")
Back to Archived Journals » Oncolytic Virotherapy » Volume 7
Oncolytic virus and PD-1/PD-L1 blockade combination therapy
Authors Chen CY, Hutzen B, Wedekind MF, Cripe TP
Received 22 February 2018
Accepted for publication 1 May 2018
Published 31 July 2018 Volume 2018:7 Pages 65—77
DOI https://doi.org/10.2147/OV.S145532
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Chun-Yu Chen,1 Brian Hutzen,1 Mary F Wedekind,1,2 Timothy P Cripe1,2
1Department of Pediatrics, Center for Childhood Cancer and Blood Diseases, Nationwide Children’s Hospital, 2Division of Hematology/Oncology/Blood and Marrow Transplantation, Nationwide Children’s Hospital, The Ohio State University, Columbus, OH, USA
Abstract: Oncolytic viruses are lytic for many types of cancers but are attenuated or replication-defective in normal tissues. Aside from tumor lysis, oncolytic viruses can induce host immune responses against cancer cells and may thus be viewed as a form of immunotherapy. Although recent successes with checkpoint inhibitors have shown that enhancing antitumor immunity can be effective, the dynamic nature of the immunosuppressive tumor microenvironment presents significant hurdles to the broader application of these therapies. Targeting one immune-suppressive pathway may not be sufficient to eliminate tumors. Here we focus on the development of the combination of oncolytic virotherapy with checkpoint inhibitors designed to target the programmed cell death protein 1 and programmed cell death ligand 1 signaling axis. We also discuss future directions for the clinical application of this novel combination therapy.
Keywords: cancer, viral oncolysis, immunotherapy, immune checkpoint blockade
Introduction
Oncolytic virotherapy
“Viral oncolysis” is the destruction of a tumor cell following viral infection. Reports of using infectious agents to induce tumor shrinkage date back at least a century, albeit with varying and largely anecdotal accounts of their success. The field of oncolytic virotherapy has steadily evolved in the decades since, and it has now entered a phase of rapid maturation as many of these so-called “oncolytic viruses” find their way into clinical use.1–4
Oncolytic virotherapy induces multiple antitumor mechanisms. As part of their lytic virus life cycle, oncolytic viruses can infect tumor cells and cause tumor lysis independent of conventional drug-resistance mechanisms.5 In addition, oncolytic viruses are capable of self-propagation and spreading to nearby tumor cells, making them potentially useful in conducting “biological surgery” for bulky disease. Tumor specificity is achieved by deleting gene(s) crucial for virus replication in normal cells or by utilizing viruses that are incapable of infecting human hosts aside from transformed cells.1 Many oncolytic viruses can also induce a form of immunogenic death in their infected target cells. This effect helps to sensitize host immunity by releasing pathogen-associated molecular patterns and damage-associated molecular patterns, which in turn facilitate dendritic cell infiltration and cross-presentation of tumor-associated antigens (TAAs) that promote antitumor immune responses.6 Immunogenic cell death can induce both innate and adaptive immune responses that contribute to antitumor efficacy directly or indirectly, making oncolytic viruses distinct from many other immunotherapies that only target one or a few immune-suppressive pathways.6,7 Virus infection may also sensitize tumor cells to external apoptotic stimuli such as chemotherapy or radiation therapy, resulting in improved therapeutic outcomes.8–17 Many oncolytic viruses can also accommodate genetic insertion of therapeutic transgenes (a process known as “arming”), that when expressed within the confines of the tumor, lead to enhanced efficacy.18,19 Although oncolytic virotherapy has vast potential, there are limits to what it can achieve as a monotherapy. As such, great efforts are now being made to find rational combination therapies that can further enhance oncolytic virus antitumor efficacy. One such method is by bolstering oncolytic virus-mediated immunogenic cell death with immune checkpoint therapy, particularly through inhibition of the programmed cell death protein 1 (PD-1)/programmed cell death ligand 1 (PD-L1) signaling axis.
PD-1 and its ligands
PD-1 is a cell-surface receptor that regulates immune cell function by delivering inhibitory signals upon engagement with its ligands, PD-L1 and PD-L2.20 PD-1 is a type I transmembrane receptor of the immunoglobulin superfamily.21 Its ligation triggers phosphorylation of a cytoplasmic immunoreceptor tyrosine-based switch motif and recruitment of an Src homology 2 domain-containing phosphatase, which in turn leads to the inhibition of T-cell receptor or B-cell receptor signaling.22–24 Although PD-1 signaling is best characterized in lymphoid cells, it also has roles in inhibiting the activities of certain myeloid cell subsets.25 For example, when PD-1 expression is induced in dendritic cells, it attenuates their ability to respond to infection by suppressing production of proinflammatory cytokines like interleukin-12 (IL-12) and tumor necrosis factor alpha.26 Likewise, expression of PD-1 by natural killer (NK) cells is associated with downregulation of both granzyme-B and interferon-gamma (IFNγ) resulting in severely impaired tumor cell-killing capability.27 Recent evidence also shows that PD-1 is found on tumor-associated macrophages, where its expression is inversely correlated with macrophage’s ability to phagocytose tumor cells.28
PD-1 has two ligands, which are both members of the B7 family of cell-surface proteins: PD-L1 (B7-H1) and PD-L2 (B7-DC).29–32 Although PD-L1 and PD-L2 show overlapping function in negative regulation of T-cell response, recent studies have revealed that each PD-1 ligand can contribute to immune suppression by interacting with distinct cell-surface receptors. PD-L1, for example, can bind the costimulatory molecule B7-1 (CD80) expressed on activated T cells and inhibit their proliferation.33 PD-L2, on the other hand, has been shown to interact with repulsive guidance molecule B (a co-receptor for bone morphogenetic proteins), where it impedes the development of lung tolerance by suppressing T-cell expansion.34
Although PD-L1 and PD-L2 expressions serve an important physiologic role in dampening T-cell activity to prevent excessive inflammation and autoimmunity, tumor cells and tumor-associated stromal cells often overexpress these ligands as a means to attenuate the host antitumor response. The importance of this signaling axis in cancer is underscored by the creation and US Food and Drug Administration (FDA) approval of antibodies designed to target PD-1 and PD-L1 as clinical cancer therapies.35 Combining PD-1 blockade with oncolytic viruses is attractive because it ameliorates a common issue with virotherapy: while these agents are effective in promoting antitumor immunity, this heightened inflammatory response often leads to upregulation of PD-1/PD-L1 on the cell types that comprise the tumor microenvironment.36–39 Tandem inhibition of the PD-1/PD-L1 signaling axis can, thus, potentially alleviate what would otherwise be a dampened antitumor immune response. This review summarizes current strategies for combining various oncolytic viruses with PD-1/PD-L1 blockade (Table 1) and discusses pertinent findings from both preclinical and clinical studies.
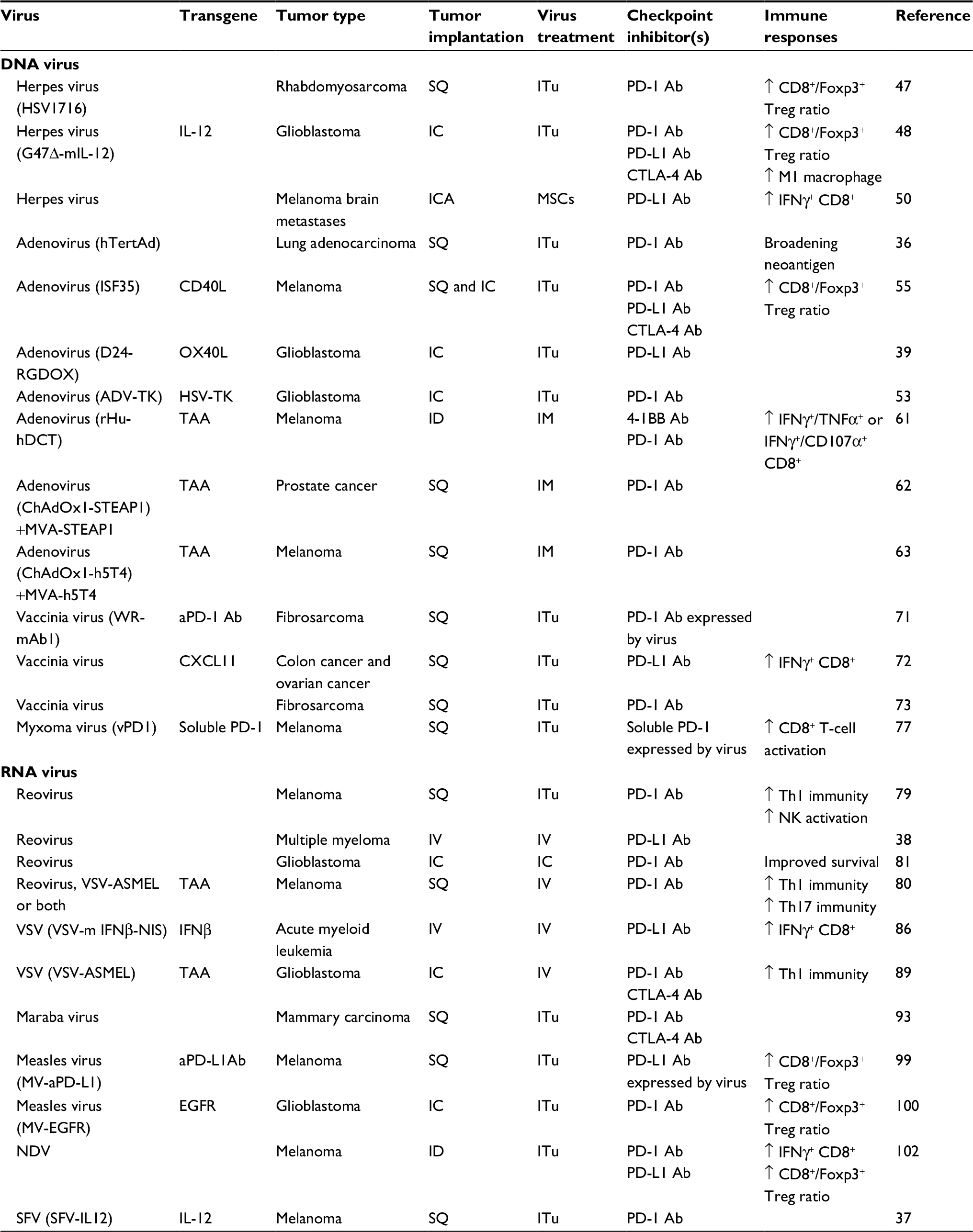 | Table 1 Preclinical studies of oncolytic virus + anti-PD-1 or anti-PD-L1 Note: “↑” represented as increased/enhanced. Abbreviations: ADV, adenovirus; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; EGFR, epidermal growth factor receptor; HSV, herpes simplex virus; IC, intracranial; ICA, internal carotid artery; ID, intradermal; IFN, interferon; IL-12, interleukin 12; IM, intramuscular; ITu, intratumoral; IV, intravenous; MSCs, mesenchymal stem cells; MVA, modified vaccinia Ankara virus; NDV; Newcastle disease virus; NK, natural killer; PD-1, programmed cell death protein 1; PD-L1, programmed cell death ligand 1; SFV, Semliki Forest virus; SQ, subcutaneous; TAA, tumor-associated antigen; TK, thymidine kinase; VSV, vesicular stomatitis virus. |
Combination of oncolytic DNA viruses with PD-1/PD-L1 checkpoint inhibitors
Oncolytic herpes simplex virus with PD-1 or PD-L1 blockade
Oncolytic herpes simplex viruses (oHSVs) are double-stranded DNA viruses of the Herpesviridae family that exhibit tropism for a broad range of cancer cell types.40 These viruses have a large genome that is amenable to genetic manipulation and the insertion of therapeutic genes, making them an attractive platform for the development of novel virus-based therapies. Several distinct oHSVs have been engineered by investigators and pharmaceutical companies,41–45 including the recently FDA-approved oHSV talimogene laherparepvec (T-VEC) for the treatment of melanoma.46 Multiple preclinical and early-phase clinical trials have shown that combining these agents with PD-1/PD-L1 blockade can be beneficial. Our lab recently conducted a preclinical study where we combined the oHSV HSV1716 with anti-PD-1 antibody to treat murine models of rhabdomyosarcoma.47 Tumor-bearing mice treated with anti-PD-1 and HSV1716 survived significantly longer than untreated mice or mice receiving individual monotherapy. We noted that positive therapeutic outcomes correlated with increased tumor infiltration of CD4+ and CD8+ T cells, but similar increases in immunosuppressive Foxp3+ T-regulatory (Treg) cells were not observed. We also noted that the efficacy of combination therapy was lost in athymic nude mice, suggesting that adaptive immunity was an essential component of the therapeutic effect.47
Similar observations were reported by Saha et al. in a syngeneic model of glioma.48 In these studies, anti-PD-1 or anti-PD-L1 antibodies were combined with G47∆-mIL-12, an oHSV that expresses murine IL-12 to promote the development of a proinflammatory Th1 response and IFNγ production by NK and T cells.49 While these combinations were more effective than their constituent monotherapies, antitumor efficacy was further enhanced by the addition of an antibody targeted against cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), an immune checkpoint molecule expressed on activated T cells and Tregs that acts to inhibit the T-cell response.48 The combination of virus, and PD-1 and CTLA-4 inhibition produced several durable cures in the treated mice, as evidenced by their resistance to subsequent tumor rechallenge. Analysis of tumor-associated immune cell infiltrates showed that triple combination therapy both reduced the presence of Foxp3+ Treg cells and increased the numbers of CD8+ T cells and antitumor “M1-like” macrophages. Conversely, depletion of CD4, CD8, or macrophage cell populations impaired antitumor efficacy. Taken together, these data suggest that the improved efficacy observed when combining PD-1 axis inhibition with oHSV may be less than optimal if compensatory T-cell suppression mechanisms are left untouched. Likewise, these data underscore the importance of the innate immune system in supporting the development of a productive adaptive immune response.
A recent report from Du et al also suggests that PD-L1 inhibition is beneficial to oHSV virotherapy. This study utilized mesenchymal stem cells as an oHSV delivery vector because of their natural tendency to home toward tumor sites.50 These infected-cell carriers, when administered through carotid artery, were able to successfully reach metastatic melanoma brain lesions, whereas the vast majority of naked oHSV virions were neutralized or lost. Because these lesions also demonstrated high levels of PD-L1 expression, the authors subsequently evaluated the effects of their oHSV construct when combined with anti-PD-L1 antibody therapy. Combination therapy greatly increased overall survival, which was concomitant with higher numbers of IFNγ-producing CD8+ T cells relative to mice receiving individual therapy.
Oncolytic adenovirus with PD-1 blockade
Adenoviruses are nonenveloped, double-stranded DNA viruses of the Adenoviridae family that normally infect a broad range of vertebrate hosts. Oncolytic variants of these viruses are similar to oHSVs in that they can accommodate the insertion of large, therapeutic transgenes.51 Moreover, they have a well-established patient safety profile and were among the first oncolytic agents to be approved by a regulatory agency for the treatment of cancer.52 Their combination with immune checkpoint inhibitors is also a fertile area of investigation.
One recent example is a study published by Speranza et al, who investigated the use of a recombinant oncolytic adenovirus expressing herpes simplex virus thymidine kinase to treat syngeneic models of glioblastoma.53 Thymidine kinase metabolizes the prodrug ganciclovir to its bioactive form, which creates double-stranded DNA breaks leading to cell death and the production of type I immune responses.53,54 While this therapy was effective, analysis of treated animals revealed that it also upregulated intratumoral expression of PD-L1. Combining these therapies with anti-PD-1 antibody increased tumor infiltration of CD8+ T cells and increased production of IFNγ, dramatically improving overall survival.
Woller et al have also recently published a study where they characterized the emergence of antitumor CD8+ T cells in the wake of oncolytic adenovirus treatment with and without PD-1 immunotherapy.36 By using transcriptomic sequencing and algorithm-based neoepitope prediction software, the authors demonstrated that virus treatment elicited a specific panel of cytotoxic CD8+ T-cell responses against tumor neoantigens in a murine lung adenocarcinoma model. Interestingly, the addition of anti-PD1-blocking antibody did not potentiate the existing pool of T-cell responses, but rather broadened the spectrum of neoepitope-specific responses. This finding correlated with superior tumor regression and the eradication of lung metastases in subsequent efficacy studies. These effects were completely abrogated in CD8 knockout mice, again highlighting the important role of CD8+ T cells in contributing to the therapeutic response.
Singh et al recently published a report wherein they used intratumoral injection of an oncolytic adenovirus expressing CD40 ligand (Ad-ISF35) to regulate T-cell activation in a murine melanoma model.55 CD40 is a T-cell co-stimulatory receptor found on antigen-presenting cells. Although CD40 agonists can be used to induce antitumor T-cell immune responses, their systemic administration is known to produce adverse side effects in patients.56–59 Ad-ISF35 was more effective than its parental virus in inhibiting tumor growth, an observation associated with increased infiltration of IFNγ-producing CD8+ T-cells, reduced Tregs, and upregulated expression of PD-1 and PD-L1 on tumor-associated T cells and myeloid cells, respectively. Combining anti-PD-L1 therapy with Ad-ISF35 was beneficial, but also led to increased expression of CTLA-4 on CD8+ T cells. The further combination of this therapy with anti-CTLA-4 antibody substantially prolonged overall survival and produced several durable complete responses. The benefits of triple combination therapy also extended to metastases and noninjected tumors on the contralateral flank of these animals, suggesting the development of systemic antitumor immunity.
Using a similar approach, Jiang et al armed an oncolytic adenovirus with the ligand for OX40, a T-cell co-stimulatory receptor whose engagement promotes T-cell survival and increased cytokine production.39,60 Coupling this novel virus with anti-PD-1 antibody therapy potentiated antitumor CD8+ T-cell proliferation, resulting in cancer-specific immunity, which increased survival in murine models of glioma.39
Modifying adenoviruses to express TAAs is another strategy used to prime antitumor T-cell immunity.61–63 McGray et al examined the use of a recombinant adenovirus expressing the antigen for human dopachrome tautomearase (an enzyme involved in melanin synthesis) to treat a murine melanoma model. Although intramuscular injection of this vector inhibited tumor growth in a prophylactic setting, it had minimal effect against established tumors.64–67 Subsequent pairing of this virus with an agonist to the T-cell costimulatory molecule 4-1BB improved efficacy, but also had the unwanted effect of stimulating PD-L1 and PD-L2 expression on the tumor cells.61 The further addition of anti-PD-1 antibody resulted in higher IFNγ production and enhanced local T-cell activity (due in part to activated NK cells and macrophages), leading to superior antitumor efficacy.
Cappuccini et al reported similar findings by combining oncolytic adenovirus and a modified vaccinia Ankara (MVA) virus to treat the murine B16 melanoma model.63 These viruses were designed to express human 5T4 oncofetal glycoprotein, an N-glycosylated transmembrane protein associated with differentiating embryonic stem cells and tumor-initiating cells of various carcinomas.68 Dual virus therapy could completely protect mice challenged with B16 tumor cells (which overexpress murine 5T4) when administered as a vaccine, but they were only modestly effective against established tumors. Combining oncolytic adenovirus, MVA and anti-PD-1 antibody resulted in stronger induction of 5T4-specific T-cell responses and significantly increased overall survival. These authors employed a similar strategy to treat a murine model of prostate cancer by using an oncolytic adenovirus and MVA that expressed a TAA known as six-transmembrane epithelial antigen of the prostate 1.62 While anti-PD-1 antibody potentiated the antitumor response, anti-PD-L1 therapy had minimal impact. These seemingly discordant results suggest that the binding of PD-1/PD-L2 might be involved. This finding also suggests that profiling the tumor response following oncolytic virotherapy may be useful to determine the best course of immune checkpoint inhibition therapy.
Vaccinia viruses with PD-1 or PD-L1 blockade
Vaccinia virus is a linear double-stranded DNA virus belonging to the Poxviridae family. Many oncolytic strains of vaccinia virus have been developed and tested in clinical trials with promising antitumor efficacy and evidence of safety.69 These viruses can also accommodate the insertion of therapeutic genes.70 To locally induce anti-PD-1 inhibition in tumor, Kleinpeter et al armed the Western Reserve strain of oncolytic vaccinia virus with soluble forms of PD-1-blocking antibodies and evaluated their expression and antitumor efficacy in a murine fibrosarcoma model.71 This construct increased the presence and persistence of an anti-PD-1 antibody in the tumor compared to conventional injection of blocking antibody, leading to significantly improved survival times. Characterization of tumor-infiltrating immune cells showed that virus treatment, regardless of arming status, promoted a similar degree of CD4+ and CD8+ T-cell infiltration. It is possible that the improved efficacy of the anti-PD-1-armed vaccinia virus treatment was due in part to enhanced activation and/or reduced suppression of the present CD8+ T cells, but as no functional assessment of these T-cells was performed, the therapeutic mechanism remains to be elucidated.
Liu et al also showed that mice given anti-PD-L1 and oncolytic vaccinia virus combination therapy displayed prolonged survival and higher intratumoral concentrations of activated CD8+ T cells than their monotherapy cohorts.72 This enhanced efficacy could be abrogated by blocking IFNγ or depleting CD4+ or CD8+ T cells, demonstrating its dependence on adaptive immunity.
Similar observations were reported by Fend et al who used oncolytic vaccinia virus in combination with anti-PD-1 and anti-CTLA-4 antibodies to treat the MCA205 model of murine sarcoma.73 The potentiation of an antitumor T-cell response was reflected in the ability of these combination therapies to impact distant, uninjected tumors. This study also examined the staging of these therapies, finding that the optimal antitumor response was generated when anti-PD-1 or anti-CTLA-4 antibodies were administered after intratumoral virus injection. This suggests that the expression of these markers in response to vaccinia therapy is time-dependent, and that proper staging of immune checkpoint inhibition should be considered to fully enable the antitumor T-cell response.
Oncolytic myxoma virus with PD-1 blockade
Myxoma virus is another family member of Poxviridae. Unlike vaccinia virus, which can infect a wide range of hosts, myxoma virus is rabbit-specific. While numerous studies suggest that myxoma virus can replicate in cancer cells and slow tumor progression, its limited efficacy necessitates rational drug combination studies.74–77 Bartee et al found that combining anti-PD-1 antibody with myxoma virus could significantly enhance overall survival in a murine model of melanoma. Nearly 30% of mice receiving combination therapy exhibited complete responses, whereas monotherapy only delayed tumor growth. This combination therapy produced a side effect, however, as it led to progressive alopecia, an autoimmune disorder resulting in hair loss.77 In an attempt to restrict PD-1 inhibition to the tumor and to reduce systemic autoimmune-like toxicity, the authors engineered an armed myxoma virus expressing a soluble PD-1-blocking antibody. Localized expression of PD-1-inhibiting antibodies not only cured ≥50% of the treated mice, but also reduced the severity of their hair loss. Compared to parental virus or parental virus and anti-PD-1 combination therapy, this armed myxoma virus also recruited more activated CD8+ T cells to tumors. Depleting CD8+ T cells, but not CD4+ T or NK cells, greatly diminished therapeutic outcomes, suggesting that CD8+ T cells contribute to the antitumor efficacy of armed myxoma virotherapy.
Combination of oncolytic RNA viruses with PD-1/PD-L1 checkpoint inhibitors
Reovirus and PD-1 blockade
Respiratory Enteric Orphan virus (reovirus) is a double-stranded RNA virus of the Reoviridae family. Reovirus does not cause human disease, but it can selectively replicate in cancer cells with active Ras signaling pathways.78 In addition to performing direct oncolysis, oncolytic reovirus can induce dendritic cell maturation as well as NK cell recruitment and activation. Rajani et al showed that combination of anti-PD-1 antibody with intratumoral reovirus treatment dramatically improved survival compared to untreated, single-therapy groups in a murine melanoma model.79 Combination therapy enhanced NK killing of virus-infected cells and reduced immune suppression mediated by Foxp3+ Treg cells. A series of studies using depletion antibodies clearly demonstrated that NK and CD8+ T cells, but not CD4+ T cells, were responsible for the antitumor efficacy.
Several studies have shown that reovirus can also be effective when administered systemically.38,80–82 Using a murine model of multiple myeloma, Kelly et al showed that systemic administration of oncolytic reovirus and PD-L1 blockade significantly enhanced antimyeloma efficacy compared to untreated or separate monotherapy groups.38 Ilett et al reported similar benefits in a murine model of melanoma. This group previously demonstrated that systemically administered reovirus rapidly associates with blood cells, which shield them from neutralization and facilitate their delivery to the tumor as “cargo” in the infected cells.82 Exploiting this concept, Ilett et al preconditioned mice with granulocyte-macrophage colony-stimulating factor (GM-CSF) to stimulate these potential carrier cells, which improved reovirus delivery and enhanced antitumor efficacy.82 Supplementing this novel therapeutic approach with anti-PD-1 antibody elicited even greater efficacy by an immune-mediated mechanism that was dependent on the activities of NK cells, monocytes/macrophages, and antitumor CD8+ T cells.80,82
Samson et al utilized a similar strategy to target a murine model of glioma, showing that systemically administered reovirus could cross the blood–brain barrier and upregulate IFN-regulated gene expression in the tumor immune microenvironment. While this promoted concomitant upregulation of PD-1/PD-L1, PD-1 blockade could ameliorate these effects, leading to significantly enhanced efficacy.81
Vesicular stomatitis virus (VSV) and PD-1 blockade
VSV is a single-stranded RNA virus of the Rhabdoviridae family that naturally infects livestock.83 VSV infection in humans is rare, but can occur in agricultural workers that come in contact with infected animals.84 Attenuated oncolytic VSVs, which no longer cause disease but still retain tumor-killing activity, are presently under investigation for the treatment of human and canine cancer patients.85 Shen et al showed that combining anti-PD-L1 antibody with systemic administration of VSV-IFNβ-NIS (a VSV construct encoding the immunostimulatory cytokine IFNβ and the sodium/iodide symporter [NIS], which can be used to uptake radioiodine in infected cells for imaging or therapeutic applications) greatly extended survival in a murine acute myeloid leukemia model.86 Imaging for NIS expression showed that these tumors were highly susceptible to VSV infection, and combination therapy significantly enhanced animal survival compared to the antibody and virus monotherapy controls. This efficacy was associated with an increase of tumor-infiltrating CD4+ and tumor-specific (and VSV-specific) CD8+ T cells. A subsequent immune cell depletion study demonstrated that the loss of NK or CD8+ T cells sharply attenuated the antitumor response, highlighting the importance of a coordinated innate and adaptive immune response in contributing to therapeutic efficacy.86
Using VSVs to express tumor antigens is another strategy used to boost antitumor T-cell immunity.87–89 Cockle et al showed that systemic delivery of complimentary DNA (cDNA) libraries encoded by VSV are able to vaccinate against a wide range of TAAs expressed by tumors of the same histologic type as the source cDNA library. They also characterized the immunogenic profile of several cancer types grown intracranially to determine if the site of tumor seeding could influence its antigenic profile. In doing so, they identified a combination of VSV-expressed TAAs from intracranial melanoma implants that were equally effective against an unrelated murine glioma model.89 Combining these selected VSVs with antibodies against PD-1 alone or anti-PD-1 in combination with anti-CTLA-4 significantly enhanced antitumor efficacy by relieving suppression of the inflammatory immune response.
Ilett et al similarly used a VSV encoding TAAs as part of a prime-boost strategy following the systemic administration of oncolytic reovirus. The rationale for using two distinct oncolytic viruses was based on observations of their differing immunostimulatory properties; reovirus preferentially stimulated CD8+ T cells, whereas the TAA-armed VSV preferentially stimulated CD4+ T-cell helper mechanisms of antitumor immunity. By leveraging the strengths of each virus, the authors were able to potentiate oncolytic virus-induced antitumor immune responses in a murine model of melanoma.80 These activities were further enhanced in mice preconditioned with GM-CSF and given PD-1-blocking antibody, which resulted in superior antitumor responses.
Maraba virus and dual PD-1 and CTLA-4 blockade
Maraba virus is another member of the Rhabdoviridae family. Like VSV, Maraba virus is a compelling oncolytic agent because of its safety profile and lack of preexisting neutralizing antibodies in the human population.90,91 A genetically modified strain of attenuated Maraba virus, designated MG1, is currently under investigation in clinical trials.92 Bourgeois-Daigneault et al examined MG1 virus in a series of “window of opportunity” studies to see if its efficacy against aggressive models of murine mammary carcinoma could be enhanced through proper staging of surgical resection.93 Administering MG1 virus prior to resection improved survival, impaired metastasis, and helped to prevent tumor recurrence through stimulation of an IFN response. However, increased PD-L1 expression and greater influx of Tregs was observed, likely leading to a dampened antitumor immune response generated by MG1 therapy. To counteract these effects, immune checkpoint inhibition therapy (consisting of anti-PD-1 and anti-CTLA-4 antibodies) was added to the treatment regimen following MG1 infection and surgical resection of the primary tumors. The combination of MG1 and immune checkpoint inhibition in this setting had a profound impact on therapeutic outcomes, resulting in several completely cured mice that were resistant to relapse.93
Measles virus and PD-1 blockade
Measles virus (MV) is an enveloped, single-stranded RNA virus of the Paramyxovirdae family.94 Attenuated MVs based on the Edmonston vaccine strain have been shown to selectively target, infect, and induce tumor oncolysis while displaying minimal cytotoxic activity against normal tissues. While oncolytic MV can naturally interact with the CD46, CD150, or nectin 4 cell-surface markers expressed by cancer cells, MV tropism can be redirected through genetic modification of its hemagglutinin gene.95–97 Enhancement of oncolytic MV therapy is also possible through integration of immune-modulatory genes into the MV genome or by combining MV with other immunotherapies.98 Engeland et al constructed MV vectors expressing antibody against PD-L1 (MV-aPD-L1), which improved antitumor efficacy in a murine melanoma model.99 Compared to parental virus, intratumoral administration of MV-aPD-L1 greatly extended survival by increasing CD8+ T-cell infiltration and IFNγ production in tumors while decreasing the presence of Foxp3+ Tregs. In a murine glioma model, Hardcastle et al also successfully improved antitumor efficacy by combining an MV retargeted to recognize the epidermal growth factor receptor (MV-EGFR) with anti-PD-1 antibody.100 While monotherapy only provided modest survival benefit, the combination of intratumoral MV-EGFR and intraperitoneal injection of anti-PD-1 therapy significantly enhanced survival with 60% of the mice remaining alive at the end of the 120-day treatment course. Combination therapy also induced a higher influx of CD8+ T cells and a higher CD8+/Foxp3+ Treg ratio in the brain, suggesting a skew in the tumor microenvironment from immunosuppressive to more immunoreactive. Survival benefits were lost in athymic nude mice, demonstrating that the antitumor effect is reliant on T-cells.
Newcastle disease virus and PD-1 blockade
Newcastle disease virus is another member of the Paramyxovirdae family. Although this virus primarily infects avian species, it can also be transmitted to humans where it causes mild influenza-like symptoms. Attenuated Newcastle disease virus replicates selectively in cancer cells, and recent reports have demonstrated that its efficacy is tied to activation of a robust type I IFN response.101 Zamarin et al recently profiled the transcriptomes of human and syngeneic mouse tumor models following Newcastle disease virus treatment and found that while virus treatment shifted the balance of T-cells from an exhausted to an effector phenotype, it was insufficient to completely eradicate the tumors.102 Further analysis showed that the virus-mediated IFN response upregulated PD-L1 expression in tumor and tumor-infiltrating immune cells. By combining Newcastle disease virus therapy with systemic anti-PD-1 or anti-PD-L1 blockade, the authors were able to markedly enhance the antitumor immune effect, leading to the rejection of directly treated and noninfected tumors alike.
Semliki Forest virus and PD-1 blockade
Semliki Forest virus is a single-stranded RNA virus belonging to the Togaviridae family whose wide host range includes humans.103 Preclinical studies with attenuated Semliki Forest viruses have shown that the antitumor efficacy of these agents can be enhanced through PD-1 inhibition. Quetglas et al combined anti-PD-1 therapy with a Semliki Forest virus expressing murine IL-12 to treat bilateral murine melanoma and colon cancer models.37 While monotherapy control groups showed little to no therapeutic benefit, mice that were given combination therapy showed significantly prolonged survival with >80% of the tumor-bearing mice remaining tumor-free on both treated and untreated flanks during the experimental course. Combining anti-PD-1 therapy with a parental Semliki Forest virus only displayed a modest antitumor effect, however, demonstrating the added benefit of arming the virus with the proinflammatory IL-12 cytokine.
Clinical study of OVs+PD-1 or PD-L1 blockade
Encouraging preclinical results have spurred the generation of multiple clinical trials to investigate the combination of oncolytic virotherapy with inhibition of the PD-1/PD-L1 signaling axis (Table 2). While the majority of these studies remain early phase and/or actively recruiting at the time of this writing, emerging preliminary results such as those outlined above speak to the potential of this therapy. Recent reports from a Phase Ib clinical trial testing the impact of T-VEC combined with anti-PD-1 therapy confirmed an objective response rate of 62% in patients with advanced melanoma, with a complete response rate of 33% per immune-related response criteria.104 Patients enrolled in the study were initially given a low dose of T-VEC to induce seroconversion and a protective antiviral immune response. This was followed by a period of high-dose virus before moving into a combination phase, where the patients were also given intravenous Keytruda® (pembrolizumab), an FDA-approved humanized anti-PD-1 antibody. Scheduled tumor biopsies collected prior to and during the course of treatment revealed broad changes in tumor inflammation, including increased CD8+ T-cell infiltration, upregulation of IFNγ signature genes, and elevated levels of PD-L1 protein expression. Overall, combination treatment resulted in a >50% reduction of 82% injected, 43% noninjected nonvisceral, and 33% of noninjected visceral lesions. No dose-limiting toxicities were reported, and treatment-related side effects were generally minor and in line with previous expectations for T-VEC or pembrolizumab treatment. Although mean progression-free and overall survival rates were not reported in this study because of its small sample size (n=21), an ongoing Phase III clinical trial is currently underway to better evaluate the efficacy of this combination therapy in patients with stage IIIB–IV melanoma (NCT02263508).
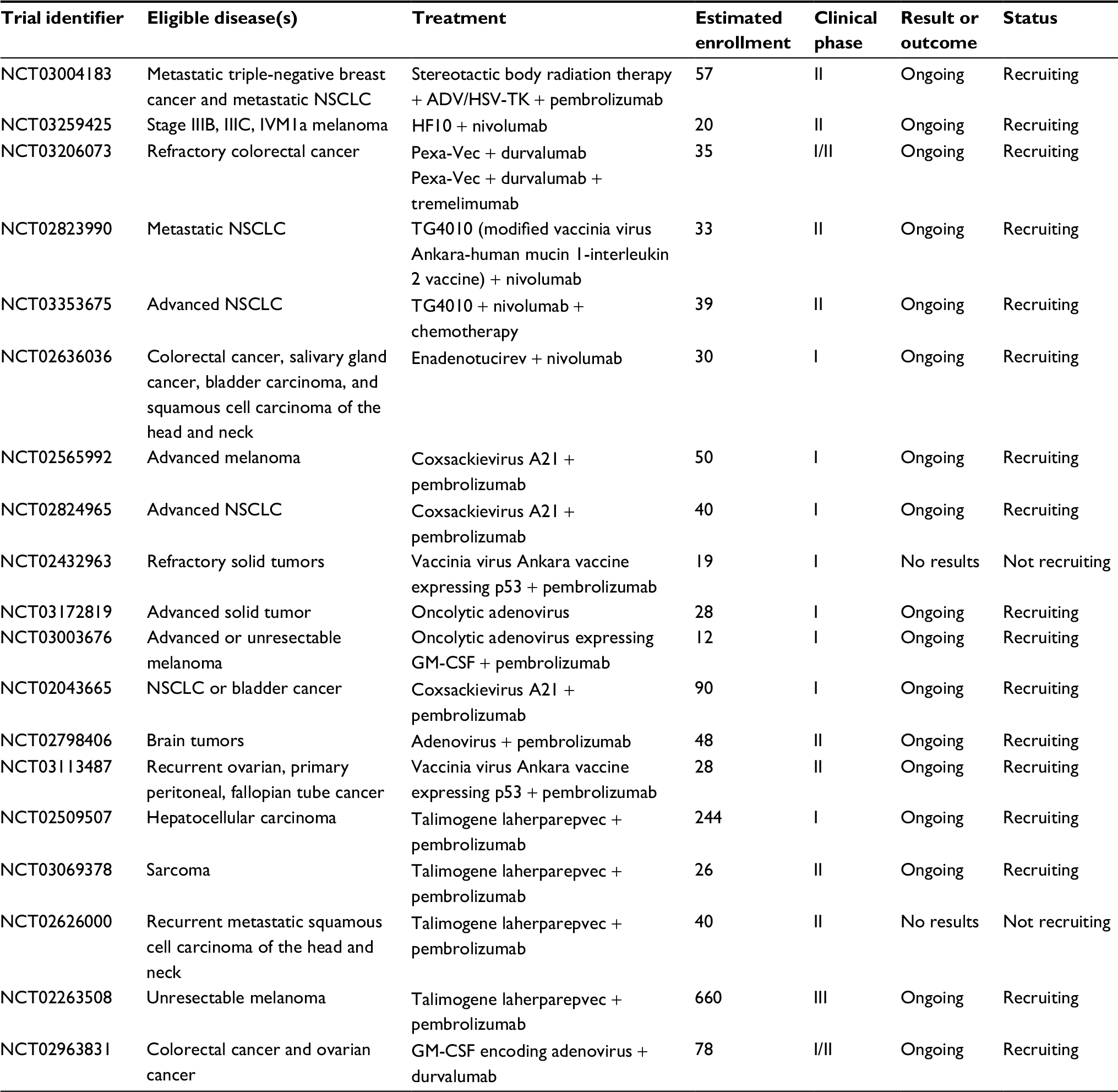 | Table 2 Clinical trials using oncolytic virus + anti-PD-1 or anti-PD-L1 Abbreviations: ADV, adenovirus; GM-CSF, granulocyte-macrophage colony-stimulating factor; HSV, herpes simplex virus; NSCLC, non-small-cell lung cancer; PD-1, programmed cell death protein 1; PD-L1, programmed cell death ligand 1; TK, thymidine kinase. |
Conclusion
The induction of an antitumor immune response is one of the central mechanisms through which oncolytic viruses achieve therapeutic efficacy. Promising results from both preclinical and clinical studies, encompassing a diverse array of viruses and tumor types, show that these antitumor activities can be further potentiated with PD-1/PD-L1 inhibition. The enhanced efficacy of this approach appears to rely on the coordinated response of innate and adaptive cell populations; however, the specific contributions of each can vary between virus and tumor type and are poorly understood in general. In many of the studies outlined here, improved therapeutic outcomes correlated with an influx of CD8+ T cells and a shift in the tumor microenvironment toward a more proinflammatory state, which may be useful prognostic factors for clinical application.
Using oncolytic viruses as a delivery vector for checkpoint inhibitors, immune-modulating cytokines, immunostimulatory ligands, or agonist antibodies in malignant tumors is not only beneficial in terms of antitumor efficacy, but may also greatly reduce adverse events induced by systemic administration of immunotherapies seen in some patients.105,106 Supplementing oncolytic virotherapy with multiple immune checkpoint inhibitors (e.g., PD-1 and CTLA-4) is also rational, as each checkpoint molecule utilizes different mechanisms to inhibit T-cell function and each targets distinct but overlapping T-cell subpopulations.107,108 Recent studies also suggest that inhibitors of the same signaling axis, such as anti-PD-1, anti-PD-L1, and anti-PD-L2, are not necessarily redundant (Figure 1).28–34 Thus, engineering viruses that can target multiple checkpoint molecules may help to fully release tumor-infiltrating T-cell function without inducing systemic toxicity.
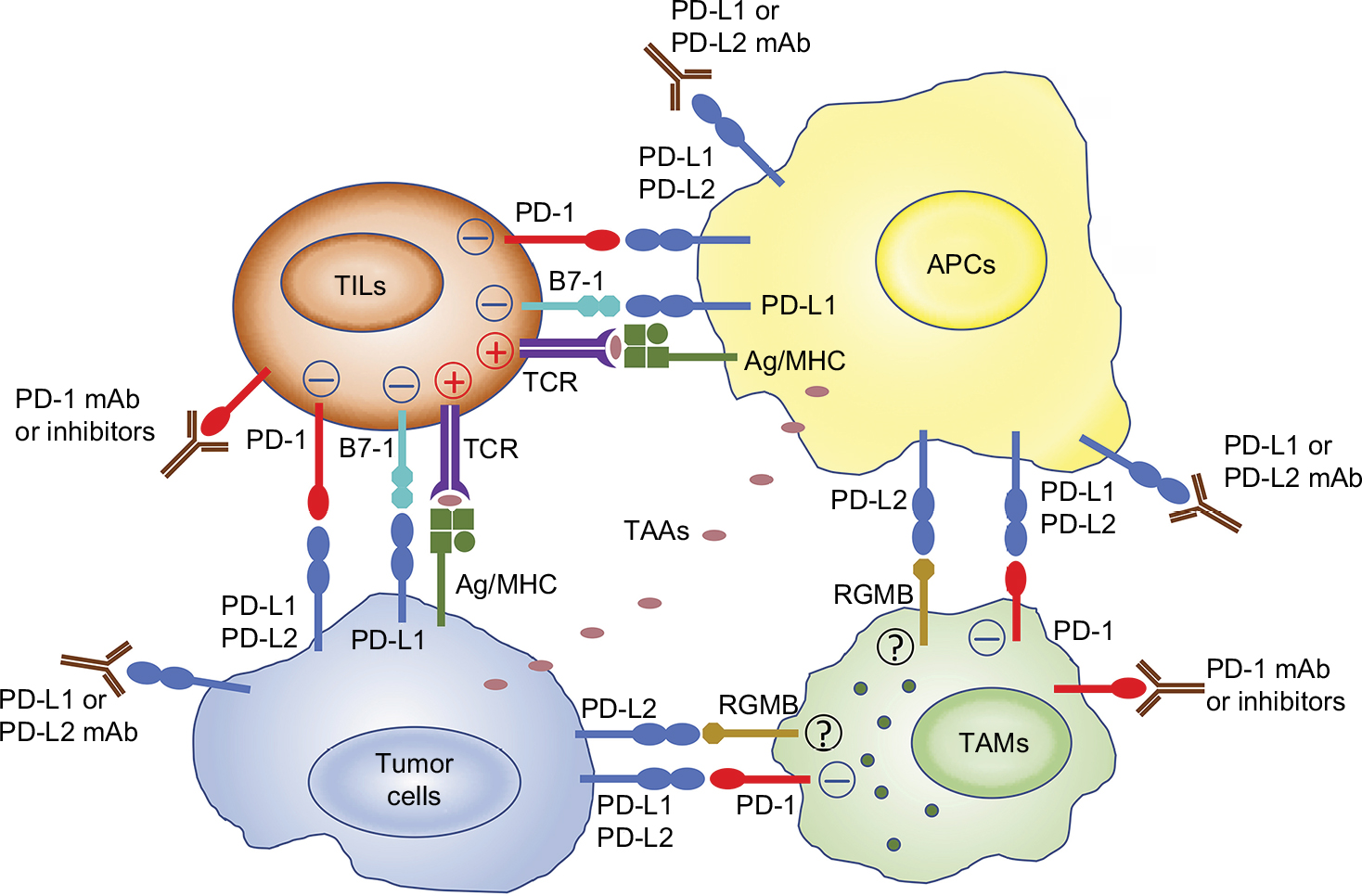 | Figure 1 The complexity of PD-1 and its ligands reveals nonredundant roles for PD-1 or PD-L blockade in cancer immunotherapy. Notes: APCs uptake TAAs from tumor cells and present them to T-cells. Tumor cells can also present TAAs to TILs and active T-cells. PD-1 is expressed on activated T-cells or TAMs and suppresses the antitumor immune responses after binding with PD-1 ligands (PD-L1 or PD-L2) expressed on the tumor cells or APCs. PD-L1 can bind to B7-1 expressed on activated T-cells and RGMB expressed on macrophages, respectively, in other models (but have not been well characterized in tumor models). Therefore, blocking either PD-1, PD-L1, or PD-L2 alone with an mAb or inhibitor may be insufficient to fully release the function of TILs and TAMs. Abbreviations: APC, antigen-presenting cell; mAb, monoclonal antibody; PD-1, programmed cell death protein 1; PD-L1, programmed cell death ligand 1; PD-L2, programmed cell death ligand 2; RGMB, repulsive guidance molecule B; TAA, tumor-associated antigen; TAM, tumor-associated macrophage; TIL, tumor-infiltrating T lymphocyte. |
Despite recent advances in virus-associated cancer immunotherapy, there are more limiting factors that need to be considered. For example, tumor cells can secrete immunosuppressive cytokines, like IL-10 and transforming growth factor beta, or recruit immunosuppressive cells such as M2-like macrophages, Foxp3+ Tregs, or myeloid-derived suppressor cells that can inhibit T-cell function and promote tumor growth.109,110 Targeting these immunosuppressive factors together with oncolytic virotherapy and checkpoint blockade may broaden the application of immunotherapy and improve antitumor efficacy. Proper staging of these therapies will need to be carefully evaluated, as factors that bolster antitumor immunity will likely enhance antiviral immunity as well. Some degree of oncolysis is critical to the therapeutic response, and premature clearance of virus could conceivably be counterproductive.
In summary, oncolytic virotherapy is a novel and safe immunotherapy that can be enhanced by anti PD-1/PD-L1 therapy. Given that immunosuppressive tumor microenvironments can vary significantly and that not all patients will respond to virotherapy and PD-1 blockade combination therapy, the future implementation of rational combinatorial therapy should focus on targeting multiple immunosuppressive pathways as well as enhancing tumor-infiltrating T-cell function to potentiate oncolytic virotherapy. Identifying biomarkers that accurately select which patients will benefit from these combinations and which combinations will be most effective is an ongoing challenge that will need to be addressed to move the field forward. We contend that the impressive preclinical and early clinical results of virotherapy with checkpoint inhibition are only a glimpse of what is possible when combining virotherapy with other emerging immunotherapies.
Disclosure
The authors report no conflicts of interest in this work.
References
Fountzilas C, Patel S, Mahalingam D. Review: oncolytic virotherapy, updates and future directions. Oncotarget. 2017;8(60):102617–102639. | ||
Kenney S, Pagano J. Viruses as oncolytic agents: a new age for “therapeutic” viruses? J Natl Cancer Inst. 1994;86:1185–1186. | ||
Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252(5007):854–856. | ||
Kirn D, Martuza R, Zwiebel J. Replication-selective virotherapy for cancer: biological principles, risk management and future directions. Nat Med. 2001;7:781–787. | ||
Coukos G, Makrigiannakis A, Kang EH, Rubin SC, Albelda SM, Molnar-Kimber KL. Oncolytic herpes simplex virus-1 lacking ICP34.5 induces p53-independent death and is efficacious against chemotherapy-resistant ovarian cancer. Clin Cancer Res. 2000;6(8):3342–3353. | ||
Yin J, Markert JM, Leavenworth JW. Modulation of the intratumoral immune landscape by oncolytic herpes simplex virus virotherapy. Front Oncol. 2017;7:136. | ||
Aurelian L. Oncolytic viruses as immunotherapy: progress and remaining challenges. Onco Targets Ther. 2016;9:2627–2637. | ||
Cook JL, Walker TA, Lewis AM Jr., Ruley HE, Graham FL, Pilder SH. Expression of the adenovirus E1A oncogene during cell transformation is sufficient to induce susceptibility to lysis by host inflammatory cells. Proc Natl Acad Sci USA. 1986;83(18):6965–6969. | ||
Cook JL, May DL, Lewis AM Jr., Walker TA. Adenovirus E1A gene induction of susceptibility to lysis by natural killer cells and activated macrophages in infected rodent cells. J Virol. 1987;61(11):3510–3520. | ||
Cook JL, May DL, Wilson BA, et al. Role of tumor necrosis factor-alpha in E1A oncogene-induced susceptibility of neoplastic cells to lysis by natural killer cells and activated macrophages. J Immunol. 1989;142(12):4527–4534. | ||
Walker TA, Wilson BA, Lewis AM Jr., Cook JL. E1A oncogene induction of cytolytic susceptibility eliminates sarcoma cell tumorigenicity. Proc Natl Acad Sci USA. 1991;88(15):6491–6495. | ||
Routes JM. Adenovirus E1A inhibits IFN-induced resistance to cytolysis by natural killer cells. J Immunol. 1993;150(10):4315–4322. | ||
Cook JL, Wilson BA, Wolf LA, Walker TA. E1A oncogene expression level in sarcoma cells: an independent determinant of cytolytic susceptibility and tumor rejection. Oncogene. 1993;8(3):625–635. | ||
Brader K, Wolf J, Hung M-C, et al. Adenovirus E1A expression enhances the sensitivity of an ovarian cancer cell line to multiple cytotoxic agents through an apoptotic mechanism. Clin Cancer Res. 1997;3:2017–2024. | ||
Sanchez-Prieto R, Quintanilla M, Cano A, et al. Carcinoma cell lines become sensitive to DNA-damaging agents by the expression of the adenovirus E1A gene. Oncogene. 1996;13:1083–1092. | ||
Ueno N, Yu D, Hung M-C. Chemosensitization of HER-2/neu-overexpressing human breast cancer cells to paclitaxel (Taxol) by adenovirus type 5 E1A. Oncogene. 1997;15:953–960. | ||
Kon T, Zhang X, Huang Q, et al. Oncolytic virus-mediated tumor radiosensitization in mice through DNA-PKcs-specific shRNA. Transl Cancer Res. 2012;1(2):4–14. | ||
Hermiston T. Gene delivery from replication-selective viruses: arming guided missiles in the war against cancer. J Clin Invest. 2000;105:1169–1172. | ||
Bauzon M, Hermiston T. Armed therapeutic viruses - a disruptive therapy on the horizon of cancer immunotherapy. Front Immunol. 2014;5:74. | ||
Riella LV, Paterson AM, Sharpe AH, Chandraker A. Role of the PD-1 pathway in the immune response. Am J Transplant. 2012;12(10):2575–2587. | ||
Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med. 2015;21(1):24–33. | ||
Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173(2):945–954. | ||
Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci USA. 2001;98(24):13866–13871. | ||
Dai S, Jia R, Zhang X, Fang Q, Huang L. The PD-1/PD-Ls pathway and autoimmune diseases. Cell Immunol. 2014;290(1):72–79. | ||
Ostrand-Rosenberg S, Horn LA, Haile ST. The programmed death-1 immune-suppressive pathway: barrier to antitumor immunity. J Immunol. 2014;193(8):3835–3841. | ||
Yao S, Wang S, Zhu Y, et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood. 2009;113(23):5811–5818. | ||
Benson DM, Jr., Bakan CE, Mishra A, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116(13):2286–2294. | ||
Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–499. | ||
Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5(12):1365–1369. | ||
Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. | ||
Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2(3):261–268. | ||
Tseng SY, Otsuji M, Gorski K, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193(7):839–846. | ||
Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27(1):111–122. | ||
Xiao Y, Yu S, Zhu B, et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med. 2014;211(5):943–959. | ||
Hsu FS, Su CH, Huang KH. A Comprehensive Review of US FDA-Approved Immune Checkpoint Inhibitors in Urothelial Carcinoma. J Immunol Res. 2017;2017:6940546. | ||
Woller N, Gurlevik E, Fleischmann-Mundt B, et al. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther. 2015;23(10):1630–1640. | ||
Quetglas JI, Labiano S, Aznar MA, et al. Virotherapy with a semliki forest virus-based vector encoding IL12 synergizes with PD-1/PD-L1 Blockade. Cancer Immunol Res. 2015;3(5):449–454. | ||
Kelly KR, Espitia CM, Zhao W, et al. Oncolytic reovirus sensitizes multiple myeloma cells to anti-PD-L1 therapy. Leukemia. 2018;32(1):230–233. | ||
Jiang H, Rivera-Molina Y, Gomez-Manzano C, et al. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res. 2017;77(14):3894–3907. | ||
Kaur B, Chiocca EA, Cripe TP. Oncolytic HSV-1 virotherapy: clinical experience and opportunities for progress. Curr Pharm Biotechnol. 2012;13(9):1842–1851. | ||
Smith ER, Chiocca EA. Oncolytic viruses as novel anticancer agents: turning one scourge against another. Expert Opin Investig Drugs. 2000;9(2):311–327. | ||
Varghese S, Rabkin S. Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther. 2002;9:967–978. | ||
Arceci R, Cripe T. Emerging cancer-targeted therapies. Pediatr Clin North Am. 2002;49:1339–1368. | ||
Cripe T, Mackall C. Exploiting genetic alterations to design novel therapies for cancer. Hematol Oncol Clin North Am. 2001;15:657–675. | ||
Sokolowski NA, Rizos H, Diefenbach RJ. Oncolytic virotherapy using herpes simplex virus: how far have we come? Oncolytic Virother. 2015;4:207–219. | ||
Johnson DB, Puzanov I, Kelley MC. Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy. 2015;7(6):611–619. | ||
Chen CY, Wang PY, Hutzen B, et al. Cooperation of oncolytic herpes virotherapy and PD-1 blockade in murine rhabdomyosarcoma models. Sci Rep. 2017;7(1):2396. | ||
Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32(2):253–267. e255. | ||
Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13(8):722–728. | ||
Du W, Seah I, Bougazzoul O, et al. Stem cell-released oncolytic herpes simplex virus has therapeutic efficacy in brain metastatic melanomas. Proc Natl Acad Sci USA. 2017;114(30):E6157–E6165. | ||
Niemann J, Kuhnel F. Oncolytic viruses: adenoviruses. Virus Genes. 2017;53(5):700–706. | ||
Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. 2006;98(5):298–300. | ||
Speranza MC, Passaro C, Ricklefs F, et al. Preclinical investigation of combined gene-mediated cytotoxic immunotherapy and immune checkpoint blockade in glioblastoma. Neuro Oncol. 2018;20(2):225–235. | ||
Aguilar LK, Guzik BW, Aguilar-Cordova E. Cytotoxic immunotherapy strategies for cancer: mechanisms and clinical development. J Cell Biochem. 2011;112(8):1969–1977. | ||
Singh M, Vianden C, Cantwell MJ, et al. Intratumoral CD40 activation and checkpoint blockade induces T cell-mediated eradication of melanoma in the brain. Nat Commun. 2017;8(1):1447. | ||
Driessens G, Kline J, Gajewski TF. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol Rev. 2009;229(1):126–144. | ||
Sotomayor EM, Borrello I, Tubb E, et al. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat Med. 1999;5(7):780–787. | ||
Vonderheide RH, Flaherty KT, Khalil M, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25(7):876–883. | ||
Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19(5):1035–1043. | ||
Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer. 2016;52:50–66. | ||
McGray AJ, Bernard D, Hallett R, et al. Combined vaccination and immunostimulatory antibodies provides durable cure of murine melanoma and induces transcriptional changes associated with positive outcome in human melanoma patients. Oncoimmunology. 2012;1(4):419–431. | ||
Cappuccini F, Stribbling S, Pollock E, Hill AV, Redchenko I. Immunogenicity and efficacy of the novel cancer vaccine based on simian adenovirus and MVA vectors alone and in combination with PD-1 mAb in a mouse model of prostate cancer. Cancer Immunol Immunother. 2016;65(6):701–713. | ||
Cappuccini F, Pollock E, Stribbling S, Hill AVS, Redchenko I. 5T4 oncofoetal glycoprotein: an old target for a novel prostate cancer immunotherapy. Oncotarget. 2017;8(29):47474–47489. | ||
Kianizad K, Marshall LA, Grinshtein N, et al. Elevated frequencies of self-reactive CD8+ T cells following immunization with a xenoantigen are due to the presence of a heteroclitic CD4+ T-cell helper epitope. Cancer Res. 2007;67(13):6459–6467. | ||
Grinshtein N, Bridle B, Wan Y, Bramson JL. Neoadjuvant vaccination provides superior protection against tumor relapse following surgery compared with adjuvant vaccination. Cancer Res. 2009;69(9):3979–3985. | ||
Grinshtein N, Ventresca M, Margl R, et al. High-dose chemotherapy augments the efficacy of recombinant adenovirus vaccines and improves the therapeutic outcome. Cancer Gene Ther. 2009;16(4):338–350. | ||
Bernard D, Ventresca MS, Marshall LA, Evelegh C, Wan Y, Bramson JL. Processing of tumor antigen differentially impacts the development of helper and effector CD4+ T-cell responses. Mol Ther. 2010;18(6):1224–1232. | ||
McGinn OJ, Krishnan S, Bourquin JP, et al. Targeting the 5T4 oncofetal glycoprotein with an antibody drug conjugate (A1mcMMAF) improves survival in patient-derived xenograft models of acute lymphoblastic leukemia. Haematologica. 2017;102(6):1075–1084. | ||
Pease DF, Kratzke RA. Oncolytic viral therapy for mesothelioma. Front Oncol. 2017;7:179. | ||
Haddad D. Genetically engineered vaccinia viruses as agents for cancer treatment, imaging, and transgene delivery. Front Oncol. 2017;7:96. | ||
Kleinpeter P, Fend L, Thioudellet C, et al. Vectorization in an oncolytic vaccinia virus of an antibody, a Fab and a scFv against programmed cell death -1 (PD-1) allows their intratumoral delivery and an improved tumor-growth inhibition. Oncoimmunology. 2016;5(10):e1220467. | ||
Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun. 2017;8:14754. | ||
Fend L, Yamazaki T, Remy C, et al. Immune checkpoint blockade, immunogenic chemotherapy or IFN-alpha blockade boost the local and abscopal effects of oncolytic virotherapy. Cancer Res. 2017;77(15):4146–4157. | ||
Chan WM, Rahman MM, McFadden G. Oncolytic myxoma virus: the path to clinic. Vaccine. 2013;31(39):4252–4258. | ||
Stanford MM, Shaban M, Barrett JW, et al. Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol Ther. 2008;16(1):52–59. | ||
Tosic V, Thomas DL, Kranz DM, et al. Myxoma virus expressing a fusion protein of interleukin-15 (IL15) and IL15 receptor alpha has enhanced antitumor activity. PLoS One. 2014;9(10):e109801. | ||
Bartee MY, Dunlap KM, Bartee E. Tumor-localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res. 2017;77(11):2952–2963. | ||
Zhao X, Chester C, Rajasekaran N, He Z, Kohrt HE. Strategic combinations: the future of oncolytic virotherapy with reovirus. Mol Cancer Ther. 2016;15(5):767–773. | ||
Rajani K, Parrish C, Kottke T, et al. Combination therapy with reovirus and anti-PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol Ther. 2016;24(1):166–174. | ||
Ilett E, Kottke T, Thompson J, et al. Prime-boost using separate oncolytic viruses in combination with checkpoint blockade improves anti-tumour therapy. Gene Ther. 2017;24(1):21–30. | ||
Samson A, Scott KJ, Taggart D, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. 2018;10(422). | ||
Ilett E, Kottke T, Donnelly O, et al. Cytokine conditioning enhances systemic delivery and therapy of an oncolytic virus. Mol Ther. 2014;22(10):1851–1863. | ||
Hastie E, Grdzelishvili VZ. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J Gen Virol. 2012;93(Pt 12):2529–2545. | ||
Bi Z, Barna M, Komatsu T, Reiss CS. Vesicular stomatitis virus infection of the central nervous system activates both innate and acquired immunity. J Virol. 1995;69(10):6466–6472. | ||
Velazquez-Salinas L, Naik S, Pauszek SJ, Peng KW, Russell SJ, Rodriguez LL. Oncolytic recombinant vesicular stomatitis virus (VSV) is nonpathogenic and nontransmissible in pigs, a natural host of VSV. Hum Gene Ther Clin Dev. 2017;28(2):108–115. | ||
Shen W, Patnaik MM, Ruiz A, Russell SJ, Peng KW. Immunovirotherapy with vesicular stomatitis virus and PD-L1 blockade enhances therapeutic outcome in murine acute myeloid leukemia. Blood. 2016;127(11):1449–1458. | ||
Pulido J, Kottke T, Thompson J, et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat Biotechnol. 2012;30(4):337–343. | ||
Kottke T, Errington F, Pulido J, et al. Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors. Nat Med. 2011;17(7):854–859. | ||
Cockle JV, Rajani K, Zaidi S, et al. Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro Oncol. 2016;18(4):518–527. | ||
Pol JG, Zhang L, Bridle BW, et al. Maraba virus as a potent oncolytic vaccine vector. Mol Ther. 2014;22(2):420–429. | ||
Koppers-Lalic D, Hoeben RC. Non-human viruses developed as therapeutic agent for use in humans. Rev Med Virol. 2011;21(4):227–239. | ||
Brun J, McManus D, Lefebvre C, et al. Identification of genetically modified Maraba virus as an oncolytic rhabdovirus. Mol Ther. 2010;18(8):1440–1449. | ||
Bourgeois-Daigneault MC, Roy DG, Aitken AS, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med. 2018;10(422). | ||
Bhattacharjee S, Yadava PK. Measles virus: background and oncolytic virotherapy. Biochem Biophys Rep. 2018;13:58–62. | ||
Bucheit AD, Kumar S, Grote DM, et al. An oncolytic measles virus engineered to enter cells through the CD20 antigen. Mol Ther. 2003;7(1):62–72. | ||
Hammond AL, Plemper RK, Zhang J, Schneider U, Russell SJ, Cattaneo R. Single-chain antibody displayed on a recombinant measles virus confers entry through the tumor-associated carcinoembryonic antigen. J Virol. 2001;75(5):2087–2096. | ||
Peng KW, Donovan KA, Schneider U, Cattaneo R, Lust JA, Russell SJ. Oncolytic measles viruses displaying a single-chain antibody against CD38, a myeloma cell marker. Blood. 2003;101(7):2557–2562. | ||
Aref S, Bailey K, Fielding A. Measles to the rescue: a review of oncolytic measles virus. Viruses. 2016;8(10). | ||
Engeland CE, Grossardt C, Veinalde R, et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther. 2014;22(11):1949–1959. | ||
Hardcastle J, Mills L, Malo CS, et al. Immunovirotherapy with measles virus strains in combination with anti-PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro Oncol. 2017;19(4):493–502. | ||
Zamarin D, Holmgaard RB, Subudhi SK, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6(226):226ra232. | ||
Zamarin D, Ricca JM, Sadekova S, et al. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J Clin Invest. 2018;128(4):1413–1428. | ||
Lundstrom K. Oncolytic alphaviruses in cancer immunotherapy. Vaccines (Basel). 2017;5(2). | ||
Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170(6):1109–1119. e1110. | ||
Tugues S, Burkhard SH, Ohs I, et al. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015;22(2):237–246. | ||
Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26(12):2375–2391. | ||
Wei SC, Levine JH, Cogdill AP, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell. 2017;170(6):1120–1133. e1117. | ||
Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39(1):98–106. | ||
Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:948098. | ||
Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208–220. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.