")
Back to Journals » Journal of Blood Medicine » Volume 5
Optimal management of hereditary hemorrhagic telangiectasia
Authors Garg N, Khunger M, Gupta A, Kumar N
Received 31 July 2014
Accepted for publication 1 September 2014
Published 15 October 2014 Volume 2014:5 Pages 191—206
DOI https://doi.org/10.2147/JBM.S45295
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Martin H Bluth
Neetika Garg,1 Monica Khunger,2 Arjun Gupta,3 Nilay Kumar4
1Department of Medicine, Beth Israel Deaconess Medical Center, Boston, MA, USA; 2Department of Medicine, All India Institute of Medical Sciences, New Delhi, India; 3Department of Medicine, UT Southwestern Medical Center, Dallas, TX, USA; 4Department of Medicine, Cambridge Health Alliance/Harvard Medical School, Cambridge, MA, USA
Abstract: Hereditary hemorrhagic telangiectasia (HHT), also known by the eponym Osler–Weber–Rendu syndrome, is a group of related disorders inherited in an autosomal dominant fashion and characterized by the development of arteriovenous malformations (AVM) in the skin, mucous membranes, and/or internal organs such as brain, lungs, and liver. Its prevalence is currently estimated at one in 5,000 to 8,000. Most cases are due to mutations in the endoglin (HHT1) or ACVRLK1 (HHT2) genes. Telangiectasias in nasal and gastrointestinal mucosa generally present with recurrent/chronic bleeding and iron deficiency anemia. Larger AVMs occur in lungs (~40%–60% of affected individuals), liver (~40%–70%), brain (~10%), and spine (~1%). Due to the devastating and potentially fatal complications of some of these lesions (for example, strokes and brain abscesses with pulmonary AVMs), presymptomatic screening and treatment are of utmost importance. However, due to the rarity of this condition, many providers lack an appreciation for the whole gamut of its manifestations and complications, age-dependent penetrance, and marked intrafamilial variation. As a result, HHT remains frequently underdiagnosed and many families do not receive the appropriate screening and treatments. This article provides an overview of the clinical features of HHT, discusses the clinical and genetic diagnostic strategies, and presents an up-to-date review of literature and detailed considerations regarding screening for visceral AVMs, preventive modalities, and treatment options.
Keywords: arteriovenous malformations, epistaxis, bevacizumab, embolization, guidelines, screening, review
Introduction
Hereditary hemorrhagic telangiectasia (HHT), also known by the eponym Osler–Weber–Rendu disease is a group of autosomal dominant disorders characterized by presence of multiple arteriovenous malformations (AVMs) in the skin, mucous membranes, and frequently internal organs such as lungs, liver, and brain. Current prevalence is estimated between one in 5,000 to 8,000. While this is thought to affect people of all races in all parts of the world, higher prevalence has been reported in specific populations, for example, one in 1,330 in Afro-Caribbean residents of Curaçao and Bonaire.1–3
AVMs in HHT are characterized by lack of intervening capillaries between arteries and veins. Small AVMs known as telangiectasias are pink to red lesions, typically 0.5–1.0 mm in diameter. They may occur anywhere in the body but are most apparent on the lips, tongue, face, fingertips, and the nasal, buccal, and gastrointestinal (GI) mucosa. Unlike petechiae, which have a similar appearance, they blanch with pressure and refill immediately after the pressure is released. Due to their thin walls and proximity to the skin or mucosal surfaces, they are prone to rupture and bleeding with minimal injury. Epistaxis and mucocutaneous telangiectasias are the most common clinical manifestations.3,4 HHT is also often complicated by the presence of larger AVMs, also referred to as arteriovenous shunts or fistulae. These can be up to several centimeters in size and are most frequently observed in the lungs, liver, and brain. While these can rupture, complications from these larger AVMs are more often a result of shunting of blood. Involvement of systemic vasculature, for example in the liver, leads to a high cardiac output state. Pulmonary AVMs (PAVMs) serve as right-to-left shunts that lead to hypoxemia or neurologic sequelae due to paradoxical embolism of cruoric or septic thrombi. It should be noted that these vascular malformations can occur in disorders besides HHT, in isolation, and as part of another syndrome, and their presence is not adequate to arrive at this diagnosis.
Since HHT affects multiple organ systems, it does not “belong to” any one subspecialty; a wide range of physicians from medical, surgical, primary care, or emergency medicine disciplines may encounter affected patients. Given the rarity of this condition, physicians may lack appreciation of its full range of manifestations and complications, resulting in missed diagnoses or significant delays before the diagnosis is established.5,6 Furthermore, due to the catastrophic nature of some of the lesions seen in this syndrome (for example, strokes and cerebral abscesses from pulmonary AVMs), screening in the presymptomatic phase is extremely important. However, many patients and families remain unaware of their diagnosis and do not receive appropriate screening and preventive treatment.7 To bridge these gaps in care for patients with HHT and their families, a group of international experts met in Canada in 2006 and formulated the “International Guidelines for Diagnosis and Management of HHT” that were published in 2011.8
Here, we provide a brief overview of the clinical manifestations in HHT, an up-to-date review of diagnostic criteria for HHT, guidelines regarding screening in asymptomatic subjects with definite or likely HHT, and optimal management of various complications. We will also discuss the literature that has become available since October 2006, when the guidelines for HHT were initially formulated. In particular, there has been much enthusiasm around the use of the vascular endothelial growth factor (VEGF) antagonist bevacizumab; it has been shown to decrease severity of epistaxis as well as reduce liver size and cardiac output in patients with large hepatic AVMs (HAVMs). Lastly, we will discuss the implications of this diagnosis in special circumstances, such as pregnancy and when anticoagulation is required.
Clinical overview
Age-related penetrance is observed in HHT. Manifestations are rarely present at birth, but develop and worsen with increasing age. Individual signs and symptoms are discussed below.
Epistaxis
Nasal bleeding is the clinical characteristic that most commonly brings patients with HHT to medical attention. In a series of 324 patients with HHT, epistaxis was reported by 96% with over half experiencing this symptom before the age of 20 years.4 In another survey of 73 patients, 93% reported epistaxis, with onset before the age of 10 years in over 50% and before the age of 20 years in over 90%; the mean frequency of nosebleeds was 18 per month.9 It is an early and clinically evident marker of disease. Occasionally, it can lead to chronic anemia and transfusion dependence.
Mucocutaneous telangiectasias
Telangiectasias typically seen on the lips, tongue, buccal mucosa, and fingertips have been reported in 75%–90% of the affected individuals.4,10 They are usually recognized at a much later age than epistaxis, likely due to the relatively subtle and nonspecific nature of these lesions. They rarely bleed and the main concern is usually cosmetic. There are rare case reports of vocal cord involvement manifesting as dysphonia.11
Gastrointestinal bleeding
Although 75% of the patients have gastric or small intestinal telangiectasias on endoscopy or capsule examination, only one-third eventually suffer from GI bleeding, usually after 40 years of age.4,12–14 Telangiectasias can occur throughout the GI tract, but are almost always more common in the stomach and duodenum. Bleeding is often slow and chronic, and may be clinically silent with no obvious melena. Symptoms may be completely absent until anemia develops. Larger AVMs or aneurysms are less commonly observed.
Pulmonary AVMs
PAVMs are more common in the general population than previously appreciated. Small pulmonary shunts are evident on transthoracic contrast echocardiography (TTCE) in 6%–7% of the individuals without HHT.15,16 In a recent study from Japan, PAVMs were identified in eight patients (0.038%) among 21,235 patients who underwent low-dose computed tomography (CT) screening for lung cancer.17 While only two patients were diagnosed with HHT in this series, other studies have shown that approximately 80%–90% of the patients with PAVMs have underlying HHT.18,19 On the other hand, depending on the imaging technique used, PAVMs are identified in 40%–60% of patients with an underlying diagnosis of HHT.20,21
PAVMs can lead to symptoms by causing complications such as systemic hypoxemia, pulmonary embolism, high output heart failure, or pulmonary hypertension. Symptoms may also manifest as a result of PAVM rupture. Dyspnea (that is sometimes noticeable only with exertion) is the most common symptom attributable to hypoxemia resulting from right-to-left shunting of blood, especially if the PAVMs are large, multiple, or bilateral.22,23 This, over time may lead to clubbing and compensatory polycythemia.24 Acute postural change to standing can increase shunting of blood through PAVMs located at the base of the lung, resulting in orthodeoxia. In a series of 257 patients with PAVMs, 29% demonstrated a fall in blood oxygen saturation of at least 2% with standing.25 Although platypnea accompanying this orthodeoxia has been reported in the literature, none of the patients in this large series reported dyspnea with standing.26 Positional oximetry, although not very sensitive, is highly specific for PAVMs and should be a part of initial evaluation in patients with definite or suspected HHT.25,27 Additionally, due to lack of a capillary filter between arterial and venous beds, these patients are at risk for paradoxical embolism of de novo in situ cruoric thrombi or cruoric thrombi/septic thrombi/air emboli developing upstream. These result in embolic cerebral events, including embolic strokes and cerebral abscesses. In one series of 76 patients undergoing embolotherapy, symptomatic strokes were reported in 18%, transient ischemic attacks in 37%, and brain abscesses in 9%.28 Notably, CT imaging of 59 patients in this series showed unequivocal evidence of stroke in 36%; cerebral AVMs (CAVMs) were documented in only 5%. Morbidity and mortality attributable to these catastrophic events is significant, hence the need for early screening and timely interventions (for example, embolization of PAVMs and antibiotic prophylaxis prior to dental procedures). Furthermore, high cardiac output state resulting from right-to-left shunting can over time lead to heart failure and/or pulmonary hypertension. According to a study of 1,143 patients undergoing PAVM embolization, the mean pulmonary arterial pressure was >20 mmHg in 9% of the patients and >25 mmHg in 6%.29 Lastly, PAVM rupture can result as a complication of pregnancy, pulmonary hypertension, or in the presence of high-pressure systemic blood supply via bronchial circulation; this may present as hemoptysis and/or hemothorax.30–33
Migraines are remarkably frequent in HHT patients with PAVMs, and there is evidence to suggest that embolization of PAVMs leads to symptom improvement.34–36 This association may be explained by passage of microthrombi and/or vasoactive substances (eg, serotonin) from the venous system to the cerebral circulation via PAVMs, or systemic hypoxemia.37 That being said, migraines are still more common in HHT patients without PAVMs than in the general population.38 A recent study by Elphick and Shovlin identified an especially strong association between epistaxis and migraines in HHT, suggesting common pathophysiologic mechanisms underlying both these manifestations.39
CAVMs
CAVMs affect approximately 10% of HHT patients.40–43 These include, in order of decreasing size, arteriovenous fistulae, classical nidal AVMs, micro-AVMs (<1 cm), and telangiectasias. Due to the potentially devastating effects of cerebral hemorrhage, symptoms suggestive of CAVMs should be investigated with magnetic resonance imaging (MRI). However, most CAVMs never bleed and complications associated with currently available diagnostic modalities are substantial. Furthermore, recent evidence for patients with sporadic CAVMs suggests that the risk of stroke or death with interventional management is significantly higher than with medical management.44 As such, the screening strategy needs to be individualized for each patient after careful discussion. It should be emphasized that most neurological events in HHT are a consequence of paradoxical embolism of clots and bacteria via PAVMs to the central nervous system, and CAVMs are detected in a small minority of the patients with definite evidence of stroke on CT imaging.28
In infants and young children, large-caliber high-flow fistulae are more common and may present with intracranial bleeds, hydrocephalus, epilepsy, cognitive deficits, or high-output cardiac failure.41,45–49 Spinal AVMs are substantially less frequent (affect 1%–2% of the HHT population), and usually present early in childhood with paralysis and/or complaint of back pain.45,50,51
HAVMs
Hepatic involvement is reported in up to three-quarters of patients with HHT, depending on the imaging technique used. In one series of 70 patients, HAVMs were detected in 74% using helical CT imaging; these led to symptoms in only 8% of these patients.52 In another series using ultrasonography, only 41% of the individuals with HHT were found to have HAVMs.53 In a study of 154 patients with HHT and HAVMs followed over a median period of 44 months, one-quarter experienced complications related to these AVMs and 5% died from their complications.54
The vascular supply of the liver is unique. Blood enters through the hepatic artery and portal vein, combines in the hepatic sinusoids and exits via the hepatic veins. Clinical manifestations depend on the direction of shunts. The most common presentation related to HAVMs in HHT is a hyperdynamic circulatory state resulting from left-to-right shunting from the hepatic artery or portal vein to the hepatic veins.55 Hepatomegaly and hepatic bruits may be appreciable on physical examination. These, over time, can lead to cardiac failure often presenting with dyspnea and dependent edema. Shunting of blood from the hepatic artery to the portal vein is less commonly observed. In this scenario, increased sinusoidal blood flow is associated with nodular transformation in the liver, also known as “pseudocirrhosis” and “arterialization” of the portal vein.56,57 The peribiliary plexus arising from the hepatic artery supplies blood to the biliary tree. Shunting of blood away from the hepatic artery to hepatic and/or portal veins may also cause ischemia of the intrahepatic or extrahepatic biliary systems, manifestations of which include severe cholestasis, recurrent cholangitis, and biliary strictures.
Juvenile polyposis
Individuals with juvenile polyposis-HHT overlap syndrome (JPHT) with mutations in Smad4 gene (see “Molecular diagnosis” section) represent 1%–2% of the total HHT population. These patients are at increased risk for colorectal cancer and require close surveillance. More recently, connective tissue defects such as enlarged aortic root, valvular insufficiency, and aortic dissection have been described as well.58,59
Venous thromboembolism
HHT patients carry a higher risk for venous thromboembolism (VTE) as compared to the general population.60 Although the precise reason(s) for this are unknown, one recent study of 609 HHT patients identified an association of deficient iron stores with high coagulation factor VIII levels and increased thromboembolic risk.61 Given the predisposition for bleeding, anticoagulation presents an obvious challenge in this group and is discussed separately in detail.62
Immunological abnormalities
In a retrospective analysis of 353 patients with HHT, 13.6% were identified to have severe infections.63 Cerebral infections accounted for one-third of all infections and were associated with the presence of PAVMs. The remaining two-thirds were extracerebral, most involved Staphylococcus aureus, and were attributed to epistaxis and need for prolonged nasal packing. In addition to these mechanical factors, defects in adaptive immunity (such as T cell and NK cell lymphopenia, increased levels of immunoglobulins G and A, and low levels of immunoglobulin M) have recently been identified in HHT.64
Malignancy risk
A prospective study in patients with over 20 years of follow-up reported increased mortality attributable to complications of HHT in patients younger than 60 years; however, mortality in individuals older than 60 years was not increased.2 Hosman et al evaluated this discrepancy and found that certain malignancies with especially poor prognosis, such as lung and liver cancers are less prevalent in HHT and could account for the better than expected life expectancy in older individuals within this population. Additionally, a modest increase in breast cancer prevalence was found, possibly related to recurrent exposure to ionizing radiation with CT and angiographic studies.65
Diagnostic approaches
Clinical diagnosis
Establishing a diagnosis of HHT is imperative for patients and their family members for them to receive the appropriate screening and preventive treatments. This diagnosis has conventionally been based on clinical grounds. Consensus clinical diagnostic criteria known as Curaçao criteria were published in 2000; they remain the mainstay of clinical diagnosis to date (Table 1).66
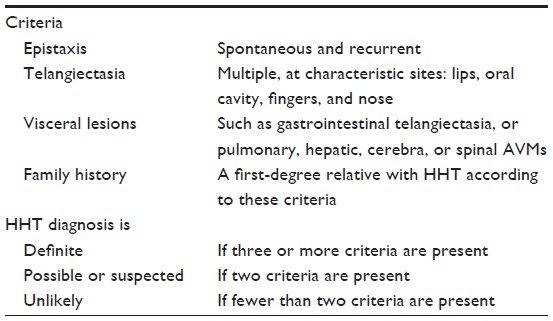 | Table 1 Curaçao criteria for clinical diagnosis of HHT |
van Gent et al recently demonstrated excellent diagnostic performance for these criteria in 263 first-degree relatives of disease-causing mutation carriers.67 Among the 186 relatives with family-specific mutations, clinical diagnosis was “definite”, “possible”, and “unlikely” in 160 (90.3%), 17 (9.1%), and one (0.5%), respectively. Among the remaining 77 family members lacking the mutation, clinical diagnosis was definite, possible, and unlikely in zero, 35 (45.5%), and 42 (54.5%), respectively. As such, the positive predictive value of a definite diagnosis was 100% and the negative predictive value of an unlikely diagnosis was 97.7%. A few notes of caution regarding employment of these criteria are worth mentioning. First, as highlighted by Govani and Shovlin, they can prove inadequate when members of a family with HHT are encountered. In an individual with positive family history and confirmed PAVM(s), the diagnosis of HHT is effectively confirmed, even though these criteria label the diagnosis as “possible” or “suspected”.68 At the same time, these criteria do not specify what counts as a PAVM. Using chest CT as the reference, TTCE (which is now recommended as the first-line examination for PAVM screening) has excellent sensitivity and negative predictive value, but lacks specificity and positive predictive value for detection of PAVMs.15,69 Velthuis et al evaluated the utility of TTCE results in establishing the diagnosis of HHT. They showed that addition of any pulmonary shunt on TTCE to the current Curaçao criteria resulted in increased sensitivity from 88% to 94%, but decreased specificity from 74% to 70%. Excluding grade 1 on TTCE as a diagnostic criterion for HHT enhanced the sensitivity to 90%, with no decrease in specificity.15 Secondly, as clinical manifestations of HHT develop with age, these criteria lack sensitivity and have limited value in children and young adults with positive family history but no other clinical manifestations. Genetic testing is required in these cases (see “Molecular diagnosis”). As commented upon in the “International Guidelines for Diagnosis and Management of HHT”, these criteria are particularly helpful in two situations:8 1) discriminating affected from non-affected older adults and 2) ruling in the diagnosis in younger adults and children.
Molecular diagnosis
HHT follows an autosomal dominant pattern of inheritance; sporadic mutations leading to this syndrome are rare. Based on underlying genetic defects, five different types of HHT have been described thus far:
- HHT type 1 results from mutations in the endoglin gene (ENG, HHT1; OMIM#187300).
- HHT type 2 results from mutations in the activin receptor-like kinase 1 gene (ACVRLK1, HHT2; OMIM 600376).
Four hundred and seventy variants in ENG and 375 variants in ACVRLK1 have been described thus far; these account for approximately 87% of the cases.70
- JPHT: If testing for ENG and ACVRLK1 is negative, sequencing of Smad4 gene (SMAD4, MADH4; OMIM 175050) identifies mutations in an additional 1%–2% of individuals.71–75
These three genes code for proteins in the transforming growth factor-beta (TGF-β) signaling pathway. The exact mechanisms by which these genetic defects influence the process of angiogenesis are not clear; they likely disturb the balance between proangiogenic and antiangiogenic signals in the blood vessels, in favor of the former. As a result, normal angiogenic stimuli lead to the development of AVMs with increasing age.
- HHT type 3 is associated with mutations in locus 5q31.3-q32 (OMIM%601101).76
- HHT type 4 is linked to mutations at location 7p14 (OMIM%610655).77
Specific genes at these two loci have not yet been identified, and the mechanisms by which they lead to HHT are not known.
- More recently, mutations in the growth differentiation factor 2 gene (GDR-2, BMP-9, HHT5; OMIM 615506) that are involved in angiogenesis were described in three individuals with a vascular-anomaly syndrome that has phenotypic similarities with HHT.78
For patients with “definite” clinical diagnosis, genetic testing is not required to confirm their diagnosis. The objective of genetic testing is often to identify the specific mutation in the index case. Once this family specific mutation is identified, targeted sequencing can be valuable in two settings: 1) to effectively establish the diagnosis in relatives of individuals with HHT who do not (yet) meet clinical diagnostic criteria, particularly in children and young adults, and 2) more commonly, to rule out the diagnosis in a branch of the family.
A directory of laboratories across the world that offer genetic testing for HHT is available on the HHT Foundation International website.79 A few caveats about diagnostic usage of genetic testing are noteworthy:
- Mutations have not been identified in 15%–20% of HHT families.80–82 In the appropriate clinical context, negative testing in the index patient does not change his/her diagnosis and management. However, this does limit being able to use targeted genetic testing to rule in or rule out the diagnosis in other family members.8
- Ten to twenty percent of the families have genetic variants (usually potential missense mutations), pathogenicity of which are not clear; this can lead to overdiagnosis. This will likely become an even bigger problem as newer sequencing technologies are applied to promoter and intronic sequences of HHT genes.80,83,84
- PAVMs and CAVMs are more common in HHT type 1, HAVMs and pulmonary arterial hypertension are observed more often in HHT type 2, and most studies have showed no significant differences in the prevalence of GI bleeding between the two HHT types.10,80,85–90 Nevertheless, all of these features are seen in both types. Knowing the underlying mutation does not alter the screening or management strategy for an individual patient. The only exception to this is patients with JPHT; they require regular colonic surveillance for GI malignancies beginning at an early age.74,75,91
Management
In a patient with “possible” and “definite” HHT, optimal management includes: 1) treatment directed towards organ-specific signs and symptoms; 2) screening and subsequent management of PAVMs and sometimes CAVMs while in the asymptomatic phase, given the disastrous and potentially fatal complications associated with them; and 3) education, with opportunity for genetic testing in index patients and in at-risk family members, whenever possible (see “Molecular diagnosis”). It should be noted that most of the current guiding principles are based on expert opinion, there are very few randomized controlled trials addressing different therapies in HHT.8
Epistaxis
Severe epistaxis has been repeatedly found to be the most significant clinical variable associated with poor quality of life among HHT patients.92–94 Treatment can be difficult and frustrating, both for the patient and providers. Modus operandi for these patients should include systematic evaluation of severity of nosebleeds, a stepwise treatment approach (including correction of anemia and iron deficiency), and regular follow-up. Hoag at al described a statistically validated epistaxis severity score (ESS) that can serve as a useful adjunct in clinical evaluation and follow-up of HHT patients.95 Given the unique characteristics of this disorder and nuances of its management, referral to an otorhinolaryngologist with expertise in HHT-related epistaxis, whenever possible, is recommended. A list of HHT treatment centers is available online at the HHT Foundation’s website (http://www.hht.org/living-with-hht/treatment-centers-2/). Various treatment options are discussed next:
- Air humidification and regular application of lubricants such as saline (to prevent crusting over nasal mucosa) and avoidance of injury to nasal mucosa (for example, with picking and blowing hard) are generally recommended for management of chronic and recurrent nosebleeds.96 Certain dietary ingredients have been reported to exacerbate nosebleeds; these include foods with high salicylate content (such as red wine, spices, chocolate, and coffee), with natural antiplatelet activity (such as ginger, garlic, ginkgo biloba, and ginseng), and with high levels of omega-3 acids (such as salmon).39,96 Identification of patient-specific triggers and their avoidance may aid with epistaxis management.
- Surgical options:
- Nasal packing is often the first-line of management for acute bleeding. Use of pneumatic packing minimizes re-bleeding after removal of packing material.
- Laser coagulation is recommended as a low-risk intervention for management of mild-to-moderate epistaxis.97,98 This is considered to be preferable to electrical and chemical cautery as it causes less damage to nasal mucosa, which would further worsen bleeding.
- For severe epistaxis that does not respond to repeated endonasal coagulation, septal dermoplasty has been shown to be effective in reducing transfusion requirements and improving quality of life measures.99 Alternatively, Young’s nasal closure procedure (in which the nostrils are sewn closed so a person no longer breathes through the nose) usually leads to complete cessation of bleeding; patient often report side effects of chronic mouth breathing including alterations in smell and taste senses.100,101 In a recent series of 43 patients undergoing this procedure, nosebleeds stopped completely in 83% of the patients and a mean increase in hemoglobin of 4.68 g/dL was observed.102
- Nasal arterial embolization is effective only in the short term, as surrounding blood vessels enlarge and lead to recurrence of bleeding. This should be reserved only for emergent management only, until more durable therapies can be started.
- Submucosal radiofrequency has recently been shown to be safe and efficacious as a therapy for epistaxis in HHT, although long-term effects remain to be evaluated.103
- Medical options: Various medical treatments (antiangiogenic, hormonal, and antifibrinolytic) have been reported for management of HHT-related epistaxis.
- Angiogenesis inhibitors: VEGF levels have been shown to be about 15-fold elevated in patients with HHT and are implicated in its pathogenesis.104 Bevacizumab is a recombinant humanized monoclonal antibody that binds to VEGF receptors on endothelial cells and serves as a competitive antagonist. In HHT, its systemic administration has been shown to improve HAVM-related symptoms as well as epistaxis.105 Several uncontrolled series reported efficacy and safety of local administration of bevacizumab in HHT-related epistaxis.106–111 In one recent double-blind, placebo-controlled trial, 15 adult HHT patients with a minimum of two epistaxis episodes per week were randomized to a single intranasal submucosal injection of 100 mg bevacizumab or placebo.112 The average daily posttreatment visual analog score decreased by 27% in the bevacizumab group and by only 3% in the placebo group (P=0.57). In another Phase I, randomized, double-blind, placebo-controlled study (ELLIPSE trial) of 40 patients, five increasing doses (12.5, 25, 50, 75, and 100 mg) of bevacizumab nasal spray were evaluated.113 The drug was well-tolerated in all patients and was not detected in their serum; however, no efficacy was observed at any dose. A Phase II study is needed to determine the effectiveness of the drug. In summary, bevacizumab represents a promising therapy for management of HHT-related epistaxis. The cost and systemic side-effect profile of this drug (hypertension, increased bleeding risk, increased thromboembolic risk, proteinuria, and less frequently, renal thrombotic microangiopathy, bowel perforation, and nasal septum perforation) are not negligible; however, the adverse effects are largely avoided with topical use.
Anecdotal positive reports of thalidomide, another antiangiogenic agent have also been presented.114,115 However, there have been no controlled trials using this drug. Additionally, nonselective beta-blockers such as propranolol and timolol may have some antiangiogenic properties as well (related to vasoconstriction and reduced VEGF expression). They have been documented to be effective in the treatment of infantile hemangiomas, and may be a potential therapeutic option for HHT in future.116–118
- Hormonal and antihormonal therapies: Combined estrogen–progesterone at doses used for oral contraception may eliminate bleeding in symptomatic HHT and could represent a reasonable option for women of childbearing age.119 Similarly, selective estrogen receptor modulators such as tamoxifen and raloxifene may have a role in management of HHT-related epistaxis in postmenopausal women.120–122 Yaniv et al reported a trial where 25 patients with HHT-related epistaxis were randomly assigned to receive oral tamoxifen 20 mg/day or placebo for 6 months. Among the 21 participants who completed the trial, nine out of ten tamoxifen-treated patients reported alleviation in the frequency and severity of their nosebleeds, as opposed to only three out of eleven in the placebo arm.121 Beneficial side effects of tamoxifen include prevention of bone loss and improvement in serum lipid profile. In addition to unpleasant effects resulting from its antiestrogenic properties (mood changes, hot flashes, etc), the most concerning adverse effect is the increased incidence of uterine malignancies.
- Antifibrinolytics: A recent randomized, double-blind and placebo-controlled trial of tranexamic acid demonstrated a 50% decrease in nosebleeds; however, no significant improvement in the primary outcome, ie, blood hemoglobin concentration was observed.123 Aminocaproic acid has also been reported to decrease epistaxis severity.124
- Antioxidants such as N-acetyl cysteine (NAC) have also been evaluated in the treatment of epistaxis. In an observational study, 43 patients taking NAC 600 mg thrice daily for 12 weeks reported a decrease in the frequency and severity of diurnal nosebleeds.125
- Angiogenesis inhibitors: VEGF levels have been shown to be about 15-fold elevated in patients with HHT and are implicated in its pathogenesis.104 Bevacizumab is a recombinant humanized monoclonal antibody that binds to VEGF receptors on endothelial cells and serves as a competitive antagonist. In HHT, its systemic administration has been shown to improve HAVM-related symptoms as well as epistaxis.105 Several uncontrolled series reported efficacy and safety of local administration of bevacizumab in HHT-related epistaxis.106–111 In one recent double-blind, placebo-controlled trial, 15 adult HHT patients with a minimum of two epistaxis episodes per week were randomized to a single intranasal submucosal injection of 100 mg bevacizumab or placebo.112 The average daily posttreatment visual analog score decreased by 27% in the bevacizumab group and by only 3% in the placebo group (P=0.57). In another Phase I, randomized, double-blind, placebo-controlled study (ELLIPSE trial) of 40 patients, five increasing doses (12.5, 25, 50, 75, and 100 mg) of bevacizumab nasal spray were evaluated.113 The drug was well-tolerated in all patients and was not detected in their serum; however, no efficacy was observed at any dose. A Phase II study is needed to determine the effectiveness of the drug. In summary, bevacizumab represents a promising therapy for management of HHT-related epistaxis. The cost and systemic side-effect profile of this drug (hypertension, increased bleeding risk, increased thromboembolic risk, proteinuria, and less frequently, renal thrombotic microangiopathy, bowel perforation, and nasal septum perforation) are not negligible; however, the adverse effects are largely avoided with topical use.
GI bleeding and malignancies
Management in these patients usually focuses on early detection and treatment of anemia and underlying iron deficiency.
Because of increased risk of GI bleeding with age, annual measurement of hemoglobin or hematocrit and serum iron studies beginning at 35 years of age is recommended.8 Direct endoscopic evaluation should be performed only if anemia is present and is disproportionate to the amount of epistaxis. Fecal occult blood testing can be falsely positive due to swallowed blood from nosebleeds, and hence, is less useful as a screening. As noted earlier, based on our current knowledge, JPHT represents the only subtype where the screening strategy is affected by the underlying genotype. Juvenile polyposis in these patients places them at increased risk for colonic malignancies. Screening colonoscopies starting at 15–18 years of age or at an age 5 years younger than at which the youngest family member developed colon cancer, whichever is earlier, and every 2 years thereafter, are recommended.91
Since most bleeding occurs in the stomach and duodenum, an upper esophagogastroduodenoscopy (EGD) is usually adequate to make the diagnosis. However, presence of telangiectasia does not rule out other sources of bleeding or anemia (for instance, epistaxis) in these patients. If upper and lower endoscopies do not adequately explain the severity of findings, or in patients where numerous lesions are expected (HHT type 1, older age, or severe anemia in the setting of limited epistaxis), video capsule endoscopy may be considered as the initial step as it provides better information about the distal part of the duodenum and small intestine.89 Iron absorption and handling are preserved in HHT, and the iron deficiency is explained by the losses due to chronic bleeding.126 Aggressive and long-term iron replacement is often required for maintenance of iron stores and to minimize the need for blood transfusions. Oral iron supplements should be attempted before switching to intravenous preparations. Evidence for use of different medical therapies to reduce chronic GI bleeding (estrogen–progesterone preparations, danazol, tamoxifen, thalidomide, lenalidomide, interferon, sirolimus, and bevacizumab) is limited to case reports and small case series.13,124,127–134 There is one small double-blind, placebo-controlled, cross-over trial of combination hormonal therapy (0.05 mg ethinyl estradiol plus 1 mg norethisterone) versus placebo; five of the six HHT patients experienced no further GI bleeding.135 Limited attempts at cauterization of large visible telangiectasias by an experienced endoscopist may be considered; however, repeated endoscopic attempts are unlikely to be effective.136–138
PAVMs
Neurological complications resulting from PAVMs are a significant cause of morbidity and mortality among HHT patients.43,139,140 Screening for PAVMs is recommended in all patients with possible or definite HHT at the time of initial evaluation, regardless of age.8,141
TTCE, also referred to as “bubble echocardiography” is recommended as the first step in screening for PAVMs. It is simple, minimally invasive, and does not require exposure to radiation. After intravenous injection of agitated saline solution, if a right-to-left pulmonary shunt is present, echoic microbubbles (referred to as contrast) can be easily visualized in the left cardiac cavities after 2–5 seconds or three to eight cardiac cycles (appearance of contrast in the left ventricle within one cycle of its appearance in the right atrium suggests an intracardiac right-to-left shunt). Depending on the extent of opacification of the left ventricle, a semi-quantitative assessment of the shunt size can be obtained.142 These grades are:
- Grade 0, no bubbles (no additional testing is required);
- Grade 1, occasional filling with less than 30 bubbles;
- Grade 3, moderate filling with 30-100 bubbles; and
- Grade 4, complete opacification with more than 100 bubbles.
Multiple prospective studies have documented excellent sensitivity (97%–100%) and negative predictive value (99%–100%) of TTCE for detection of PAVMs, making it an excellent screening test.16,69 However, it lacks specificity (49%–77%) and the positive predictive value is low (32%–51%). Additionally, as noted earlier, a study by Velthuis et al showed limited diagnostic significance of grade 1 shunts detected by TTCE.15 A recent cross-sectional study by the same group that included over 1,000 patients with HHT showed that a pulmonary shunt grade 1 was not associated with increased prevalence of cerebral complications (odds ratio [OR]: 0.44; 95% confidence interval [CI]: 0.05–4.13; P=0.47), whereas pulmonary shunts grade 2 and grade 3 carried an OR of 4.78 (95% CI: 1.14–20.0; P=0.03) and 10.4 (95% CI: 2.4–45.3; P=0.002), respectively, for cerebral ischemic event or brain abscess.143 Another analysis demonstrated that among 772 HHT patients, none of the 201 individuals with grade 1 TTCE results had PAVMs on chest CT that were amenable to embolization.144 Based on these data and concerns about potential long-term effects of repeated exposure to radiation, it appears reasonable to state that CT can safely be withheld in asymptomatic individuals with grade 1 pulmonary shunt on TTCE.145 On the other hand, follow-up imaging is definitely warranted in all patients with grade 2 and grade 3 results. Low-dose CT techniques allow an approximately 50% reduction in radiation dose without any significant difference in detection of abnormalities, and should be preferred over standard CT protocols.145 Use of magnetic resonance angiography (MRA) has not been evaluated for PAVM screening; however, it may have a role in preembolization planning.146 Its biggest advantage over CT is avoidance of ionizing radiation exposure.
Management of PAVMs is largely interventional. The “International Guidelines for Diagnosis and Management of HHT” recommended transcatheter embolization for PAVMs with feeding artery diameter greater than 2–3 mm.8 In 2008, Shovlin et al documented that the risk of cerebral events from PAVMs in HHT is independent of the feeding artery diameter; their study also demonstrated effectiveness of embolization in treating PAVMs in reducing strokes.147 These data imply that all PAVMs, where embolization is considered technically feasible and safe (irrespective of size), should be treated. Once the decision to proceed with embolization is made, a conventional diagnostic pulmonary angiogram is required to fully characterize the AVM, followed by catheter-directed placement of embolic material (such as metallic coils, detachable balloons, and Amplatzer vascular plugs) into the feeding artery until the blood flow through the PAVM stops. Whenever possible, both these steps should be done in the same session to minimize total radiation exposure. Amplatzer vascular plugs allow better management of larger AVMs, complex AVMs, and multiple AVMs in a single session and are being increasingly used.148–150 Various studies have documented safety and short- and long-term effectiveness of the procedure.18,148,149,151–154 Pleuritic chest pain, usually self-limited, is the most common complication. Recanalization of the occluded feeding artery was identified as the most common reason for treatment failure, often requiring reembolization.154,155 As a result, CT is recommended 3–6 months after the procedure, and then another 3 years later.155 Recent evidence suggests that MRA may be a reasonable alternative to CT for follow-up examinations and detection of recanalization.156 Of note, TTCE remains positive in ~90% of the patients despite complete occlusion of all angiographically visible PAVMs (likely reflective of other smaller PAVMs), and is not helpful in follow-up.157
The role of surgery is limited to management of life-threatening hemorrhage from a ruptured AVM, when embolization is not available. Lung transplantation is occasionally pursued for patients who have diffuse bilateral disease refractory to all other treatment and are therefore at considerable risk for death from their underlying PAVMs.158 There are no data regarding efficacy of any pharmacologic options discussed earlier in management of PAVMs.
Other measures that should be taken for prevention of complications related to PAVMs, treated or untreated, include:
- Lifelong antibiotic prophylaxis: Strong association between oral microorganisms and brain abscesses in HHT is well-documented.147 Antibiotic prophylaxis is recommended for all patients with PAVMs (treated or untreated) prior to procedures with risk of bacteremia, in particular dental procedures.159
- Careful precautions should be taken in these patients to avoid introduction of intravenous air during administration of intravenous medications.
- Avoidance of scuba diving is generally recommended; however, the concern regarding increased risk of complications from decompression in patients with PAVMs is only theoretical.8 Interestingly, with the exception of epistaxis, in-flight complications in patients with HHT are relatively uncommon.160
CAVMs
Due to the debilitating and potentially fatal effects of CAVM rupture and the lack of evidence regarding effectiveness of treatment for asymptomatic CAVMs, the “International Guidelines for Diagnosis and Management of HHT” recommended screening for them in all patients (including children in the first 6 months of life) with possible or definite HHT.8 However, this has been a subject of significant debate for many reasons. First of all, most CAVMs never bleed. In fact, the bleeding risk of CAVMs associated with HHT (~0.5% per year) is much lower than with sporadic CAVMs (2%–4% per year).161 However, there remains concern regarding selection bias and survivor bias leading to better CAVM rupture rates in HHT. Secondly, the techniques employed for establishing the diagnosis carry more than minimal risk. While MRI itself poses minimal risk, sedation or anesthesia is often required in infants and children undergoing this procedure. Diagnostic angiography, which is the gold standard for diagnosis and required for treatment planning, is associated with a 0.5% risk of permanent stroke.162 Thirdly and most importantly, the evidence that has become available since 2006 when these guidelines were initially prepared suggests that the benefits of managing these CAVMs are outweighed by the risks of interventional therapies. In the ARUBA trial, where 223 patients with non-HHT–related CAVMs were randomized to interventional therapy vs medical management alone, the risk of stroke or death in the interventional arm was three-fold higher (30.7% vs 10.1%), warranting premature termination of the study.44 Such studies evaluating the risk–benefit considerations have not been conducted in the HHT population (where the true incidence of CAVM rupture may be higher). Considering all these reasons, we suggest that instead of routinely screening all asymptomatic individuals with definite or possible HHT, the possibility that CAVMs will be identified in about 10% of the individuals with HHT and the concern that the risks of therapy may outweigh its benefits should be discussed explicitly with the patients and their families. The decision to proceed or not proceed with imaging should be made only after careful discussions. History of cerebral hemorrhage in a relative may be a considerable source of worry for the other family members with HHT. Whether the predisposition to cerebral hemorrhage related to CAVM is familial or related to a particular genotype is not known; however, a negative imaging study may help alleviate their anxiety. Another scenario where this may be useful is prior to making a decision regarding initiation of anticoagulation (see “Antiplatelet therapy and anticoagulation”).
When the decision is made to proceed with screening or in patients with symptoms suggestive of CAVMs, brain MRI with and without contrast is the appropriate initial investigation.163–166 Angiography is required for confirmation of diagnosis and treatment planning. The size and number of lesions, venous drainage pattern, and involvement of the eloquent cortex determine the feasibility and risks of intervention (most importantly the risk of hemorrhage).167 Referral to a center with expertise in managing these AVMs is recommended. Open microsurgical excision provides the best chance for cure. For lesions that are inoperable, stereotactic radiosurgery is another option; however, the cure rate is much lower and depends on the size of the AVM.168 Unlike with PAVMs, embolization alone is rarely effective in the management of CAVMs but may be a useful adjunct to surgery or radiation therapy.169
HAVMs
Screening for HAVMs in asymptomatic individuals is not done routinely, because 1) HAVMs are symptomatic in <10% of the cases; 2) when symptomatic, their presentation is not sudden and catastrophic (as with PAVMs and CAVMs); and 3) usefulness of treatment of asymptomatic lesions is not known. That said, screening for HAVMs can be used for clarification of diagnosis in patients meeting one or two Curaçao criteria. When screening is undertaken, it is advisable to start with Doppler ultrasound.8,170 It is accurate, noninvasive, highly available, and inexpensive.52,53,55,71,171–175 Multi-slice CT and MRI provide better characterization of hepatic lesions and may be considered if expertise in Doppler ultrasound for liver AVMs is not available. Mesenteric angiography is almost never required. Liver biopsy carries a high risk of hemorrhage in these patients and should be avoided.
High-output cardiac failure is treated with conventional medical therapy, including correction of anemia (often requiring blood transfusions), salt and fluid restriction, diuretics, and beta-blockers. Portal hypertension is managed according to present guidelines for cirrhotic patients with salt and fluid restriction and diuretics, therapeutic large-volume paracenteses with plasma expansion with albumin may be required.
Orthotopic liver transplantation (OLT) offers a definite cure.176–178 In the largest series of 40 HHT patients undergoing OLT, 17.5% patients died in the early postoperative period mainly due to bleeding complications; however, the actuarial 5- and 10-year survival rates were over 80%.178 Interestingly, two of these patients developed multiple recurrent AVMs in the graft 8 and 10 years later, possibly due to circulating proangiogenic factors. Given the significant risk from surgery and chronic immunosuppression, OLT is reserved for: 1) intractable heart failure, 2) intractable portal hypertension, and 3) ischemic biliary necrosis.8
Hepatic artery embolization frequently improves heart failure and portal hypertension; however, the benefit is usually transient and the rate of severe complications (hepatic necrosis, cholangitis, and cholecystitis) and death is extremely high.179–182 As a result, this procedure is limited to patients with intractable heart failure or portal hypertension who are not candidates for transplantation, after exclusion of those at extremely high risk for complications (ie, those with cirrhosis, severe biliary disease, or portovenous shunts).183
Systemic bevacizumab is a promising therapeutic option for HAVMs. In a recent Phase II trial, 25 adult HHT patients with severe liver involvement and a high cardiac index were treated with bevacizumab (5 mg/kg every 14 days for a total of six injections).105 Six-month follow-up data was available for 23 patients; it showed complete response (ie, normalization of cardiac index) in five cases and partial response in 15 cases. Blinded cardiac index at 6 months (mean 4.06 L/min/m2; 95% CI: 3.80–4.33) was significantly lower than at the beginning of treatment (mean 5.01 L/min/m2; 95% CI: 4.72–5.29). Hypertension and proteinuria are common side effects of this drug. Additionally, VEGF inhibition has been implicated in glomerular injury and development of a renal thrombotic microangiopathy, requiring withdrawal of treatment.184 While OLT remains the therapeutic option of choice, bevacizumab may be useful as bridging therapy for patients who are awaiting transplantation or an alternative for patients who are not eligible for transplantation.
Special considerations
Pregnancy
Pregnancy in HHT is high risk. While a majority of women with HHT have an uneventful pregnancy, it is associated with an increased risk of life-threatening complications. In a study of 484 pregnancies in 199 women, 1.0% resulted in a major PAVM bleed, 1.2% in stroke, and 1.0% in maternal death.30 Screening and management recommendations specific to pregnancy include:
- Ideally, PAVMs should be screened for and maximally treated prior to a planned pregnancy.8
- If a woman with HHT who has not had screening for PAVMs presents when she is already pregnant, screening should still be considered. A small series of seven patients showed that transcatheter embolization by an experienced interventionalist, with modifications to minimize fetus exposure to radiation may be safely undertaken in the second and third trimesters.185
- In a recent retrospective analysis, no increased risk of hemorrhage was found in patients with sporadic CAVMs during pregnancy and puerperium.186 Given the low risk of bleeds and high risk of any interventional therapy, the role of screening (even in women with family history of cerebral hemorrhage) is unclear.
- Although frequently performed, current evidence does not support routine screening in all pregnant women with HHT for spinal AVMs prior to administration of epidural anesthesia. Spinal AVMs are rare, affecting only 1%–2% of the patients and usually present in early childhood.50,51 In one study, spinal or epidural anesthesia was used in 92 out of 185 deliveries in women with HHT, none of whom had undergone screening for spinal AVMs; no complications were observed.187
- Parents should be made aware of the 50% likelihood of disease in the offspring. There is no evidence for or against antenatal testing for HHT at this time.
Antiplatelet therapy and anticoagulation
Patients with HHT (who carry an increased risk for VTE) are frequently advised to avoid antiplatelet therapy and anticoagulation.62 There are reports where withholding treatment for myocardial infarction due to diagnosis of HHT contributed to a patient’s death; there are also reports where treatment for myocardial infarction led to hemorrhages and worsened outcomes.62 While epistaxis worsens with these therapies in about 60% of the patients, it is notable that nearly 40% report no change in the frequency or severity of nosebleeds.62 The side-effect profile with respect to bleeding associated with use of antiplatelet and anticoagulant therapies in this population is quite variable.188 While data are limited to guide use of antiplatelet and anticoagulant therapies in individual patients, we want to emphasize that HHT by itself should not be considered an absolute contraindication. If faced with a compelling indication, these medications may be prescribed, at the lowest possible doses and with careful monitoring.
Radiation risk
HHT patients, particularly those with PAVMs, are repeatedly exposed to ionizing radiation from interventional procedures and CTs. In a recent analysis, 246 HHT patients with PAVMs underwent 3,309 procedures requiring ionizing radiation and cumulative effective dose exceeded 100 mSv (a level at which there is considered to be good evidence for risk of significant harm) in 11%.145 Physicians need to be cognizant of these risks. MRI or low-dose protocols for CTs definitely warrant consideration in patients who may be expected to undergo multiple procedures over the course of their lifetime, especially children.189–191
Summary
Optimal management in HHT comprises:
- Early diagnosis: The diagnosis of HHT typically relies on clinical findings. Genetic testing is not required to confirm the diagnosis in an individual case. However, if a family-specific pathogenic mutation is identified, it can be used to establish the diagnosis in other family members, particularly children and young adults who may not meet the clinical diagnostic criteria;
- Screening for visceral AVMs: Decisions regarding screening and treatment of visceral AVMs (especially CAVMs) should be made after explicit and careful discussions with patients and their families, in view of their age, educational level, and cultural background;
- Treatment of symptomatic and certain presymptomatic lesions such as PAVMs: Our understanding of the efficacy and safety of existing therapies in HHT is rapidly evolving and many new treatment options are becoming available. Whenever appropriate, patients should be encouraged to participate in research studies, and;
- Education: Effective education and counseling of affected individuals regarding the implications of diagnosis of “possible” or “definite” HHT and the preventive and therapeutic options available for them is critical not only for their management, but also for identification of at-risk family members. Establishing a diagnosis in them while still in the presymptomatic phase can allow appropriate screening and treatment measures to be done in a timely manner.
Disclosure
The authors report no conflicts of interest in this work.
References
Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat. 2002;19(2):140–148. | |
Kjeldsen AD, Vase P, Green A. Hereditary haemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. J Intern Med. 1999;245(1):31–39. | |
Westermann CJ, Rosina AF, De Vries V, de Coteau PA. The prevalence and manifestations of hereditary hemorrhagic telangiectasia in the Afro-Caribbean population of The Netherlands Antilles: a family screening. Am J Med Genet A. 2003;116A(4):324–328. | |
Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet. 1989;32(3):291–297. | |
Pierucci P, Lenato GM, Suppressa P, et al. A long diagnostic delay in patients with hereditary haemorrhagic telangiectasia: a questionnaire-based retrospective study. Orphanet J Rare Dis. 2012;7:33. | |
Grosse SD, Boulet SL, Grant AM, Hulihan MM, Faughnan ME. The use of US health insurance data for surveillance of rare disorders: hereditary hemorrhagic telangiectasia. Genet Med. 2014;16(1):33–39. | |
Bernhardt BA, Zayac C, Pyeritz RE. Why is genetic screening for autosomal dominant disorders underused in families? The case of hereditary hemorrhagic telangiectasia. Genet Med. 2011;13(9):812–820. | |
Faughnan ME, Palda VA, Garcia-Tsao G, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48(2):73–87. | |
AAssar OS, Friedman CM, White RI Jr. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope. 1991;101(9):977–980. | |
Berg J, Porteous M, Reinhardt D, et al. Hereditary haemorrhagic telangiectasia: a questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. J Med Genet. 2003;40(8):585–590. | |
Chang J, Yung KC. Dysphonia and vocal fold telangiectasia in hereditary hemorrhagic telangiectasia. Ann Otol Rhinol Laryngol. Epub June 9, 2014. | |
Kjeldsen AD, Kjeldsen J. Gastrointestinal bleeding in patients with hereditary hemorrhagic telangiectasia. Am J Gastroenterol. 2000;95(2):415–418. | |
Longacre AV, Gross CP, Gallitelli M, Henderson KJ, White RI Jr, Proctor DD. Diagnosis and management of gastrointestinal bleeding in patients with hereditary hemorrhagic telangiectasia. Am J Gastroenterol. 2003;98(1):59–65. | |
Ingrosso M, Sabba C, Pisani A, et al. Evidence of small-bowel involvement in hereditary hemorrhagic telangiectasia: a capsule-endoscopic study. Endoscopy. 2004;36(12):1074–1079. | |
Velthuis S, Vorselaars VM, van Gent MW, et al. Role of transthoracic contrast echocardiography in the clinical diagnosis of hereditary hemorrhagic telangiectasia. Chest. 2013;144(6):1876–1882. | |
Gazzaniga P, Buscarini E, Leandro G, et al. Contrast echocardiography for pulmonary arteriovenous malformations screening: does any bubble matter? Eur J Echocardiogr. 2009;10(4):513–518. | |
Nakayama M, Nawa T, Chonan T, et al. Prevalence of pulmonary arteriovenous malformations as estimated by low-dose thoracic CT screening. Intern Med. 2012;51(13):1677–1681. | |
Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI Jr. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radiol. 2006;17(1):35–44; quiz 45. | |
Wong HH, Chan RP, Klatt R, Faughnan ME. Idiopathic pulmonary arteriovenous malformations: clinical and imaging characteristics. Eur Respir J. 2011;38(2):368–375. | |
van Gent MW, Post MC, Snijder RJ, Westermann CJ, Plokker HW, Mager JJ. Real prevalence of pulmonary right-to-left shunt according to genotype in patients with hereditary hemorrhagic telangiectasia: a transthoracic contrast echocardiography study. Chest. 2010;138(4):833–839. | |
Cottin V, Plauchu H, Bayle JY, Barthelet M, Revel D, Cordier JF. Pulmonary arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia. Am J Respir Crit Care Med. 2004;169(9):994–1000. | |
Cottin V, Plauchu H, Dupuis-Girod S, Cordier JF. Pulmonary arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia: follow-up and pathophysiologic considerations. J Vasc Interv Radiol. 2007;18(7):938–939; author reply 939. | |
Gossage JR, Kanj G. Pulmonary arteriovenous malformations. A state of the art review. Am J Respir Crit Care Med. 1998;158(2):643–661. | |
Santhirapala V, Williams LC, Tighe HC, Jackson JE, Shovlin CL. Arterial oxygen content is precisely maintained by graded erythrocytotic responses in settings of high/normal serum iron levels, and predicts exercise capacity: an observational study of hypoxaemic patients with pulmonary arteriovenous malformations. PLoS One. 2014;9(3):e90777. | |
Santhirapala V, Chamali B, McKernan H, et al. Orthodeoxia and postural orthostatic tachycardia in patients with pulmonary arteriovenous malformations: a prospective 8-year series. Thorax. Epub April 8, 2014. | |
Kumar N, Kraemer RR, Murthy RK, Hartig JR. Platypnea-orthodeoxia syndrome as a presentation of hereditary hemorrhagic telangiectasia. Circulation. 2012;126(22):2645–2647. | |
Thompson RD, Jackson J, Peters AM, Dore CJ, Hughes JM. Sensitivity and specificity of radioisotope right-left shunt measurements and pulse oximetry for the early detection of pulmonary arteriovenous malformations. Chest. 1999;115(1):109–113. | |
White RI Jr, Lynch-Nyhan A, Terry P, et al. Pulmonary arteriovenous malformations: techniques and long-term outcome of embolotherapy. Radiology. 1988;169(3):663–669. | |
Shovlin CL, Tighe HC, Davies RJ, Gibbs JS, Jackson JE. Embolisation of pulmonary arteriovenous malformations: no consistent effect on pulmonary artery pressure. Eur Respir J. 2008;32(1):162–169. | |
Shovlin CL, Sodhi V, McCarthy A, Lasjaunias P, Jackson JE, Sheppard MN. Estimates of maternal risks of pregnancy for women with hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): suggested approach for obstetric services. BJOG. 2008; 115(9):1108–1115. | |
Chadha D, Handa A, Kumar A. Pulmonary hypertension in a patient with hereditary haemorrhagic telangiectasia. BMJ Case Rep. 2013;2013. pii: bcr2012008352. | |
Montani D, Price LC, Girerd B, et al. Fatal rupture of pulmonary arteriovenous malformation in hereditary haemorrhagic telangiectasis and severe PAH. Eur Respir Rev. 2009;18(111):42–46. | |
Sharma KB, Forkert L. Massive pulmonary hemorrhage from dual circulation pulmonary arteriovenous malformations in hereditary hemorrhagic telangiectasia. Can Respir J. 2004;11(8):597–598. | |
Post MC, van Gent MW, Plokker HW, et al. Pulmonary arteriovenous malformations associated with migraine with aura. Eur Respir J. 2009;34(4):882–887. | |
Post MC, Thijs V, Schonewille WJ, et al. Embolization of pulmonary arteriovenous malformations and decrease in prevalence of migraine. Neurology. 2006;66(2):202–205. | |
Post MC, Letteboer TG, Mager JJ, Plokker TH, Kelder JC, Westermann CJ. A pulmonary right-to-left shunt in patients with hereditary hemorrhagic telangiectasia is associated with an increased prevalence of migraine. Chest. 2005;128(4):2485–2489. | |
Post MC, van Gent MW, Snijder RJ, et al. Pulmonary arteriovenous malformations and migraine: a new vision. Respiration. 2008;76(2):228–233. | |
Marziniak M, Jung A, Guralnik V, Evers S, Prudlo J, Geisthoff UW. An association of migraine with hereditary haemorrhagic telangiectasia independently of pulmonary right-to-left shunts. Cephalalgia. 2009;29(1):76–81. | |
Elphick A, Shovlin CL. Relationships between epistaxis, migraines, and triggers in hereditary hemorrhagic telangiectasia. Laryngoscope. 2014;124(7):1521–1528. | |
Haitjema T, Disch F, Overtoom TT, Westermann CJ, Lammers JW. Screening family members of patients with hereditary hemorrhagic telangiectasia. Am J Med. 1995;99(5):519–524. | |
Fulbright RK, Chaloupka JC, Putman CM, et al. MR of hereditary hemorrhagic telangiectasia: prevalence and spectrum of cerebrovascular malformations. AJNR Am J Neuroradiol. 1998;19(3):477–484. | |
Bharatha A, Faughnan ME, Kim H, et al. Brain arteriovenous malformation multiplicity predicts the diagnosis of hereditary hemorrhagic telangiectasia: quantitative assessment. Stroke. 2012;43(1):72–78. | |
Maher CO, Piepgras DG, Brown RD Jr, Friedman JA, Pollock BE. Cerebrovascular manifestations in 321 cases of hereditary hemorrhagic telangiectasia. Stroke. 2001;32(4):877–882. | |
Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383(9917):614–621. | |
Krings T, Ozanne A, Chng SM, Alvarez H, Rodesch G, Lasjaunias PL. Neurovascular phenotypes in hereditary haemorrhagic telangiectasia patients according to age. Review of 50 consecutive patients aged 1 day-60 years. Neuroradiology. 2005;47(10):711–720. | |
Morgan T, McDonald J, Anderson C, et al. Intracranial hemorrhage in infants and children with hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome). Pediatrics. 2002;109(1):E12. | |
Weon YC, Yoshida Y, Sachet M, et al. Supratentorial cerebral arteriovenous fistulas (AVFs) in children: review of 41 cases with 63 non choroidal single-hole AVFs. Acta Neurochir (Wien). 2005;147(1):17–31. | |
Yoshida Y, Weon YC, Sachet M, et al. Posterior cranial fossa single-hole arteriovenous fistulae in children: 14 consecutive cases. Neuroradiology. 2004;46(6):474–481. | |
Krings T, Chng SM, Ozanne A, Alvarez H, Rodesch G, Lasjaunias PL. Hereditary hemorrhagic telangiectasia in children: endovascular treatment of neurovascular malformations: results in 31 patients. Neuroradiology. 2005;47(12):946–954. | |
Poisson A, Vasdev A, Brunelle F, Plauchu H, Dupuis-Girod S; French Italian HHT network. Acute paraplegia due to spinal arteriovenous fistula in two patients with hereditary hemorrhagic telangiectasia. Eur J Pediatr. 2009;168(2):135–139. | |
Cullen S, Alvarez H, Rodesch G, Lasjaunias P. Spinal arteriovenous shunts presenting before 2 years of age: analysis of 13 cases. Childs Nerv Syst. 2006;22(9):1103–1110. | |
Ianora AA, Memeo M, Sabba C, Cirulli A, Rotondo A, Angelelli G. Hereditary hemorrhagic telangiectasia: multi-detector row helical CT assessment of hepatic involvement. Radiology. 2004;230(1):250–259. | |
Buscarini E, Danesino C, Olivieri C, et al. Doppler ultrasonographic grading of hepatic vascular malformations in hereditary hemorrhagic telangiectasia – results of extensive screening. Ultraschall Med. 2004;25(5):348–355. | |
Buscarini E, Leandro G, Conte D, et al. Natural history and outcome of hepatic vascular malformations in a large cohort of patients with hereditary hemorrhagic teleangiectasia. Dig Dis Sci. 2011;56(7):2166–2178. | |
Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2000;343(13):931–936. | |
Cooney T, Sweeney EC, Coll R, Greally M. ‘Pseudocirrhosis’ in hereditary haemorrhagic telangiectasia. J Clin Pathol. 1977;30(12):1134–1141. | |
Schwartz SI, Morton JH, McGovern GR. Experimental arterialization of the liver. Surgery. 1961;49:611–617. | |
Wain KE, Ellingson MS, McDonald J, et al. Appreciating the broad clinical features of SMAD4 mutation carriers: a multicenter chart review. Genet Med. 2014;16(8):588–593. | |
Teekakirikul P, Milewicz DM, Miller DT, et al. Thoracic aortic disease in two patients with juvenile polyposis syndrome and SMAD4 mutations. Am J Med Genet A. 2013;161A(1):185–191. | |
Shovlin CL, Sulaiman NL, Govani FS, Jackson JE, Begbie ME. Elevated factor VIII in hereditary haemorrhagic telangiectasia (HHT): association with venous thromboembolism. Thromb Haemost. 2007;98(5):1031–1039. | |
Livesey JA, Manning RA, Meek JH, et al. Low serum iron levels are associated with elevated plasma levels of coagulation factor VIII and pulmonary emboli/deep venous thromboses in replicate cohorts of patients with hereditary haemorrhagic telangiectasia. Thorax. 2012;67(4):328–333. | |
Devlin HL, Hosman AE, Shovlin CL. Antiplatelet and anticoagulant agents in hereditary hemorrhagic telangiectasia. N Engl J Med. 2013;368(9):876–878. | |
Dupuis-Girod S, Giraud S, Decullier E, et al. Hemorrhagic hereditary telangiectasia (Rendu-Osler disease) and infectious diseases: an underestimated association. Clin Infect Dis. 2007;44(6):841–845. | |
Guilhem A, Malcus C, Clarivet B, Plauchu H, Dupuis-Girod S. Immunological abnormalities associated with hereditary haemorrhagic telangiectasia. J Intern Med. 2013;274(4):351–362. | |
Hosman AE, Devlin HL, Silva BM, Shovlin CL. Specific cancer rates may differ in patients with hereditary haemorrhagic telangiectasia compared to controls. Orphanet J Rare Dis. 2013;8:195. | |
Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000;91(1):66–67. | |
van Gent MW, Velthuis S, Post MC, et al. Hereditary hemorrhagic telangiectasia: how accurate are the clinical criteria? Am J Med Genet A. 2013;161A(3):461–466. | |
Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009;17(7):860–871. | |
van Gent MW, Post MC, Luermans JG, et al. Screening for pulmonary arteriovenous malformations using transthoracic contrast echocardiography: a prospective study. Eur Respir J. 2009;33(1):85–91. | |
HHT Mutation Database [homepage on the Internet]. Available from: http://arup.utah.edu/database/HHT/. Accessed July 7, 2014. | |
Buscarini E, Buscarini L, Danesino C, et al. Hepatic vascular malformations in hereditary hemorrhagic telangiectasia: Doppler sonographic screening in a large family. J Hepatol. 1997;26(1):111–118. | |
McDonald JE, Miller FJ, Hallam SE, Nelson L, Marchuk DA, Ward KJ. Clinical manifestations in a large hereditary hemorrhagic telangiectasia (HHT) type 2 kindred. Am J Med Genet. 2000;93(4):320–327. | |
Olivieri C, Mira E, Delù G, et al. Identification of 13 new mutations in the ACVRL1 gene in a group of 52 unselected Italian patients affected by hereditary haemorrhagic telangiectasia. J Med Genet. 2002;39(7):E39. | |
Gallione CJ, Repetto GM, Legius E, et al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004;363(9412):852–859. | |
Gallione CJ, Richards JA, Letteboer TG, et al. SMAD4 mutations found in unselected HHT patients. J Med Genet. 2006;43(10):793–797. | |
Cole SG, Begbie ME, Wallace GM, Shovlin CL. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005;42(7):577–582. | |
Bayrak-Toydemir P, McDonald J, Akarsu N, et al. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A. 2006;140(20):2155–2162. | |
Wooderchak-Donahue WL, McDonald J, O’Fallon B, et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am J Hum Genet. 2013; 93(3):530–537. | |
HHT Foundation International, Inc. [webpage on the Internet]. Genetic testing for HHT. Available from: http://www.hht.org/living-with-hht/genetic-testing/. Accessed September 5, 2014. | |
Bossler AD, Richards J, George C, Godmilow L, Ganguly A. Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): correlation of genotype with phenotype. Hum Mutat. 2006;27(7):667–675. | |
Prigoda NL, Savas S, Abdalla SA, et al. Hereditary haemorrhagic telangiectasia: mutation detection, test sensitivity and novel mutations. J Med Genet. 2006;43(9):722–728. | |
Letteboer TG, Zewald RA, Kamping EJ, et al. Hereditary hemorrhagic telangiectasia: ENG and ALK-1 mutations in Dutch patients. Hum Genet. 2005;116(1–2):8–16. | |
McDonald J, Gedge F, Burdette A, et al. Multiple sequence variants in hereditary hemorrhagic telangiectasia cases: illustration of complexity in molecular diagnostic interpretation. J Mol Diagn. 2009;11(6):569–575. | |
Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 2010;24(6):203–219. | |
Sabba C, Pasculli G, Lenato GM, et al. Hereditary hemorrhagic telangiectasia: clinical features in ENG and ALK1 mutation carriers. J Thromb Haemost. 2007;5(6):1149–1157. | |
Lesca G, Olivieri C, Burnichon N, et al; French-Italian-Rendu-Osler Network. Genotype-phenotype correlations in hereditary hemorrhagic telangiectasia: data from the French-Italian HHT network. Genet Med. 2007;9(1):14–22. | |
Letteboer TG, Mager JJ, Snijder RJ, et al. Genotype-phenotype relationship in hereditary haemorrhagic telangiectasia. J Med Genet. 2006;43(4):371–377. | |
Letteboer TG, Mager HJ, Snijder RJ, et al. Genotype-phenotype relationship for localization and age distribution of telangiectases in hereditary hemorrhagic telangiectasia. Am J Med Genet A. 2008; 146A(21):2733–2739. | |
Canzonieri C, Centenara L, Ornati F, et al. Endoscopic evaluation of gastrointestinal tract in patients with hereditary hemorrhagic telangiectasia and correlation with their genotypes. Genet Med. 2014;16(1):3–10. | |
Kjeldsen AD, Moller TR, Brusgaard K, Vase P, Andersen PE. Clinical symptoms according to genotype amongst patients with hereditary haemorrhagic telangiectasia. J Intern Med. 2005;258(4):349–355. | |
Dunlop MG; British Society for Gastroenterology; Association of Coloproctology for Great Britain and Ireland. Guidance on gastrointestinal surveillance for hereditary non-polyposis colorectal cancer, familial adenomatous polypolis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut. 2002;51 Suppl 5:V21–V27. | |
Geirdal AO, Dheyauldeen S, Bachmann-Harildstad G, Heimdal K. Quality of life in patients with hereditary hemorrhagic telangiectasia in Norway: a population based study. Am J Med Genet A. 2012;158A(6):1269–1278. | |
Geisthoff UW, Heckmann K, D’Amelio R, et al. Health-related quality of life in hereditary hemorrhagic telangiectasia. Otolaryngol Head Neck Surg. 2007;136(5):726–733; discussion 734–725. | |
Pasculli G, Resta F, Guastamacchia E, Di Gennaro L, Suppressa P, Sabbà C. Health-related quality of life in a rare disease: hereditary hemorrhagic telangiectasia (HHT) or Rendu-Osler-Weber disease. Qual Life Res. Dec 2004;13(10):1715–1723. | |
Hoag JB, Terry P, Mitchell S, Reh D, Merlo CA. An epistaxis severity score for hereditary hemorrhagic telangiectasia. Laryngoscope. 2010; 120(4):838–843. | |
Silva BM, Hosman AE, Devlin HL, Shovlin CL. Lifestyle and dietary influences on nosebleed severity in hereditary hemorrhagic telangiectasia. Laryngoscope. 2013;123(5):1092–1099. | |
Mahoney EJ, Shapshay SM. Nd-YAG laser photocoagulation for epistaxis associated with hereditary hemorrhagic telangiectasia. Laryngoscope. 2005;115(2):373–375. | |
Karapantzos I, Tsimpiris N, Goulis DG, Van Hoecke H, Van Cauwenberge P, Danielides V. Management of epistaxis in hereditary hemorrhagic telangiectasia by Nd:YAG laser and quality of life assessment using the HR-QoL questionnaire. Eur Arch Otorhinolaryngol. 2005;262(10):830–833. | |
Fiorella ML, Ross D, Henderson KJ, White RI Jr. Outcome of septal dermoplasty in patients with hereditary hemorrhagic telangiectasia. Laryngoscope. 2005;115(2):301–305. | |
Lund VJ, Howard DJ. Closure of the nasal cavities in the treatment of refractory hereditary haemorrhagic telangiectasia. J Laryngol Otol. 1997;111(1):30–33. | |
Gluckman JL, Portugal LG. Modified Young’s procedure for refractory epistaxis due to hereditary hemorrhagic telangiectasia. Laryngoscope. 1994;104(9):1174–1177. | |
Richer SL, Geisthoff UW, Livada N, et al. The Young’s procedure for severe epistaxis from hereditary hemorrhagic telangiectasia. Am J Rhinol Allergy. 2012;26(5):401–404. | |
Mortuaire G, Boute O, Hatron PY, Chevalier D. Pilot study of submucosal radiofrequency for epistaxis in hereditary hemorrhagic telangiectasia. Rhinology. 2013;51(4):355–360. | |
Sadick H, Riedel F, Naim R, et al. Patients with hereditary hemorrhagic telangiectasia have increased plasma levels of vascular endothelial growth factor and transforming growth factor-beta1 as well as high ALK1 tissue expression. Haematologica. 2005;90(6):818–828. | |
Dupuis-Girod S, Ginon I, Saurin JC, et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012;307(9):948–955. | |
Simonds J, Miller F, Mandel J, Davidson TM. The effect of bevacizumab (Avastin) treatment on epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope. 2009;119(5):988–992. | |
Karnezis TT, Davidson TM. Treatment of hereditary hemorrhagic telangiectasia with submucosal and topical bevacizumab therapy. Laryngoscope. 2012;122(3):495–497. | |
Guldmann R, Dupret A, Nivoix Y, Schultz P, Debry C. Bevacizumab nasal spray: noninvasive treatment of epistaxis in patients with Rendu-Osler disease. Laryngoscope. 2012;122(5):953–955. | |
Karnezis TT, Davidson TM. Efficacy of intranasal Bevacizumab (Avastin) treatment in patients with hereditary hemorrhagic telangiectasia-associated epistaxis. Laryngoscope. 2011;121(3):636–638. | |
Rohrmeier C, Sachs HG, Kuehnel TS. A retrospective analysis of low dose, intranasal injected bevacizumab (Avastin) in hereditary haemorrhagic telangiectasia. Eur Arch Otorhinolaryngol. 2012;269(2):531–536. | |
Chen St, Karnezis T, Davidson TM. Safety of intranasal Bevacizumab (Avastin) treatment in patients with hereditary hemorrhagic telangiectasia-associated epistaxis. Laryngoscope. 2011;121(3):644–646. | |
Riss D, Burian M, Wolf A, Kranebitter V, Kaider A, Arnoldner C. Intranasal submucosal bevacizumab for epistaxis in hereditary hemorrhagic telangiectasia: a double-blind, randomized, placebo-controlled trial. Head Neck. Epub March 4, 2014. | |
Dupuis-Girod S, Ambrun A, Decullier E, et al. ELLIPSE Study: a Phase 1 study evaluating the tolerance of bevacizumab nasal spray in the treatment of epistaxis in hereditary hemorrhagic telangiectasia. MAbs. 2014;6(3):794–799. | |
Franchini M, Frattini F, Crestani S, Bonfanti C. Novel treatments for epistaxis in hereditary hemorrhagic telangiectasia: a systematic review of the clinical experience with thalidomide. J Thromb Thrombolysis. 2013;36(3):355–357. | |
Lebrin F, Srun S, Raymond K, et al. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med. 2010;16(4):420–428. | |
Storch CH, Hoeger PH. Propranolol for infantile haemangiomas: insights into the molecular mechanisms of action. Br J Dermatol. 2010;163(2):269–274. | |
Olitsky SE. Topical timolol for the treatment of epistaxis in hereditary hemorrhagic telangiectasia. Am J Otolaryngol. 2012;33(3):375–376. | |
Morais D, Millás T, Zarrabeitia R, Botella LM, Almaraz A. Local sclerotherapy with polydocanol (Aethoxysklerol®) for the treatment of epistaxis in Rendu-Osler-Weber or hereditary hemorrhagic telangiectasia (HHT): 15 years of experience. Rhinology. Mar 2012;50(1):80–86. | |
Jameson JJ, Cave DR. Hormonal and antihormonal therapy for epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope. 2004;114(4):705–709. | |
Yaniv E, Preis M, Shevro J, Nageris B, Hadar T. Anti-estrogen therapy for hereditary hemorrhagic telangiectasia – a long-term clinical trial. Rhinology. 2011;49(2):214–216. | |
Yaniv E, Preis M, Hadar T, Shvero J, Haddad M. Antiestrogen therapy for hereditary hemorrhagic telangiectasia: a double-blind placebo-controlled clinical trial. Laryngoscope. 2009;119(2):284–288. | |
Albiñana V, Bernabeu-Herrero ME, Zarrabeitia R, Bernabéu C, Botella LM. Estrogen therapy for hereditary haemorrhagic telangiectasia (HHT): Effects of raloxifene, on Endoglin and ALK1 expression in endothelial cells. Thromb Haemost. 2010;103(3):525–534. | |
Geisthoff UW, Seyfert UT, Kübler M, Bieg B, Plinkert PK, König J. Treatment of epistaxis in hereditary hemorrhagic telangiectasia with tranexamic acid – a double-blind placebo-controlled cross-over phase IIIB study. Thromb Res. 2014;134(3):565–571. | |
Korzenik JR, Topazian MD, White R. Treatment of bleeding in hereditary hemorrhagic telangiectasia with aminocaproic acid. N Engl J Med. 1994;331(18):1236. | |
de Gussem EM, Snijder RJ, Disch FJ, Zanen P, Westermann CJ, Mager JJ. The effect of N-acetylcysteine on epistaxis and quality of life in patients with HHT: a pilot study. Rhinology. 2009;47(1):85–88. | |
Finnamore H, Le Couteur J, Hickson M, Busbridge M, Whelan K, Shovlin CL. Hemorrhage-adjusted iron requirements, hematinics and hepcidin define hereditary hemorrhagic telangiectasia as a model of hemorrhagic iron deficiency. PLoS One. 2013;8(10):e76516. | |
Haq AU, Glass J, Netchvolodoff CV, Bowen LM. Hereditary hemorrhagic telangiectasia and danazol. Ann Intern Med. 1988; 109(2):171. | |
Fleagle JM, Bobba RK, Kardinal CG, Freter CE. Iron deficiency anemia related to hereditary hemorrhagic telangiectasia: response to treatment with bevacizumab. Am J Med Sci. 2012;343(3):249–251. | |
Lupu A, Stefanescu C, Treton X, Attar A, Corcos O, Bouhnik Y. Bevacizumab as rescue treatment for severe recurrent gastrointestinal bleeding in hereditary hemorrhagic telangiectasia. J Clin Gastroenterol. 2013;47(3):256–257. | |
Wang XY, Chen Y, Du Q. Successful treatment of thalidomide for recurrent bleeding due to gastric angiodysplasia in hereditary hemorrhagic telangiectasia. Eur Rev Med Pharmacol Sci. 2013;17(8):1114–1116. | |
Suppressa P, Liso A, Sabbà C. Low dose intravenous bevacizumab for the treatment of anaemia in hereditary haemorrhagic telangiectasia. Br J Haematol. 2011;152(4):365. | |
Bowcock SJ, Patrick HE. Lenalidomide to control gastrointestinal bleeding in hereditary haemorrhagic telangiectasia: potential implications for angiodysplasias? Br J Haematol. 2009;146(2):220–222. | |
Skaro AI, Marotta PJ, McAlister VC. Regression of cutaneous and gastrointestinal telangiectasia with sirolimus and aspirin in a patient with hereditary hemorrhagic telangiectasia. Ann Intern Med. 2006;144(3):226–227. | |
Wheatley-Price P, Shovlin C, Chao D. Interferon for metastatic renal cell cancer causing regression of hereditary hemorrhagic telangiectasia. J Clin Gastroenterol. 2005;39(4):344–345. | |
van Cutsem E, Rutgeerts P, Vantrappen G. Treatment of bleeding gastrointestinal vascular malformations with oestrogen-progesterone. Lancet. 1990;335(8695):953–955. | |
Bown SG, Swain CP, Storey DW, et al. Endoscopic laser treatment of vascular anomalies of the upper gastrointestinal tract. Gut. 1985; 26(12):1338–1348. | |
Sargeant IR, Loizou LA, Rampton D, Tulloch M, Bown SG. Laser ablation of upper gastrointestinal vascular ectasias: long term results. Gut. 1993;34(4):470–475. | |
Gostout CJ, Bowyer BA, Ahlquist DA, Viggiano TR, Balm RK. Mucosal vascular malformations of the gastrointestinal tract: clinical observations and results of endoscopic neodymium: yttrium-aluminum-garnet laser therapy. Mayo Clin Proc. 1988;63(10):993–1003. | |
Moussouttas M, Fayad P, Rosenblatt M, et al. Pulmonary arteriovenous malformations: cerebral ischemia and neurologic manifestations. Neurology. 2000;55(7):959–964. | |
Angriman F, Ferreyro BL, Wainstein EJ, Serra MM. Pulmonary arteriovenous malformations and embolic complications in patients with hereditary hemorrhagic telangiectasia. Arch Bronconeumol. 2014;50(7):301–304. | |
Verkerk MM, Shovlin CL, Lund VJ. Silent threat? A retrospective study of screening practices for pulmonary arteriovenous malformations in patients with hereditary haemorrhagic telangiectasia. Rhinology. 2012;50(3):277–283. | |
Feinstein JA, Moore P, Rosenthal DN, Puchalski M, Brook MM. Comparison of contrast echocardiography versus cardiac catheterization for detection of pulmonary arteriovenous malformations. Am J Cardiol. 2002;89(3):281–285. | |
Velthuis S, Buscarini E, van Gent MW, et al. Grade of pulmonary right-to-left shunt on contrast echocardiography and cerebral complications: a striking association. Chest. 2013;144(2):542–548. | |
Velthuis S, Buscarini E, Mager JJ, et al. Predicting the size of pulmonary arteriovenous malformations on chest computed tomography: a role for transthoracic contrast echocardiography. Eur Respir J. 2014;44(1):150–159. | |
Hanneman K, Faughnan ME, Prabhudesai V. Cumulative radiation dose in patients with hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations. Can Assoc Radiol J. 2014;65(2):135–140. | |
Schneider G, Uder M, Koehler M, et al. MR angiography for detection of pulmonary arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia. AJR Am J Roentgenol. 2008;190(4):892–901. | |
Shovlin CL, Jackson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax. 2008;63(3):259–266. | |
Hart JL, Aldin Z, Braude P, Shovlin CL, Jackson J. Embolization of pulmonary arteriovenous malformations using the Amplatzer vascular plug: successful treatment of 69 consecutive patients. Eur Radiol. 2010;20(11):2663–2670. | |
Kucukay F, Ozdemir M, Senol E, Okten S, Ereren M, Karan A. Large pulmonary arteriovenous malformations: long-term results of embolization with AMPLATZER vascular plugs. J Vasc Interv Radiol. 2014;25(9):1327–1332. | |
Hundt W, Kalinowski M, Kiessling A, et al. Novel approach to complex pulmonary arteriovenous malformation embolization using detachable coils and Amplatzer vascular plugs. Eur J Radiol. 2012;81(5):e732–e738. | |
Mager JJ, Overtoom TT, Blauw H, Lammers JW, Westermann CJ. Embolotherapy of pulmonary arteriovenous malformations: long-term results in 112 patients. J Vasc Interv Radiol. 2004;15(5):451–456. | |
Gupta P, Mordin C, Curtis J, Hughes JM, Shovlin CL, Jackson JE. Pulmonary arteriovenous malformations: effect of embolization on right-to-left shunt, hypoxemia, and exercise tolerance in 66 patients. AJR Am J Roentgenol. 2002;179(2):347–355. | |
Hsu CC, Kwan GN, Thompson SA, Evans-Barns H, van Driel ML. Embolisation for pulmonary arteriovenous malformation. Cochrane Database Syst Rev. 2012;8:CD008017. | |
Remy-Jardin M, Dumont P, Brillet PY, Dupuis P, Duhamel A, Remy J. Pulmonary arteriovenous malformations treated with embolotherapy: helical CT evaluation of long-term effectiveness after 2–21-year follow-up. Radiology. 2006;239(2):576–585. | |
Woodward CS, Pyeritz RE, Chittams JL, Trerotola SO. Treated pulmonary arteriovenous malformations: patterns of persistence and associated retreatment success. Radiology. 2013;269(3):919–926. | |
Kawai T, Shimohira M, Kan H, et al. Feasibility of time-resolved MR angiography for detecting recanalization of pulmonary arteriovenous malformations treated with embolization with platinum coils. J Vasc Interv Radiol. 2014;25(9):1339–1347. | |
Lee WL, Graham AF, Pugash RA, et al. Contrast echocardiography remains positive after treatment of pulmonary arteriovenous malformations. Chest. 2003;123(2):351–358. | |
Fukushima H, Mitsuhashi T, Oto T, et al. Successful lung transplantation in a case with diffuse pulmonary arteriovenous malformations and hereditary hemorrhagic telangiectasia. Am J Transplant. 2013;13(12):3278–3281. | |
Shovlin C, Bamford K, Wray D. Post-NICE 2008: antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J. 2008;205(10):531–533. | |
Mason CG, Shovlin CL. Flight-related complications are infrequent in patients with hereditary haemorrhagic telangiectasia/pulmonary arteriovenous malformations, despite low oxygen saturations and anaemia. Thorax. 2012;67(1):80–81. | |
Willemse RB, Mager JJ, Westermann CJ, Overtoom TT, Mauser H, Wolbers JG. Bleeding risk of cerebrovascular malformations in hereditary hemorrhagic telangiectasia. J Neurosurg. 2000;92(5):779–784. | |
Willinsky RA, Taylor SM, TerBrugge K, Farb RI, Tomlinson G, Montanera W. Neurologic complications of cerebral angiography: prospective analysis of 2,899 procedures and review of the literature. Radiology. 2003;227(2):522–528. | |
Gauvrit JY, Oppenheim C, Nataf F, et al. Three-dimensional dynamic magnetic resonance angiography for the evaluation of radiosurgically treated cerebral arteriovenous malformations. Eur Radiol. 2006;16(3):583–591. | |
Lundby B, Gordon P, Hugo F. MRI in children given gadodiamide injection: safety and efficacy in CNS and body indications. Eur J Radiol. 1996;23(3):190–196. | |
Mori H, Aoki S, Okubo T, et al. Two-dimensional thick-slice MR digital subtraction angiography in the assessment of small to medium-size intracranial arteriovenous malformations. Neuroradiology. 2003;45(1):27–33. | |
Mukherji SK, Quisling RG, Kubilis PS, Finn JP, Friedman WA. Intracranial arteriovenous malformations: quantitative analysis of magnitude contrast MR angiography versus gradient-echo MR imaging versus conventional angiography. Radiology. 1995;196(1):187–193. | |
Spetzler RF, Martin NA. A proposed grading system for arteriovenous malformations. J Neurosurg. 1986;65(4):476–483. | |
Kano H, Kondziolka D, Flickinger JC, et al. Stereotactic radiosurgery for arteriovenous malformations after embolization: a case-control study. J Neurosurg. 2012;117(2):265–275. | |
Brown RD Jr, Flemming KD, Meyer FB, Cloft HJ, Pollock BE, Link ML. Natural history, evaluation, and management of intracranial vascular malformations. Mayo Clin Proc. 2005;80(2):269–281. | |
Buscarini E, Plauchu H, Garcia Tsao G, et al. Liver involvement in hereditary hemorrhagic telangiectasia: consensus recommendations. Liver Int. 2006;26(9):1040–1046. | |
Caselitz M, Bahr MJ, Bleck JS, et al. Sonographic criteria for the diagnosis of hepatic involvement in hereditary hemorrhagic telangiectasia (HHT). Hepatology. 2003;37(5):1139–1146. | |
Garcia-Tsao G. Liver involvement in hereditary hemorrhagic telangiectasia (HHT). J Hepatol. 2007;46(3):499–507. | |
Ocran K, Rickes S, Heukamp I, Wermke W. Sonographic findings in hepatic involvement of hereditary haemorrhagic telangiectasia. Ultraschall Med. 2004;25(3):191–194. | |
Ravard G, Soyer P, Boudiaf M, et al. Hepatic involvement in hereditary hemorrhagic telangiectasia: helical computed tomography features in 24 consecutive patients. J Comput Assist Tomogr. 2004;28(4):488–495. | |
Buonamico P, Suppressa P, Lenato GM, et al. Liver involvement in a large cohort of patients with hereditary hemorrhagic telangiectasia: echo-color-Doppler vs multislice computed tomography study. J Hepatol. 2008;48(5):811–820. | |
Dupuis-Girod S, Chesnais AL, Ginon I, et al. Long-term outcome of patients with hereditary hemorrhagic telangiectasia and severe hepatic involvement after orthotopic liver transplantation: a single-center study. Liver Transpl. 2010;16(3):340–347. | |
Cag M, Audet M, Saouli AC, et al. Successful liver transplantation for Rendu-Weber-Osler disease, a single centre experience. Hepatol Int. 2011;5(3):834–840. | |
Lerut J, Orlando G, Adam R, et al. Liver transplantation for hereditary hemorrhagic telangiectasia: report of the European liver transplant registry. Ann Surg. 2006;244(6):854–862; discussion 862–854. | |
Chavan A, Caselitz M, Gratz KF, et al. Hepatic artery embolization for treatment of patients with hereditary hemorrhagic telangiectasia and symptomatic hepatic vascular malformations. Eur Radiol. 2004;14(11):2079–2085. | |
Chavan A, Galanski M, Wagner S, et al. Hereditary hemorrhagic telangiectasia: effective protocol for embolization of hepatic vascular malformations – experience in five patients. Radiology. 1998;209(3):735–739. | |
Miller FJ Jr, Whiting JH, Korzenik JR, White RI. Caution with use of hepatic embolization in the treatment of hereditary hemorrhagic telangiectasia. Radiology. 1999;213(3):928–930. | |
Chavan A, Luthe L, Gebel M, et al. Complications and clinical outcome of hepatic artery embolisation in patients with hereditary haemorrhagic telangiectasia. Eur Radiol. 2013;23(4):951–957. | |
Sabba C, Pompili M. Review article: the hepatic manifestations of hereditary haemorrhagic telangiectasia. Aliment Pharmacol Ther. 2008;28(5):523–533. | |
Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358(11):1129–1136. | |
Gershon AS, Faughnan ME, Chon KS, et al. Transcatheter embolotherapy of maternal pulmonary arteriovenous malformations during pregnancy. Chest. 2001;119(2):470–477. | |
Liu XJ, Wang S, Zhao YL, et al. Risk of cerebral arteriovenous malformation rupture during pregnancy and puerperium. Neurology. 2014;82(20):1798–1803. | |
de Gussem EM, Lausman AY, Beder AJ, et al. Outcomes of pregnancy in women with hereditary hemorrhagic telangiectasia. Obstet Gynecol. 2014;123(3):514–520. | |
Edwards CP, Shehata N, Faughnan ME. Hereditary hemorrhagic telangiectasia patients can tolerate anticoagulation. Ann Hematol. 2012;91(12):1959–1968. | |
Brenner D, Elliston C, Hall E, Berdon W. Estimated risks of radiation-induced fatal cancer from pediatric CT. AJR Am J Roentgenol. 2001;176(2):289–296. | |
Chodick G, Ronckers CM, Shalev V, Ron E. Excess lifetime cancer mortality risk attributable to radiation exposure from computed tomography examinations in children. Isr Med Assoc J. 2007;9(8):584–587. | |
Thierry-Chef I, Simon SL, Land CE, Miller DL. Radiation dose to the brain and subsequent risk of developing brain tumors in pediatric patients undergoing interventional neuroradiology procedures. Radiat Res. 2008;170(5):553–565. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.