")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 7
Optimal management of hyperphosphatemia in end-stage renal disease: an Indian perspective
Authors Reddy Y, Sundaram V, Abraham G, Nagarajan P, Reddy YNV
Received 14 July 2014
Accepted for publication 23 August 2014
Published 23 October 2014 Volume 2014:7 Pages 391—399
DOI https://doi.org/10.2147/IJNRD.S49933
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Pravin Singhal
Yogesh NV Reddy,1 Varun Sundaram,2 Georgi Abraham,3 Prethivee Nagarajan,3 Yuvaram NV Reddy3
1Department of Cardiology, Mayo Clinic, Rochester, MN, USA; 2Department of Cardiology, University Hospitals Case Medical Center, Cleveland, OH, USA; 3Department of Nephrology, Madras Medical Mission, Chennai, India
Abstract: There has been an exponential increase in the incidence of diabetes and hypertension in India in the last few decades, with a proportional increase in chronic kidney disease (CKD). Preventive health care and maintenance of asymptomatic chronic disease such as CKD are often neglected by patients until they become symptomatic with fluid retention and uremia. Management of hyperphosphatemia in CKD remains one of the challenges of nephrology in India for this reason, as it is almost completely asymptomatic but contributes to renal osteodystrophy, metastatic vascular calcification, and acceleration of cardiovascular disease. Lack of understanding of the dangers of asymptomatic hyperphosphatemia, the huge pill burden of phosphate binders, difficulty with dietary and dialysis compliance, and most importantly, the added expense of the drugs places additional road blocks in the treatment of hyperphosphatemia at a population level in developing countries like India. In this review we seek to address the contribution of hyperphosphatemia to adverse outcomes and discuss economic, cultural, and societal factors unique to the management of phosphate levels in Indian patients with advanced CKD.
Keywords: dialysis, chronic kidney disease, vascular calcification
The economic and cultural challenges of managing chronic kidney disease in India
As with many other developing countries, India continues to show rapid economic and financial growth in many sections of its society. As a result of economic reforms introduced in the early 1990s, the prevalence of Indians living in extreme poverty has decreased from 45% in 1994 to 22% in 2012.1 This halving of the poverty rate coincides with the period of fastest economic growth in India’s history.2 As part of this development, there has been a huge surge in noncommunicable diseases like type 2 diabetes mellitus and hypertension, in part related to an increase in the incidence of metabolic syndrome approaching levels seen in the Western hemisphere. This has led to an increase in the incidence of chronic kidney disease (CKD) from diabetic and hypertensive nephropathy.
For many years the true incidence of CKD and end-stage renal disease (ESRD) in India was not known, as the majority of patients with CKD are asymptomatic until the late stages and therefore do not present to the health care system. Health care in India is primarily a cash for service business, with the majority of hospitals requiring payment out of pocket for services provided, apart from the limited number of government-funded hospitals. With increasing financial security, more patients are approaching health care establishments for primary care and being diagnosed with asymptomatic CKD, leading to an increase in the apparent incidence of CKD. One population-based study showed that the incidence of ESRD in India was 152 per million population.3 The Indian CKD Registry initiative was undertaken to try and establish the characteristics of CKD in the country and is still ongoing (http://ckdri.org/). A preliminary report has shown that patients in India develop ESRD at a younger age, and also that diabetic nephropathy is the most common cause of ESRD in India.4
In spite of rapid economic growth and the halving of the poverty rate in the last couple of decades, the majority of India’s population still struggle to fulfil all their family’s health care and educational needs. The Empowerment Line reveals that 56% of India’s population lacks the means for a acceptable minimum standard of living. The Mckinsey report calculated India’s Empowerment Line at 1,336 rupees per capita per month, or almost 6,700 rupees for a family of five per month. As of 2012, the consumption levels of almost 680 million people across both urban and rural areas of the country fell short of this mark. The monthly cost of the common dialysis prescriptions, two hemodialysis sessions/week and three peritoneal dialysis exchanges/day, was estimated at 29,852 rupees ($609) and 28,763 rupees ($585), respectively.5 This statistic clearly shows that despite economic growth there remains a large gap in the proportion of the population who have the means for additional expenditures beyond what is needed for daily living. However, among the proportion of people with access to dialysis, their prognosis and outcomes are similar to most worldwide statistics.
Unfortunately, preventive health care and maintenance of asymptomatic chronic disease such as CKD are often neglected by patients until they become symptomatic with ESRD requiring dialysis or advanced CKD with fluid retention and uremia. Management of hyperphosphatemia in CKD remains one of the challenges of nephrology in India for this reason, as it is almost completely asymptomatic but contributes to renal osteodystrophy, metastatic vascular calcification, and acceleration of cardiovascular disease. Many staples of the Indian diet, including rice and grain-based foods such as rotis, are very high in phosphorus content. Rice and rice-based meals make up the vast majority of most Indian food. A cup of rice has about 160 mg of phosphorus, which becomes challenging for Indian patients with advanced CKD, who are often restricted to <500 mg of phosphorus a day. Diet control in India is again challenging given the personal expense to a family of preparing a special diet for one member of the family with CKD and a regular diet for the remainder of the household. Malnutrition and protein energy malnutrition in CKD patients in India is a large problem, and, in general, protein restriction is not advisable in Indian CKD patients despite some controversial evidence that it slows progression of CKD.6–8 Prescription of special diets and dietary restrictions often leads to worsening malnutrition in patients in the Indian subcontinent who live below the poverty line. Poor nutrition also contributes to calcium and vitamin D deficiency, which further exacerbates renal osteodystrophy associated with hyperphosphatemia and secondary hyperparathyroidism. Vitamin D deficiency is ubiquitous in India, with levels approaching 70%. The true prevalence of mineral bone disease in India is unknown, and the only data available come from small cross-sectional studies. Larger population-based studies are needed, and the Indian CKD Registry should provide conclusive population-based answers in the future.
Physiology of phosphate metabolism in ESRD
About 85% of the 600 g of total body phosphorus is present in bone.9 The remainder exists as a major intracellular anion and as a component of proteins, enzymes, intermediates in carbohydrate and lipid metabolism, nucleic acids, and high energy stores like adenosine triphosphate and nucleic acids.10 Unlike calcium, which has a much higher concentration intracellularly than in the serum, phosphorus exists intracellularly at concentrations close to that of the extracellular fluid (around 1–2 mmol/L). Normal serum phosphorus ranges in adults from 0.75 mmol/L to 1.45 mmol/L (2.5–4.5 mg/dL). Since the volume of intracellular fluid is about twice that of extracellular fluid, measurement of serum phosphorus may not accurately reflect total phosphate stores and phosphate availability to cells. Phosphorus is absorbed efficiently in the small intestine even in the absence of vitamin D (65%), but this efficiency is increased to 85%–90% via 1,25-dihydroxy vitamin D-induced active transport.11,12 The total amount of phosphate absorbed daily is therefore highly dependent on dietary phosphate intake and absorption, and therefore the majority of drugs for hyperphosphatemia work by binding phosphate in the gut and preventing its passive and active absorption.
The physiology of phosphorus metabolism is intricately linked with calcium homeostasis. Parathyroid hormone (PTH) governs the primary hormonal regulation of serum calcium and phosphorus levels and maintains them in a very tight range. Calcium exists in both a bound form to albumin (about 45%) and phosphate (about 10%), and a free or ionized form (50%) that is physiologically active.13 PTH regulation of phosphorus and calcium allows this physiologically active ionized calcium to remain regulated within a tight range in normal physiology. Changes in ionized calcium of even 0.1 mg/dL lead to changes in PTH secretion to compensate. PTH’s primary site of action for phosphate metabolism is in the proximal tubule where it inhibits phosphate reabsorption leading to increased urinary phosphate excretion. PTH also regulates increased calcium reabsorption of the 10% of the filtered load of calcium delivered to the distal convoluted tubule.14 PTH also helps stimulate bone turnover by activating osteoclasts and causing release of calcium and phosphorus from the mineral component of bone. Through actions on the kidney, it also activates production of 1-alpha-hydroxylase, leading to increased 1,25-dihydroxy vitamin D. This 1,25-dihydroxy vitamin D activates both phosphorus and calcium reabsorption in the intestine, and thus PTH indirectly stimulates both calcium and phosphorus reabsorption. Through the aforementioned effects on the kidney, there is a preferential increase in ionized calcium through urinary excretion of phosphate and reabsorption of calcium in the kidney. This helps control the increase in phosphorus related bone breakdown by osteoclasts and reabsorption in the intestine. This homeostatic action is lost in patients with advanced CKD and ESRD.
In addition to the effects of PTH, there is additional PTH-independent regulation of phosphate levels. In response to a phosphate load, urinary excretion of phosphate increases within hours preceding any change in serum phosphorus concentrations. This led to the nearly decade-long search for the so-called phosphatonins – substances leading to phosphorus excretion independent of PTH. Fibroblast growth factor 23 (FGF23) is widely considered to be the long-sought-after phosphatonin. It works to lower phosphorus both by inducing phosphaturia (by independently lowering the tubular maximum of filtered phosphate in the proximal tubule) and also by decreasing 1,25(OH)2D levels.15 FGF23 requires klotho, a transmembrane protein, for its binding to FGF receptors. Elevation of FGF23 is associated with certain rare congenital causes of rickets such as X-linked and autosomal dominant vitamin D-resistant rickets and the acquired tumor-induced or oncogenic osteomalacia.16 Accumulating evidence suggests that FGF23 is associated with increasing mortality and left ventricular hypertrophy in dialysis patients. FGF23 has also been associated with adverse outcomes in patients with CKD and may be a novel biomarker for adverse outcomes in early-stage disease.17–19
With progressive CKD, there is loss of the 1-alpha-hydroxylase activity in the proximal tubule, leading to a decrease in the activated 1,25-dihydroxy form of vitamin D.20 This decrease in 1,25(OH)2 vitamin D decreases calcium absorption in the intestine, and also has a direct effect on the parathyroid gland to stimulate PTH through loss of negative feedback (through a direct vitamin D receptor on the parathyroid gland).21 Metabolic acidosis from renal disease also exacerbates hyperphosphatemia through efflux of phosphates from the intracellular to extracellular compartment to maintain ionic equilibrium. The decrease in ionized calcium resulting from decreased phosphate clearance in the kidney and hyperphosphatemia also results in an appropriate increase in PTH to counteract these changes and restore homeostasis. This secondary hyperparathyroidism is compensatory initially, but prolonged elevation of PTH has been associated with adverse outcomes in patients with ESRD, leading to metastatic calcification and increased cardiovascular mortality.22,23 There is also a chronic effect on bone density, and chronic osteoclastic activation leads to osteolysis and fibrosis. This bone disease resulting from secondary hyperparathyroidism is one form of renal osteodystrophy and greatly increases the risk of fractures.24
Adverse effects of hyperphosphatemia in ESRD
The symptoms of hyperphosphatemia in renal failure tend to be related to the rapidity at which they develop. Patients with acute hyperphosphatemia from release of large intracellular stores such as in rhabdomyolysis and tumor lysis syndrome become dramatically symptomatic. The hyperphosphatemia-induced decrease in ionized calcium results in tetany, hypotension, seizures, and arrhythmias. The high phosphate levels in the acute setting can also lead to metastatic calcification in soft tissues, including the kidney, and can contribute to renal failure.25
In contrast, the chronic hyperphosphatemia of ESRD tends to be much less symptomatic, and patients primarily present with the consequences of hyperparathyroidism and heterotopic calcification. Hyperparathyroidism is associated with bone pain, fractures, brown tumors, and erythropoietin resistance from bone marrow fibrosis.26 Accumulating evidence suggests that PTH by itself is likely a uremic toxin contributing to cardiac muscle fibrosis, muscle weakness, and constitutional symptoms.27 The other important complication of hyperphosphatemia is calcification of coronary and other vasculature by deposition of calcium phosphate products. In patients with CKD or ESRD, soft tissue or metastatic calcification occurs when the product of the concentrations of serum calcium and phosphorus exceeds 6.5 mg/dL, but it can occur with lower levels. The more important determinant appears to be the serum phosphorus as opposed to calcium levels, and the deposition of crystals seems to be linked most closely to an increase in the local concentration of phosphates.28,29 The devastating complication of calciphylaxis was long considered to be related to abnormalities in calcium and phosphate metabolism. It leads to pathological changes of calcific uremic arteriolopathy with patches of ischemic necrosis in fat-rich areas. However, it is being increasingly reported in the absence of hyperphosphatemia and significant hyperparathyroidism, and thus its association to high phosphate levels is unclear at this time.30
Epidemiological evidence suggests a strong association between elevated phosphate levels and cardiovascular mortality in patients with stage five CKD and even in earlier stages, with an incremental risk ranging from ten- to 200-fold.31,32 The exact reason for the excessive mortality is unclear, but one hypothesized mechanism is related to hyperphosphatemia-induced calcification of the tunica media of coronary arteries and even heart valves.33 The serum calcium phosphorus product is widely followed as a marker for risk of metastatic calcification. Risk factors for vascular calcification apart from hyperphosphatemia include age, low PTH levels, and low bone turnover. It is possible that with low uptake of calcium in bone from adynamic bone disease, there is increased calcium uptake in extraosseous sites like the vascular bed.34 Hyperphosphatemia by itself can also independently induce changes in gene expression in vascular cells to an osteoblast profile, further accelerating vascular calcification.35 This pathophysiology led to enthusiasm toward treating high phosphate levels in patients with CKD and ESRD in the hope that cardiovascular mortality could be reduced. It remains unclear if hyperphosphatemia is simply a marker of more advanced kidney disease or if it truly is independently causative of adverse cardiovascular events in patients with CKD and ESRD. Overall, coronary disease in patients with ESRD has fundamental differences from the general population, and randomized trials have failed to show a cardiovascular benefit to statin therapy or even to alteration of bone mineral disease with the calcimimetic cinacalcet (EVOLVE study), vitamin D (PRIMO study), or calcium- and noncalcium-based phosphate binders.36,37 These data support the notion that the medial calcification form of coronary artery disease seen in CKD does not respond to the typically accepted therapies for the intimal form.
However, on the basis of retrospective and nonrandomized cohorts of patients demonstrating increased mortality with higher phosphate levels, the National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF KDOQI) and Kidney Disease Improving Global Outcomes (KDIGO) guidelines do recommend treatment of hyperphosphatemia.38 Phosphate binders are the primary treatment tool for hyperphosphatemia, but there is some evidence that these drugs may actually accelerate vascular calcification, despite their lowering of serum phosphorus and attenuation of secondary hyperparathyroidism.37,39 This requires confirmation in additional larger studies but is an interesting observation nonetheless, particularly in the absence of high-quality randomized clinical trials proving that treating hyperphosphatemia actually improves hard clinical end points. Retrospective registry studies and nonrandomized trials have suggested benefit from phosphate binders, but due to concern over the ethics of not treating the control group’s hyperphosphatemia, there has been no large randomized trial to date. This is an area that requires resolution given the extreme pill burden and cost of phosphate-binding drugs superimposed on the already intense physical and emotional expectations of patients on dialysis in developing countries.
Contemporary management strategies of hyperphosphatemia
Dietary restriction of phosphorus
Serum concentrations of phosphorus are typically not elevated in patients with CKD stage two and three, but are clearly increased when CKD stage four or five was reached.40,41 It has been shown that dietary phosphorus is an important determinant of secondary hyperparathyroidism even in mild and moderate renal insufficiency (CKD stage two and three) in spite of normal serum phosphorus concentrations. Hence, dietary phosphorus restriction becomes crucial even in early stages of CKD.42 Dietary intake of phosphorus is largely derived from foods with a high protein content and additives. Because proteins and phosphorus are closely related in foods, it becomes difficult to restrict phosphorus without restricting proteins. This complicates the management, as earlier studies showed poor quality of life and increased mortality with protein restriction in patients with advanced CKD (stage four or five).43
Besides absolute phosphorus, it is also essential to understand the type of dietary phosphorus intake (organic vs inorganic) and source (animal vs plant), as it has implications in gastrointestinal absorption by altering the bioavailability of phosphorus. Organic phosphorus is mostly bound in vivo to proteins and hence is found in food that is rich in proteins (animal and plant sources).44 Organic protein is hydrolyzed in the intestinal tract, and close to 60% of it is absorbed. There is considerable variation in absorption based on the source of phosphorus. Unlike phosphorus in meat, which is present as organic phosphates that are readily absorbed, phosphorus in plants is mostly in the form of phytic acid. Humans do not express the degrading enzyme phytase, and hence the bioavailability of phosphorus from plant-derived food is relatively low, usually <50%. Therefore, plant foods are less likely to increase phosphorous burden when compared with that of animal foods.45 Unlike organic phosphates, inorganic phosphates such as additives are not bound to protein, as a result of which they are easily dissociated, and it is estimated that >90% of it is effectively absorbed. On average, phosphorus additives contribute close to 1,000 mg/day to the average American diet. Some of the food items that have high amounts of phosphate additives are soft drinks and cheese.46,47
According to the Food and Nutrition Board of the Institute of Medicine, the recommended dietary reference intake (DRI) is 700 mg/day in healthy adults and 1,200 mg/day in older children and pregnant women. Since dietary restrictions of phosphate can reduce the severity of hyperparathyroidism, the KDOQI guidelines recommend phosphorus intake to be decreased to DRI in patients with early stages of CKD (stage two and three) even with normal serum phosphorus concentrations and to 80% of DRI in CKD stages four and five.48
Drugs
Phosphate binders used in the management of hyperphosphatemia are categorized as calcium-containing phosphate binders and noncalcium-containing phosphate binders. Phosphate binders should be initiated when serum phosphorus levels are above target range despite dietary restriction or if dietary restriction precludes intake of critically important nutrients like proteins. Though there is evidence that serum phosphorus levels >5.5 mg/dL are associated with increased mortality, as mentioned, there is no prospective randomized evidence proving that maintaining phosphorus levels within this target range is associated with improved outcomes. These target ranges are part of the KDOQI guidelines and are opinion based, as shown in Table 1.38
 | Table 1 Target phosphorous levels in patients with chronic kidney disease (CKD) |
There has been no clear consensus on the choice of phosphate binders to be used for patients with CKD. In general, we suggest that patients with low serum calcium concentrations can be initiated on calcium-containing phosphate binders, and patients with hypercalcemia or normocalcemia on vitamin D replacement therapy should be started on noncalcium-containing phosphate binders (Figure 1).
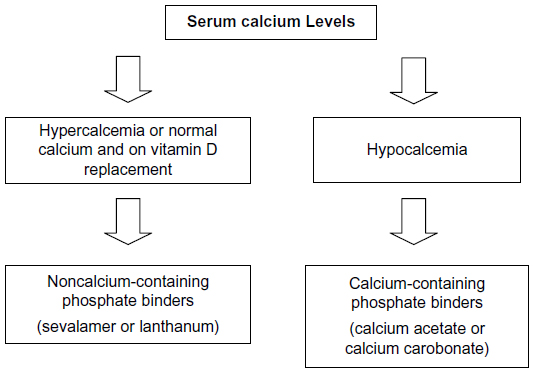 | Figure 1 Algorithm for choice of phosphate binder. |
Calcium-containing phosphate binders
The first phosphate binder that was approved in CKD patients was aluminum hydroxide. But after many years, aluminum intoxication was reported and was thought to be due to the gradual accumulation of aluminium in the tissue, which was poorly cleared by the diseased kidney. This led to the development of calcium salt-containing phosphate binders.49,50 The calcium salts that are used in current clinical practice are calcium acetate and calcium carbonate. Calcium acetate is preferred, as calcium carbonate dissolves only at acidic pH, and many patients with CKD have achlorhydria. Hypercalcemia is a common complication of calcium salts, especially when given with vitamin D supplements. Another known complication is that of coronary arterial calcification when intake exceeds 1.5 g/day of elemental calcium. Due to these complications, serum calcium should be carefully monitored when these drugs are used.51 The 2003 KDOQI guidelines also suggest that the dialysate calcium concentration in hemodialysis or peritoneal dialysis should be 2.5 mEq/L (1.25 mmol/L).38
Noncalcium phosphate binders
Sevelamer (hydrochloride and carbonate) and lanthanum are the two noncalcium phosphate binders that are currently available. There are a number of trials that have shown that sevelamer is effective in reducing serum phosphorus levels without altering calcium concentrations. This drug is usually well tolerated, but studies have shown an increased incidence of metabolic acidosis with sevelamer hydrochloride. Though sevelamer has been effective in reducing serum phosphorus levels, the data on reducing mortality and vascular calcification have been inconsistent.52 The drug, however, is more expensive, and this is a significant factor in a country like India, where cost is a major deciding factor in patient management and often dictates compliance. For patients in India, we often lean more in favor of starting patients on calcium-containing phosphate binders unless hypercalcemia is a serious issue.
Lanthanum is a rare earth element and has been shown to significantly reduce phosphate levels. One advantage of lanthanum is that it can potentially increase compliance by reducing total pill burden. There have been no significant adverse effects reported with lanthanum.
Newer drugs
Sucroferric oxyhydroxide is a chewable phosphate binder that was found to have similar efficacy to standard phosphate binders and significantly decreases the pill burden to almost one-third of the typical number of pills. It has received approval from the US Food and Drug Administration for treatment of hyperphosphatemia in CKD stage five and may be an important option in the future once costs decrease and long-term data are available.53
CKD mineral bone disease in India: what is new?
There are a paucity of data on mineral bone disease in maintenance hemodialysis patients from India. A retrospective analysis was undertaken on 858 patients (males:599; females:259) from two medical centers on maintenance hemodialysis from 1998 to 2010.54 Overall, 191 patients died (22%) during the observation period. There was an 86% patient survival rate at 1 year on dialysis and an overall predicted 3-year survival rate of 78% (Figure 2). A relatively higher intact parathyroid hormone (P=0.012), a need for vitamin D supplementation (P=0.003), fewer hours on dialysis per session (P=0.046), and a nonvegetarian diet (P=0.022) were significantly associated with mortality.
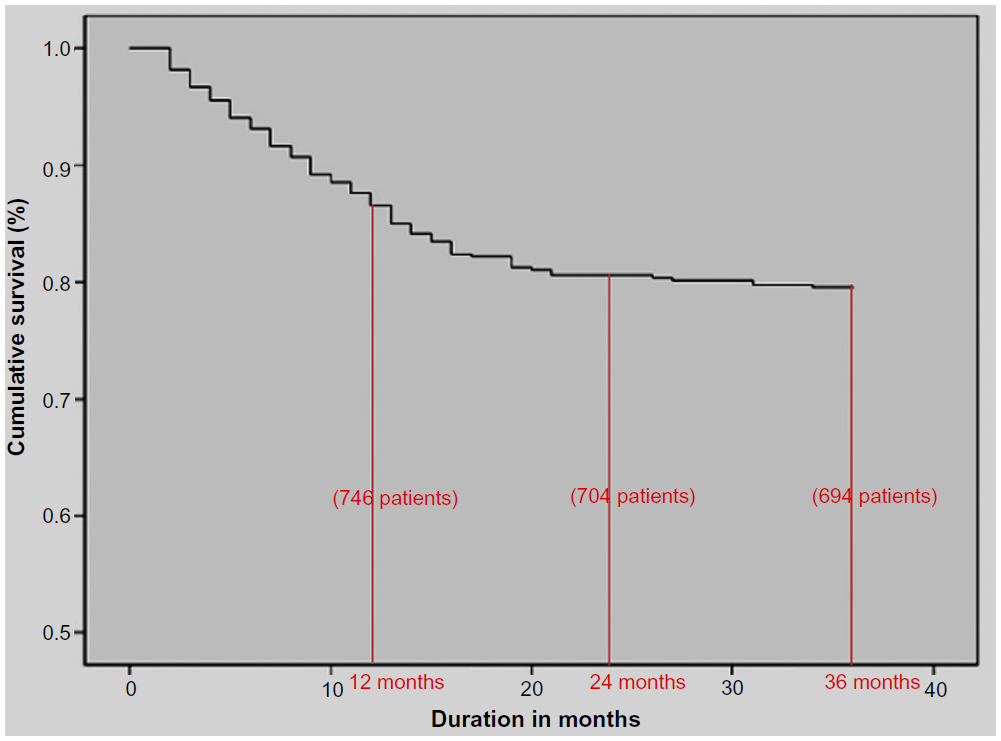 | Figure 2 Patient survival on hemodialysis among a retrospective cohort from South India. |
A randomized multicenter clinical trial was done to compare the efficacy and tolerability of two sevelamer formulations, sevelamer carbonate and sevelamer hydrochloride, in the treatment of hyperphosphatemia in Indian ESRD patients.55 A total of 97 ESRD patients on hemodialysis were enrolled and randomized to receive either sevelamer carbonate or sevelamer hydrochloride. All patients were evaluated weekly over 6 weeks for efficacy and safety variables, and there were similar reductions in mean serum phosphorus and the calcium phosphorus product in both the groups (P<0.001). There was no significant difference between the groups for adverse events and the laboratory parameters. Previous studies have shown that both are largely equivalent in terms of phosphate-lowering effects, but that sevelamer hydrochloride can induce metabolic acidosis, which is not seen with sevelamer carbonate.56 Our study did show that sevelamer carbonate can be safely used in the Indian ESRD population.
Another study looked at the abdominal aortic calcification (AAC) score in predicting cardiac valvular calcification (CVC) and atherosclerotic carotid plaque (CP) in ESRD patients in India.57 AAC, CVC, and atherosclerotic CP are known cardiovascular risk factors and are strongly linked to hyperphosphatemia. Twenty-two existing dialysis patients (group 1) and 26 consecutive nondialysis stage 5 CKD patients (group 2) were assessed for their demographic and laboratory variables. A lateral radiograph of the lumbosacral spine was used to assess the AAC score. CP and CVC were assessed using carotid sonography and echocardiogram, respectively. Prevalence of AAC, CP, and CVC in groups 1 and 2 was, respectively, 72.7%, 81.8%, and 72.7% and 76.9%, 80.8%, and 57.7%. AAC was strongly associated with CP and CVC in both groups (P<0.001). Tests of accuracy for the AAC score as a predictor of CP and CVC showed moderate accuracy with regards to sensitivity, specificity, and positive and negative predictive value. The AAC score can thus predict CP and CVC with moderate accuracy in Indian ESRD patients and may hold promise as a low-cost intervention for risk stratification. However, as the study was underpowered, the findings need validation in larger, adequately powered studies.
The Indian perspective: focus on cost and drug adherence
As mentioned earlier, financial factors play a major role in decision making in patients with ESRD in India. The pharmaceutical industry has taken great initiative in locally manufacturing and lowering the price of phosphate binders, vitamin D analogs, and the calcimimetic cinacalcet. The price of these drugs in the Indian market is listed in Table 2 and is significantly lower than the price in the US. This lower price has helped improve compliance, but adherence to the intensive pill burden from phosphate-lowering drugs remains far from ideal. Calcium acetate and sevelamer need to be taken three times a day and may include two to three pills per dose. This complex regimen further complicates drug adherence in patients who may not have extensive understanding of the dangers of their medical condition and asymptomatic hyperphosphatemia. Many patients living below the poverty line still have difficulty affording the subsidized rates on these drugs, and this remains a great challenge in providing care for ESRD patients in India.
 | Table 2 Cost of phosphate binders in India |
More so than in other parts of the developed world, hyperphosphatemia in ESRD continues to remain a challenging problem in India. Lack of understanding of the dangers of asymptomatic hyperphosphatemia, the huge pill burden of phosphate binders, difficulty with dietary and dialysis compliance, and, most importantly, the added expense of the drugs place additional road blocks in the treatment of hyperphosphatemia at a population level. The majority of patients live in villages and have difficulty accessing health care. The grossly inadequate number of nephrologists in India places an added burden on primary care providers to manage CKD risk factors, and hyperphosphatemia often gets less priority than more obvious and easily treatable risk factors such as hypertension and diabetes management. The recommendation to treat hyperphosphatemia, however, remains largely opinion based given the lack of high-quality randomized controlled trial data. The field is in desperate need of a definitive placebo-controlled randomized trial to determine if lowering high phosphate levels in ESRD patients improves morbidity, cardiovascular outcomes, and mortality. This will greatly help determine whether we should be allocating as many resources to management and control of this problem in developing countries. In the absence of evidence to the contrary, and given the lack of definitive harm with lowering phosphate levels, we should continue to aggressively manage and counsel our patients regarding this difficult to control problem.
Disclosure
The authors report no conflicts of interest in this work.
References
Jha V. Current status of end-stage renal disease care in India and Pakistan. Kidney Int Suppl. 2013;3:157–160. | |
Gupta R, Sankhe S, Dobbs R, Woetzel J, Madgavkar A, Hasyagar A. India’s path from poverty to empowerment [webpage on the Internet]. New York, NY: McKinsey Global Institute. Available from: http://www.mckinsey.com/insights/asia-pacific/indias_path_from_poverty_to_empowerment. Accessed September 9, 2014. | |
Modi G, Jha V. Incidence of ESRD in India. Kidney Int. 2011;79:573. | |
Rajapurkar MM, John GT, Kirpalani AL, et al. What do we know about chronic kidney disease in India: first report of the Indian CKD Registry. BMC Nephrol. 2012;13:10. | |
Jeloka TK, Upase S, Chitikeshi S. Monthly cost of three sessions a day peritoneal dialysis is same as of thrice a week hemodialysis in self-paying Indian patients. Indian J Nephrol. 2012;22:39–41. | |
kizler TA. Dietary protein restriction in CKD: the debate continues. Am J Kidney Dis. 2009;53(2):189–191. | |
Menon V, Kopple JD, Wang X, et al. Effect of a very low-protein diet on outcomes: long-term follow-up of the Modification of Diet in Renal Disease (MDRD) Study. Am J Kidney Dis. 2009;53(2):208–217. | |
Levey AS, Greene T, Sarnak MJ, et al. Effect of dietary protein restriction on the progression of kidney disease: long-term follow-up of the Modification of Diet in Renal Disease (MDRD) Study. Am J Kidney Dis. 2006;48(6):879–888. | |
Wagner CA. Novel insights into the regulation of systemic phosphate homeostasis and renal phosphate excretion. J Nephrol. 2007;20:130–134. | |
Golub EE. Biomineralization and matrix vesicles in biology and pathology. Semin Immunopathol. 2011;33:409–417. | |
Murer H, Forster I, Biber J. The sodium phosphate cotransporter family SLC34. Pflugers Arch. 2004;447:763–767. | |
Marks J, Debnam ES, Unwin RJ. The role of the gastrointestinal tract in phosphate homeostasis in health and chronic kidney disease. Curr Opin Nephrol Hypertens. 2013;22(4):481–487. | |
Michigami T. Regulation and disorders of calcium and phosphate metabolism. Clin Calcium. 2014;24(2):169–175. | |
Manghat P, Sodi R, Swaminathan R. Phosphate homeostasis and disorders. Ann Clin Biochem. Epub February 28, 2014. | |
Wahl P, Wolf M. FGF23 in chronic kidney disease. Adv Exp Med Biol. 2012;728:107–125. | |
Pavone V, Testa G, Gioitta Iachino S, et al. Hypophosphatemic rickets: etiology, clinical features and treatment. Eur J Orthop Surg Traumatol. Epub June 24, 2014. | |
Kestenbaum B, Sachs MC, Hoofnagle AN, et al. Fibroblast growth factor-23 and cardiovascular disease in the general population: the Multi-Ethnic Study of Atherosclerosis. Circ Heart Fail. 2014;7(3):409–417. | |
Quarles D. Reducing cardiovascular mortality in chronic kidney disease: something borrowed, something new. J Clin Invest. 2013;123(2):542–543. | |
Fliser D, Kollerits B, Neyer U, et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J Am Soc Nephrol. 2007;18(9):2600–2608. | |
Saliba W, El-Haddad B. Secondary hyperparathyroidism: pathophysiology and treatment. J Am Board Fam Med. 2009;22(5):574–581. | |
Hruska KA, Mathew S, Lund R, et al. Hyperphosphatemia of chronic kidney disease. Kidney Int. 2008;74(2):148–157. | |
Lishmanov A, Dorairajan S, Pak Y, et al. Elevated serum parathyroid hormone is a cardiovascular risk factor in moderate chronic kidney disease. Int Urol Nephrol. 2012;44(2):541–547. | |
Eddington H, Kalra PA. The association of chronic kidney disease-mineral bone disorder and cardiovascular risk. J Ren Care. 2010; 36 Suppl 1:61–67. | |
Sprague SM. Renal bone disease. Curr Opin Endocrinol Diabetes Obes. 2010;17(6):535–539. | |
Alfrey AC. The role of abnormal phosphorus metabolism in the progression of chronic kidney disease and metastatic calcification. Kidney Int Suppl. 2004;90:S13–S17. | |
Brancaccio D, Cozzolino M, Gallieni M. Hyperparathyroidism and anemia in uremic subjects: a combined therapeutic approach. J Am Soc Nephrol. 2004;15 Suppl 1:S21–S24. | |
Rodriguez M, Lorenzo V. Parathyroid hormone, a uremic toxin. Semin Dial. 2009;22(4):363–368. | |
Cozzolino M, Dusso AS, Slatopolsky E. Role of calcium-phosphate product and bone-associated proteins on vascular calcification in renal failure. J Am Soc Nephrol. 2001;12:2511–2516. | |
Hujairi NM, Afzali B, Goldsmith DJ. Cardiac calcification in renal patients: what we do and don’t know. Am J Kidney Dis. 2004;43:234–243. | |
Brandenburg VM, Sinha S, Specht P, Ketteler M. Calcific uraemic arteriolopathy: a rare disease with a potentially high impact on chronic kidney disease-mineral and bone disorder. Pediatr Nephrol. Epub January 29, 2014. | |
Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of serum phosphorus and calcium phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis. 1998;31:607–617. | |
Ganesh SK, Stack AG, Levin NW, Hulbert-Shearon T, Port FK. Association of elevated serum PO(4), Ca PO(4) product, and parathyroid hormone with cardiac mortality risk in chronic hemodialysis patients. J Am Soc Nephrol. 2001;12:2131–2138. | |
Rufino M, García S, Jiménez A, et al. Heart valve calcification and calcium x phosphorus product in hemodialysis patients: analysis of optimum values for its prevention. Kidney Int Suppl. 2003;85:S115–S118. | |
Cozzolino M, Brancaccio D, Gallieni M, Slatopolsky E. Pathogenesis of vascular calcification in chronic kidney disease. Kidney Int. 2005; 68(2):429–436. | |
Jakoby MG 4th, Semenkovich CF. The role of osteoprogenitors in vascular calcification. Curr Opin Nephrol Hypertens. 2000;9(1):11–15. | |
Thadhani R, Appelbaum E, Pritchett Y, et al. Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: the PRIMO randomized controlled trial. JAMA. 2012;307(7):674–684. | |
Block GA, Wheeler DC, Persky MS, et al. Effects of phosphate binders in moderate CKD. J Am Soc Nephrol. 2012;23(8):1407–1415. | |
National Kidney Foundation. K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis. 2003;42(4 Suppl 3):S1. | |
Russo D, Miranda I, Ruocco C, et al. The progression of coronary artery calcification in pre-dialysis patients on calcium carbonate or sevelamer. Kidney Int. 2007;72:1255–1261. | |
Malluche HH, Ritz E, Lange HP, et al. Bone histology in incipient and advanced renal failure. Kidney Int. 1976;9:355–362. | |
Coburn JW, Koppel MH, Brickman AS, Massry SG. Study of intestinal absorption of calcium in patients with renal failure. Kidney Int. 1973;3:264–272. | |
Burnett S-A, Gunawardene S, Bringhurst FR, Juppner H, Lee H, Finkelstein JS. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res. 2006;21:1187–1196. | |
Collins AJ, Kasiske B, Herzog C, et al. Experts from the United States Renal Data System 2004 annual data report: atlas of end stage renal disease in the United States. Am J Kidney Dis. 2005;45:S1–S280. | |
Ramirez JA, Emmett M, White MG, et al. The absorption of dietary phosphorus and calcium in hemodialysis patients. Kidney Int. 1986;30:753–759. | |
Lei XG, Porres JM. Phytase enzymology, applications, and biotechnology. Biotechnol Lett. 2003;25:1787–1794. | |
Sullivan C, Sayre SS, Leon JB, et al. Effect of food additives on hyperphosphatemia among patients with end-stage renal disease: a randomized controlled trial. JAMA. 2009;301:629–635. | |
Sullivan CM, Leon JB, Sehgal AR. Phosphorus-containing food additives and the accuracy of nutrient databases: implications for renal patients. J Ren Nutr. 2007;17:350–354. | |
Food and Nutrition Board: Phosphorus. In: Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Institute of Medicine, editor. Washington, DC: National Academy Press; 1997:146–189. | |
Alfrey AC, Legendre GR, Kaehny WD. Dialysis encephalopathy syndrome: possible aluminum intoxications. N Engl J Med. 1976;294:184–188. | |
Hewitt CD, Savory J, Wills MR. Aspects of aluminum toxicity. Clin Lab Med. 1990;10:403–422. | |
Emmett M. A comparison of clinically useful phosphorus binders for patients with chronic kidney failure. Kidney Int Suppl. 2004;90:S25–S32. | |
Jamal SA, Vandermeer B, Raggi P, et al. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: an updated systematic review and meta-analysis. Lancet. 2013;382:1268. | |
Floege F, Covic AC, Ketteler M, et al. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int. 2014;86:638–647. | |
Reddy Y, Abraham G, Reddy Y, et al. Mineral bone disease in maintenance hemodialysis patients: association with morbidity and mortality. Indian J Nephrol. Epub cited July 13, 2014. Available from: http://www.indianjnephrol.org/preprintarticle.asp?id=132988. Accessed September 9, 2104. | |
Abraham G, Kher V, Saxena S, et al. Sevelamer carbonate experience in Indian end stage renal disease patients. Indian J Nephrol. 2012;22(3):189–192. | |
Pai AB, Shepler BM. Comparison of sevelamer hydrochloride and sevelamer carbonate: risk of metabolic acidosis and clinical implications. Pharmacotherapy. 2009;29(5):554–561. | |
Shantha GP, Kumar AA, Mancha A, et al. Is abdominal aortic calcification score a cost-effective screening tool to predict atherosclerotic carotid plaque and cardiac valvular calcification in patients with end-stage renal disease? Indian J Nephrol. 2012;22(6):431–437. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.