2a titer is sensitive to vaccination route, whereas IgG1 titer is somewhat insensitive to vaccination route of the particulate delivery systems.33 In contrast, our results showed that both IgG1 and IgG2a were sensitive to immunization route. Since the composition of a subunit vaccine affected the type of immune responses, this might be the reason behind this difference.
Owing to its intracellular survival, efficient immune responses against Brucella include cell-mediated immunity. Additionally, T helper 1 (Th1) and cytotoxic T lymphocyte (CTL) responses are crucial components involved in anti-Brucella protection. Proinflammatory cytokines such as TNF-α, IFN-γ and IL-12 produced at the beginning of infection have shown to play a key role in the battle against this illness. Principally, IFN-γ, which activates the bactericidal function of macrophages, is generally considered crucial in anti-Brucella immunity.40,41 Our results showed that the significant IFN-γ and IL-12 production was achieved in Omp31-IFA in the ip route and TMC/Omp31 NPs in orally and intraperitoneally immunized mice (Figure 5). Furthermore, the significant IL-17 titer was determined in orally immunized mice. By contrast, IL-4 production was significantly higher only in the group immunized with TMC/Omp31 in the ip route but not in the other groups. It has been reported that Omp31 DNA vaccination stimulates partial protection against B. ovis and B. melitensis infections. This protection was related to the induction of Omp31-specific CD8+ T cells that eradicate Brucella-infected cells via the perforin pathway, a low humoral response, and an absence of Th1 response.42 On the other hand, plasmids encode the Omp31 priming followed by rOmp31 boosting resulting in moderately improved degree of protection against a challenge with B. ovis or B. melitensis.43 Cassataro et al showed that ip immunization with rOmp31-IFA elicits a strong IgG response. After rOmp31 in vitro stimulation, spleen cells from ip rOmp31-vaccinated mice produced significant amounts of the IFN-γ and IL-2 cytokines, but not IL-10 or IL-4, which suggests the elicitation of a Th1 response. The immunization induced partial protection against B. ovis and B. melitensis infections.14 Hence, our data were in accordance with the report of Cassataro et al that had suggested that ip immunization with Omp31-IFA induces Th1 immune response. We showed that oral administration of TMC/Omp31 NPs induces a significant cellular-mixed Th1–Th17 immune response that is in accordance with the observations by Pasquevich et al36 and Abkar et al44 that had shown that oral administration of another antigen (plant-expressed Omp19) and TMC/Omp19 elicits a significant cellular-mixed Th1–Th17 immune response. In a study conducted by Abkar et al, oral vaccination with TMC/Omp19 induced immune responses and a high level of protection against systemic infection. Similar to our report, the researchers did not examine the protection against oral challenge.44
The results obtained in this study showed the importance of administration route in Omp31 protective efficiency. Compared with the Rev.1 vaccine strain, oral immunization of TMC/Omp31 conferred equivalent protection in mice against B. melitensis 16M challenge at 4 weeks post-challenge. The obtained protection after oral immunization was higher than that obtained by ip immunization of Omp31. It has been indicated that an IL-4-dependent Th2 response does not develop or play a negative role during the Brucella pathogenesis, whereas pathogen-specific Th17 cells may boost or work synergistically with Th1 cells for high protection against Brucella infection.36,45 Hence, high levels of Th17 immune responses in oral administration can be one of the reasons behind the high level of protection obtained against B. melitensis challenges compared to the protection level attained in ip immunization.
Indeed, the cell proliferative response observed in Omp31-vaccinated mice represents the activation of cellular immune responses, which is considered to be important for controlling Brucella infections. Data obtained from the cell proliferation assay showed that the vaccination with Omp31 stimulates vigorous production of antigen-specific cytokines secreting cells in spleen, which could be further increased after oral vaccination with TMC/Omp31 NPs.
Conclusion
Our data suggest that administration route plays a main role in determining the immune response type. Furthermore, the protection units obtained show that Omp31 when administered orally confers more protection, which can be due to the elicited Th17 response. TMC/Omp31 may bind and activate a specific cell population present at the gut that positively regulated the differentiation of IL-17-producing T helper cells. It is an ongoing project, and further investigations focusing on increasing efficacy of Omp31 and other Brucella antigen-based vaccines using various adjuvants or evaluation of protection against oral challenges are underway in our laboratory.
Acknowledgments
The authors would like to thank Dr Bram Slutter and Dr Nima Khoramabadi for their scientific advice. The authors also wish to thank the Research Consultation Center (RCC) at Shiraz University of Medical Sciences for their invaluable assistance in editing this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Cloeckaert A, Verger JM, Grayon M, et al. Classification of Brucella spp. isolated from marine mammals by DNA polymorphism at the omp2 locus. Microbes Infect. 2001;3(9):729–738. | ||
Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV. The new global map of human brucellosis. Lancet Infect Dis. 2006;6(2):91–99. | ||
Luna-Martinez JE, Mejia-Teran C. Brucellosis in Mexico: current status and trends. Vet Microbiol. 2002;90(1–4):19–30. | ||
Ariza J, Gudiol F, Pallares R, et al. Treatment of human brucellosis with doxycycline plus rifampin or doxycycline plus streptomycin. A randomized, double-blind study. Ann Intern Med. 1992;117(1):25–30. | ||
Montejo JM, Alberola I, Glez-Zarate P, et al. Open, randomized therapeutic trial of six antimicrobial regimens in the treatment of human brucellosis. Clin Infect Dis. 1993;16(5):671–676. | ||
Schurig GG, Sriranganathan N, Corbel MJ. Brucellosis vaccines: past, present and future. Vet Microbiol. 2002;90(1–4):479–496. | ||
Ashford DA, di Pietra J, Lingappa J, et al. Adverse events in humans associated with accidental exposure to the livestock brucellosis vaccine RB51. Vaccine. 2004;22(25):3435–3439. | ||
Perkins SD, Smither SJ, Atkins HS. Towards a Brucella vaccine for humans. FEMS Microbiol Rev. 2010;34(3):379–394. | ||
Abkar M, Amani J, Sahebghadam Lotfi A, Nikbakht Brujeni G, Alamian S, Kamali M. Subcutaneous immunization with a novel immunogenic candidate (urease) confers protection against Brucella abortus and Brucella melitensis infections. APMIS. 2015;123(8):667–675. | ||
Fu S, Xu J, Li X, et al. Immunization of mice with recombinant protein CobB or AsnC confers protection against Brucella abortus infection. PLoS One. 2012;7(2):e29552. | ||
Goel D, Bhatnagar R. Intradermal immunization with outer membrane protein 25 protects Balb/c mice from virulent B. abortus 544. Mol Immunol. 2012;51(2):159–168. | ||
Abkar M, Lotfi A, Amani J, Ghorashi S, Brujeni G, Kamali M. Design of a chimeric DNA vaccine against Brucella spp. Minerva Biotecnol. 2014;26(4):223–233. | ||
Lalsiamthara J, Lee JH. Brucella lipopolysaccharide reinforced Salmonella delivering Brucella immunogens protects mice against virulent challenge. Vet Microbiol. 2017;205:84–91. | ||
Cassataro J, Estein SM, Pasquevich KA, et al. Vaccination with the recombinant Brucella outer membrane protein 31 or a derived 27-amino-acid synthetic peptide elicits a CD4+ T helper 1 response that protects against Brucella melitensis infection. Infect Immun. 2005;73(12):8079–8088. | ||
Reed SG, Bertholet S, Coler RN, Friede M. New horizons in adjuvants for vaccine development. Trends Immunol. 2009;30(1):23–32. | ||
Lima KM, dos Santos SA, Rodrigues JM Jr, Silva CL. Vaccine adjuvant: it makes the difference. Vaccine. 2004;22(19):2374–2379. | ||
Gaucher G, Satturwar P, Jones MC, Furtos A, Leroux JC. Polymeric micelles for oral drug delivery. Eur J Pharm Biopharm. 2010;76(2):147–158. | ||
Yamanaka YJ, Leong KW. Engineering strategies to enhance nanoparticle-mediated oral delivery. J Biomater Sci Polym Ed. 2008;19(12):1549–1570. | ||
Kostrzak A, Cervantes Gonzalez M, Guetard D, et al. Oral administration of low doses of plant-based HBsAg induced antigen-specific IgAs and IgGs in mice, without increasing levels of regulatory T cells. Vaccine. 2009;27(35):4798–4807. | ||
Baumann U. Mucosal vaccination against bacterial respiratory infections. Expert Rev Vaccines. 2008;7(8):1257–1276. | ||
Mahapatro A, Singh DK. Biodegradable nanoparticles are excellent vehicle for site directed in-vivo delivery of drugs and vaccines. J Nanobiotechnology. 2011;9:55. | ||
Jabbal-Gill I, Watts P, Smith A. Chitosan-based delivery systems for mucosal vaccines. Expert Opin Drug Deliv. 2012;9(9):1051–1067. | ||
Garg NK, Mangal S, Khambete H, Tyagi RK. Mucosal delivery of vaccines: role of mucoadhesive/biodegradable polymers. Recent Pat Drug Deliv Formul. 2010;4(2):114–128. | ||
Slutter B, Plapied L, Fievez V, et al. Mechanistic study of the adjuvant effect of biodegradable nanoparticles in mucosal vaccination. J Control Release. 2009;138(2):113–121. | ||
Singha H, Mallick AI, Jana C, et al. Escheriosomes entrapped DNA vaccine co-expressing Cu-Zn superoxide dismutase and IL-18 confers protection against Brucella abortus. Microbes Infect. 2008;10(10–11):1089–1096. | ||
Chen F, Zhang ZR, Yuan F, Qin X, Wang M, Huang Y. In vitro and in vivo study of N-trimethyl chitosan nanoparticles for oral protein delivery. Int J Pharm. 2008;349(1–2):226–233. | ||
Chen W, Patel GB, Yan H, Zhang J. Recent advances in the development of novel mucosal adjuvants and antigen delivery systems. Hum Vaccin. 2010;6(9):11561. | ||
Fasihi-Ramandi M, Ghobadi-Ghadikolaee H, Ahmadi-Renani S, Taheri RA, Ahmadi K. Vibrio cholerae lipopolysaccharide loaded chitosan nanoparticle could save life by induction of specific immunoglobulin isotype. Artif Cells Nanomed Biotechnol. 2017;28:1–6. | ||
Garcia-Fuentes M, Alonso MJ. Chitosan-based drug nanocarriers: where do we stand? J Control Release. 2012;161(2):496–504. | ||
Sandri G, Bonferoni MC, Rossi S, et al. Nanoparticles based on N-trimethylchitosan: evaluation of absorption properties using in vitro (Caco-2 cells) and ex vivo (excised rat jejunum) models. Eur J Pharm Biopharm. 2007;65(1):68–77. | ||
Subbiah R, Ramalingam P, Ramasundaram S, et al. N,N,N-Trimethyl chitosan nanoparticles for controlled intranasal delivery of HBV surface antigen. Carbohydr Polym. 2012;89(4):1289–1297. | ||
Amidi M, Romeijn SG, Verhoef JC, et al. N-trimethyl chitosan (TMC) nanoparticles loaded with influenza subunit antigen for intranasal vaccination: biological properties and immunogenicity in a mouse model. Vaccine. 2007;25(1):144–153. | ||
Mohanan D, Slutter B, Henriksen-Lacey M, et al. Administration routes affect the quality of immune responses: a cross-sectional evaluation of particulate antigen-delivery systems. J Control Release. 2010;147(3):342–349. | ||
Bal SM, Slutter B, Verheul R, Bouwstra JA, Jiskoot W. Adjuvanted, antigen loaded N-trimethyl chitosan nanoparticles for nasal and intradermal vaccination: adjuvant- and site-dependent immunogenicity in mice. Eur J Pharm Sci. 2012;45(4):475–481. | ||
Bal SM, Slutter B, Jiskoot W, Bouwstra JA. Small is beautiful: N-trimethyl chitosan-ovalbumin conjugates for microneedle-based transcutaneous immunisation. Vaccine. 2011;29(23):4025–4032. | ||
Pasquevich KA, Coria LM, Samartino CG, et al. An oral vaccine based on U-Omp19 induces protection against B. abortus mucosal challenge by inducing an adaptive IL-17 immune response in mice. PLoS One. 2011;6(1):e16203. | ||
Golding B, Scott DE, Scharf O, et al. Immunity and protection against Brucella abortus. Microbes Infect. 2001;3(1):43–48. | ||
Boontha S, Junginger HE, Waranuch N, Polnok A, Pitaksuteepong T. Chitosan and trimethyl chitosan particles as oral vaccine delivery systems: comparison of the potential to initiate immune responses. Journal of Metals, Materials and Minerals. 2011;21(1):43–47. | ||
Zhan Y, Cheers C. Endogenous gamma interferon mediates resistance to Brucella abortus infection. Infect Immun. 1993;61(11):4899–4901. | ||
Vitry MA, De Trez C, Goriely S, et al. Crucial role of gamma interferon-producing CD4+ Th1 cells but dispensable function of CD8+ T cell, B cell, Th2, and Th17 responses in the control of Brucella melitensis infection in mice. Infect Immun. 2012;80(12):4271–4280. | ||
Saraiva M, Christensen JR, Veldhoen M, Murphy TL, Murphy KM, O’Garra A. Interleukin-10 production by Th1 cells requires interleukin-12-induced STAT4 transcription factor and ERK MAP kinase activation by high antigen dose. Immunity. 2009;31(2):209–219. | ||
Cassataro J, Velikovsky CA, De La Barrera S, et al. A DNA vaccine coding for the Brucella outer membrane protein 31 confers protection against B. melitensis and B. ovis infection by eliciting a specific cytotoxic response. Infect Immun. 2005;73(10):6537–6546. | ||
Cassataro J, Velikovsky CA, Bruno L, et al. Improved immunogenicity of a vaccination regimen combining a DNA vaccine encoding Brucella melitensis outer membrane protein 31 (Omp31) and recombinant Omp31 boosting. Clin Vaccine Immunol. 2007;14(7):869–874. | ||
Abkar M, Lotfi AS, Amani J, et al. Survey of Omp19 immunogenicity against Brucella abortus and Brucella melitensis: influence of nanoparticulation versus traditional immunization. Vet Res Commun. 2015;39(4):217–228. | ||
Kumar P, Chen K, Kolls JK. Th17 cell based vaccines in mucosal immunity. Curr Opin Immunol. 2013;25(3):373–380. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.