")
Back to Archived Journals » Oncolytic Virotherapy » Volume 3
Oncolytic viral therapy for pancreatic cancer: current research and future directions
Authors Ady J, Heffner J, Klein E, Fong Y
Received 1 September 2013
Accepted for publication 2 December 2013
Published 17 February 2014 Volume 2014:3 Pages 35—46
DOI https://doi.org/10.2147/OV.S53858
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Justin W Ady, Jacqueline Heffner, Elizabeth Klein, Yuman Fong
Department of Surgery, Memorial Sloan-Kettering Cancer Center, New York, NY, USA
Abstract: The development of targeted agents and chemotherapies for pancreatic cancer has only modestly affected clinical outcome and not changed 5-year survival. Fortunately the genetic and molecular mechanisms underlying pancreatic cancer are being rapidly uncovered and are providing opportunities for novel targeted therapies. Oncolytic viral therapy is one of the most promising targeted agents for pancreatic cancer. This review will look at the current state of the development of these self-replicating nanoparticles in the treatment of pancreatic cancer.
Keywords: pancreatic cancer, oncolytic virus, review
Introduction
After 40 years of cancer research, pancreatic adenocarcinoma remains a highly lethal disease that is extremely difficult to detect and treat. Five-year survival rates have only slightly improved to 6%, and nearly all diagnosed patients will recur and succumb to the disease.1 It is the fourth-leading cause of cancer-related death in the United States, with an estimated 45,220 new cases being diagnosed in 2013 and 38,460 associated deaths.2,3 Complete surgical resection remains the only chance for long-term disease-free survival. Unfortunately 80% of patients present with locally advanced or metastatic disease and are unresectable at diagnosis. For those patients that are unresectable, prognosis is poor with an expected survival of just 6 months. Most patients who do undergo surgery have only a slightly improved survival, with tumors recurring in virtually all patients.4,5
Due to the high rate of recurrence, adjuvant therapy has been suggested following surgery. However, the long-term impact of adjuvant radiation therapy has proven to be inconclusive.6–8 The high incidence of systemic recurrence after surgery and radiation has led to the notion that metastasis may occur early in the course of pancreatic tumorogenesis.9 For this reason, the treatment of pancreatic cancer has most recently focused on multimodality treatment with surgery, chemotherapy, and radiation.
Gemcitabine and erlotinib are the only two agents approved for use in advanced disease, but both have only a modest benefit. Gemcitabine was shown to prolong survival of patients by 5.6 months compared to 4.4 months with 5-fluorouracil.10 The addition of erlotinib to gemcitabine saw a miniscule increase of median survival from 5.9 to 6.2 months.11 Thus, development of targeted agents and chemotherapies over the last five decades has only modestly affected clinical outcome and not changed 5-year survival. Fortunately, the genetic and molecular mechanisms underlying pancreatic cancer are being rapidly uncovered and are providing opportunities for novel targeted therapies. Oncolytic viral therapy is of the most promising targeted agents for pancreatic cancer. This review will look at the current state of the development of these self-replicating nanoparticles in the treatment of pancreatic cancer.
Oncolytic viruses
Due to its tumor selectivity and ability to cause cancer cell lysis, oncolytic virotherapy is becoming increasingly popular for the treatment of many different forms of cancer. Oncolytic viruses selectively target tumor cells through engineered mutations that prevent the binding to and replication of virus in normal cells and by expressing foreign genes that cause cell death both directly and/or indirectly.12 These viruses are also usually replication competent. Thus, replication in tumor cells generates infectious progeny that may further spread through and kill the tumor mass.13 The efficacy of oncolytic viruses depends upon the direct tumor lysis, the ability of the virus to spread to surrounding cancer cells, and the virus’s ability to direct an immune response to the tumor.
Optimal oncolytic viruses need to be extremely selective in the cells that they infect and replicate in. There are a number of ways in which their selectivity is improved: 1) by deleting viral genes that are required for replication in normal cells but are unnecessary in cancer cells; 2) by transcriptional targeting, where viral replication is controlled by tissue specific promoters; and/or 3) transductional targeting, where the virus is retargeted specifically to tumor cells. One of the important advantages oncolytic viruses have over other cancer therapies is the great potential to genetically manipulate the candidate virus to assume greater potency against a specific cancer. These manipulations help both to increase the potency of viral therapy against a tumor and to abrogate the adverse effects of cancer therapy (Figure 1).
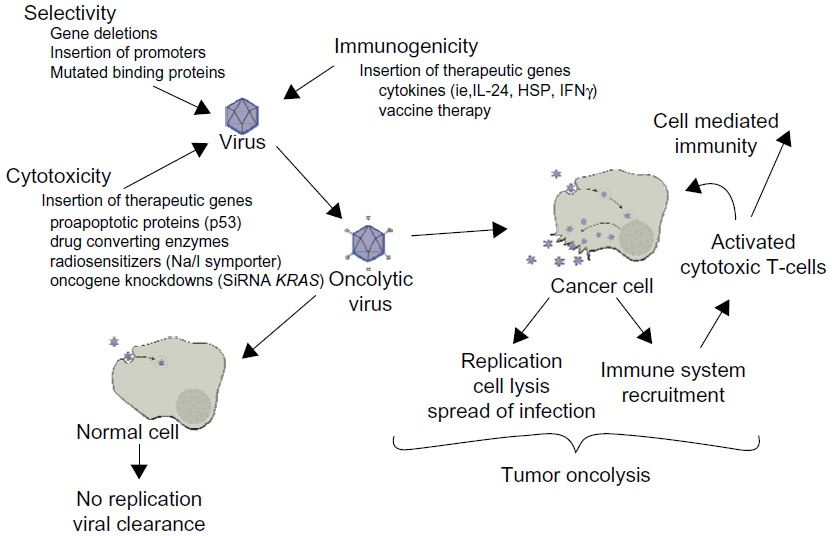 | Figure 1 Oncolytic viral therapy. |
Microbiology and tumor microenvironment of pancreatic cancer
The microbiology of pancreatic cancer provides unique challenges in designing and implementing effective oncolytic vectors and gene therapy. Oncogenic transformation in the pancreas is currently understood to be a multistage process that involves the accumulation of inherited and acquired mutations of specific cancer-associated genes in preneoplastic lesions. The literature describes three types of pancreatic ductal adenocarcinoma (PDAC) precursor lesions: pancreatic intraepithelial neoplasia, intraductal papillary neoplasia, and mucinous cystic neoplasms. They all follow a multistep progression from preneoplastic to invasive cancer, characterized by increasing degrees of morphological and cytological atypia.14,15 Most commonly this starts with an activating mutation in the KRAS gene, which is then followed by a somatic mutation in one or more of the tumor suppressor genes TP53, p16/CDKN2A, and SMAD4.16 Approximately 10% of cases can be partially ascribed to one of several germline mutations including BRCA2, STK11/LKB1, or p16/CDKN2A.17 This model for the stepwise development of PDAC has been supported in animal models by genetically engineered mice with a single point mutation in the KRAS oncogene. These mice were found to develop pancreatic preneoplastic lesions similar to those found in humans.18 Progression to metastatic carcinoma required both the KRAS mutation and the loss of a tumor suppressor gene.18–20 Researchers have found a total of 12 core signaling pathways that are commonly dysregulated in PDAC, of which KRAS, TP53, p16/CDKN2A, and SMAD4 are found in two thirds of tumors studied.21 Targeting of these molecular pathways is just one of the ways that oncolytic viruses and targeted therapies are being tailored to pancreatic cancer (Table 1).
 | Table 1 Genetic mutations in pancreatic cancer |
The high resistance to conventional and targeted therapies in PDAC may in part be explained by the diverse influences that are being exerted by the pancreatic microenviroment on the cancer cells. The dense extracellular matrix in pancreatic cancer distorts the normal architecture of the tissue and causes an abnormal configuration of blood and lymphatic vessels, resulting in a hypoxic tumor mass, which is one reason why many systemically delivered therapies, such as systemic chemotherapy and viral therapy work poorly in pancreatic cancer.22 In fact, many strategies are being studied that target the stromal matrix in an effort to increase the effect of systemic therapies. Inhibition of hedgehog signaling, which is involved in desmoplasia, has been shown to increase the penetration of gemcitabine into pancreatic xenografts. Also, the use of hyaluronidase to enzymatically remodel the stromal matrix has shown an increase in the delivery of systemic therapies to the tumor tissue. Initial preclinical success with these therapies suggests that the dense extracellular matrix in pancreatic cancer is partly responsible for the innate chemoresistance of pancreatic tumors, by creating barriers that protect tumor cells from systemically circulating therapeutic compounds.23–26 The tumor extracellular matrix is only one potential target to improve systemic and molecular therapies.
It is becoming increasingly clear that a crucial component of the tumor microenvironment is the pancreatic stellate cells (PaSCs). These rare stromal cells have been shown to both increase pancreatic cancer cell proliferation and migration by release of growth factors and cytokines.27 This has been confirmed in in vivo studies in which co-injection of PaSCs with tumor cells in orthotopic models of PDAC resulted in increased tumor size and caused a higher incidence of metastasis.28 Strikingly, PaSCs were also detected in metastatic foci in the liver of nude mice, suggesting co-migration of PaSCs with cancer cells to establish a potentially favorable microenvironment for tumor at distant sites.28 Recent studies also demonstrated that PaSCs in vitro increase the stem cell phenotype of pancreatic cancer cells, providing a therapeutic target that may decrease the metastatic potential of early PDAC.29
Inflammation is an important factor in the development of PDAC. Some studies suggest that over 50% of the tumor mass is made up of inflammatory cells and numerous studies point to long-term inflammation of the pancreas as a driving force in the development of PDAC.30,31 The PDAC tumor microenvironment is significantly pro-tumorigenic, with the majority of the cells being made up of immunosuppressive cells such as regulatory T-cells and myeloid-derived suppressor cells.30 Successful immunotherapy requires cytotoxic T-cells to have high affinity for cancer cell antigens without causing autoimmunity. Unfortunately, cytotoxic T-cells make up a significant minority of the immune cells in the tumor, severely restricting the immunogenicity that results from infection with oncolytic viruses, suggesting that virus augmented with cytokines that are systemically or locally delivered to increase the immune response would be beneficial.
Another important factor in the development of chemoresistance and the propensity for metastasis of pancreatic cancer is that the cancer can undergo epithelial to mesenchymal transition (EMT).32,33 EMT is characterized by decreased expression of E-cadherin and increased expression of mesenchymal markers such as vimentin, N-cadherin, and zinc finger transcription factors (Snail, Slug, Zeb1, and Twist).34,35 Gene expression profiling has shown that EMT cells are significant contributors to chemoresistance in PDAC.33,36 EMT plays a role in modulating resistance to targeted biological therapies as well. Cells with mutated E cadherin or expressing mesenchymal markers showed significantly decreased growth inhibition with the epidermal growth factor receptor inhibitor erlotinib than cells with an epithelial phenotype.37
The microenvironment of pancreatic cancer makes it difficult for systemic therapies to access cancer cells. Breaching this stromal barrier is a promising strategy to improve the delivery and efficacy of cytotoxic drugs and oncolytic vectors. Significant therapeutic benefit could be obtained by using strategies that aim to deplete the desmoplastic stroma, activate the immune system to target tumor cells, and target cells that increase the metastatic potential of PDAC.
Adenovirus
One of the most commonly used viral vectors being designed for treatment of cancer is the adenovirus. Gendicine and Oncorine are two adenoviruses that are currently approved for the treatment of cancer in China.38 Adenoviruses are double-stranded DNA viruses that are encompassed by an icosahedral protein capsid. When they come in contact with a cell, the virus binds to a receptor on a cell with high affinity.39 Once inside the cell, the virion moves to the nuclear pore complex, where its genome is released into the nucleoplasm and gene expression and genome replication occur. In a final step, the progeny viruses are released by disintegration of the cell membrane and infection of neighboring tumor cells occurs.
In order to achieve cancer cell selectivity in adenoviruses, two major strategies have been employed (Table 2). The first was to delete essential viral genes needed for replication of normal cells. These deletional mutations take advantage of the dysregulation of particular cell signaling pathways in cancer cells that allow viral replication in cancer cells even after key genes are deleted. One such virus is ONXY-15, the first replication-competent oncolytic virus used in a clinical trial for pancreatic cancer.40 It has been engineered to lack the E1B gene that coded for a 55 kDa protein that in normal cells binds to tumor supressor p53 and causes progression of the cell cycle and viral replication.41 E1B-deleted viruses do not generally replicate in normal cells. It is known that 50%–75% of pancreatic tumors lack p53, thus allowing replication of E1B-deleted viruses. ONXY-15 was shown to be effective in a murine model of human xenografts with antitumor efficacy and increased survival.42 In Phase I/II clinical trials, it was found that ONYX-15, when combined with gemcitabine, was found to be a feasible and well-tolerated therapy in patients with pancreatic cancer.40
 | Table 2 Modifications of the adenovirus for cancer therapy |
Oncorine is another virus in clinical use that has a deletion of the E1B gene and a partial deletion of the E3 gene. It has been approved for the treatment of head and neck cancers in China and has been shown to be well-tolerated and have good efficacy when combined with or without chemotherapy.38,40 It is currently in clinical trial in the United States for pancreatic cancer.
Another deletional mutation in adenoviruses for pancreatic cancer is the E1A gene. The E1A protein binds to retinoblastoma protein (pRb), a protein in normal cells that forces progression of the cell cycle from G1 to S, which allows E2F1, a transcription factor important in viral reproduction, to function. Many of the genes in pancreatic cancer commonly mutated, such as CDKN2A, are involved in the regulation of the G1–S cell cycle. These mutations lead to cell cycle dysregulation, making E1A binding to pRb unnecessary for E2F1 to support viral replication in pancreatic cancer. Thus, oncolytic adenoviruses that utilize this gene deletion for tumor selectivity are great candidates for use in pancreatic cancer.43 E1B 19 kDa is another good candidate for creating tumor selectivity in adenoviruses. The E1B 19 kDa gene codes for the antiapoptotic B-cell lymphoma 2 homologue, which in the aberrant apoptotic environment of pancreatic cancer is unnecessary.21
Deletion or mutation of multiple genes further improves selectivity of virus for cancer and prevents reversion to wild type. When both the E1A gene and the E1B 19 kDa gene are deleted there is a significant increase in tumor selectivity. Importantly, the potency of the virus was retained when compared to the wild type adenovirus without any gene deletions. Efficacy of the virus was shown to further increase when it was in combination with gemcitabine and other chemotherapies.44,45 Importantly, these studies show that oncolytic viruses can be engineered to be selective while still retaining their potency.
The second way that adenoviruses have been engineered to improve selectivity in pancreatic cancer is by placing important proteins behind tumor-specific promoters. At least five tumor specific promoters have been used to construct targeted adenoviruses for pancreatic cancer therapy. The promoters used have been cyclooxygenase-2 promoter, urokinase-type plasminogen activator receptor promoter, telomerase reverse transcriptase promoter, hypoxia-responsive promoter, KRAS promoter, and human carcinoembryonic antigen promoter.46–54 The promoters have been shown to increase the selectivity of the virus without negatively impacting the infectivity.
One of the major disadvantages of adenoviruses as an oncolytic vector is the poor intratumoral spread and infectivity natively inherent to the virus. This is because most oncolytic adenoviruses are constructed from the wild type adenovirus serotype 5. The adenovirus serotype 5 viruses bind to the coxsackie adenovirus receptor, which has little or no expression in pancreatic cancer, leading to the requirement of high doses of virus to overcome the low infectivity. One way researchers have overcome this is by substituting the fiber knob with the adenoviral subtype 3 fiber knob. Oncolytic adenoviruses with this new binding motif were found to infect tumor tissue independent of coxsackie virus receptor levels. This new mutant adenovirus uses the desmoglein-2 protein as a binding site and in result, had enhanced viral infectivity in pancreatic cancer.50,55,56
With improved infectivity and selectivity, oncolytic adenoviruses are now being made more potent by arming them with therapeutic genes that help to prime the immune system against pancreatic cancer and improve oncolysis. Some of these therapeutic genes are presented in Table 1. Interleukin 24 (IL-24) is a good example of a potential therapeutic protein that can both improve immune response against tumor antigens and abate potential side effects. IL-24 is known to increase the immune system’s recognition for pancreatic cancer. Unfortunately, there can be severe side effects when given systemically, thus limiting its usefulness as a cancer treatment. An adenovirus, ZD55-IL-24, was engineered to manufacture IL-24 locally in the tumor cells, thus avoiding the systemic effects. When compared with the parental virus ZD55 (E1B 55-kDa-deleted oncolytic adenovirus) in immune-competent mice, there was significant decrease in tumor growth and there was a stronger immune response measured by T-cell response to pancreatic cancer and the levels of interleukin 6 instead (IL-6) and interferon gamma.57 In addition to delivering cytokines, therapeutic genes can be used to inhibit differentiation of tumors, silence oncogenes, activate prodrugs, and image the presence of virus. Using adenovirus with a thymidine kinase gene inserted, researchers took micro-positron-emission-tomography images of pancreatic flank tumors using 18F-FEAU (2’-[18F]fluoro-5-ethyl-1-beta-D-arabinofuranosyluracil) radiotracer, demonstrating the presence of virus.58 Quantification of the thymidine kinase activity in the tumors correlated with the antitumor effects of the virus.58
Oncolytic herpes virus
The herpes simplex viruses (HSV) HSV-1 and HSV-2 are large enveloped viruses with double-stranded DNA. In the wild they cause self-limiting infection, often causing sores in the mouth, lips, or genitalia, and can establish latent infection in the neural ganglia. As an oncolytic virus, HSV has been genetically modified to replicate only in cancer cells and has demonstrated itself as a promising agent in the virotherapy of pancreatic cancer.
Compared to other oncolytic viruses, HSV has a number of distinct advantages and disadvantages. The genome is large compared to other viruses, with many nonessential genes that can be mutated and replaced with therapeutic genes. Several anti-HSV medications are available (acyclovir, ganciclovir) for the treatment of patients with adverse reactions to virotherapy. HSV does not integrate itself into the cellular DNA, thus reducing the risk of foreign genes being inserted into the patient’s DNA. Compared to other families of viruses, HSV is known to infect and kill tumor cells more quickly. The hope is that this will prevent the immune system from restricting the spread of the virus prior to tumor oncolysis. HSV has the ability to infect many different cancer cell types, making it a viable candidate for many different forms of cancer.59 Finally, HSV exhibits strong T-cell-mediated tumor reactivity, and can indirectly cause an immune response to cancer and tumor regression from cytotoxic T-cells- and natural killer (NK) cells-mediated mechanism. This likely plays an essential role in the antitumor abilities of HSV.
One potential disadvantage is that exposure to HSV is common in the general population. So, there is a risk that people may carry preformed immunity to HSV and clear the virus before it can have an oncolytic effect. However, studies in murine animals have not demonstrated that this anti-HSV immunity has significant deleterious effects on oncolysis.60,61
There are a number of different HSV oncolytic viruses that have been studied in pancreatic cancer (Table 3). They use two major strategies to engineer selectivity for cancer cells: 1) the deletion of essential viral genes for replication (ie, γ34.5 gene); and 2) deletion of genes that regulate the protein kinase response (PKR) pathway (ie, ICP6 gene). A number of HSV-based oncolytic viruses have produced encouraging results in preclinical and clinical studies.
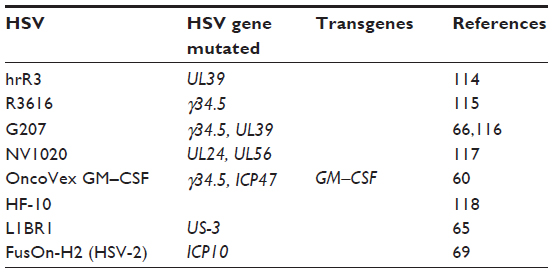 | Table 3 HSVs designed for cancer therapy |
The oncolytic viruses R3616 and HSV1716 have a deletion of the γ34.5 gene, resulting in the inability of virally infected cells to turn off PKR phosphorylation. This results in inactivation of eukaryotic translation initiation factor 2 alpha (eIF2α), preventing translation within normal cells. In pancreatic cancer cells, eIF2α is constituently activated due to mutations in glycogen synthase kinase-3, an alternate pathway that is independent of the PKR pathway, allowing for viral replication. Interestingly, replication and oncolysis of R3616 in pancreatic cancer cells is due to dysregulation and activation of the phosphatidylinositide 3-kinase pathway and not dysregulation of PKR or enhanced Ras signaling, as was once thought.62
The virus hrR3 lacks the UL39 gene, which codes for ICP6 protein, a viral homologue of the cellular ribonucleotide reductase; in pancreatic cells this protein is upregulated, allowing for viral replication. This virus also has an active HSV thymidine kinase, which allows concurrent treatment of cancer cells with ganciclovir. Ganciclovir disrupts cellular and viral DNA replication when activated by the viral thymidine kinase and has been shown to augment tumorlysis. In a xenograft model of pancreatic carcinomatosis in which mice were given hrR3 virus and ganciclovir together, there was a 30% increase in survival at 150 days when compared to treatment with virus alone.63
The resistant nature of pancreatic cancer to systemic chemotherapy has made finding adjuvant treatments that improve the outcomes of chemotherapy important.22 Using oncolytic HSVs is one of the more promising options. In one study, R3616 (γ34.5 deletion) was compared to hrR3 (UL39 deletion) with and without gemcitabine. It was found that treatment with R3616 had better long-term survival than hrR3 with or without gemcitabine. Of note though, when combined with gemcitabine, R3616 showed improved long-term survival, whereas hrR3 combined with gemcitabine showed decreased long-term survival. These data suggest that chemotherapy may be affecting viral replication and cancer cell killing differently depending on the targeting mechanism of the virus.64 Another study looking at L1BR1 (US3 locus deficient HSV-2) showed synergy with the anticancer drugs fluorouracil and cisplatin.65
G207 virus is a mutated HSV-1 virus with deletions at both γ34.5 loci and a lacZ insertion interrupting the ICP6 gene that encodes the HSV ribonucleotide reductase. Its activity has been shown in a wide variety of solid tumors including pancreatic cancer.66,67 NV1020, a closely related virus with only one copy of the γ34.5 loci deleted, has a higher proliferative rate then G207 in pancreatic cancer. NV1023 and NV1066, both derivatives of NV1020, are being actively studied in pancreatic cancer. NV1066 and hyperthermia significantly increased killing of pancreatic cancer cells secondary to enhanced replication through a heat shock protein.59 NV1020 in combination with radiation demonstrated synergistic effects when it came to tumor oncolysis.68
One especially promising HSV oncolytic virus is FusON-H2, another HSV-2 virus. This virus has a deletion of the ICP10 gene that encodes a serine/threonine protein kinase activity that is involved in activation of the Ras/mitogen-activated protein kinase pathway. When used to treat pancreatic cancer, intratumoral injections of FusON-H2 showed complete eradication of subcutaneous pancreatic xenografts. Intravenous administration showed significant antitumor effects. When the virus was given through intraperitoneal administration, there was eradication of 75% of tumors and prevention of metastasis in animals.69
There are two oncolytic HSV-1 viruses in clinical trial for pancreatic cancer, HF10 and OncoVex granulocyte–macrophage colony-stimulating factor (GM–CSF). HF10 is a naturally occurring replication-competent HSV-1 mutant that has been found to replicate and spread efficiently in cancer cells.70 A Phase I clinical trial has been completed in which patients were treated with three doses of HF10.71 No adverse reactions were noted in the trial. Of the six patients treated, three had stable disease, one patient had regression, and two patients had progression. This is a safe treatment with potential and Phase II and Phase III studies are currently in planning. A Phase I trial looking at giving HF10 via injection from endoscopic ultrasound in unresectable disease is currently being run with no data reported at this point.71
The second HSV oncolytic virus in clinical trial is OncoVex GM–CSF, a γ34.5 and ICP47 deleted mutant that expresses human GM–CSF. The γ34.5 deletion provides for tumor selective replication. ICP47 encodes for a protein, US11, that inhibits PKR activation, further increasing selectivity of replication. Most importantly, this virus is also engineered to express human GM–CSF. Human GM–CSF is associated with the recruitment and differentiation of activating dendritic cells in the tumor microenvironment.72,73 The hope is that this virus, by recruiting and stimulating dendritic cells, will subsequently prime antigen specific T-cells immunity in the body. In Phase I/II clinical trials in solid tumors (head and neck, squamous cell cancer, breast cancer, gastrointestinal cancer, and malignant melanoma), OncoVex GM–CSF was found to be well-tolerated at high and repeated doses.74,75 There is currently a Phase I clinical trial in pancreatic cancer underway with no interim results at this point.73
Poxviruses
The family Poxviridae consists of enveloped double-stranded DNA viruses, of which there are many members including vaccinia virus, myxoma virus (MYXV), and raccoon pox. The most widely studied is vaccinia virus, which has had a crucial role in one of the greatest achievements in medicine: the eradication of smallpox. The highly immunogenic nature of vaccinia infection, which produces a strong cytotoxic T lymphocyte response and circulating neutralizing antibodies that can be detected many decades later, was crucial to the successful use of vaccinia in smallpox eradication and has led to its continued use in immunotherapy today.76,77 The ability of poxviruses to express foreign antigens and stimulate an immune response is one of the reasons why it is now being studied as an oncolytic virus for the treatment of cancer.78 In pancreatic cancer, vaccinia has been genetically modified to increase the tumor cytotoxicity against pancreatic cancer. One such modified vaccinia virus expresses the endostatin-angiostatin fusion protein, which has been documented in pancreatic cancer to inhibit angiogenesis. Current studies with this endostatin-angiostatin fusion protein expressing vaccinia virus have shown significant antitumor potency in vivo against pancreatic cancer.79
GLV-1h68 is a replication-competent virus with attenuation mutations in F14.5L, J2R (encodes thymidine kinase), and A56R (encodes hemagglutinin). It has been reported to infect, replicate, and lyse pancreatic cancer cells both in vivo and in vitro. Importantly though, when combined with gemcitabine or cisplatin, the tumors showed enhanced and accelerated therapeutic effects.
The protein survivin has been associated with resistance to chemotherapy in pancreatic cancer and is found to be overexpressed in 70%–80% of tumors. A modified vaccinia Ankara virus expressing murine survivin produces significant tumor regression and increased survival in mice when combined with gemcitabine.80
One of the factors in the resistance of pancreatic cancer to systemic therapies is the hypoxic nature of the tumor. It was found that the Lister strain of vaccinia, when allowed to infect pancreatic cells under hypoxic conditions, showed no degradation in viral replication, suggesting that vaccinia may be uniquely suited as a viral vector to the hypoxic conditions of pancreatic cancer.81
MYXV is a rabbit-specific poxvirus that has been shown to replicate in human cancer cells. In pancreatic cancer, when combined with gemcitabine, MYXV was able to show excellent replication and significant oncolysis. In a disseminated pancreatic cancer model in immune-compromised mice, MYXV combined with gemcitabine resulted in 100% long-term survival. These data suggest that MYXV is an effective oncolytic virus for pancreatic cancer and can be combined with gemcitabine to enhance survival, particularly in the presence of an intact host immune system.82 A newly engineered replication-competent MYXV, vMyxgfp, has the green fluorescent protein construct placed within it. Investigators were able to show replication and oncolysis of chemotherapy-resistant cell lines, making this a promising new virus in the treatment of pancreatic cancer.83
There is one poxvirus currently in clinical trial as a vaccine therapy. It is a vaccinia virus that expresses the tumor antigens carcinoembryonic antigen and mucin-2, two molecules known to be expressed by pancreatic cancer cells. It is packaged with three costimulatory molecules, B7.1 (cluster of differentiation 80), ICAM-1 (intracellular adhesion molecule-1), and LFA-3 (leukocyte function-associated antigen-3) (TRICOM)(PAN-VAC-V), to help increase the immune response to the vaccine. To further enhance the vaccine therapy interferon alpha is also given. The results are promising with a significant overall increase in patient survival with mild injection site reactions as the most common adverse event. This suggests that in pancreatic cancer vaccinia virus is capable of creating antigen-specific immune responses.84,85
Parvovirus
Parvoviruses, such as H-1 (H-1PV), are small, non-enveloped icosahedral particles containing a single-stranded DNA genome that require actively dividing cells to replicate, making them innately oncolytic.86 They have no pathogenicity in animals and humans, but have been shown to have tumor suppressive effects in animal models.87 Parvoviruses cause both direct oncolysis and immunomodulatory effects, which have been shown to prime the immune systems of animals against infected tumors.88 When given after the administration of gemcitabine, H-1PV in vivo showed reduced tumor growth, prolonged animal survival, and absence of metastasis on computed tomography scans. This suggests that parvoviruses can be a useful adjunct in multimodal therapy for pancreatic cancer.89
Studies with parvovirus suggest that infection can increase the NK tumor-mediated cell killing in pancreatic ductal carcinoma.90 When the virus is armed with IL-2 or the chemokine ligand 7, there was improved antitumor response of NK cells and monocytes to pancreatic ductal carcinoma.91
Measles virus
As oncolytic viruses, measles viruses have shown promising results against many different tumor models. They depend upon the overexpression of CD46, a viral entry receptor found in many cancer cells.92 An engineered measles virus expressing the sodium iodine symporter gene (MV-NIS) has shown oncolytic activity in pancreatic tumor xenografts in mice with tumor regression and increased survival. Unfortunately, when this was combined with radiotherapy, no synergy was found.93 In multiple studies, the authors were able to use iodinated contrast and micro-single-photon emission computed tomography to accurately image initial viral distribution within a solid tumor.94,95
Additionally, another modified measles virus was created to target prostate stem cell antigen (PSCA), which is a protein expressed in pancreatic cancer. To enhance the virus’ efficacy, it was armed with the suicide gene, convertase purine nucleoside phosphorylase (PNP), which activates the prodrug fludarabine. The authors succeeded in demonstrating that MV-PNP-anti-PSCA exhibited beneficial therapeutic effects in pancreatic cancer. Importantly, they were able to show that gemcitabine resistant pancreatic adenocarcinoma was susceptible to both the virus and activated prodrug.96
Reovirus
Reoviruses are double-stranded RNA viruses. The replication of this virus is dependent upon cellular activity of Ras. KRAS mutations in pancreatic cancer are high (85%–90%), making reovirus a promising candidate as an oncolytic virus in pancreatic carcinoma.47 In immunocompetent animal models, reovirus administration intraportally resulted in decreased metastatic tumor volumes in the liver.97,98 It also resulted in decreased tumor mass and decreased ascites when administered intraperitoneally.97 Reolysin® (Oncolytics Biotech Inc., Calgary, AB, Canada) is the name of the reovirus currently in a Phase II clinical trial in pancreatic cancer.99–101
Future directions
With the current treatment modalities of surgery, chemotherapy, and radiotherapy, the prognosis of pancreatic cancer remains poor. The microenvironment of pancreatic cancer presents many unique challenges that will need to be overcome if we are going to successfully treat pancreatic cancer. Fortuitously, there are a number of different oncolytic viruses that have been shown to have real value in the treatment of pancreatic cancer in preclinical animal models. Clinical trials of oncolytic viruses have shown that oncolytic virotherapy is both a safe and feasible option for pancreatic cancer treatment in humans. However, there is still a great deal of opportunity for optimizing oncolytic viruses in the treatment of pancreatic cancer. In the future, viruses targeted to disrupt EMT may help to reduce the population of cancer stem cells that are contributing to treatment resistance and tumor metastasis. Also, combining viruses with systemic therapies that aim to disrupt the desmoplastic matrix and allow better penetration of viral therapy into tumors should lead to more successful viral therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. | |
Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7(3):163–172. | |
American Cancer Society. Cancer Facts and Figures 2013. Atlanta: American Cancer Society; 2013. Available from: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-036845.pdf. Accessed August 1, 2013. | |
Bilimoria KY, Bentrem DJ, Ko CY, et al. Validation of the 6th edition AJCC Pancreatic Cancer Staging System: report from the National Cancer Database. Cancer. 2007;110(4):738–744. | |
Allison DC, Piantadosi S, Hruban RH, et al. DNA content and other factors associated with ten-year survival after resection of pancreatic carcinoma. J Surg Oncol. 1998;67(3):151–159. | |
Neoptolemos JP, Stocken DD, Bassi C, et al; European Study Group for Pancreatic Cancer. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA. 2010;304(10):1073–1081. | |
Zygogianni GA, Kyrgias G, Kouvaris J, et al. Intraoperative radiation therapy on pancreatic cancer patients: a review of the literature. Minerva Chir. 2011;66(4):361–369. | |
Ruano-Ravina A, Almazán Ortega R, Guedea F. Intraoperative radiotherapy in pancreatic cancer: a systematic review. Radiother Oncol. 2008;87(3):318–325. | |
Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1–2):349–361. | |
Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–2413. | |
Moore MJ, Goldstein D, Hamm J, et al; National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–1966. | |
Vacchelli E, Eggermont A, Sautès-Fridman C, et al. Trial watch: Oncolytic viruses for cancer therapy. Oncoimmunology. 2013;2(6):e24612. | |
Zeyaullah M, Patro M, Ahmad I, et al. Oncolytic viruses in the treatment of cancer: a review of current strategies. Pathol Oncol Res. 2012;18(4):771–781. | |
Koorstra JB, Hustinx SR, Offerhaus GJ, Maitra A. Pancreatic carcinogenesis. Pancreatology. 2008;8(2):110–125. | |
Gnoni A, Licchetta A, Scarpa A, et al. Carcinogenesis of pancreatic adenocarcinoma: precursor lesions. Int J Mol Sci. 2013;14(10):19731–19762. | |
Iacobuzio-Donahue CA, Velculescu VE, Wolfgang CL, Hruban RH. Genetic basis of pancreas cancer development and progression: insights from whole-exome and whole-genome sequencing. Clin Cancer Res. 2012;18(16):4257–4265. | |
Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006;20(2):197–209. | |
Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003;63(9):2016–2019. | |
Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17(24):3112–3126. | |
Bardeesy N, Aguirre AJ, Chu GC, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A. 2006;103(15):5947–5952. | |
Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. | |
Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–4276. | |
Thompson CB, Shepard HM, O’Connor PM, et al. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol Cancer Ther. 2010;9(11):3052–3064. | |
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–429. | |
Tian H, Callahan CA, DuPree KJ, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci U S A. 2009;106(11):4254–4259. | |
Bailey JM, Swanson BJ, Hamada T, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008;14(19):5995–6004. | |
Erkan M, Adler G, Apte MV, et al. StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut. 2012;61(2):172–178. | |
Vonlaufen A, Joshi S, Qu C, et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 2008;68(7):2085–2093. | |
Lonardo E, Frias-Aldeguer J, Hermann PC, Heeschen C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle. 2012;11(7):1282–1290. | |
Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–9527. | |
Momi N, Kaur S, Krishn SR, Batra SK. Discovering the route from inflammation to pancreatic cancer. Minerva Gastroenterol Dietol. 2012;58(4):283–297. | |
Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol. 2007;14(12):3629–3637. | |
Wang Z, Li Y, Kong D, et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69(6):2400–2407. | |
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | |
Yang AD, Camp ER, Fan F, et al. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006;66(1):46–51. | |
Yang AD, Fan F, Camp ER, et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. 2006;12(14 Pt 1):4147–4153. | |
Buck E, Eyzaguirre A, Barr S, et al. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol Cancer Ther. 2007;6(2):532–541. | |
Garber K. China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst. 2006;98(5):298–300. | |
Cerullo V, Koski A, Vähä-Koskela M, Hemminki A. Chapter eight – Oncolytic adenoviruses for cancer immunotherapy: data from mice, hamsters, and humans. Adv Cancer Res. 2012;115:265–318. | |
Hecht JR, Bedford R, Abbruzzese JL, et al. A phase I/II trial of intratumoral endoscopic ultrasound injection of ONYX-015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin Cancer Res. 2003;9(2):555–561. | |
Bischoff JR, Kirn DH, Williams A, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274(5286):373–376. | |
Heise C, Sampson-Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997;3(6):639–645. | |
Heise C, Hermiston T, Johnson L, et al. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med. 2000;6(10):1134–1139. | |
Oberg D, Yanover E, Adam V, et al. Improved potency and selectivity of an oncolytic E1ACR2 and E1B19K deleted adenoviral mutant in prostate and pancreatic cancers. Clin Cancer Res. 2010;16(2):541–553. | |
Cherubini G, Kallin C, Mozetic A, et al. The oncolytic adenovirus AdΔΔ enhances selective cancer cell killing in combination with DNA-damaging drugs in pancreatic cancer models. Gene Ther. 2011;18(12):1157–1165. | |
Xu C, Sun Y, Wang Y, et al. CEA promoter-regulated oncolytic adenovirus-mediated Hsp70 expression in immune gene therapy for pancreatic cancer. Cancer Lett. 2012;319(2):154–163. | |
Mihaljevic AL, Michalski CW, Friess H, Kleeff J. Molecular mechanism of pancreatic cancer – understanding proliferation, invasion, and metastasis. Langenbecks Arch Surg. 2010;395(4):295–308. | |
Bortolanza S, Bunuales M, Otano I, et al. Treatment of pancreatic cancer with an oncolytic adenovirus expressing interleukin-12 in Syrian hamsters. Mol Ther. 2009;17(4):614–622. | |
Hu Y, Ou Y, Wu K, Chen Y, Sun W. miR-143 inhibits the metastasis of pancreatic cancer and an associated signaling pathway. Tumour Biol. 2012;33(6):1863–1870. | |
Ramirez PJ, Vickers SM, Ono HA, et al. Optimization of conditionally replicative adenovirus for pancreatic cancer and its evaluation in an orthotopic murine xenograft model. Am J Surg. 2008;195(4):481–490. | |
Nelson AR, Davydova J, Curiel DT, Yamamoto M. Combination of conditionally replicative adenovirus and standard chemotherapies shows synergistic antitumor effect in pancreatic cancer. Cancer Sci. 2009;100(11):2181–2187. | |
Huch M, Gros A, José A, González JR, Alemany R, Fillat C. Urokinase-type plasminogen activator receptor transcriptionally controlled adenoviruses eradicate pancreatic tumors and liver metastasis in mouse models. Neoplasia. 2009;11(6):518–528. | |
Onimaru M, Ohuchida K, Nagai E, et al. Combination with low-dose gemcitabine and hTERT-promoter-dependent conditionally replicative adenovirus enhances cytotoxicity through their crosstalk mechanisms in pancreatic cancer. Cancer Lett. 2010;294(2):178–186. | |
Onimaru M, Ohuchida K, Mizumoto K, et al. hTERT-promoter-dependent oncolytic adenovirus enhances the transduction and therapeutic efficacy of replication-defective adenovirus vectors in pancreatic cancer cells. Cancer Sci. 2010;101(3):735–742. | |
Wang H, Li ZY, Liu Y, et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat Med. 2011;17(1):96–104. | |
Chu QD, Sun G, Pope M, et al. Virotherapy using a novel chimeric oncolytic adenovirus prolongs survival in a human pancreatic cancer xenograft model. Surgery. 2012;152(3):441–448. | |
He B, Huang X, Liu X, Xu B. Cancer targeting gene-viro-therapy for pancreatic cancer using oncolytic adenovirus ZD55-IL-24 in immune-competent mice. Mol Biol Rep. 2013;40(9):5397–5405. | |
Abate-Daga D, Andreu N, Camacho-Sanchez J, et al. Oncolytic adenoviruses armed with thymidine kinase can be traced by PET imaging and show potent antitumoural effects by ganciclovir dosing. PloS One. 2011;6(10):e26142. | |
Eisenberg DP, Carpenter SG, Adusumilli PS, et al. Hyperthermia potentiates oncolytic herpes viral killing of pancreatic cancer through a heat shock protein pathway. Surgery. 2010;148(2):325–334. | |
Liu BL, Robinson M, Han ZQ, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303. | |
Varghese S, Rabkin SD, Nielsen GP, MacGarvey U, Liu R, Martuza RL. Systemic therapy of spontaneous prostate cancer in transgenic mice with oncolytic herpes simplex viruses. Cancer Res. 2007;67(19):9371–9379. | |
Sarinella F, Calistri A, Sette P, Palù G, Parolin C. Oncolysis of pancreatic tumour cells by a g34.5- deleted HSV-1 does not rely upon Ras-activation, but on the PI 3-kinase pathway. Gene Ther. 2006;13(14):1080–1087. | |
Kasuya H, Nishiyama Y, Nomoto S, Hosono J, Takeda S, Nakao A. Intraperitoneal delivery of hrR3 and ganciclovir prolongs survival in mice with disseminated pancreatic cancer. J Surg Oncol. 1999; 72(3):136–141. | |
Watanabe I, Kasuya H, Nomura N, et al. Effects of tumor selective replication-competent herpes viruses in combination with gemcitabine on pancreatic cancer. Cancer Chemother Pharmacol. 2008;61(5):875–882. | |
Kasuya H, Nishiyama Y, Nomoto S, et al. Suitability of a US3-inactivated HSV mutant (L1BR1) as an oncolytic virus for pancreatic cancer therapy. Cancer Gene Ther. 2007;14(6):533–542. | |
Varghese S, Rabkin SD. Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther. 2002;9(12):967–978. | |
Kanai R, Tomita H, Shinoda A, et al. Enhanced therapeutic efficacy of G207 for the treatment of glioma through Musashi1 promoter retargeting of gamma34.5-mediated virulence. Gene Ther. 2006;13(2):106–116. | |
Dai MH, Zamarin D, Gao SP, et al. Synergistic action of oncolytic herpes simplex virus and radiotherapy in pancreatic cancer cell lines. Br J Surg. 2010;97(9):1385–1394. | |
Fu X, Tao L, Li M, Fisher WE, Zhang X. Effective treatment of pancreatic cancer xenografts with a conditionally replicating virus derived from type 2 herpes simplex virus. Clin Cancer Res. 2006;12(10):3152–3157. | |
Nawa A, Luo C, Zhang L, et al. Non-engineered, naturally oncolytic herpes simplex virus HSV1 HF-10: applications for cancer gene therapy. Curr Gene Ther. 2008;8(3):208–221. | |
Nakao A, Kasuya H, Sahin TT, et al. A phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancer. Cancer Gene Ther. 2011;18(3):167–175. | |
Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. 2011;19(6):1008–1016. | |
Campadelli-Fiume G, De Giovanni C, Gatta V, Nanni P, Lollini PL, Menotti L. Rethinking herpes simplex virus: the way to oncolytic agents. Rev Med Virol. 2011;21(4):213–226. | |
Hu JC, Coffin RS, Davis CJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12(22):6737–6747. | |
Harrington KJ, Hingorani M, Tanay MA, et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res. 2010;16(15):4005–4015. | |
Pütz MM, Midgley CM, Law M, Smith GL. Quantification of antibody responses against multiple antigens of the two infectious forms of Vaccinia virus provides a benchmark for smallpox vaccination. Nat Med. 2006;12(11):1310–1315. | |
Miller JD, van der Most RG, Akondy RS, et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28(5):710–722. | |
Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9(1):64–71. | |
Tysome JR, Briat A, Alusi G, et al. Lister strain of vaccinia virus armed with endostatin-angiostatin fusion gene as a novel therapeutic agent for human pancreatic cancer. Gene Ther. 2009;16(10):1223–1233. | |
Ishizaki H, Manuel ER, Song GY, et al. Modified vaccinia Ankara expressing survivin combined with gemcitabine generates specific antitumor effects in a murine pancreatic carcinoma model. Cancer Immunol Immunother. 2011;60(1):99–109. | |
Hiley CT, Yuan M, Lemoine NR, Wang Y. Lister strain vaccinia virus, a potential therapeutic vector targeting hypoxic tumours. Gene Ther. 2010;17(2):281–287. | |
Wennier ST, Liu J, Li S, Rahman MM, Mona M, McFadden G. Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Mol Ther. 2012;20(4):759–768. | |
Woo Y, Kelly KJ, Stanford MM, et al. Myxoma virus is oncolytic for human pancreatic adenocarcinoma cells. Ann Surg Oncol. 2008;15(8):2329–2335. | |
Petrulio CA, Kaufman HL. Development of the PANVAC-VF vaccine for pancreatic cancer. Expert Rev Vaccines. 2006;5(1):9–19. | |
Kaufman HL, Kim-Schulze S, Manson K, et al. Poxvirus-based vaccine therapy for patients with advanced pancreatic cancer. J Transl Med. 2007;5:60. | |
Tattersall P. The evolution of parvoviral taxonomy. In: Kerr JR, Cotmore S, Bloom ME, Linden RM, Parrish CR, editors. The Parvoviruses. London: Hodder Arnold; 2006:5–14. | |
Rommelaere J, Geletneky K, Angelova AL, et al. Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev. 2010;21(2–3):185–195. | |
Grekova S, Aprahamian M, Giese N, et al. Immune cells participate in the oncosuppressive activity of parvovirus H-1PV and are activated as a result of their abortive infection with this agent. Cancer Biol Ther. 2010;10(12):1280–1289. | |
Angelova AL, Aprahamian M, Grekova SP, et al. Improvement of gemcitabine-based therapy of pancreatic carcinoma by means of oncolytic parvovirus H-1PV. Clin Cancer Res. 2009;15(2):511–519. | |
Bhat R, Dempe S, Dinsart C, Rommelaere J. Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int J Cancer. 2011;128(4):908–919. | |
Dempe S, Lavie M, Struyf S, et al. Antitumoral activity of parvovirus-mediated IL-2 and MCP-3/CCL7 delivery into human pancreatic cancer: implication of leucocyte recruitment. Cancer Immunol Immunother. 2012;61(11):2113–2123. | |
Galanis E. Therapeutic potential of oncolytic measles virus: promises and challenges. Clin Pharmacol Ther. 2010;88(5):620–625. | |
Penheiter AR, Wegman TR, Classic KL, et al. Sodium iodide symporter (NIS)-mediated radiovirotherapy for pancreatic cancer. AJR Am J Roentgenol. 2010;195(2):341–349. | |
Penheiter AR, Dingli D, Bender CE, Russell SJ, Carlson SK. Monitoring the initial delivery of an oncolytic measles virus encoding the human sodium iodide symporter to solid tumors using contrast-enhanced computed tomography. J Gene Med. 2012;14(9–10):590–597. | |
Carlson SK, Classic KL, Hadac EM, et al. Quantitative molecular imaging of viral therapy for pancreatic cancer using an engineered measles virus expressing the sodium-iodide symporter reporter gene. AJR Am J Roentgenol. 2009;192(1):279–287. | |
Bossow S, Grossardt C, Temme A, et al. Armed and targeted measles virus for chemovirotherapy of pancreatic cancer. Cancer Gene Ther. 2011;18(8):598–608. | |
Himeno Y, Etoh T, Matsumoto T, Ohta M, Nishizono A, Kitano S. Efficacy of oncolytic reovirus against liver metastasis from pancreatic cancer in immunocompetent models. Int J Oncol. 2005;27(4):901–906. | |
Etoh T, Himeno Y, Matsumoto T, et al. Oncolytic viral therapy for human pancreatic cancer cells by reovirus. Clin Cancer Res. 2003;9(3):1218–1223. | |
Maitra R, Ghalib MH, Goel S. Reovirus: a targeted therapeutic – progress and potential. Mol Cancer Res. 2012;10(12):1514–1525. | |
Galanis E, Markovic SN, Suman VJ, et al. Phase II trial of intravenous administration of Reolysin(®) (Reovirus Serotype-3-dearing Strain) in patients with metastatic melanoma. Mol Ther. 2012;20(10):1998–2003. | |
Morris DG, Feng X, DiFrancesco LM, et al. REO-001: A phase I trial of percutaneous intralesional administration of reovirus type 3 dearing (Reolysin®) in patients with advanced solid tumors. Invest New Drugs. 2013;31(3):696–706. | |
Fueyo J, Gomez-Manzano C, Alemany R, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19(1):2–12. | |
Rocha-Lima CM, Raez LE. Erlotinib (tarceva) for the treatment of non-small-cell lung cancer and pancreatic cancer. P T. 2009;34(10):554–564. | |
Armstrong L, Arrington A, Han J, et al. Generation of a novel, cyclooxygenase-2-targeted, interferon-expressing, conditionally replicative adenovirus for pancreatic cancer therapy. Am J Surg. 2012;204(5):741–750. | |
Shan YF, Fang YF, Wang XQ, Jin R, Zhang QY, Andersson R. Experimental studies on treatment of pancreatic cancer with double-regulated duplicative adenovirus AdTPHre-hEndo carrying human endostatin gene. Pancreatology. 2013;13(4):393–400. | |
Lisiansky V, Naumov I, Shapira S, et al. Gene therapy of pancreatic cancer targeting the KRas oncogene. Cancer Gene Ther. 2012;19(12):862–869. | |
Xie FJ, Zhao P, Zhang YP, et al. Adenovirus-mediated interferon-gamma gene therapy induced human pancreatic carcinoma Capan-2 cell apoptosis in vitro and in vivo. Anat Rec (Hoboken). 2013;296(4):604–610. | |
Hasegawa N, Abei M, Yokoyama KK, et al. Cyclophosphamide enhances antitumor efficacy of oncolytic adenovirus expressing uracil phosphoribosyltransferase (UPRT) in immunocompetent Syrian hamsters. Int J Cancer. 2013;133(6):1479–1488. | |
Guo J, Gao J, Li Z, et al. Adenovirus vector-mediated Gli1 siRNA induces growth inhibition and apoptosis in human pancreatic cancer with Smo-dependent or Smo-independent Hh pathway activation in vitro and in vivo. Cancer Lett. 2013;339(2):185–194. | |
Zhang YA, Nemunaitis J, Samuel SK, Chen P, Shen Y, Tong AW. Antitumor activity of an oncolytic adenovirus-delivered oncogene small interfering RNA. Cancer Res. 2006;66(19):9736–9743. | |
Freytag SO, Barton KN, Brown SL, et al. Replication-competent adenovirus-mediated suicide gene therapy with radiation in a preclinical model of pancreatic cancer. Mol Ther. 2007;15(9):1600–1606. | |
Wang H, Pei W, Luan Q, et al. A feasibility study on gene therapy of pancreatic carcinoma with Ad-PUMA. Cancer Biol Ther. 2012;13(9):712–719. | |
Lupu-Meiri M, Geras-Raaka E, Lupu R, et al. Knock-down of plasminogen-activator inhibitor-1 enhances expression of E-cadherin and promotes epithelial differentiation of human pancreatic adenocarcinoma cells. J Cell Physiol. 2012;227(11):3621–3628. | |
Yoon SS, Carroll NM, Chiocca EA, Tanabe KK. Cancer gene therapy using a replication-competent herpes simplex virus type 1 vector. Ann Surg. 1998;228(3):366–374. | |
He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A. 1997;94(3):843–848. | |
Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1(9):938–943. | |
Meignier B, Longnecker R, Roizman B. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020: construction and evaluation in rodents. J Infect Dis. 1988;158(3):602–614. | |
Kimata H, Takakuwa H, Goshima F, et al. Effective treatment of disseminated peritoneal colon cancer with new replication-competent herpes simplex viruses. Hepatogastroenterology. 2003;50(52):961–966. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.