")
Back to Journals » Cancer Management and Research » Volume 12
Oncogenic Roles Of A Histone Methyltransferase SETDB2 In AML1-ETO Positive AML
Received 13 August 2019
Accepted for publication 4 November 2019
Published 4 February 2020 Volume 2020:12 Pages 783—792
DOI https://doi.org/10.2147/CMAR.S227036
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chien-Feng Li
Guangfu Mu, Fangping Chen
Department of Hematology, The Third Xiangya Hospital, Central South University, Changsha, Hunan 410013, People’s Republic of China
Correspondence: Fangping Chen
Department of Hematology, The Third Xiangya Hospital, Central South University, Tongzipo Road No. 138, Changsha, Hunan 410013, People’s Republic of China
Tel/Fax +86 073 1861 8841
Email [email protected]
Introduction: AML1-ETO produced by t(8;21) abnomality has multiple effects on the leukemogenesis of acute myeloid leukemia (AML). SET domain, bifurcated 2 (SETDB2) can mediate gene silencing by trimethylation of the ninth lysine residue of histone H3 protein (H3K9) of the promoter and has been confirmed as an oncogene in many cancers. The role of SETDB2 in AML1-ETO positive AML is not clear.
Methods: Quantitative reverse transcription PCR was performed to measure SETDB2 expression in bone marrow from AML patients and healthy people. Kaplan-Meier analysis was performed to investigate the effect of SETDB2 on prognosis of AML patients. Dual luciferase reporter gene assay, chromatin immunoprecipitation were performed to investigate the regulatory mechanism of AML1-ETO on SETDB2. CCK-8 and colony formation assay were performed to measure the effect of SETDB2 on leukemic cells.
Results: SETDB2 is highly expressed in AML1-ETO positive AML. The overall survival, event-free and relapse-free survival rate of patients with high SETDB2 expression was lower than those of patients with low SETDB2 expression. SETDB2 is epigenetically upregulated by AML1-ETO fusion protein. Downregulation of SETDB2 expression significantly inhibits the proliferation and clonality of leukemic cells and promotes the sensitivity of leukemic cells to an epigenetic inhibitor JQ1.
Conclusion: AML1-ETO/SETDB2 is a novel epigenetic pathway of leukemogenesis and SETDB2 is a potential therapeutic target of t(8;21) AML.
Keywords: acute myeloid leukemia, AML1-ETO, SETDB2, epigenetic, clinical biomarker
Introduction
Due to the high degree of heterogeneity, there is still an insurmountable barrier to targeted therapy of t(8; 21) acute myeloid leukemia (AML).1 Studies have shown that gene expression regulated by epigenetic mechanisms plays an important role in controlling cellular behavior.2 In cancer cells, normal DNA methylation distribution is disrupted and persists during malignant transformation.3 Abnormal distribution of promoter DNA methylation of multiple genes has been found in t(8; 21) AML, such as THAP domain-containing protein 10 (THAP10) and brain acid soluble protein 1 (BASP1).4–6 However, the leukemogenic mechanism of t(8; 21) AML is not yet clear.
There are about 50 proteins containing the SET domain, which have the function of histone lysine methyltransferase and are important regulators of epigenetic regulation mechanisms.7 SET domain, bifurcated 2 (SETDB2) belongs to the KMT1 subfamily of the SET domain protein family, and other members include SUV39H1, G9a and SETDB1. SETDB2 mediates gene silencing by trimethylation of the ninth lysine residue of histone H3 protein (H3K9) of the promoter.8 Studies have shown that SETDB2 can silence target genes by epigenetic regulation, thereby regulating early embryonic development of zebrafish and inhibiting inflammatory responses.9 In addition, genomic sequencing data from the TCGA database indicates that SETDB2 has mutations and copy number changes in various cancer subgroups.10 Recent studies have shown that in acute lymphoblastic leukemia, E2A-PBX1 directly regulates SETDB2, while SETDB2 inhibits cyclin-dependent kinase 4 inhibitor C (CDKN2C) expression by histone H3K9 trimethylation, thereby promoting the progression of ALL.11 However, the role and mechanism of SETDB2 in t(8; 21) acute myeloid leukemia (AML) is not clear.
In this study, we investigated the regulation of AML1-ETO on SETDB2 in t(8; 21) AML and the role of SETDB2 in t(8; 21) AML. Our results illustrate the epigenetic mechanism of AML1-ETO/SETDB2 in t(8; 21) AML, and SETDB2 may be a new clinical biomarker and therapeutic target for patients with AML1-ETO positive AML.
Materials And Methods
Human Sample Collection
There are three independent sample cohorts in this study. The first sample cohort included bone marrow tissue from 7 normal healthy individuals and 10 AML patients collected from March 2016 to April 2017. The CD34+CD38-cells were used to detect the expression of SETDB2. The second sample cohort included bone marrow tissue of 13 normal healthy individuals and 96 AML patients collected from June 2017 to June 2018 (including t(8; 21), n = 56; pML-RARa, n = 25; inv(16), n = 15), used to isolate mononuclear cells for detection of SETDB2 expression; the third sample cohort included 42 patients with AML1-ETO negative and 50 patients with AML1-ETO positive AML from June 2013 to June 2018. These cases contain patient survival data. Mononuclear cells of bone marrow samples were isolated using Ficoll-Hypaque (Sigma-Aldrich, St Louis, MO). The isolated mononuclear cells were used to detect the expression of SETDB2. Cytochemistry and cytogenetics were performed in all cases. Cases were classified according to the 2008 revision of the World Health Organization classification of myeloidneoplasms and acute leukemia. The study was approved by the ethics committees of The Third Xiangya Hospital, Central South University, and all patients and healthy individuals provided written informed consent prior to entering this study under the Helsinki Declaration.
Cell Culture
Jurkat, MM6, U937, NB4, Kasumi-1, SKNO-1, K562 cells were purchased from the Chinese Academy of Sciences Cell bank (Shanghai, China). These cells were cultured in RPMI 1640 medium supplemented with 50 μg/mL streptomycin, 50 IU penicillin and 10% FBS.
Cell Transfection
To knock down AML1-ETO, the expression of AML1-ETO was silenced using a siRNA sequence directed against the AML1-ETO mRNA fusion site (siA/E-RNA) with reference to published literature.12
For shRNA-mediated knockdown of SETDB2 gene, 1×106 Kasumi-1 cells or SKNO-1 cells were seeded in 6-well plates to a 80% confluence, and then transfected with shRNA lentivirus. The shRNA sequence used in this study (shRNA1#, CCGGTCTGACGTGGATATTAGTAATCTCGAGATTACTAATATCCACGTCAGATTTTTG; shRNA2#, CCGGCCAAGCAATGAATCTAGTAAACTCGAGTTTACTAGATTCATTGCTTGGTTTTTG) was synthesized by GenePharma (Shanghai, China) and packaged into hU6-MCS-PGK-EGFP lentiviral vector (GenePharma, Shanghai, China). The cells were infected with MOI=100 virus. Transfection efficiency was determined by FACScalibur flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA) with a transfection efficiency of over 90%.
Quantitative Reverse Transcription-PCR
Total RNA was extracted using the Trizol reagent. After DNAse I (Thermo Scientific) treatment, RNA was reverse transcribed with reverse transcriptase (Thermo Scientific). QRT-PCR for mature miRNAs was measured by All-in-One™ miRNA qRT-PCR (GeneCopoeia, Shanghai, China) following the manufacturer recommended protocol on a CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). The sequences of SETDB2 primers were as following: forward, GTGGACCAAAAGATGGAGTTGG; reverse, TCAGACAAGCACCAGAGCAG.
Dual Luciferase Reporter Gene Assay
SETDB2 promoter fragments were amplified from human genomic DNA by PCR. Primer sequences are shown in Supplementary Table 1. All fragments were inserted into the pGL3-LUC reporter vector (Promega). Corresponding mutants were generated using the Site-directed Mutagenesis Kit (Sangon Biotech (Shanghai) Co., Ltd., Shanghai, China) according to the manufacturer’s instructions. All constructs were verified by DNA sequencing. 2×105 HEK293T cells were seeded in a 24-well plate for 12 hrs, and 100 ng of the blank pcDNA3 vector or the pcDNA3 vector containing the AML1-ETO cDNA and the 400 ng LUC reporter vector were co-transfected into the cells for 48 hrs. Co-transfection with the pRL-TK Renilla Luciferase Reporter Vector (Promega) was used as a control. After transfection, cells were harvested and analyzed using Dual Luciferase Assay (Promega) according to the manufacturer’s instructions. Luciferase activity was measured on SpectraMax® L (Molecular Devices, Sunnyvill, CA, USA) and analyzed with SoftMax® Pro (Molecular Devices).
Chromatin Immunoprecipitation (ChIP)
To detect the AML1-ETO binding region on the SETDB2 promoter, a ChIP-seq experiment was performed. First, after knocking down AML1-ETO in Kasumi-1 cells, cells were harvested, and cross-linked chromatin was prepared, which was then broken up into fragments of about 200 bp in size using a Bioruptor UCD-200 sonicator (Diagenode). These fragmented chromatins was incubated with specific antibodies ((AML1 (ab23980, Abcam), ETO (ab124269, Abcam), HDAC1 (ab7028, Abcam), HDAC3a (ab13537, Abcam), HDAC3b (ab13888, Abcam), HDAC3c (ab13604, Abcam), p300 (ab14984, Abcam) overnight at 4 degrees. Rabbit serum was used as a control. RNA elution and purification were performed using the High-Sensitivity ChIP Kit (ab185913, Abcam) according to the manufacturer’s instructions. The expression of the sub-AML1 binding region in SETDB2 promoter was detected by qPCR. GAPDH was used as a control.
Western Blot Analysis
After treatment, the cells were lysed in RIPA buffer containing 1 mM DTT, 11 μg/mL DNase I and protease inhibitor cocktail (Roche) and incubated on ice for 30 mins. Sixty micrograms of protein sample were separated by electrophoresis on a denatured 10% SDS-PAGE gel and blotted onto a PVDF membrane (Millipore). Immunoblotting was performed using primary antibody (Goat polyclonal to SETDB2 antibody (1:1000, ab13712, Abcam)); Rabbit monoclonal to GAPDH (1:2000, EPR16891, Abcam) was used as a loading control. Proteins were visualized by the ECL method (Beyotime Biotechnology, Shanghai, China).
Cell Proliferation Measurement
cell proliferation was assessed using a CCK-8 kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions. Briefly, after transfection, the cells (3000 cells/well) were seeded into 96-well plates and further cultured for 24 hr. The medium was replaced with 100 μl fresh medium. Ten μl CCK-8 solution was added and incubated for 1 hr. Absorbance at 450 nm was measured on SpectraMax® L (Molecular Devices, Sunnyvill, CA, USA) and analyzed with SoftMax® Pro (Molecular Devices).
For IC50 determination, the transfected cells were seeded into 96-well plates (3000 cells/well), culture was continued for 24 hrs, and then 10 μl of CCK-8 solution was added to the cell culture using a different concentration of JQ1 or C646. After incubation for another 1 hr, the absorbance at 450 nm was measured on SpectraMax® L (Molecular Devices) and analyzed with SoftMax® Pro (Molecular Devices).
Colony Formation Analysis
Colony formation assays were performed using a methylcellulose culture system (Cat. M6385, Sigma) according to the manufacturer’s instructions. After incubation for 10 days in a humid environment at 37 °C and 5% CO2, the number of clones > 50 cells was counted.
Statistical Analyses
All statistical analyses were performed using Graphpad Prism (version 7.04). Continuity variables are expressed as mean ± standard deviation of at least three independent experiments. The expression of SETDB2 greater or equal to the mean was defined as high expression, otherwise were defined as low expression. The overall survival rate, event-free survival and relapse-free survival estimates over time were calculated using the Kaplan-Meier method with log rank test. Student’s t test was used for comparison between the two groups, one-way ANOVA was used for three or more comparisons, and if there were differences between groups, the Tukey method was used for post hoc comparison. All statistical analyses were performed using a two-sided test. P <0.05 was statistically significant.
Results
Expression Of SETDB2 In AML1-ETO Positive AML Patients
There has been no report on the expression and function of SETDB2 in AML, so we determined the expression of SETDB2 in AML and normal bone marrow CD34+ cells by qPCR. The expression of SETDB2 in CD34+/CD38- cells isolated from AML1-ETO positive cases (n = 10) was significantly higher than that in healthy human CD34+/CD38 cells (Figure 1A). In addition, SETDB2 expression in t(8;21) positive AML was significantly higher than that in normal bone marrow CD34+ cells, PML-RARa, inv(16) and FLT3-ITD positive AML cases (Figure 1B). In the AML cell lines, we also found that SETDB2 expression was significantly higher in t(8;21) positive AML cells than in other t(8;21) negative AML cells (Figure 1C). Importantly, the SETDB2 expression of AML1-ETO positive AML cases was significantly higher than that of AML1-ETO negative AML cases (Figure 1D). We analyzed the mRNA levels of SETDB2 in bone marrow mononuclear cells before and after chemotherapy in patients with AML1-ETO+ AML. Compared with the expression level at the time of diagnosis, the mRNA level of SETDB2 was significantly decreased in AML1-ETO + AML patients who achieved complete remission after chemotherapy, while the mRNA level of SETDB2 was significantly increased in the relapsed period (Figure 1E).
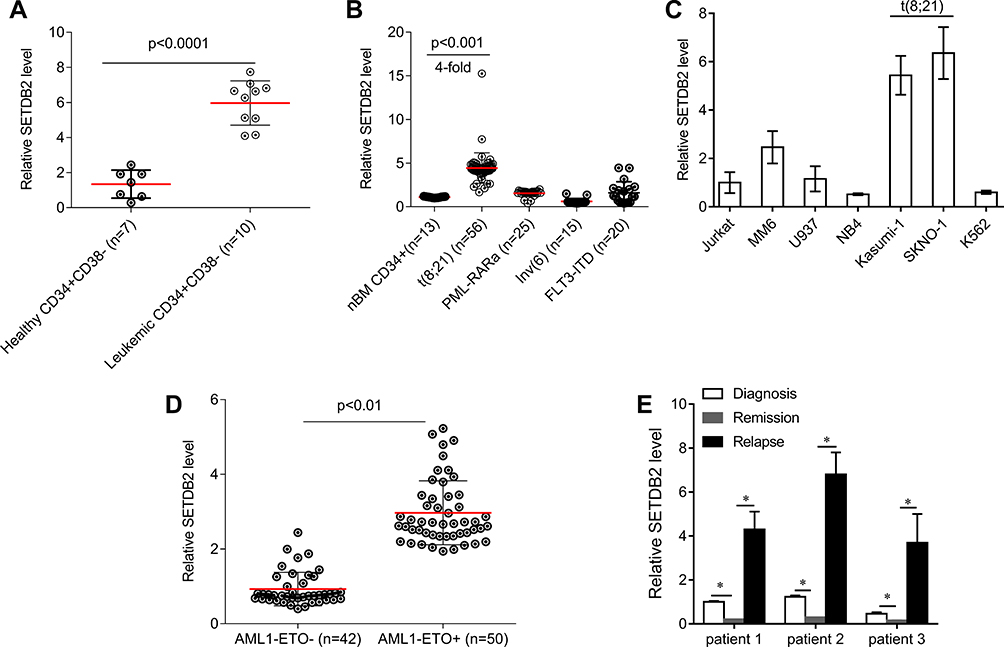 |
Figure 1 The expression of SETDB2 in AML. Notes: (A) Quantification of SETDB2 expression in patients with AML1-ETO-positive AML and normal BM subpopulations by qRT-PCR. (B) Quantification of SETDB2 expression in AML patients with AE, PML-RARa fusions, or Inv(16), and normal human BM CD34+ cells by qRT-PCR. (C) Quantification of SETDB2 expression in AML cell lines by qRT-PCR. (D) Quantification of SETDB2 expression in patients with AML1-ETO -positive AML or AML1-ETO -negative AML by qRT-PCR. (E) Sequential analyses of SETDB2 mRNA levels in mononuclear cells isolated from bone marrow samples of three individual patients with AML1-ETO -positive AML at different stages of disease, including newly diagnosed, remission and relapse. Expression values are shown as mean ± SEM. *P<0.05. |
Relationship Between Expression Of SETDB2 And Survival Of AML Patients
We analyzed the association between the expression level of SETDB2 and AML1-ETO, and the prognosis of AML patients with different SETDB2 expression. The results showed that SETDB2 mRNA levels were positively correlated with AML1-ETO (Pearson R=0.63, p<0.01, Figure 2A). Fifty patients with AML1-ETO-positive AML were divided into SETDB2 high expression (n = 34) and SETDB2 low expression patients (n = 16) according to the mean expression level of SETDB2. The overall survival rate of patients with SETDB2 low expression was higher than that of patients with SETDB2 high expression(Figure 2B), and the event-free and relapse-free survival time of patients with SETDB2 low expression (Figure 2C and D) was longer than that of patients with high SETDB2 expression. Taken together, these results indicate that high expression of the SETDB2 gene is associated with poor prognosis in patients with AML1-ETO positive AML.
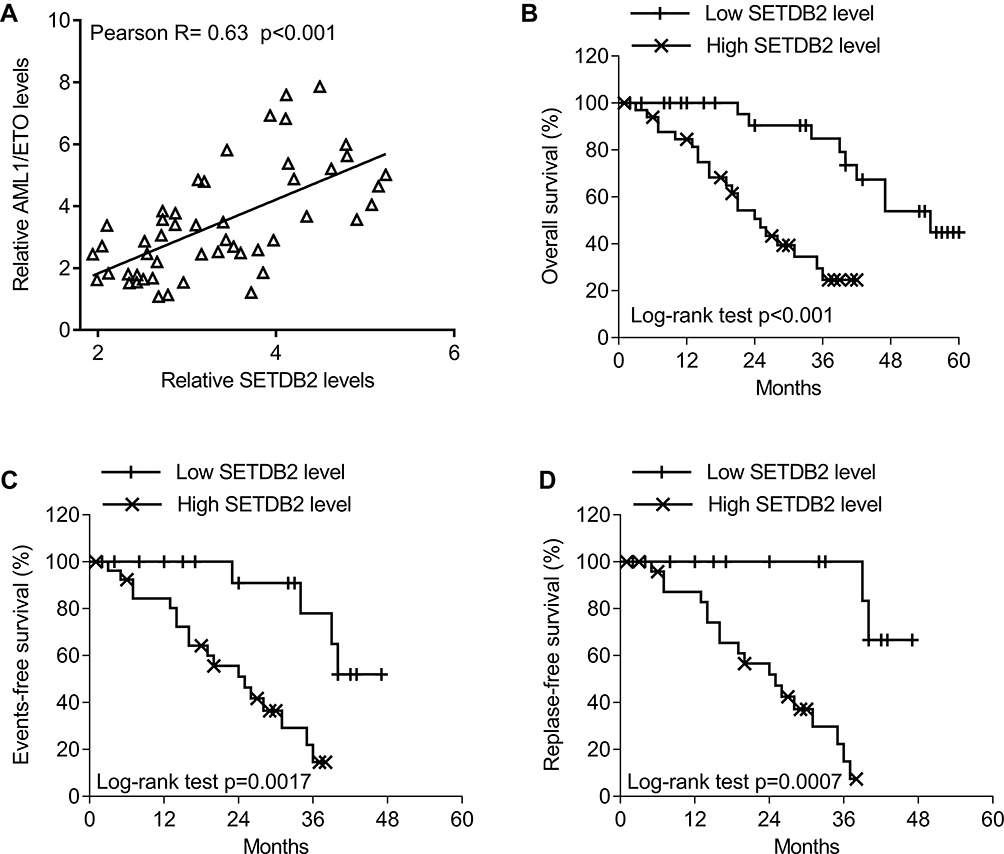 |
Figure 2 The association between SETDB2 expression and clinical outcome in patients with AML1-ETO -positive AML. Notes: (A) Correlations in gene expression between SETDB2 and AML1-ETO (Pearson test, R = 0.63, P<0.001). (B) The log rank test was used for the survival analysis. Correlations of SETDB2 expression with overall survival (P<0.001). (C) Correlations of SETDB2 expression with event-free survival (P=0.0017). (D) Correlations of SETDB2 expression with relapse-free survival (P=0.0007). |
AML1-ETO Epigenetically Enhances The Expression Of SETDB2
The promoter region methylation site of SETDB2 and the possible AML1 binding sites were analyzed by bioinformatics (Figure 3A). Therefore, we constructed a luciferase reporter gene containing the wild type (SETDB2-full, SETDB2-P1 to SETDB2-P4) or mutation (SETDB2-P1-M to SETDB2-P4-M) sequences of the SETDB2 promoter region (Figure 3A). Each reporter gene and AML1-ETO or empty vector were co-transfected into 293T cells to detect luciferase activity. The results showed that overexpression of AML1-ETO activates luciferase activity leading to SETDB2-full, SETDB2-P1- SETDB2-P4, SETDB2-P1-M, SETDB2-P2-M and SETDB2-P3-M, but does not increase SETDB2 -P4-M luciferase activity (Figure 3B), indicating that AML1-ETO binds to the promoter region of SETDB2 (−500 nt to. +1 nt) and may be involved in AML1-ETO-induced expression of SETDB2.
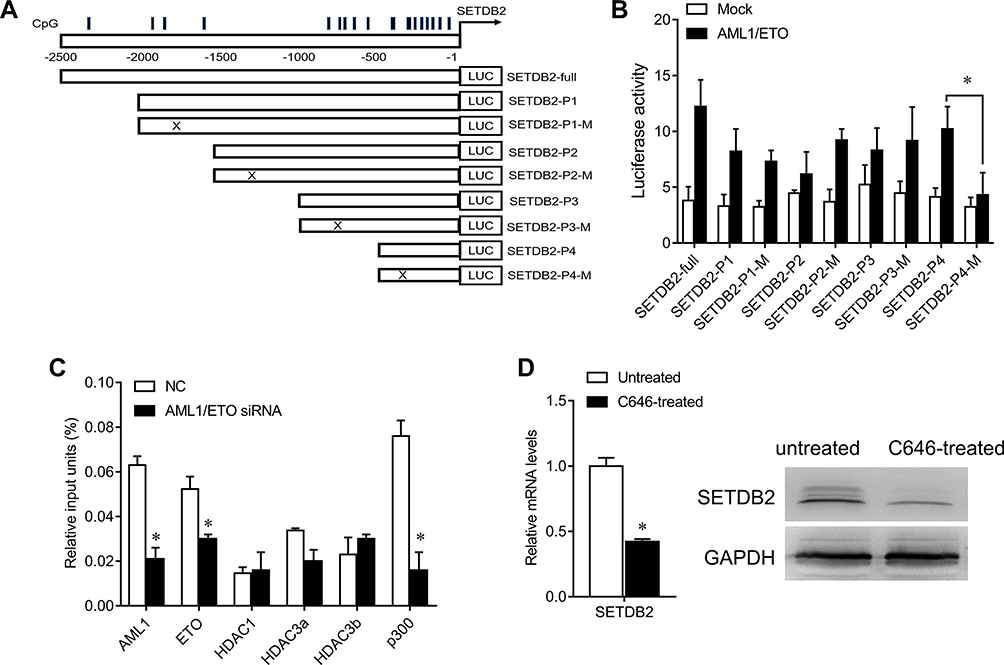 |
Figure 3 AML1-ETO epigenetically activa SETDB2 expression. Notes: (A) Schematic diagrams of the CpG islands along the SETDB2 promoter. Numbers indicate the nucleotides relative to SETDB2 (+1 nt). Vertical lines indicate CpG dinucleotides. Lower panels: A series of constructs and their mutants. (B) Luciferase reporter activities of human 293T cells transiently co-transfected for 48 h with luciferase reporter constructs containing the wild-type sequence of SETDB2 promoter or its mutant counterparts, together with AML1-ETO or mock cDNA. (C) After ChIP using the indicated antibodies or IgG, qRT–PCR was performed to evaluate the specificity of protein binding in the region containing the predicted AML1-binding site. (D) After Kasumi-1 cells were treated with C646 (30 nM) for 24 h, qRT–PCR and Western blot were performed to quantify SETDB2 levels. Expression values are shown as mean ± SEM. *P<0.05. |
Furthermore, studies have confirmed that the NHR1 domain of AML1-ETO has a binding site for p300.13 We further analyzed whether AML1-ETO can recruit related epigenetic regulatory molecules to the promoter region of SETDB2 by ChIP experiments. The results show that the SETDB2 promoter region is capable of binding both AML1-ETO and p300, but not HDAC1, HDAC3a, HDAC3b and HDAC3c (Figure 3C), suggesting that AML1-ETO can recruit p300 to the promoter region of SETDB2 and activate its transcription. To further demonstrate this effect, we used the p300 small molecule inhibitor C646 to treat Kasumi-1 cells and tested the expression of SETDB2 to determine the function of p300 in acetylation of the SETDB2 promoter region. C646 treatment significantly reduced the mRNA and protein levels of SETDB2 (Figure 3D). Therefore, these results indicate that AML-ETO recruits p300 to the SETDB2 promoter region and activates SETDB2 transcription, thereby enhancing SETDB2 expression.
Impact Of SETDB2 In AML-ETO Positive AML Cells
We further analyzed the function of SETDB2 in AML. We designed two shRNA sequences to reduce the expression of SETDB2. Transfection of shRNA in SKNO-1 and Kasumi-1 cells significantly reduced the expression of SETDB2 both in mRNA and protein levels, and shRNA 2# was more efficient (Figure 4A). Interference with SETDB2 significantly reduced the colony-forming ability of SKNO-1 and Kasumi-1 cells (Figure 4B) and inhibited the proliferation of SKNO-1 and Kasumi-1 cells compared to the negative control. The cell survival rate after SETDB2 expression inhibition was reduced by nearly 20% and 45%, respectively (Figure 4C). Therefore, inhibition of expression of SETDB2 can inhibit proliferation in AML-ETO positive AML cells.
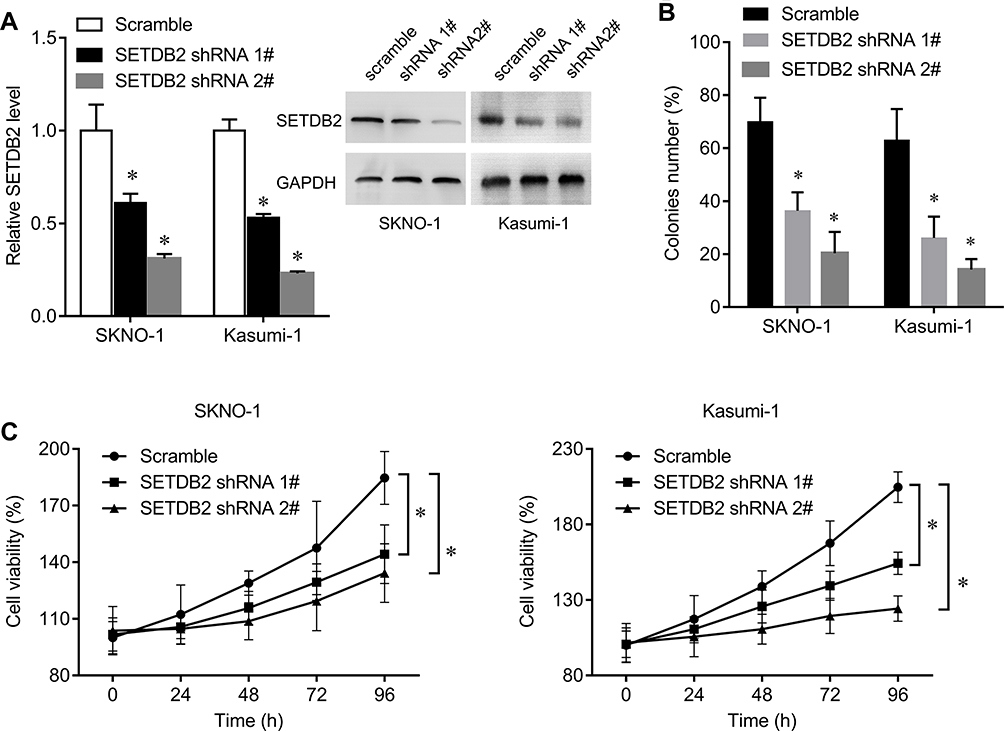 |
Figure 4 Impact of shRNA-mediated knockdown of SETDB2 on AML1-ETO -positive AML cell lines. Notes: (A) After SKNO-1 and Kasumi-1 cells were infected with shRNA lentivirus for 48 h, qRT–PCR and Western blot were performed to quantify SETDB2 levels. (B) Effect of SETDB2 knockdown compared to scramble control on colony formation. (C) Effect of SETDB2 knockdown compared to scramble control on proliferation. Expression values are shown as mean ± SEM. *P<0.05. |
Depletion Of SETDB2 Enhances Sensitivity To Epigenetic Inhibitors
SETDB2 is an epigenetic regulatory molecule. To further evaluate the role of SETDB2 in AML chemotherapy, we knocked down SETDB2 and simultaneously treated SKNO-1 and Kasumi-1 cells with epigenetic inhibitor JQ1 and observed cell proliferation rate. JQ1 is a potent inhibitor of the BET family of bromodomain proteins, which has broad activity in a variety of acute leukemia subtypes.14 The results showed that SETDB2 knockdown significantly increased the cytotoxicity of JQ1 in SKNO-1 and Kasumi-1 cells evaluated by reducing the half-inhibitory concentration of JQ1 (IC50) (SETDB2 shRNA2# 63.36 nM vs scramble 197.2 nM for SKNO-1; SETDB2 shRNA2# 29.78 nM vs scramble 84.9 nM for Kasumi-1; Figure 5A). In addition, SETDB2 knockdown also enhanced the cytotoxicity of C646 in SKNO-1 and Kasumi-1 cells (SETDB2 shRNA2# 244.7 nM vs scramble 916.6 nM for SKNO-1; SETDB2 shRNA2# 154.7 nM vs scramble 510.5 nM for Kasumi-1; Figure 5B). These results indicate that combination inhibiting SETDB2 with other epigenetic inhibitor can improve the therapeutic effect of AML-ETO positive AML.
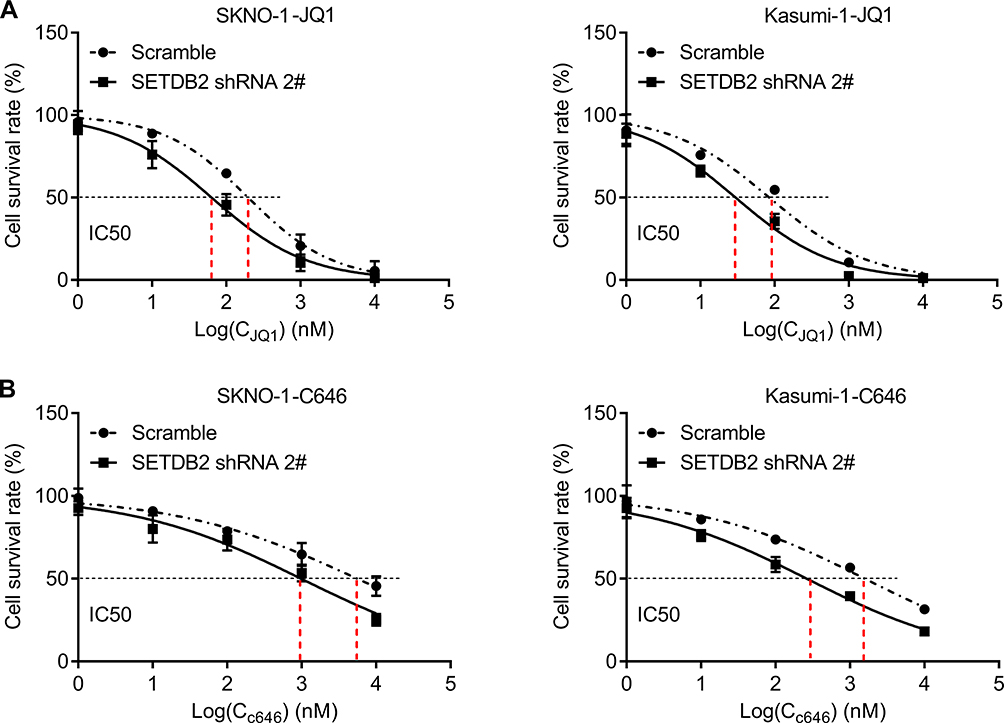 |
Figure 5 SETDB2 knockdown increases sensitivity to epigenetic inhibitors. Notes: Dose-response curves are shown for control or SETDB2 shRNA-treated SKNO-1 and Kasumi-1 cells cultured in increasing concentrations of JQ1 (A) and C646 (B). IC50 for JQ1 in combination with control or SETDB2 shRNA is 197.2 versus 63.36 nM (for SKNO-1 cells) and 84.9 versus 29.78 nM (for Kasumi-1 cells), respectively. IC50 for C646 in combination with control or SETDB2 shRNA is 916.6 versus 244.7 nM (for SKNO-1 cells) and 510.5 versus 154.7 nM (for Kasumi-1 cells), respectively. |
Discussion
In this study, we elucidated the leukemogenic effects of SETDB2 in AML1-ETO positive AML. SETDB2 is highly expressed in AML1-ETO positive AML and is directly targeted by AML1-ETO. Interference with the expression of SETDB2 significantly inhibits the proliferation and clonality of AML cells and promotes the sensitivity of AML cells to epigenetic inhibitors. This study provides a new therapeutic target for AML treatment and provides an evidence base for the combined use of epigenetic inhibitors to treat AML.
AML1-ETO positive, ie t(8; 21) AML, accounted for 15% of all AML cases.15 In this study, we found that SETDB2 was directly regulated by AML1-ETO fusion protein. Moreover, the expression of SETDB2 in AML1-ETO positive AML cases was significantly higher than that of normal hematopoietic stem cells; and it was confirmed in a small sample cohort that SETDB2 was positively correlated with AML1-ETO expression. SETDB2 high expression was negatively correlated with overall survival and event-free survival time. SETDB2 decreased with the remission of AML and increased with relapse. We will confirm this in a larger sample cohort.
Two CpG islands were found in the promoter region of SETDB2 (−1000-1) by bioinformatics analysis. The luciferase reporter gene assay demonstrated that AML1-ETO binds to the fragment of −500-1 in SETDB2 promoter, indicating the promoter activity of SETDB2 is directly regulated by AML1-ETO. AML1-ETO can silence the target gene by recruiting methylation transferase to the promoter, and can also activate target gene transcription by recruiting acetylation cofactors. Through Chip analysis, we found that AML1-ETO may enhance transcription and expression by recruiting a histone acetyltransferase p300 to the promoter of SETDB2. The inhibition of SETDB2 expression by histone acetylation inhibitor C646 treatment also demonstrates this view. However, in other subtypes of AML, it is unclear whether other fusion proteins directly regulate the expression of SETDB2, and further research is needed.
This study clarified the function of SETDB2 in AML1-ETO positive AML. Interference with SETDB2 significantly inhibited the proliferation and clonality of AML cells. Previous studies have shown that SETDB2 is an oncogene in gastric and renal cancer.16,17 SETDB2 plays a role in promoting the development of renal cancer by transcriptional inhibition of WW domain-containing oxidoreductase (WWOX) and Cell adhesion molecule 1 (CADM1).17 In addition, SETDB2 expression is maintained as a direct target gene of the chimeric transcription factor E2A-PBX1 and suppresses expression of the cell-cycle inhibitor CDKN2C through histone H3K9 tri-methylation in ALL.11 Analysis of various tumor tissue genome sequencing data in the TCGA database revealed that SETDB2 has various mutations and copy number changes in various cancers (data not shown). However, in this study, we have not identified any target gene for SETDB2 in AML1-ETO-positive AML. Given that the downstream target genes of SETDB2 are tissue specific, we will continue to screen out direct target genes of SETDB2 through the integration of ChIP-seq and transcriptome datasets.
In addition, combination with the epigenetic inhibitor JQ1 or C646 significantly enhanced SETDB2 silencing-mediated proliferation inhibition in AML cells. JQ1 is an inhibitor of the BET family and competes with acetylated histones for binding to the BET-derived bromodomain (BD), which inhibits bromodomain-containing protein 4 (BRD4) from recruiting c-myc to the promoter region of the gene, thereby inhibiting transcription.18 JQ1 can effectively treat leukemia, colon cancer, and prostate cancer, so JQ1 is considered to be a potential effective drug for clinical treatment of cancer.19–24 This study found that AML1-ETO increased the expression of SETDB2 by enhancing the histone acetylation of the promoter of SETDB2, and that JQ1 treatment and silencing of SETDB2 significantly enhanced the inhibition of AML cells. Whether BRD4 also has transcriptional activation on SETDB2 requires further study. Although there is no selective SETDB2 small molecule inhibitor so far, the synergistic relationship between SETDB2 and epigenetic inhibitors has opened up a new way for the development of drugs for the treatment of AML1-ETO positive AML.
In conclusion, this study demonstrates that AML1-ETO/SETDB2 is a novel epigenetic pathway for the development of t(8; 21) AML. These findings also indicate that SETDB2 is a novel biomarker and potential therapeutic target in the t(8; 21) AML.
Ethics Approval And Informed Consent
This study was approved by Ethics Committee of the Third Xiangya Hospital, Central South University.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. van der Kouwe E, Staber PB. RUNX1-ETO: attacking the epigenome for genomic instable leukemia. Int J Mol Sci. 2019;20(2). doi:10.3390/ijms20020350
2. Alvarez S, Suela J, Valencia A, et al. DNA methylation profiles and their relationship with cytogenetic status in adult acute myeloid leukemia. PLoS ONE. 2010;5(8):e12197. doi:10.1371/journal.pone.0012197
3. Figueroa ME, Lugthart S, Li Y, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17(1):13–27. doi:10.1016/j.ccr.2009.11.020
4. Rasmussen KD, Jia G, Johansen JV, et al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015;29(9):910–922. doi:10.1101/gad.260174.115
5. Li Y, Ning Q, Shi J, et al. A novel epigenetic AML1-ETO/THAP10/miR-383 mini-circuitry contributes to t(8;21) leukaemogenesis. EMBO Mol Med. 2017;9(7):933–949. doi:10.15252/emmm.201607180
6. Zhou L, Fu L, Lv N, et al. Methylation-associated silencing of BASP1 contributes to leukemogenesis in t(8;21) acute myeloid leukemia. Exp Mol Med. 2018;50(4):44. doi:10.1038/s12276-018-0067-4
7. Torrano J, Al EA, Hammerlindl H, Schaider H. Emerging roles of H3K9me3, SETDB1 and SETDB2 in therapy-induced cellular reprogramming. Clin Epigenetics. 2019;11(1):43. doi:10.1186/s13148-019-0644-y
8. Huang Y, Liu C, Shen WH, Ruan Y. Phylogenetic analysis and classification of the Brassica rapa SET-domain protein family. BMC Plant Biol. 2011;11:175. doi:10.1186/1471-2229-11-175
9. Du TT, Xu PF, Dong ZW, et al. Setdb2 controls convergence and extension movements during zebrafish gastrulation by transcriptional regulation of dvr1. Dev Biol. 2014;392(2):233–244. doi:10.1016/j.ydbio.2014.05.022
10. Hung MH, Chen KF. Reprogramming the oncogenic response: SET protein as a potential therapeutic target in cancer. Expert Opin Ther Targets. 2017;21(7):685–694. doi:10.1080/14728222.2017.1336226
11. Lin CH, Wong SH, Kurzer JH, et al. SETDB2 links E2A-PBX1 to cell-cycle dysregulation in acute leukemia through CDKN2C repression. Cell Rep. 2018;23(4):1166–1177. doi:10.1016/j.celrep.2018.03.124
12. Li Y, Gao L, Luo X, et al. Epigenetic silencing of microRNA-193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood. 2013;121(3):499–509. doi:10.1182/blood-2012-07-444729
13. Wang L, Gural A, Sun XJ, et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science. 2011;333(6043):765–769. doi:10.1126/science.1201662
14. Da CD, Agathanggelou A, Perry T, et al. BET inhibition as a single or combined therapeutic approach in primary paediatric B-precursor acute lymphoblastic leukaemia. Blood Cancer J. 2013;3:e126. doi:10.1038/bcj.2013.24
15. Sun XJ, Wang Z, Wang L, et al. A stable transcription factor complex nucleated by oligomeric AML1-ETO controls leukaemogenesis. Nature. 2013;500(7460):93–97. doi:10.1038/nature12287
16. Ferreira MJ, Pires-Luis AS, Vieira-Coimbra M, et al. SETDB2 and RIOX2 are differentially expressed among renal cell tumor subtypes, associating with prognosis and metastization. Epigenetics. 2017;12(12):1057–1064. doi:10.1080/15592294.2017.1385685
17. Nishikawaji T, Akiyama Y, Shimada S, et al. Oncogenic roles of the SETDB2 histone methyltransferase in gastric cancer. Oncotarget. 2016;7(41):67251–67265. doi:10.18632/oncotarget.v7i41
18. Zhou Y, Zhou J, Lu X, Tan TZ, Chng WJ. BET bromodomain inhibition promotes de-repression of TXNIP and activation of ASK1-MAPK pathway in acute myeloid leukemia. BMC Cancer. 2018;18(1):731. doi:10.1186/s12885-018-4661-6
19. Pericole FV, Lazarini M, de Paiva LB, et al. BRD4 inhibition enhances azacitidine efficacy in acute myeloid leukemia and myelodysplastic syndromes. Front Oncol. 2019;9:16. doi:10.3389/fonc.2019.00016
20. Stewart H, Chaudry S, Crichlow A, Luiling FF, Chevassut T. BET inhibition suppresses S100A8 and S100A9 expression in acute myeloid leukemia cells and synergises with daunorubicin in causing cell death. Bone Marrow Res. 2018;2018:5742954. doi:10.1155/2018/5742954
21. Kang C, Kim CY, Kim HS, Park SP, Chung HM. The bromodomain inhibitor JQ1 enhances the responses to all-trans retinoic acid in HL-60 and MV4-11 leukemia cells. Int J Stem Cells. 2018;11(1):131–140. doi:10.15283/ijsc18021
22. Knoechel B, Roderick JE, Williamson KE, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46(4):364–370. doi:10.1038/ng.2913
23. Roderick JE, Tesell J, Shultz LD, et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood. 2014;123(7):1040–1050. doi:10.1182/blood-2013-08-522698
24. Stewart HJ, Horne GA, Bastow S, Chevassut TJ. BRD4 associates with p53 in DNMT3A-mutated leukemia cells and is implicated in apoptosis by the bromodomain inhibitor JQ1. Cancer Med. 2013;2(6):826–835. doi:10.1002/cam4.146
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.