")
Back to Journals » International Journal of Nanomedicine » Volume 14
Oral absorption and lymphatic transport of baicalein following drug–phospholipid complex incorporation in self-microemulsifying drug delivery systems
Authors Liao H , Gao Y, Lian C , Zhang Y, Wang B, Yang Y, Ye J, Feng Y, Liu Y
Received 8 May 2019
Accepted for publication 4 August 2019
Published 6 September 2019 Volume 2019:14 Pages 7291—7306
DOI https://doi.org/10.2147/IJN.S214883
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Hengfeng Liao1,2, Yue Gao1,2, Chunfang Lian1,2, Yun Zhang1,2, Bangyuan Wang1,2, Yanfang Yang1,2, Jun Ye1,2, Yu Feng1,2, Yuling Liu1,2
1State Key Laboratory of Bioactive substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, 100050, People’s Republic of China; 2Beijing Key Laboratory of Drug Delivery Technology and Novel Formulation, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, People’s Republic of China
Correspondence: Yuling Liu
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, 1 Xiannongtan Street, Beijing 100050, People’s Republic of China
Tel +86 108 928 5188
Fax +86 108 928 5190
Email [email protected]
Purpose: The aims of this study were to prepare a baicalein self-microemulsion with baicalein-phospholipid complex as the intermediate (BAPC-SMEDDS) and to compare its effects with those of conventional baicalein self-microemulsion (CBA-SMEDDS) on baicalein oral absorption and lymphatic transport.
Methods: Two SMEDDS were characterized by emulsifying efficiency, droplet size, zeta potential, cloud point, dilution stability, physical stability, and in vitro release and lipolysis. Different formulations of 40 mg/kg baicalein were orally administered to Sprague-Dawley rats to investigate their respective bioavailabilities. The chylomicron flow blocking rat model was used to evaluate their lymphatic transport.
Results: The droplet sizes of BAPC-SMEDDS and CBA-SMEDDS after 100x dilution were 9.6±0.2 nm and 11.3±0.4 nm, respectively. In vivo experiments indicated that the relative bioavailability of CBA-SMEDDS and BAPC-SMEDDS was 342.5% and 448.7% compared to that of free baicalein (BA). The AUC0–t and Cmax of BAPC-SMEDDS were 1.31 and 1.87 times higher than those of CBA-SMEDDS, respectively. The lymphatic transport study revealed that 81.2% of orally absorbed BA entered the circulation directly through the portal vein, whereas approximately 18.8% was transported into the blood via lymphatic transport. CBA-SMEDDS and BAPC-SMEDDS increased the lymphatic transport ratio of BA from 18.8% to 56.2% and 70.2%, respectively. Therefore, self-microemulsion not only significantly improves oral bioavailability of baicalein, but also increases the proportion lymphatically transported. This is beneficial to the direct interaction of baicalein with relevant immune cells in the lymphatic system and for proper display of its effects.
Conclusion: This study demonstrates the oral absorption and lymphatic transport characteristics of free baicalein and baicalein SMEDDS with different compositions. This is of great significance to studies on lymphatic targeted delivery of natural immunomodulatory compounds.
Keywords: SMEDDS, phospholipid complex, baicalein, oral bioavailability, lymphatic transport
Introduction
Proper absorption of most orally administered drugs involves dissolution in gastrointestinal fluid, diffusion into intestinal epithelial cells, and eventual passage into the bloodstream through the portal vein. Only minute quantities of drugs that enter the epithelial cells can bind to lipoproteins and enter the bloodstream through the intestinal lymphatic system. The advantage of lymphatic transport over portal vein absorption is its ability to enable drugs avoid the first-pass effect of the liver, enhance immunomodulator or chemotherapeutic lymphatic targeting, and facilitate the direct action of immunomodulatory drugs on immune cells.1,2 There are 3 main drug transport routes through the intestinal lymphatic system: the cell bypass pathway, the gut-associated lymphoid tissue (GALT), and transmembrane transport (or intestinal lipid transport system). After entering the central lacteal (in the small intestine villi), the drugs flow into the cisterna chyli through the capillary lymph duct, mesenteric lymphatic duct, and lymph nodes. They finally flow into blood through the thoracic duct, at the junction of the left jugular and the left subclavian vein. The degree of drug-lymphatic transport is often associated with the carrier structure and drug lipophilicity. It has been reported that the lymphatic transport of a digestible carrier is greater than that of a non-digestible carrier. More so, a longer fatty acid chain or higher degree of unsaturation of the lipid in the carrier increases the degree of drug-lymphatic transport. Drug-lymphatic transport has been shown to increase with increasing carrier dispersion: micelles>emulsion>lipid solution.3 In one such study, Caliph et al used a lymphatic duct intubation model to study the lymphatic transport of halofantrine® in short (C4), medium (C8–10), and long (C18) chain triglyceride vehicles. Their results (expressed as a percentage of the dose) indicated the respective lymphatic transport values as: C18 (15.8%)>C8–10 (5.5%)>C4 (2.22%)>C0 (0.34%).4
Self-emulsifying drug delivery systems (SEDDS) are defined as isotropic mixtures of oils, surfactants, and cosurfactants. They can form fine oil−in−water (o/w) emulsions (SEDDS), microemulsions (SMEDDS), or nanoemulsions (SNEDDS), when diluted under mild agitation with aqueous media such as GI fluids.5–7 SEDDS have higher physical stability and drug loading capacity than other lipid carriers. They are also easier to prepare. However, changes in the composition of SEDDS have a significant impact on lymphatic transport. Using fasted lymph cannulated canines, Rene et al8 showed that SMEDDS containing triglycerides with different structures exhibited different degrees of halofantrine-lymphatic transport. The lymphatic transport through the thoracic duct was 17.9% and 27.4% of the administered dose for animals which received SMEDDS containing 1, 3-dioctanoyl-2-linoleyl-sn-glycerol (C8:0-C18:2- C8:0) and 1, 3-dilinoyl-2-octanoyl-sn-glycerol (C18:2-C8:0-C18:2), respectively. Sun et al9 used the immunosuppressant sirolimus, as a model drug to investigate the effects of SMEDDS containing different proportions of oil phase and emulsifier on the lymphatic transport of the drug. The resulting lymphatic transport of the SMEDDS with 0%, 15%, 25%, and 35% MCT (medium-chain triglyceride) achieved 4.8%, 35.1%, 68.7%, and 74.0% of oral absorption of sirolimus, respectively. However, the proportion of emulsifier in the formulation did not significantly affect drug-lymphatic transport. Hence, self-emulsifying drug delivery systems have recently received increasing attention and are widely used to improve the oral bioavailability of poorly soluble natural drugs or the lymphatic absorption of immunomodulatory drugs.7,10–14 Jin Qiao et al15 prepared SMEDDS containing puerarin flavonoids. Their in vivo rat experiments showed that SMEDDS can increase the oral bioavailability of pueraria flavone 2.57 times relative to that of puerarin suspension. Their chylomicron flow blocking rat model also showed that SMEDDS increased the lymphatic transport ratio of oral absorption of pueraria flavone from 20.12–29.12%.
Baicalein is a natural flavonoid with various pharmacological properties such as anti-bacterial, anti-oxidation, anti-cancer, and anti-viral. Baicalein can also inhibit the biosynthesis of leukotrienes in lymphocytes, and the activities of 5-lipoxygenase and phospholipase A2; its main anti-inflammatory and anti-allergic mechanism.16–18 Baicalein can promote lymphocyte apoptosis through the mitochondrial pathway for the treatment of autoimmune hepatitis.19 The oral bioavailability of baicalein is low because of its poor water solubility and lipophilicity, hence, no product containing baicalein monomer exists on the market. We therefore postulated that, SEDDS could improve the oral bioavailability and lymphatic transport of baicalein, to enable its direct action on the relevant immune cells in the lymphatic system, which is beneficial to baicalein efficacy.
For poorly soluble drugs such as baicalein, lipophilicity of drugs is the key to encapsulate them in SEDDS.20 It has recently been found that when some poorly absorbed drugs and phospholipid molecules form relatively stable compounds or complexes by charge transfer, their physical and chemical properties, as well as biological activities will change greatly and exhibit many different characteristics from the parent drug. Therefore, the formation of drug-phospholipid complexes can enhance the liposolubility of drugs, which is more conducive to the encapsulation of the drug in SEDDS.21,22 So far, there has been no report on the self-microemulsion of the baicalein-phospholipid complex or the lymphatic transport of baicalein in SEDDS. Therefore, the overall aim of this study was to prepare SMEDDS with the baicalein-phospholipid complex and investigate its characteristics in vitro and in vivo. We also compared the effect of SMEDDS containing the baicalein-phospholipid complex with that of conventional SMEDDS on baicalein oral absorption and lymphatic transport, using rat pharmacokinetic studies and the chylomicron flow blocking rat model.
Materials and methods
Materials
Baicalein (98% purity) was purchased from Nanjing Zelang Biological Technology Co. Ltd. (Nanjing, China). Soybean phospholipids were purchased from Shanghai Tywei Pharmaceutical Co. Ltd (Shanghai, China). Ethylis oleas, Tween 80, and ascorbic acid were supplied by Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). Trizma maleate, sodium taurodeoxycholate (NaTDC), pancreatin from porcine pancreas (8×USP specifications), and 6-Hydroxyflavone were purchased from Sigma-aldrich (Saint Louis, MO, USA). Baicalin reference substances, 4-bromophenylboronic acid (4-BPB), cycloheximide, and heparin sodium were obtained from J&K Chemicals (Beijing, China). Baicalein reference substances were purchased from Shanghai Standard Technology Co. Ltd. (Shanghai, China). Transcutol HP was kindly donated by Gattefossé (Saint-Priest, France). Acetonitrile, methanol, and formic acid were purchased from Fisher Chemical (Shanghai, China).
Methods
Preparation of SMEDDS
The baicalein-phospholipid complex (BAPC) was prepared with baicalein (BA) and phospholipids at a mass ratio of 1:3.5 (w/w) using the solvent evaporation method. Briefly, 1.0 g of baicalein and 3.5 g of soybean phospholipid were transferred into a 500 mL round-bottomed flask, and dissolved in 20 mL of tetrahydrofuran at room temperature. The mixture was stirred for 15–30 min. Subsequently, tetrahydrofuran was evaporated by a rotary evaporator under vacuum at 40 °C. The residue was collected and vacuum dried. The resultant BAPC was stored in a desiccator at room temperature until use.
Ethyl oleate (EO), Tween-80 (T-80), and Transcutol HP (HP) were used as oil, surfactant, and co-surfactant, respectively, for preparing SMEDDS. Both formulations are shown in Table 1. EO, T-80, and HP were accurately weighted into a glass bottle and homogenized to form a single phase. BA and BAPC were then added. These mixtures were sonicated at 25 °C for 60 min for BA, or stirred on an air bath oscillator for BAPC (25 °C, 200 rpm), until a transparent solution was obtained. The mixtures were then centrifuged at 4000 rpm for 15 min to collect the supernatant. Finally, conventional baicalein self-microemulsions (CBA-SMEDDS) and baicalein-phospholipid complex self-microemulsions (BAPC-SMEDDS) were prepared.
 |
Table 1 The formulations of two SMEDDS |
HPLC analyses
The content of baicalein in BAPC-SMEDDS and CAB-SMEDDS was determined using the Agilent 1260 HPLC system from Agilent Technologies (Santa Clara, CA, USA), equipped with an Agilent ZORBAX SB C18 column (250×4.6 mm, 5 μm), at room temperature. The mobile phase was composed of methanol and 0.05% H3PO4 at a volumetric ratio of 65:35. The flow rate was 1.0 mL/min. A 10 μL volume was injected into the column and the wavelength for detection was 275 nm.23
Assessing the efficiency of self-emulsification
SMEDDS were added in drops at a ratio of 1:100 (v/v) to purified water undergoing magnetic stirring, and at a temperature of 37.0±0.5°C. The rate of emulsification and final appearance of the SMEDDS were assessed visually, based on Khoo’s classification24 with slight modifications. The efficiency of self-emulsification was visually assessed using the following grading system: I, denoting a rapidly forming (within 1 min) microemulsion, which was clear or slightly blue in appearance; II, denoting a rapidly forming (within 1 min) bright white emulsion (similar in appearance to milk); III, denoting a rapidly forming microemulsion, slightly less clear than I, which had a small amount of insoluble colloidal particles; IV, denoting a rapidly forming microemulsion, less clear than III, which had a large number of insoluble colloidal particles; and V, denoting a formulation, which exhibited either poor or minimal emulsification, with large oil droplets present on the surface.
Determination of droplet size and zeta potential
The droplet size of BAPC-SMEDDS and CBA-SMEDDS diluted in water was determined immediately after the self-emulsification study, using the NICOMPTM380ZLS zeta potential/particle sizer from Particle Sizing Systems (PSS NICOMP, Santa Barbara, CA, USA), based on the laser light scattering phenomenon. The zeta potential of the resulting microemulsion was also determined using the NICOMPTM380ZLS zeta potential/particle sizer from Particle Sizing Systems (PSS NICOMP, Santa Barbara, CA, USA). Each study was carried out in triplicates to ensure reproducibility.25,26
Cloud point measurement
The cloud point values of BAPC-SMEDDS and CBA-SMEDDS were determined and compared. Each formulation was added to water (1:100) under magnetic stirring at 37.0±0.5 °C, then, the temperature was gradually increased. The temperature at which there was a sudden appearance of cloudiness was measured as cloud point.27,28
Dilution stability
The dilution stability of BAPC-SMEDDS and CBA-SMEDDS were assessed by exposing them to 5-, 10-, 20-, 50-, 100-, 500-, and 1000-folds dilution in purified water. The droplet size of the diluted microemulsion was determined using the NICOMPTM380ZLS zeta potential/particle sizer from Particle Sizing Systems (PSS NICOMP, Santa Barbara, CA, USA).29
Transmission electron microscopy (TEM)
BAPC-SMEDDS and CBA-SMEDDS were diluted in water (1:200) and converted into microemulsions. The samples were dropped on a carbon film copper mesh and blotted dry with filter paper after 5 min. After staining with phosphotungstic acid for 5 mins, they were again blotted with a filter paper and dried under an infrared lamp for 5 min. The dried samples were taken out and put into the transmission electron microscope (TEM) observation room for photo observation.30
Storage and colloidal stability
BAPC-SMEDDS and CBA-SMEDDS were stored at 4 °C, 25 °C, and 40 °C. Visual turbidity and phase separation of different formulations were observed and recorded in the 0th, 1st, and 2nd month. Colloidal stability of the microemulsions formed by BAPC-SMEDDS and CBA-SMEDDS were assessed using Turbiscan Tower from Formulaction (L’Union, France). BAPC-SMEDDS and CBA-SMEDDS were diluted in water (1:100) and turned into microemulsions. The samples were placed into cylindrical glass tubes and submitted to Turbiscan Tower. The analysis of stability was carried out at 37 °C for 12 h. The results were expressed as a variation of delta transmission (△t) and turbiscan stability index (TSI) profiles.31,32
Drug release in vitro
Drug release (in vitro) from BAPC-SMEDDS and CBA-SMEDDS were tested by dialysis with phosphate buffer at pH 6.8 and hydrochloric acid solution at pH 1.0 containing 0.5% Tween-80 as release media. A 100 μL volume of BAPC-SMEDDS and CBA-SMEDDS concentrations were respectively put in 5 mL of distilled water to form their respective microemulsions. Diluted solution (1 mL) was placed in the dialysis bag (27 mm, MW: 14,000) from BIOSHARP (USA), and introduced into 100 mL of release media at 37 °C, with a rotation speed of 100 rpm during the release period, to compare the release profile with that of free baicalein (BA). For the release studies, 2 mL of sample was taken out and replaced with 2 mL fresh medium at 0.5, 1, 2, 4, 8, 12, and 24 h. Each study was carried out in triplicate and the concentrations of baicalein were analyzed with HPLC.33,34
Lipolysis study in vitro
The lipolysis medium was composed of 2.00 mM Trizma maleate, 150.00 mM NaCl, 5.00 mM CaCl2, 5.00 mM NaTDC, and 1.25 mM phospholipid. PH was adjusted to 6.5 with 0.1 M NaOH. Pancreatin (3.2 g) from porcine pancreas (8×USP specifications) purchased from sigma (Saint Louis, MO, USA), was suspended in 20 mL of lipolysis medium and homogenized thoroughly. Blank SMEDDS with phospholipid (K-PC-SMEDDS) and blank SMEDDS without phospholipid (K-SMEDDS) (1 mL each) were emulsified in 45 mL of lipolysis medium in a thermostatically controlled vessel (37 °C), and the pH was adjusted to 6.5 using 0.1 M NaOH, and kept constant throughout the study. The lipolysis process was initiated by adding 5 mL of freshly prepared pancreatic lipase suspension. The rate of SMEDDS lipolysis was calculated from NaOH consumption.35,36
BAPC-SMEDDS and CBA-SMEDDS (1 mL each) were emulsified in 45 mL of lipolysis medium, in a thermostatically controlled vessel (37 °C), and pH was adjusted to 6.5 using 0.1 M NaOH. The lipolysis process was initiated by adding 5 mL of freshly prepared pancreatic lipase suspension. 5 mL of sample was withdrawn, and the lipolysis was inhibited immediately with 25 μL of 4-bromophenylboronic acid (4-BPB, 1.0 M) at 1, 15, 30, 45, 60, 90 and 180 min. The samples were centrifuged at 4000 rpm for 10 min. The droplet size and baicalein concentration of the aqueous phase were determined as previously described.37,38
Bioavailability study in vivo
The pharmacokinetic studies of free baicalein (BA), BAPC-SMEDDS, and CBA-SMEDDS were performed using male Sprague-Dawley rats, and all animal protocols were approved by the Institutional Animal Care and Use Committee of Peking Union Medical College. The care of laboratory animal and animal experimental operation were performed in accordance with the Beijing Administration Rule of Laboratory Animal. Male Sprague-Dawley rats (250±20 g) were purchased from Beijing Huafukang Bioscience co.inc (Beijing, China). Seven days prior to the experiment, the animals were maintained in air-conditioned standard cages with alternating 12-h light/dark cycles at a room temperature of 25±2 °C, and fed with standard animal chow with free access to water. The animals were allowed to fast for 12 h, with free access to water before the experiment. Fifteen male rats were divided randomly into 3 groups. Rats in each group were intragastrically administered 2~3 mL solution or suspension with a single dose of 40 mg/kg baicalein. One group received BA, and the others received BAPC-SMEDDS and CBA-SMEDDS. All formulations were dispersed in water. And BA suspension was grinded and vortexed again before use to make sure the homogeneity and the exact amount of dose. After oral administration, 0.5 mL of blood sample was collected by retro-orbital bleeding at 5, 15, 30, 45, 60, 75, 180, 360, 480, 600, 720, and 1440 min. And blood samples were placed in tubes containing heparin sodium. Plasma was harvested by centrifuging blood for 15 min at 12,000 rpm and stored at −80 °C until analyzed.39–41
Chylomicron flow blocking rat model
In order to investigate the lymphatic transport of baicalein in BAPC-SMEDDS and CBA-SMEDDS in vivo, we chose the chylomicron flow blocking rat model, and the animals were housed as described above. The animals were intraperitoneally (i.p) administered 3.0 mg/kg cycloheximide dissolved in saline (0.6 mg/mL, w/v). Post-injection (1 h), the animals were administered (by oral gavage) BA, BAPC-SMEDDS, or CBA-SMEDDS (a single dose of 40.0 mg/kg baicalein). After oral administration, 0.5 mL of blood samples were collected by retro-orbital bleeding at 5, 15, 30, 45, 60, 75, and 180 min. Plasma was harvested by centrifuging blood for 15 min at 12,000 rpm and stored at −80 °C until analyzed.42–45
Quantitative analyses of plasma baicalein
Since baicalein is easily oxidatively degraded in solution, it is necessary to add ascorbic acid to increase its storage stability. Blood plasma (100 μL), 10 μL of ascorbic acid (200 mg/mL), and 20 μL of internal standard solution (6-Hydroxyflavone, 5 μg/mL) were added into a centrifuge tube. The mixture was homogenized with 300 μL of methanol by vortex-mixing for 2 min, and centrifuged for 10 min at 12, 000 rpm. After centrifugation, the supernatant was withdrawn and the solvent evaporated using a centrifugal thickener (Christ, RVC2-331R, Germany). The dryness was dissolved with 200 μL of mobile phase, and then 20 μL of sample was subjected to LC-MS/MS analysis.46–48
The LC–MS/MS system was made up of a 6410 triple quadrupole mass spectrometer from Agilent Technologies (Santa Clara, CA, USA), equipped with an electrospray ionization (ESI) source, and an Agilent 1200 HPLC system from Agilent Technologies (Santa Clara, CA, USA) consisting of a vacuum degasser, a binary pump and an auto-sampler. Separation was performed on an Agilent ZORBAX SB C18 column (250×4.6 mm, 5 µm) at 25 °C. The mobile phase was composed of a mixture of 0.1% formic acid in water (A) and acetonitrile (B), which was run in a gradient elution program at a flow-rate of 1 mL/min. The HPLC gradient program was set as follows: 20%→30% B at 0.0–5.0 min; 30%→50% B at 5.0–20.0 min; 50%→20% B at 20.0–25.0 min; 20%→20% B at 25.0–30.0 min. The quantification of the baicalein (BA), baicalin (BG), and 6-Hydroxyflavone (IS) were performed using the MRM data acquisitions and 3 pairs of MRM transitions were selected, namely m/z 271.1→122.8, 447.0→271.1 and 239.0→137.0 for BA, BG, and IS, respectively.49,50
Statistical analyses
Pharmacokinetic analyses were performed using the DAS 2.0 software. All results are expressed as the mean ± standard deviation (SD). Statistical analyses were carried out using independent-samples t-test at the SPSS 17.0 software. Differences were considered to be statistically significant at P<0.05.
Results
Baicalein content in SMEDDS
Lipophilicity of drugs is crucial for SMEDDS as highly lipophilic drugs are required for proper dissolution in the SMEDDS. Hence, some low lipophilic drugs such as baicalein, face difficulties being incorporated into SMEDDS. However, the baicalein-phospholipid complex can increase the saturation solubility of baicalein in oil from 0.3 mg/g to 60.0 mg/g, and the saturation solubility of baicalein in SMEDDS concentrate from 20.0 mg/g to 100.0 mg/g. A dose of 20.0 mg/g was selected as the drug loading of CBA-SMEDDS and BAPC-SMEDDS, to enable proper comparison of the performances of these 2 formulations in vitro and in vivo.
Efficiency of self-emulsification and TEM
The efficiency of self-emulsification of BAPC-SMEDDS and CBA-SMEDDS was assessed. The results demonstrated that both formulations rapidly formed microemulsions within 1 min when diluted in water; clear and slightly yellow in appearance with no phase separation or drug precipitation within 24 h. The TEM pictures of BAPC-SMEDDS and CBA-SMEDDS are shown in Figure 1. Spherical microemulsion droplets of BAPC-SMEDDS and CBA-SMEDDS were observed and equally distributed. There was no significant difference in the appearance of both SMEDDS.
 |
Figure 1 The TEM pictures of BAPC-SMEDDS and CBA-SMEDDS. Abbreviations: BAPC-SMEDDS, baicalein-phospholipid complex self-microemulsions; CBA-SMEDDS, conventional baicalein self-microemulsions. |
Droplet size and zeta potential
Emulsion droplet size is one of the most important factors in self-emulsification performance as it greatly influences the rate and degree of drug release or absorption. The smaller the droplet size, the faster the drug release. It provides a larger surface area for drug dissolution. Zeta potential is also a very essential factor in the characterization of SMEDDS as it can identify the charge on the surface of the emulsion droplet. Greater electrostatic repulsive forces between the globules can prevent the coalescence of microemulsions. However, the decrease in the electrostatic repulsion forces may lead to phase separation.27
The values obtained for the droplet size and zeta potential are shown in Table 2; the distribution widths are expressed as the polydispersity index. The droplet sizes of BAPC-SMEDDS and CBA-SMEDDS were 9.6±0.2 nm and 11.3±0.4 nm, respectively. BAPC-SMEDDS, which contained the phospholipid, had significantly higher zeta potential than CBA-SMEDDS, suggesting after self-emulsification, SMEDDS with the baicalein-phospholipid complex would produce a more stable emulsion compared than conventional SMEDDS.
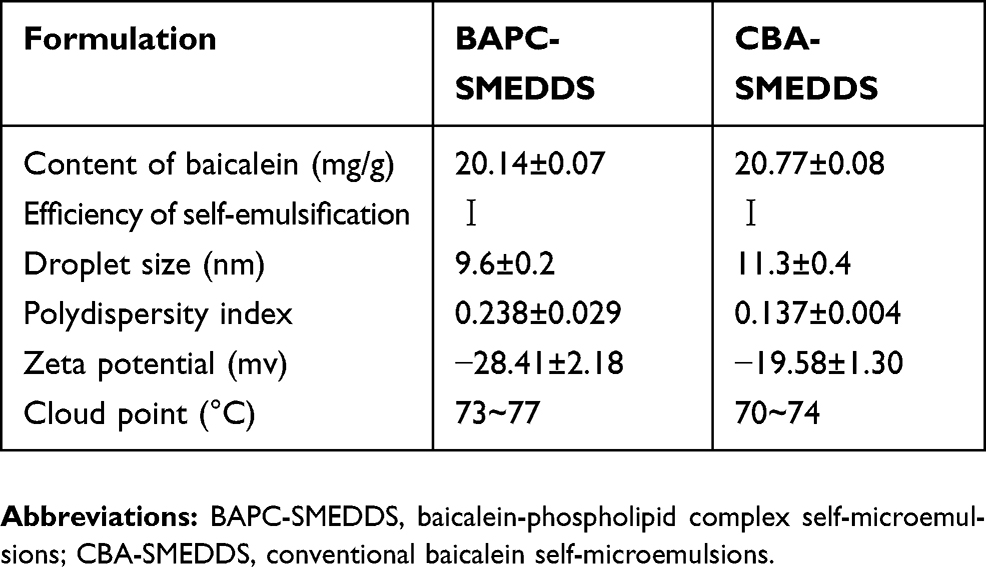 |
Table 2 Characterization of BAPC-SMEDDS and CBA-SMEDDS (n=3) |
Cloud point
The cloud point is the temperature above which an irreversible phase separation will occur, and clear microemulsion consisting of nonionic surfactants turns cloudy with the dehydration of the polyethylene oxide moiety. Any phase separation in the gastrointestinal tract (GIT) will negatively affect drug absorption. To avoid the occurrence of cloudiness and phase separation, the cloud point for SMEDDS should be above 37 °C, which is essential for the stability of microemulsion at the GIT physiological temperature.27–29 In this study, both SMEDDS had similar cloud points; 70–77 °C as reported in Table 2.
Robustness to dilution
BAPC-SMEDDS and CBA-SMEDDS were exposed to different folds of dilution in an attempt to mimic the in vivo conditions of gradual dilution, which may affect self-emulsification of SMEDDS and precipitate the drug at high dilutions.28 As shown in Figure 2, there was no significant change in the emulsion droplet size of both SMEDDS within the range of 20–100 fold dilution. When the dilution ratio was<20 or>100x, the droplet size was slightly increased but still less than 25 nm. All dispersions of BAPC-SMEDDS and CBA-SMEDDS showed no signs of separation, cloudiness, or precipitation for 12 h, which confirmed their robustness to different dilution regimens in water.
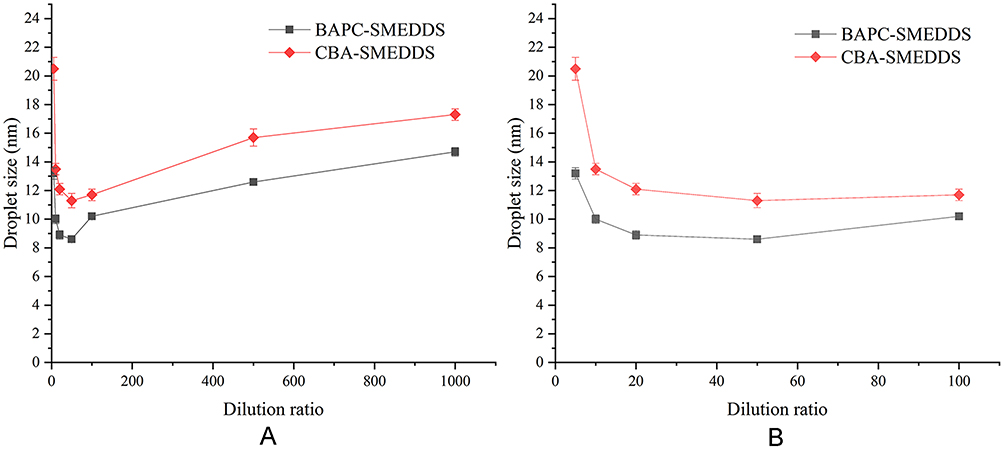 |
Figure 2 The size distribution of BAPC-SMEDDS and CBA-SMEDDS, which were subjected to different folds of dilution with purified water (n=3). Notes: A, dilution ratio (1:5–1:1000); B, dilution ratio (1:5–1:100).Abbreviations: BAPC-SMEDDS, baicalein-phospholipid complex self-microemulsions; CBA-SMEDDS, conventional baicalein self-microemulsions. |
Storage and colloidal stability
The results indicated that SMEDDS containing the baicalein-phospholipid complex could maintain clarity irrespective of the temperature. However, drug precipitation occurred in CBA-SMEDDS in the 1st month at 4 °C, 25 °C, and 45 °C.
The results of the colloidal stability analyses of BAPC-SMEDDS and CBA-SMEDDS are shown in Figure 3. Microemulsion migration or changes in droplet size trigger a change in the transmission signal, graphically reported as a positive (transmission increase) or negative (transmission decrease) peak. It is generally considered that when the ΔT profile is varied within a range of±2%, there is no significant change in the particle size of the carrier. A ΔT of>±10% indicates carrier instability.31 The calculation of TSI is based directly on the raw data acquired from the instrument, including backscattering and transmission signals. It combines all physical instable phenomena of samples, giving a number that evaluates sample instability. TSI reflects the extent of instability of the sample. The larger the TSI value, the more unstable the sample. Therefore, from the △t and TSI profiles of BAPC-SMEDDS and CBA-SMEDDS, it was observed that the colloidal stability of BAPC-SMEDDS was better than that of CBA-SMEDDS. The variation of △t for BAPC-SMEDDS was<±2%, demonstrating the stability of BAPC-SMEDDS at 37 °C within 12 h.
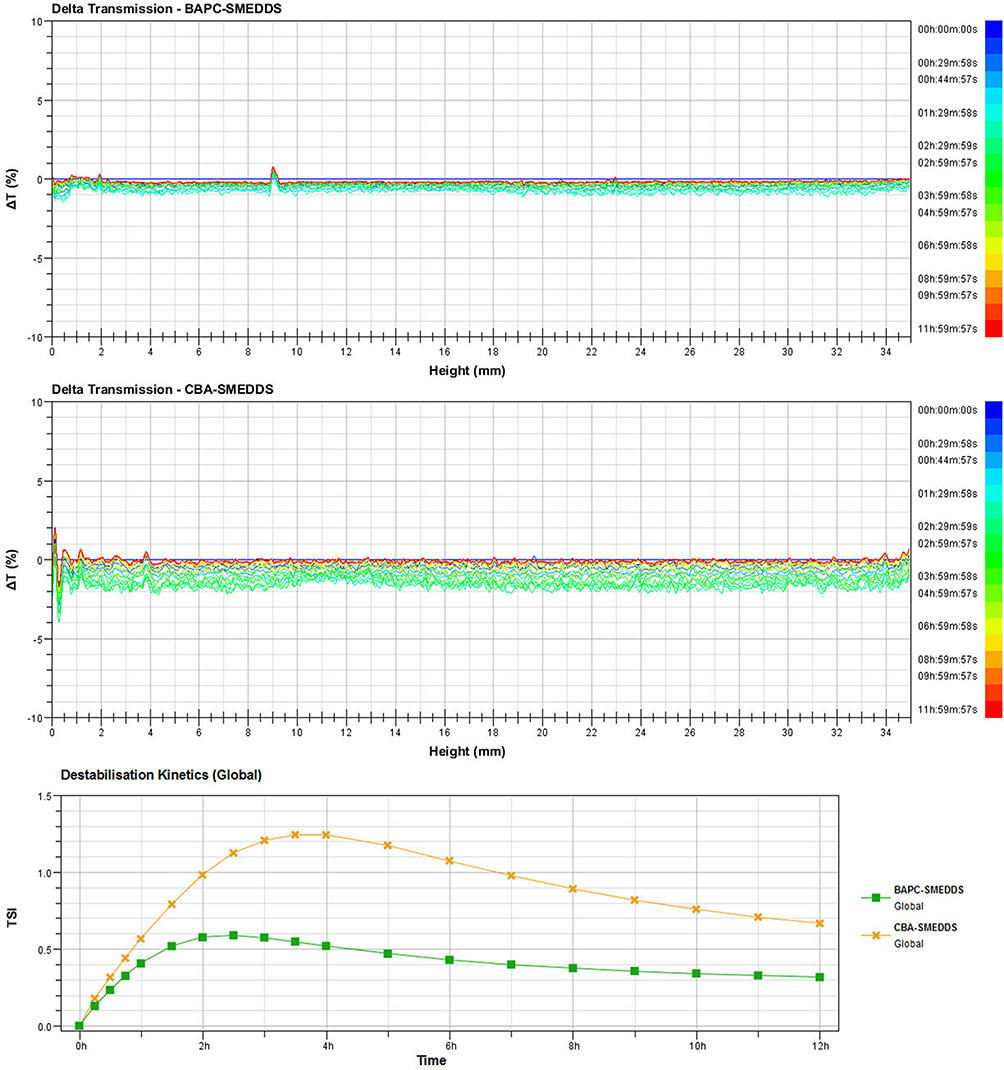 |
Figure 3 Delta transmission (△t) and turbiscan stability index (TSI) profiles of BAPC-SMEDDS and CBA-SMEDDS by using Turbiscan TOWER. Abbreviations: BAPC-SMEDDS, baicalein-phospholipid complex self-microemulsions; CBA-SMEDDS, conventional baicalein self-microemulsions. |
Drug release in vitro
In order to determine free drug molecules released from SMEDDS, encompassed by drugs in the microemulsion globules or micelles, the dialysis bag method was adopted. The release patterns of baicalein from BAPC-SMEDDS, CBA-SMEDDS, and BA were assessed in simulated gastric fluid (pH 1.0) and intestinal fluid (pH 6.8). The release profiles (Figures 4 and 5) indicated that the accumulated amount of baicalein released from BAPC-SMEDDS and CBA-SMEDDS was>90% within 12 h, with no significant difference, while only 40% and 60% baicalein was released within 24 h from BA in dissolve medium with pH 6.8 and 1.0, respectively. The drug release of both SMEDDS was higher than that of BA.
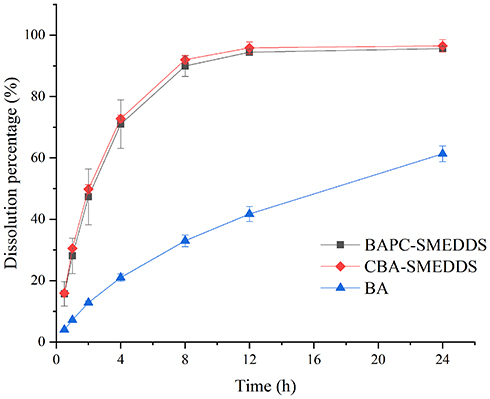 |
Figure 4 Release profile of baicalein from BAPC-SMEDDS, CBA-SMEDDS and BA (pH1.0, n=3). Abbreviations: BAPC-SMEDDS, baicalein-phospholipid complex self-microemulsions; CBA-SMEDDS, conventional baicalein self-microemulsions; BA, free baicalein. |
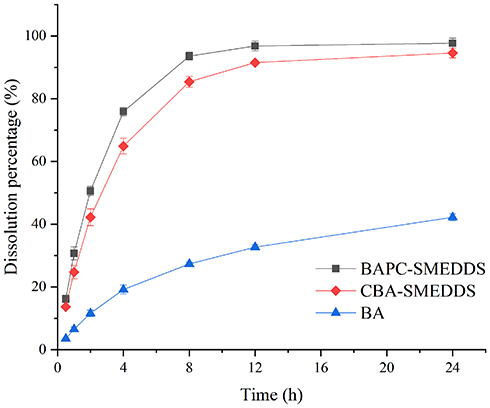 |
Figure 5 Release profile of baicalein from BAPC-SMEDDS, CBA-SMEDDS and BA (pH6.8, n=3). Abbreviations: BAPC-SMEDDS, baicalein-phospholipid complex self-microemulsions; CBA-SMEDDS, conventional baicalein self-microemulsions; BA, free baicalein. |
Lipolysis studies
In the GIT, SMEDDS lipids are primarily digested by lipase and colipase into free fatty acids or monoglycerides which may lead to drug precipitation, form lamellar phase structures, or affect drug absorption. However, lipid digestion is also a prerequisite for the production of fatty acids necessary to drive the production of chylomicrons, directly related to the degree of drug-lymphatic transport.3 In this study, lipolysis experiments were conducted for K-SMEDDS, K-PC-SMEDDS, BAPC-SMEDDS, and CBA-SMEDDS. The liberated fatty acids were titrated with NaOH (0.1 mol/L) to measure the rate of lipolysis.35,51 As seen in Figure 6, the lipolysis rate of SMEDDS containing phospholipid (K-PC-SMEDDS) was significantly faster in the first 30 mins, than that of SMEDDS without phospholipid (K-SMEDDS), while there was no significant difference in the lipolysis rate and degree between both SMEDDS after 30 mins. This showed that phospholipids could significantly promote the lipolysis rate of SMEDDS in the initial stage of lipolysis, which may accelerate the release and lymphatic transport of drugs in SMEDDS.
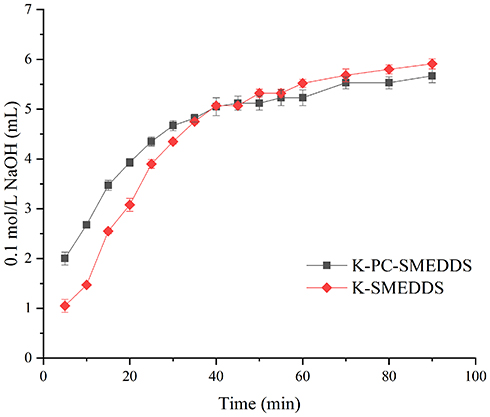 |
Figure 6 Consumption of 0.1 M NaOH during lipolysis of blank-SMEDDS with or without phospholipid (n=3). Abbreviations: K-PC-SMEDDS, blank SMEDDS with phospholipid; K-SMEDDS, blank SMEDDS without phospholipid. |
Figure 7 shows that the droplet size of BAPC-SMEDDS increased significantly after lipolysis commenced, and attained a maximum value at 15 min. After 30 min, the droplet size remained stable. The droplet size change of CBA-SMEDDS was higher than that of BAPC-SMEDDS. Therefore, formation of the baicalein-phospholipids complex reduced the droplet size change of SMEDDS, which may be caused by the zeta potential increase on the surface of microemulsion droplets. The distribution of baicalein in the aqueous phase during BAPC-SMEDDS and CBA-SMEDDS lipolysis is shown in Figure 8. Considering the whole lipolysis process, the aqueous phase baicalein content of both SMEDDS were maintained at>90% of the initial amount, with minute amounts involved in drug precipitation.
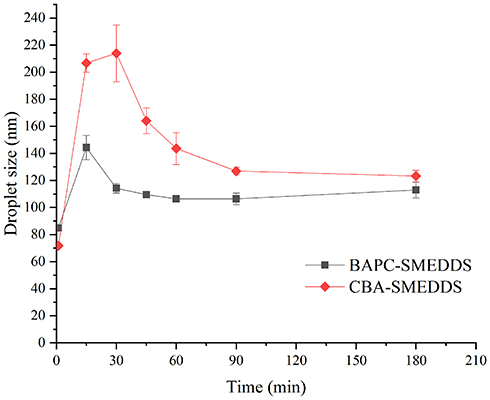 |
Figure 7 Mean droplet size of microemulsion globules or micelles formed during the in vitro lipolysis (n=3). Abbreviations: BAPC-SMEDDS, baicalein-phospholipid complex self-microemulsions; CBA-SMEDDS, conventional baicalein self-microemulsions. |
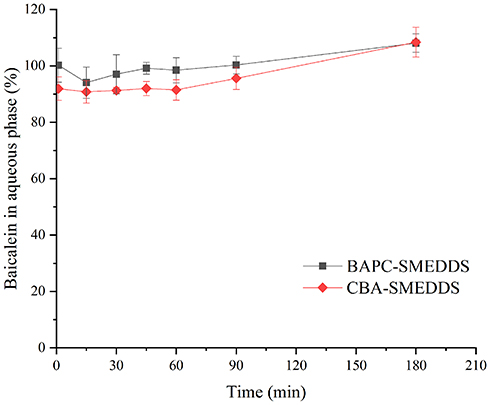 |
Figure 8 Distribution of baicalein in aqueous phase during lipolysis of BAPC-SMEDDS and CBA-SMEDDS (n=3). Abbreviations: BAPC-SMEDDS, baicalein-phospholipid complex self-microemulsions; CBA-SMEDDS, conventional baicalein self-microemulsions. |
Pharmacokinetic study
Baicalin, one of the mono-glucuronidation metabolites of baicalein, is reportedly the main plasma conjugated metabolite of orally administered baicalein. The conjugation metabolism of baicalein occurred very rapidly and extensively in vivo. The pharmacological effects of baicalin are similar to those of baicalein. Therefore, we chose baicalein and baicalin as the targets for the bioavailability study. Pharmacokinetic parameters were calculated from the plasma concentrations of baicalein and baicalin.
A LC-MS/MS analytic method using 6-Hydroxyflavone (6-HF) as the internal standard was established for the determination of baicalein and baicalin in rat plasma samples. Baicalein, baicalin, and the internal standard could be baseline separated from endogenous impurities in plasma with retention times of 16.4, 8.9, and 18.4 min, respectively. In the concentration range of 0.002–1.000 μg/mL, the corresponding peak area ratio of baicalein/6-HF (Y1) properly correlates with the plasma concentration of baicalein: (X1): Y1=0.0026X1+0.0026 (R2=0.9987, n=7). Good linearity was also obtained with the baicalin (X2) concentration range of 0.020–10.000 μg/mL: Y2=0.0032X2+0.0029 (R2=0.9947, n=7). Y2 is the corresponding peak area ratio of baicalin and 6-HF. The limits of quantification for baicalein and baicalin were 2 and 20 ng/mL, respectively.
The plasma concentrations of baicalin and baicalein versus time profiles are shown in Figures 9 and 10, respectively. The plasma concentrations of baicalin and baicalein of BAPC-SMEDDS and CBA-SMEDDS were significantly higher than those of BA. Compared with BA, the Cmax of baicalin for BAPC-SMEDDS and CBA-SMEDDS were improved by 7.7 and 4.1 times, respectively. The Cmax of BAPC-SMEDDS was approximately 1.9 times higher than that of CBA-SMEDDS. Table 3 indicates that the relative bioavailability of BAPC-SMEDDS and CBA-SMEDDS calculated from baicalin were 448.7% and 342.5%, respectively, compared with that of BA. The baicalin area under concentration-time curve (AUC0-t) of BAPC-SMEDDS was approximately 1.3-fold that of CBA-SMEDDS. Figures 9 and 10 also show that baicalein was rapidly metabolized to baicalin after oral administration, hence, baicalein plasma concentration was much lower than that of baicalin. According to Figure 11, the plasma baicalein, Cmax magnitudes and AUC0-t of various formulations are as follows: BAPC-SMEDDS>CBA-SMEDDS>BA. The results showed that SMEDDS improved the oral bioavailability of baicalein. It also showed that SMEDDS with the baicalein-phospholipid complex as intermediate not only facilitated the entrapment of the drug in the SMEDDS, but had better bioavailability and plasma concentration than the conventional SMEDDS.
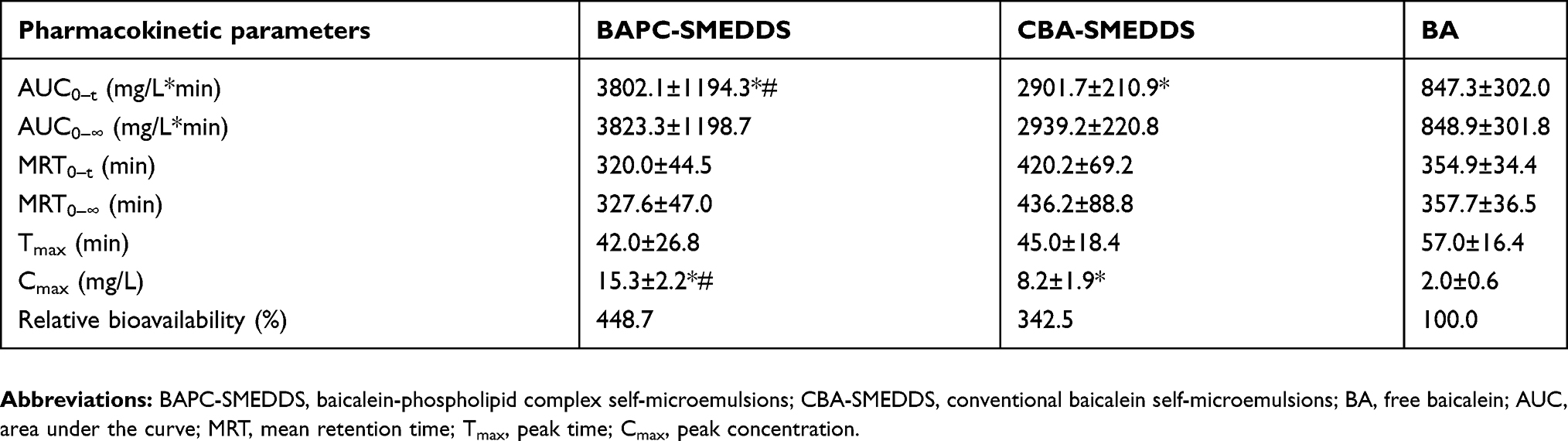 |
Table 3 Pharmacokinetic parameters of baicalin after oral administration of BAPC-SMEDDS, CBA-SMEDDS and BA (n=5) |
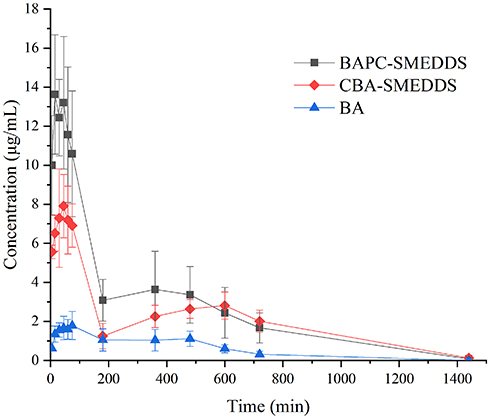 |
Figure 9 Mean plasma concentration–time curves of baicalin in rats after oral administration of BAPC-SMEDDS, CBA-SMEDDS and BA (n=5). Notes: The data were presented as mean ± SD (n=5).Abbreviations: BAPC-SMEDDS, baicalein-phospholipid complex self-microemulsions; CBA-SMEDDS, conventional baicalein self-microemulsions; BA, free baicalein. |
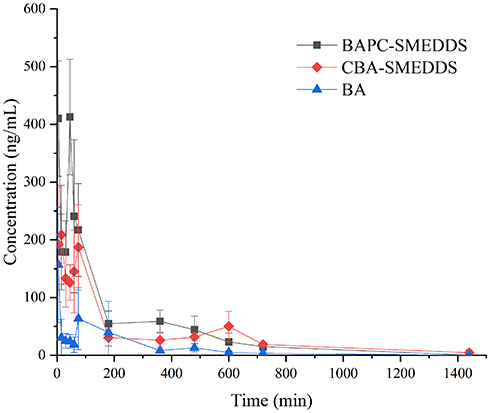 |
Figure 10 Mean plasma concentration–time curves of baicalein in rats after oral administration of BAPC-SMEDDS, CBA-SMEDDS and BA (n=5). Notes: The data were presented as mean ± SD (n=5).Abbreviations: BAPC-SMEDDS, baicalein-phospholipid complex self-microemulsions; CBA-SMEDDS, conventional baicalein self-microemulsions; BA, free baicalein. |
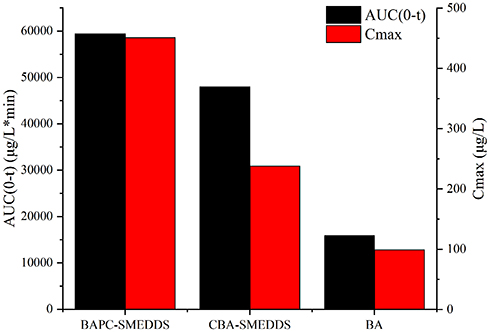 |
Figure 11 The average AUC0-t and Cmax of baicalein after oral administration of BAPC-SMEDDS, CBA-SMEDDS and BA (n=5). Abbreviations: BAPC-SMEDDS, baicalein-phospholipid complex self-microemulsions; CBA-SMEDDS, conventional baicalein self-microemulsions; BA, free baicalein; AUC, area under the curve; Cmax, peak concentration. |
Lymphatic transport of different formulations
Lipophilic drugs absorbed by the enterocyte can reach systemic blood circulation via the lymphatic transport and the portal blood. The lymphatic transport pathway is entirely dependent on the formation and transport of chylomicrons within the intestinal epithelial cells.42,52 According to previous reports, the secretion of chylomicron can be completely blocked by cycloheximide. In other words, the lymphatic transport pathway can be completely blocked by cycloheximide, which has the same effect as lymphatic vessel cannulation.42,43,52
According to the results of bioavailability study in vivo, it was found that baicalein was quickly metabolized to baicalin in plasma. And the absorption and metabolism processes of baicalein and baicalin mainly occurred within 3 hrs. Therefore, in this experiment, the systemic exposure of bacalin in plasma within 3 hrs after oral administration of baicalein was used to conduct lymphatic transport studies of baicalein. The data in Table 4 is derived from the bioavailability study in vivo. Thus, the AUC0–t of baicalin in Table 4 reflected the baicalein absorbed from all pathways (include the lymphatic transport). After intraperitoneal injection of cycloheximide, the AUC0–t of baicalin in Table 5 reflected the baicalein fraction absorbed from other pathways (except the lymphatic transport), which was transported directly into the systemic blood circulation. Therefore, the fraction transported via the lymphatic system was calculated as the difference between the AUC0–t determined from rats without pretreatment (Table 4) and the AUC0–t from rats pretreated with cycloheximide (Table 5). The results were shown in Table 6.
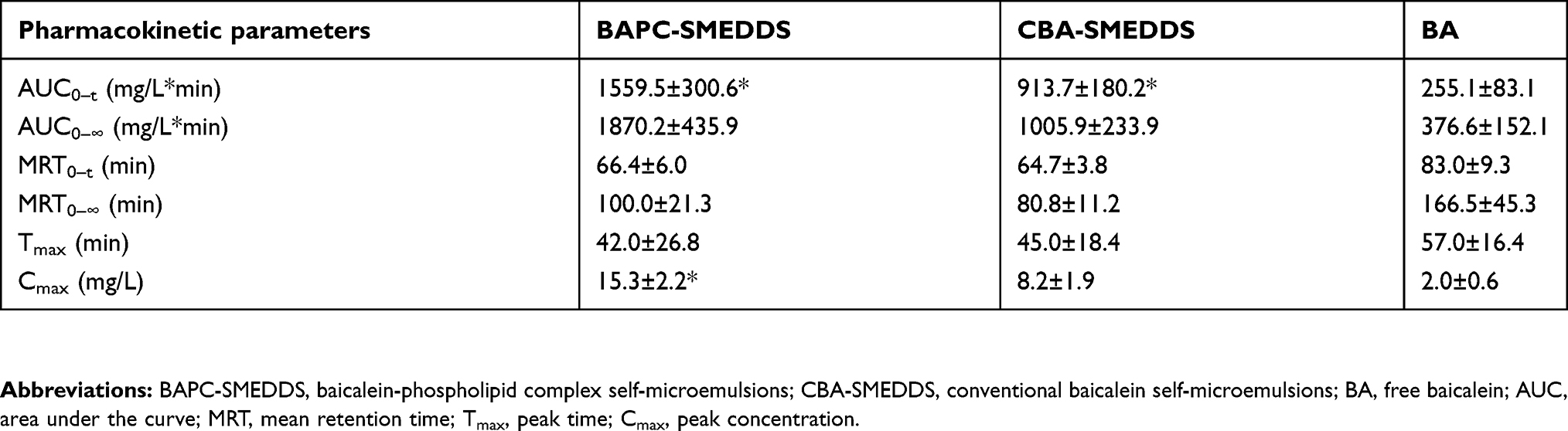 |
Table 4 Pharmacokinetic parameters of baicalin within 3 hrs after oral administration of BAPC-SMEDDS, CBA-SMEDDS and BA (n=5) |
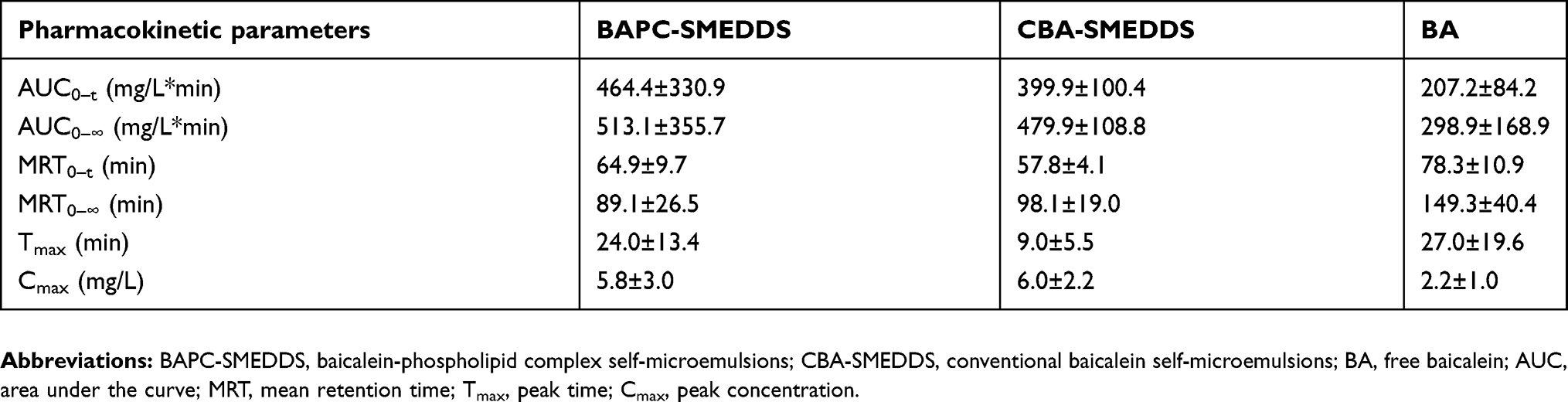 |
Table 5 Pharmacokinetic parameters of baicalin within 3 hrs after intraperitoneal pretreatment with 3.0 mg/kg cycloheximide following oral administration of BAPC-SMEDDS, CBA-SMEDDS and BA (n=5) |
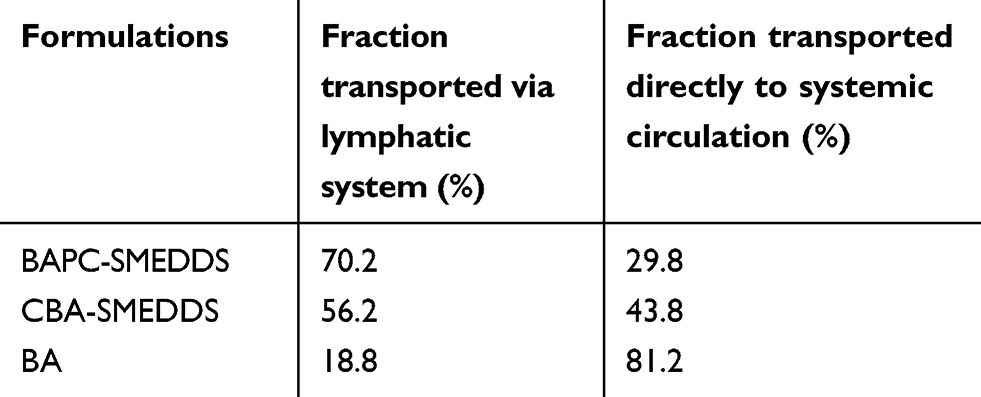 |
Table 6 Fraction of baicalein transported directly to the systemic blood circulation and fraction of baicalein transported indirectly to the systemic blood circulation via intestinal lymphatic system after oral administration of baicalein in different formulations to rats |
As shown in Figure 12, after intraperitoneal injection of cycloheximide, the mean plasma concentrations of baicalin of BAPC-SMEDDS and CBA-SMEDDS decreased significantly, while that of BA did not significantly change. This suggests that only minute quantities of BA can enter the bloodstream through lymphatic transport. However, after lymphatic transport was inhibited, there was no significant difference in the AUC0–t of BAPC-SMEDDS and CBA-SMEDDS, but they were greater than the AUC0–t of BA. It can therefore be inferred that SMEDDS containing baicalein-phospholipid complexes may further enhance drug oral bioavailability by improving baicalein lymphatic transport, compared with conventional SMEDDS. It also indicates that SMEDDS can enhance the oral absorption of drugs by other means besides lymphatic transport. The results of pharmacokinetic parameters (Table 4, 5, and 6) showed that CBA-SMEDDS and BAPC-SMEDDS increased the lymphatic transport ratio of oral absorption of baicalein from 18.8% to 56.2% and 70.2%, respectively, compared with the BA ratio. In conclusion, the magnitudes of the extent of lymphatic transport of various formulations were as follows: BAPC-SMEDDS>CBA-SMEDDS>BA.
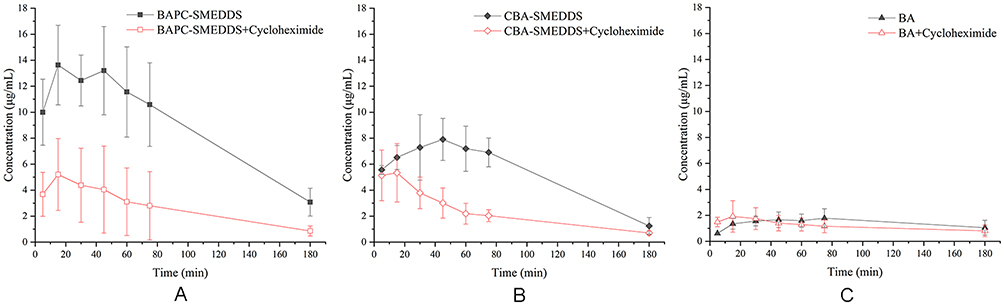 |
Figure 12 Mean plasma concentration–time curves of baicalin in rats within 3 hrs after intraperitoneal pretreatment with or without 3.0 mg/kg cycloheximide following oral administration of BAPC-SMEDDS, CBA-SMEDDS and BA (n=5). Notes: A, intraperitoneal pretreatment with or without 3.0 mg/kg cycloheximide following oral administration of BAPC-SMEDDS; B, intraperitoneal pretreatment with or without 3.0 mg/kg cycloheximide following oral administration of CBA-SMEDDS; C, intraperitoneal pretreatment with or without 3.0 mg/kg cycloheximide following oral administration of BA. The data were presented as mean ± SD (n=5).Abbreviations: BAPC-SMEDDS, baicalein-phospholipid complex self-microemulsions; CBA-SMEDDS, conventional baicalein self-microemulsions; BA, free baicalein. |
Discussion
This study showed that formation of the baicalein-phospholipid complex increased the saturation solubility of baicalein in oil from 0.3–60.0 mg/g, and that of baicalein in SMEDDS from 20.0–100 0.0 mg/g. This indicates that SMEDDS with an intermediary drug-phospholipid complex are more conducive to the entrapment of drugs in SMEDDS. Compared with CBA-SMEDDS, the presence of the baicalein-phospholipid complex in BAPC-SMEDDS increased the Zeta potential on the surface of microemulsion droplets, which improved the stability of the droplets in gastric and intestinal fluids. BAPC-SMEDDS could be completely emulsified within 1 min of mixing with water. The microemulsion was clear and transparent, and the droplet size was approximately 10 nm. The cloud point of BAPC-SMEDDS was in the range of 73–77 °C, far above the normal body temperature of 37 °C. Therefore, BAPC-SMEDDS can be quickly emulsified into nano-sized emulsion droplets after entering the human body, and there is no risk of phase transition. Diluting BAPC-SMEDDS in distilled water (5-1000x), had no significant effect on the microemulsion droplet size. The microemulsion was clear and transparent 12 h after emulsification, without any drug precipitation. Physical stability studies showed that BAPC-SMEDDS had better stability than conventional SMEDDS, and showed no drug precipitation or turbidity after 2 months at 4 °C, 25 °C, and 40 °C.
The pharmacokinetic study in rats indicated the oral bioavailability of different formulations were as follows: BAPC-SMEDDS>CBA-SMEDDS>BA. CBA-SMEDDS and BAPC-SMEDDS increased the proportion of lymphatic transport in the oral absorption of baicalein from 18.8% to 56.2% and 70.2%, respectively, compared with the BA. However, the bioavailability of BAPC-SMEDDS and CBA-SMEDDS were significantly reduced after lymphatic transport was inhibited. In addition, there was no significant difference in the AUC0–t of BAPC-SMEDDS and CBA-SMEDDS, but they were greater than that of BA. Therefore, it can be inferred that another reason why the self-emulsifying drug delivery system significantly improved the oral bioavailability of baicalein was to improve baicalein lymphatic transport. This also suggests that SMEDDS containing baicalein-phospholipid complexes may further enhance oral bioavailability of baicalein by improving its lymphatic transport, compared with conventional SMEDDS. In vitro release tests equally indicated no significant difference in the release curves of BAPC-SMEDDS and CBA-SMEDDS at pH 1.0 and 6.8. However, the release rate and degree of both SMEDDS were much higher than that of the pure drug group. Therefore, apart from increasing baicalein lymphatic transport, SMEDDS can enhance the oral absorption of drugs by accelerating their release and increasing their dissolution. The results of in vitro lipolysis tests indicated that although phospholipids could accelerate the microemulsion lipolysis rate at the initial stage of lipolysis (<30 min), the water phase distribution of baicalein in both SMEDDS did not significantly change within 3 h; no drug precipitation was generated. Nevertheless, an increase in the rate of lipolysis can accelerate the production of fatty acids, to drive the production of chylomicrons, which is directly related to the increase of lymphatic transport of BAPC-SMEDDS. The drop size change of BAPC-SMEDDS was smaller, with both being approximately 100 nm, which was beneficial to their direct absorption by intestinal epithelial cells and gut-associated lymphoid tissue (GALT).
According to results from previous studies3,6,7,13,53 and ours, part of the mechanism for improving the bioavailability of baicalein by preparing BAPC-SMEDDS and CBA-SMEDDS can be preliminarily speculated. After oral administration, baicalein SMEDDS can be rapidly emulsified in gastrointestinal fluid to form microemulsion droplets, which are widely distributed in the GIT. This can greatly improve the distribution ratio of drugs in the aqueous phase, facilitate the passage of drugs through the non-liquid water layer on the surface of small intestinal villi, and increase their gastrointestinal absorption. Surfactants such as Tween 80 contained in SMEDDS can open the tight connection of cell bypass, facilitating the bypass absorption of a drug-phospholipid complex molecules and nano-size emulsion droplets, to increase the lymphatic transport of drugs. The digestion of lipids in SMEDDS can drive the production of chylomicrons, facilitating drug-lymphatic transport. The presence of phospholipids in SMEDDS not only accelerates the production of chylomicrons but also promotes lipid transporter-mediated microemulsion endocytosis. However, further studies that fully elucidate the absorption mechanisms are required.
Conclusion
SMEDDS with an intermediary baicalein-phospholipid complex were successfully prepared. The droplet size of SMEDDS was as low as 9.6 nm, which significantly increased the release rate and degree of baicalein in vitro. More so, the SMEDDS with baicalein-phospholipid complex was more stable than conventional SMEDDS at 4 °C, 25 °C, and 40 °C. And this study is the first to demonstrate the oral absorption and lymphatic transport characteristics of free baicalein and baicalein SMEDDS with different compositions. It was found that BAPC-SMEDDS could not only significantly improve baicalein oral bioavailability, but also increase the proportion of lymphatically transported orally absorbed baicalein. These are of great significance to lymphatic targeted delivery studies of natural immunomodulatory compounds.
Acknowledgment
This work was financially supported by the National Science and Technology Major Project (No. 2018ZX09721003) of the Chinese government.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Swartz MA. The physiology of the lymphatic system. Adv Drug Deliv Rev. 2001;50(1):3–20.
2. Porter C, Pouton CJ, Charman W, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60(6):673–691. doi:10.1016/j.addr.2007.10.014
3. O’Driscoll CM. Lipid-based formulations for intestinal lymphatic delivery. Eur J Hosp Pharm. 2002;15(5):405–415.
4. Caliph SM, Charman WN, Porter CJH. Effect of short-, medium-, and long-chain fatty acid-based vehicles on the absolute oral bioavailability and intestinal lymphatic transport of halofantrine and assessment of mass balance in lymph-cannulated and non-cannulated rats. J Pharm Sci. 2000;89(8):1073–1084. doi:10.1002/1520-6017(200008)89:8<1073::aid-jps12>3.0.co;2-v
5. Pouton CW. Formulation of self-emulsifying drug delivery systems. Adv Drug Deliv Rev. 1997;25(1):47–58. doi:10.1016/S0169-409X(96)00490-5
6. Aboulfotouh K, Allam AA, El-Badry M, El-Sayed AM. Role of self-emulsifying drug delivery systems in optimizing the oral delivery of hydrophilic macromolecules and reducing interindividual variability. Colloids Surf B Biointerfaces. 2018;167(2018):82–92. doi:10.1016/j.colsurfb.2018.03.034
7. Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58(3):173–182. doi:10.1016/j.biopha.2004.02.001
8. René H, Porter CJH, Edwards GA, Anette M, Kristensen HG, Charman WN. Examination of oral absorption and lymphatic transport of halofantrine in a triple-cannulated canine model after administration in self-microemulsifying drug delivery systems (SMEDDS) containing structured triglycerides. Eur J Hosp Pharm. 2003;20(1):91–97.
9. Minghui S, Xuezhen Z, Kewen X, et al. Intestinal absorption and intestinal lymphatic transport of sirolimus from self-microemulsifying drug delivery systems assessed using the single-pass intestinal perfusion (SPIP) technique and a chylomicron flow blocking approach: linear correlation with or. Saudi Pharm J. 2011;43(3):132–140. doi:10.1016/j.ejps.2011.04.011
10. Zhao L, Zhang L, Meng L, Wang J, Zhai G. Design and evaluation of a self-microemulsifying drug delivery system for apigenin. Drug Dev Ind Pharm. 2013;39(5):662–669. doi:10.3109/03639045.2012.687378
11. Borhade V, Nair H, Hegde D. Design and evaluation of Self-Microemulsifying Drug Delivery System (SMEDDS) of tacrolimus. AAPS PharmSciTech. 2008;9(1):13–21. doi:10.1208/s12249-007-9014-8
12. Mandawgade SD, Sharma S, Pathak S, Patravale VB. Development of SMEDDS using natural lipophile: application to β-artemether delivery. Int J Pharm. 2008;362(1):179–183. doi:10.1016/j.ijpharm.2008.06.021
13. Zhang L, Zhang L, Zhang M, et al. Self-emulsifying drug delivery system and the applications in herbal drugs. Drug Deliv. 2013;22(4):475–486. doi:10.3109/10717544.2013.861659
14. Wu X, Xu J, Huang X, Wen C. Self-microemulsifying drug delivery system improves curcumin dissolution and bioavailability. Drug Dev Ind Pharm. 2011;37(1):15. doi:10.3109/03639045.2010.489560
15. Qiao J, Ji D, Sun S, et al. Oral bioavailability and lymphatic transport of pueraria flavone-loaded self-emulsifying drug-delivery systems containing sodium taurocholate in rats. Pharmaceutics. 2018;10(3):147. doi:10.3390/pharmaceutics10030147
16. Kimura Y, Okuda H, Arichi S. Effects of baicalein on leukotriene C4 biosynthesis in human lymphocytes. Phytother Res. 1987;1(1):48–49. doi:10.1002/(ISSN)1099-1573
17. Yin J, Xiang C, Wang P, Yin Y, Hou Y. Biocompatible nanoemulsions based on hemp oil and less surfactants for oral delivery of baicalein with enhanced bioavailability. Int J Nanomedicine. 2017;12:2923–2931. doi:10.2147/IJN.S131167
18. Pi J, Wang S, Wen L, et al. A nano-cocrystal strategy to improve the dissolution rate and oral bioavailability of baicalein. Asian J Pharm Sci. 2019;14(2):154-164. S1818087617309492-.
19. Yan Z, Lei S, Yaping H, et al. Baicalein selectively induces apoptosis in activated lymphocytes and ameliorates concanavalin a-induced hepatitis in mice. PLoS One. 2013;8(7):e69592. doi:10.1371/journal.pone.0069592
20. Lam HT, Le-Vinh B, Phan TNQ, Bernkop-Schnürch A. Self-emulsifying drug delivery systems and cationic surfactants: do they potentiate each other in cytotoxicity? J Pharm Pharmacol. 2019;71(2):156–166. doi:10.1111/jphp.13021
21. Zhou H, Wan J, Wu L, et al. A new strategy for enhancing the oral bioavailability of drugs with poor water-solubility and low liposolubility based on phospholipid complex and supersaturated SEDDS. PLoS One. 2013;8(12):e84530. doi:10.1371/journal.pone.0084530
22. Wu H, Long X, Yuan F, et al. Combined use of phospholipid complexes and self-emulsifying microemulsions for improving the oral absorption of a BCS class IV compound, baicalin. Acta Pharm Sin B. 2014;4(3):217–226. doi:10.1016/j.apsb.2014.03.002
23. Zhou Y, Dong W, Ye J, et al. A novel matrix dispersion based on phospholipid complex for improving oral bioavailability of baicalein: preparation, in vitro and in vivo evaluations. Drug Deliv. 2017;24(1):720–728. doi:10.1080/10717544.2017.1311968
24. Khoo S-M, Humberstone AJ, Porter CJH, Edwards GA, Charman WN. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int J Pharm. 1998;167(1):155–164. doi:10.1016/S0378-5173(98)00054-4
25. Kadu PJ, Kushare SS, Thacker DD, Gattani SG. Enhancement of oral bioavailability of atorvastatin calcium by self-emulsifying drug delivery systems (SEDDS). Pharm Dev Technol. 2011;16(1):65–74. doi:10.3109/10837450903499333
26. Singh B, Khurana L, Bandyopadhyay S, Kapil R, Katare OO. Development of optimized self-nano-emulsifying drug delivery systems (SNEDDS) of carvedilol with enhanced bioavailability potential. Drug Deliv. 2011;18(8):599–612. doi:10.3109/10717544.2011.604686
27. Avachat AM, Patel VG. Self nanoemulsifying drug delivery system of stabilized ellagic acid–phospholipid complex with improved dissolution and permeability. Saudi Pharm J. 2015;23(3):276–289. doi:10.1016/j.jsps.2014.11.001
28. Zhang P, Liu Y, Feng N, Xu J. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int J Pharm. 2008;355(1):269–276. doi:10.1016/j.ijpharm.2007.12.026
29. Elnaggar YSR, El-Massik MA, Abdallah OY. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: design and optimization. Int J Pharm. 2009;380(1):133–141. doi:10.1016/j.ijpharm.2009.07.015
30. Shen H, Zhong M. Preparation and evaluation of self-microemulsifying drug delivery systems (SMEDDS) containing atorvastatin. J Pharm Pharmacol. 2010;58(9):1183–1191. doi:10.1211/jpp.58.9.0004
31. Marianecci C, Paolino D, Celia C, Fresta M, Carafa M, Alhaique F. Non-ionic surfactant vesicles in pulmonary glucocorticoid delivery: characterization and interaction with human lung fibroblasts. J Control Release. 2010;147(1):127–135. doi:10.1016/j.jconrel.2010.06.022
32. Celia C, Trapasso E, Cosco D, Paolino D, Fresta M. Turbiscan lab expert analysis of the stability of ethosomes and ultradeformable liposomes containing a bilayer fluidizing agent. Colloids Surf B Biointerfaces. 2009;72(1):155–160. doi:10.1016/j.colsurfb.2009.03.007
33. Kang BK, Lee Jschon SK, Jeong SY, et al. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm. 2004;274(1):65–73. doi:10.1016/j.ijpharm.2003.12.028
34. Wenli L, Rui T, Wenjing H, et al. Preparation and evaluation of self-microemulsifying drug delivery system of baicalein. Fitoterapia. 2012;83(8):1532–1539. doi:10.1016/j.fitote.2012.08.021
35. Zupančič O, Partenhauser A, Lam HT, Rohrer J, Bernkop-Schnürch A. Development and in vitro characterisation of an oral self-emulsifying delivery system for daptomycin. Eur J Hosp Pharm. 2016;81:129–136. doi:10.1016/j.ejps.2015.10.005
36. Zangenberg NH, Müllertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model. I. Controlling the rate of lipolysis by continuous addition of calcium. Eur J Hosp Pharm. 2001;14(2):115–122.
37. Dahan A, Hoffman A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J Controlled Release. 2008;129(1):1–10. doi:10.1016/j.jconrel.2008.03.021
38. Christensen JØ, Schultz K, Mollgaard B, Kristensen HG, Mullertza A. Solubilisation of poorly water-soluble drugs during in vitro lipolysis of medium- and long-chain triacylglycerols. Eur J Hosp Pharm. 2004;23(3):287–296. doi:10.1016/j.ejps.2004.08.003
39. Zhang J, Lv H, Jiang K, Gao Y. Enhanced bioavailability after oral and pulmonary administration of baicalein nanocrystal. Int J Pharm. 2011;420(1):180–188. doi:10.1016/j.ijpharm.2011.08.023
40. He X, Pei L, Tong HHY, Ying Z. Comparison of spray freeze drying and the solvent evaporation method for preparing solid dispersions of baicalein with pluronic F68 to improve dissolution and oral bioavailability. AAPS PharmSciTech. 2011;12(1):104–113. doi:10.1208/s12249-010-9560-3
41. Zhang Y, Luo R, Chen Y, Ke X, Hu D, Han M. Application of carrier and plasticizer to improve the dissolution and bioavailability of poorly water-soluble baicalein by hot melt extrusion. AAPS PharmSciTech. 2014;15(3):560–568. doi:10.1208/s12249-013-0071-x
42. Fan Z, Wu J, Fang X, Sha X. A new function of vitamin E-TPGS in the intestinal lymphatic transport of lipophilic drugs: enhancing the secretion of chylomicrons. Int J Pharm. 2013;445(1–2):141–147. doi:10.1016/j.ijpharm.2013.01.070
43. Lind ML, Jette J, René H, Anette M. Intestinal lymphatic transport of halofantrine in rats assessed using a chylomicron flow blocking approach: the influence of polysorbate 60 and 80. Saudi Pharm J. 2008;35(3):211–218. doi:10.1016/j.ejps.2008.07.003
44. Vivek M, Rashmi J, Komal P, Manish N, Amita J. Solid lipid nanoparticles (SLN) of efavirenz as lymph targeting drug delivery system: elucidation of mechanism of uptake using chylomicron flow blocking approach. Int J Pharm. 2015;495(1):439–446. doi:10.1016/j.ijpharm.2015.09.014
45. Dahan A, Hoffman A. Use of a dynamic in vitro lipolysis model to rationalize oral formulation development for poor water soluble drugs: correlation with in vivo data and the relationship to intra-enterocyte processes in rats. Pharm Res. 2006;23(9):2165–2174. doi:10.1007/s11095-006-9054-x
46. Chen M, Wu Q, Jiang J, et al. Preparation, characterization and in vivo evaluation of a formulation of dantrolene sodium with hydroxypropyl-β-cyclodextrin. J Pharm Biomed Anal. 2017;135:153–159. doi:10.1016/j.jpba.2016.12.022
47. Ju L, Wenlan W, Qiuwei L, Shan C. Long-circulating nanoliposomes (LCNs) sustained delivery of baicalein (BAI) with desired oral bioavailability in vivo. Drug Deliv. 2013;20(8):319–323. doi:10.3109/10717544.2013.834420
48. Huang Y, Zhang B, Gao Y, Zhang J, Shi L. Baicalein–nicotinamide cocrystal with enhanced solubility, dissolution, and oral bioavailability. J Pharm Sci. 2014;103(8):2330–2337. doi:10.1002/jps.24048
49. Feng J, Xu W, Tao X, et al. Simultaneous determination of baicalin, baicalein, wogonin, berberine, palmatine and jatrorrhizine in rat plasma by liquid chromatography–tandem mass spectrometry and application in pharmacokinetic studies after oral administration of traditional Chinese. J Pharm Biomed Anal. 2010;53(3):591–598. doi:10.1016/j.jpba.2010.04.002
50. Jun-Xiao Y, Wang W, Li-Hui Q, Chun-Yu L, Qi C, Yong-Hong L. An LC-MS/MS method for the simultaneous determination of chlorogenic acid, forsythiaside A and baicalin in rat plasma and its application to pharmacokinetic study of shuang-huang-lian in rats. J Pharm Biomed Anal. 2010;52(4):625–630. doi:10.1016/j.jpba.2010.01.035
51. Ali H, Nazzal M, Zaghloul AAA, Nazzal S. Comparison between lipolysis and compendial dissolution as alternative techniques for the in vitro characterization of α-tocopherol self-emulsified drug delivery systems (SEDDS). Int J Pharm. 2008;352(1):104–114. doi:10.1016/j.ijpharm.2007.10.023
52. Dahan A, Hoffman A. Evaluation of a chylomicron flow blocking approach to investigate the intestinal lymphatic transport of lipophilic drugs. Eur J Pharm Sci. 2005;24(4):381–388. doi:10.1016/j.ejps.2004.12.006
53. Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘Self-microemulsifying’ drug delivery systems. Eur J Hosp Pharm. 2000;11(2,supplement):S93–S98. doi:10.1016/S0928-0987(00)00167-6
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.