")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 13
On the Role of Artificial Intelligence in Genomics to Enhance Precision Medicine
Authors Álvarez-Machancoses Ó, DeAndrés Galiana EJ, Cernea A, Fernández de la Viña J, Fernández-Martínez JL
Received 2 December 2019
Accepted for publication 17 February 2020
Published 19 March 2020 Volume 2020:13 Pages 105—119
DOI https://doi.org/10.2147/PGPM.S205082
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Óscar Álvarez-Machancoses,1,2 Enrique J DeAndrés Galiana,1 Ana Cernea,1 J Fernández de la Viña,1 Juan Luis Fernández-Martínez2
1Group of Inverse Problems, Optimization and Machine Learning, Department of Mathematics, University of Oviedo, Oviedo 33007, Spain; 2DeepBiosInsights, NETGEV (Maof Tech), Dimona 8610902, Israel
Correspondence: Juan Luis Fernández-Martínez
Group of Inverse Problems, Optimization and Machine Learning, Department of Mathematics, University of Oviedo, C. Federico García Lorca, 18, Oviedo 33007, Spain
Email [email protected]
Abstract: The complexity of orphan diseases, which are those that do not have an effective treatment, together with the high dimensionality of the genetic data used for their analysis and the high degree of uncertainty in the understanding of the mechanisms and genetic pathways which are involved in their development, motivate the use of advanced techniques of artificial intelligence and in-depth knowledge of molecular biology, which is crucial in order to find plausible solutions in drug design, including drug repositioning. Particularly, we show that the use of robust deep sampling methodologies of the altered genetics serves to obtain meaningful results and dramatically decreases the cost of research and development in drug design, influencing very positively the use of precision medicine and the outcomes in patients. The target-centric approach and the use of strong prior hypotheses that are not matched against reality (disease genetic data) are undoubtedly the cause of the high number of drug design failures and attrition rates. Sampling and prediction under uncertain conditions cannot be avoided in the development of precision medicine.
Keywords: artificial intelligence, big data, genomics, precision medicine, drug design
Introduction
As biomedical research has become more data-intensive, with a higher throughput of studies, cases and assays, technology has advanced in order to create toolkits capable of analyzing, interpreting, and integrating a vast amount of data.1 This trend is understood within the medical sector as a paradigm change; since medical practice in essence relied on making predictions about the patient’s health or disease with a limited amount of data, levering diagnosis on their experience, judgement, and personal problem-solving skills.2
This change of paradigm is accompanied by a healthcare industry transformation, in which disruptive technologies have emerged to accommodate healthcare “big data” and Artificial Intelligence (AI) techniques in the biomedical sector, benefiting medical professionals and their patients.3 This change was also provoked by the fact that looking for solutions of complex diseases relies more on disciplines such as molecular biology, biochemistry, applied mathematics and computer science. The clearer example is looking for solutions in cancer, neurodegenerative and rare diseases, among a vast range of pathologies that currently have no solution. As the Broad Institute stated on its corporate website:
This generation has a historic opportunity and responsibility to transform medicine by using systematic approaches in the biological sciences to dramatically accelerate the understanding and treatment of disease.
In this process, the advanced interpretation of genomics through artificial intelligence and machine learning approaches plays a crucial role in the search for solutions. The use of these techniques is compulsory since the physical model that controls these processes is unknown.
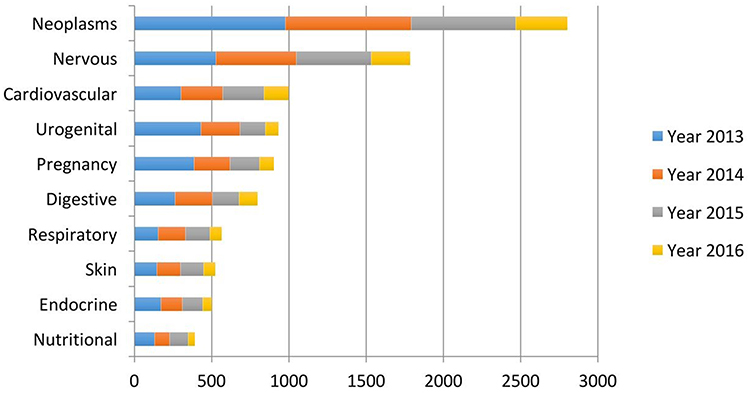 |
Figure 1 Leading diseases where AI is considered. Despite the vast amount of AI literature in healthcare, the research mainly concentrates around a few disease types: cancer and neurodegenerative diseases. Reproduced from: Jiang et al. Artificial intelligence in healthcare: past, present and future. Stroke Vascular Neurol. 2017;2:e000101.4 |
The conclusions of “big data” analysis through AI relating to medicine reveal two major problems:1 the limited amount of samples with respect to the number of control variables (genes for example), that provokes high uncertainty in medical decision-making problems. Besides, the data have an inherent level of noise that falsifies the predictions.2,5 The great heterogeneity existing in the processes that contribute to disease and health, suggests a need for tailoring medical care.6,7 Consequently, instead of making diagnostics according to classical medicine in which decisions are taken based on disease and patient’s similar characteristics; precision medicine aims to shift medicine toward prevention, personalization, and precision through genomics, AI, and biotechnology. Provided how important these toolkits are in elucidating appropriate intervention targets and medical strategies for treating individual patients, AI can play an important role in the development of personalized medicines and treatments.7
The definition of Personalized Medicine, according to the Precision Medicine Initiative, considers it “an emerging approach for disease treatment and prevention that takes into account individual variability in genes, environment, and lifestyle for each person.” Nowadays, there are available tools that are capable of collecting a large amount of genomic data, alongside with cutting-edge data analytics for interpretation, which aid in our understanding of genomics, disease mechanisms, and treatments (Figure 1).8–10
Current Trends in AI and Precision Medicine
Past research trends were strongly based on evaluating medical diagnosis based on AI in contrast to human practitioners,11,12 however, AI should be deemed as an additional tool to aid in medical care; not to replace medical doctors. Later research trends intended to use AI techniques to generate more accurate methods of diagnosis based on the compilation of standardized hospital data13–15 in order to improve the detection of diseases such as cancer or cardiovascular diseases.16–19 However, in recent years, AI is generally used for a variety of purposes in medical care, which ranges from medical diagnosis, preventive medicine, palliative medicine to drug design and development (Figure 2).
The common point to all these problems is that the mathematical model that serves to relate the input and the output, 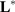 , is unknown. If we write the system
, is unknown. If we write the system 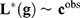 , where
, where 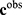 are observed class of the samples,
are observed class of the samples,  are the variables of control (genes for instance in a genetic prediction problem) and
are the variables of control (genes for instance in a genetic prediction problem) and 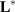 () is called the forward model, that serves to achieve the predictions, then it can be concluded that in most of these problems, only the second member
() is called the forward model, that serves to achieve the predictions, then it can be concluded that in most of these problems, only the second member 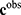 is known: the forward model
is known: the forward model 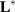 has to be constructed, and the set of discriminatory variables
has to be constructed, and the set of discriminatory variables  (a subset of
(a subset of 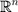 ) has to be found. The first problem is called model construction, and the second one, feature selection. The whole process is called learning or training. Once this process has been achieved, the model
) has to be found. The first problem is called model construction, and the second one, feature selection. The whole process is called learning or training. Once this process has been achieved, the model 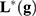 has to be validated, if possible, with an independent cohort.
has to be validated, if possible, with an independent cohort.
Summarizing, most of the prediction and decision problems involved in precision medicine involve solving a discrete system of equations system 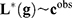 where only the second term
where only the second term 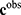 is known. The dimensions of the model space (dimension of
is known. The dimensions of the model space (dimension of 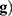 and those of the data space (dimension of
and those of the data space (dimension of 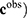 will greatly influence the dimension of the corresponding uncertainty space. Typically, these problems are highly underdetermined. Nevertheless, it has to be pointed out that over-determined problems, where the number of data exceeds the number of model parameters also have an uncertainty space20,21 that has to be considered in the corresponding decision problem.
will greatly influence the dimension of the corresponding uncertainty space. Typically, these problems are highly underdetermined. Nevertheless, it has to be pointed out that over-determined problems, where the number of data exceeds the number of model parameters also have an uncertainty space20,21 that has to be considered in the corresponding decision problem.
Preventive Medicine
One of the highest potentialities of AI in precision medicine is its use in preventing diseases using techniques that could assess the risk of disease development. Cardiovascular medicine represents an area in which AI has been vastly influential, where several studies such as the one carried out by Li et al assessed the risk of heart attack utilizing an artificial neuron network.22 Other studies employed machine learning in order to identify the risk of developing other diseases, such as colorectal cancer,23 respiratory virus affinity,24 melanoma,25 mortality in smokers,26 depression1 or HIV transmission,27 among others.
Palliative Medicine
Not only does AI have a potential use when disease strikes, but it may also be very useful in palliative applications by mitigating the disease progression. In this sense, there are remarkable studies such as the one by Dente et al28 which employed machine learning approaches to identify predictive profiles of pneumonia bacteria; Dagliati et al29 who used AI to tackle diabetes complications or in predicting focal epilepsy outcomes and ischemic stroke and thromboembolism.30,31
Monitoring Medical Care
AI appears to be predominantly beneficial in enhancing medical doctors’ work. Several works have been published to report how diagnosis performance is enhanced with the use of computer-aided detection of diseases, such as breast cancer thermography detection using deep neural networks32 or three-dimensional brain magnetic resonance.33 Other uses in medical care monitoring include the assessment of surgery tolerance or chemotherapy34 or metabolic disorders.35 Some research studies suggest that reducing the number of false positives in disease detection is possible through medical imaging,36,37 biopsies,38 by monitoring the disease progression39 and health status.40
Drug Design and AI
Drug design is considered a very capital-intensive process. Therefore, it is necessary to re-think the current paradigm of one disease – one target – one drug.41 The current understanding of drug design is that a drug must be capable of re-establishing homeostasis; the drug hits the targets causing the disease by re-establishing the equilibrium. The reason why most compounds fail in clinical trials is because mechanisms of action are not fully understood.42 Also, the target-centric approach is commonly used, and it is not well connected with an advanced interpretation of the genomic data at disposal.
Bioinformatics and chemo-informatics utilize AI tools in order to rationalize the development of new compounds to target diseases. The idea in these fields is to develop multi-scale models that are capable of considering simultaneously the activity of the infection agents and the parameters of absorption, distribution, metabolism, and toxicity (known as ADMET profile).43,44 Further models utilize large datasets of chemical compounds with different composition, sizes, surface activity, in order to generate a meta-structure. This meta-structure is modeled in order to elucidate the relationships with the disease agents utilizing perturbation models.45 These models have been proven to be very versatile when applied to infectious diseases,46 immunological disorders,47 neurological pathologies,48 and cancer.49 Furthermore, the new approach in drug discovery is known as de novo multi-scale approach in which a drug is designed within the chemical subspace where it could be deemed beneficial. Several studies have been carried out following this paradigm in order to elucidate the relationships between the drugs and the targets, alongside the ADMET profile.50,51
Since a target-centric approach is mainly responsible of the high attrition rates and low productivity in pharmaceutical research and development,52 global models such as the ones described previously are required. In addition, network system biology is of utmost importance in order to understand the impact of genetic and epigenetic factors on drug actions, where AI plays a crucial role.53,54
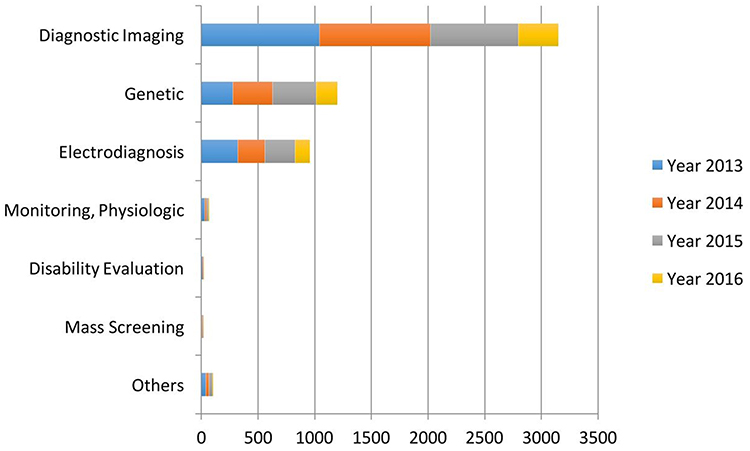 |
Figure 2 Main applications of AI in healthcare. Reprdoduced from: Jiang et al. Artificial intelligence in healthcare: past, present and future. Stroke Vascular Neurol. 2017;2:e000101.4 |
AI for Precision Medicine: Data Quality and Relevance
Despite the recent advances in AI toolkits and their performance in medical studies, there are still some gaps and margin for improvement, suggesting that AI needs either more data or better algorithms to improve the precision medicine accuracy. In general, current trends in AI are focused on developing an algorithm, which requires large amounts of data in order to maximize the performance. This is because current ML approaches are tremendously data-hungry.55 It was found that vast amounts of data with simpler algorithms perform better than complex algorithms with less data. This suggests that advanced AI techniques work well only in low variability problems, which is not the case in medical care.56 Therefore, deep sampling approaches, as suggested by Fernández-Martínez et al,5,57,58 are a plausible alternative.
The relevance and accuracy of data is one of the most important factors in order to ensure that the trained model accurately represents the reality. The quality of the predictions made will never exceed the quality of the dataset utilized to train the machine-learning model.59 While conventional statistical methods could be used to filter the data bags; removing outliers, these methods have limited effectiveness. Their lack of effectiveness is due to the fact that these methods do not assess the quality and relevance of the data. Therefore, inaccuracy of data points may influence the trained model by introducing biases.60,61 Consequently, advanced methods in high-dimensional data analysis,62 sensitivity analysis and feature selection and model reduction techniques are required63 to tackle these problems.
As pointed out before, not only data accuracy is important, but also data relevance to the disease of interest. Many recent attempts to use Machine learning techniques rely on the use of complex machine learning tools which are trained utilizing a wide range of disease attributes, without performing an analysis in order to elucidate which of them are predictors. Carrying out this approach, the model is trained with an excess of attributes, with the associated excess of noise and inducing biases.64 To be more specific with regards to healthcare, the majority of precision medicine solutions have been based on large DNA sequence data,65 however, the majority of common diseases are believed to be caused by both epigenetic and environmental factors. Due to this, AI Genomics is the tool required to elucidate these dependencies by filtering and treating appropriately the genetic pathways, since it links DNA sequencing, environmental factors, and disease mechanisms.66 The major drawback of genomics in precision medicine is the lack of available data from patients that are directly related to the disease of interest.67
AI in Modern Genomics: Gathering Data from Large-Scale Genomics
Application of massively parallel or Next Generation Sequencing (NGS) to large-scale genomics, which has caused an increasing level of precision and success in the medical practice, has been observed over the past 8 years.68 NGS is generally combined with machine learning or any other analytical approaches in order to better inform the clinical care of patients. The obtained “big data” in combination with the machine learning analytics could improve not only the clinical practice but also provide a deeper understanding of cancer and rare diseases. However, despite the opportunities that NGS and machine learning offer, there are several challenges in the clinical interpretation of data. Every small alteration in samples, NGS data, variant/mutation would require a different computational approach.
Challenges with Samples
As already pointed out in the previous section, data gathering from samples is one of the major challenges. Gathering information from tissue samples in order to provide insight into cancer or rare diseases could be harnessed by the introduction of bias from non-malignant cells (such as immune cells, stromal cells …) which leads to a loss of signal and decreases the detection sensitivity.69 To avoid these issues, the Cancer Genome Atlas restricted tissues to large, high-quality, high-purity and frozen tumor samples.70
Challenges Associated with NGS
Several comprehensive approaches are available in order to predict the entire range of alterations, such as whole-genome, whole-exome, and whole-transcriptome.71 However, these approaches typically have higher computational requirements and longer turnaround times, therefore also incurring higher costs than their more-targeted alternatives. Among bulk-NGS analyses, hundreds of thousands to millions of cells are analyzed at once, providing a global idea of a set of cells. Consequently, the majority of our knowledge of individual cells comes from the analysis of bulk-NGS, which does not address the real heterogeneity of cells. Consequently, advanced methods in high-dimensional data analysis,61 sensitivity analysis and feature selection and model reduction techniques are required62 to tackle these problems. Currently, smaller panels of hundreds of genes are utilized to infer pathologies; however, this approach is not robust enough as the discriminatory power of the genes utilized is not evaluated beforehand.72 By utilizing this approach, the targeted panels can be used to screen larger sets of patients who might potentially benefit from the detection of clinically actionable mutations (the most discriminatory ones). Furthermore, single-cell NGS does endeavour to address the issues of data generalization of bulk-NGS and provides information on genetic, transcriptomic and epigene for a given cell.
Challenges in Interpreting Alleles and Variants
The major limitation in the interpretation of alleles and variants is the fact that the response of allele variants whose implications, resulting in protein miss-function or miss-response to a targeted therapy, have not yet been identified.
Data sharing is of utmost importance in order to bypass this issue. It will improve the understanding and dramatically increase patients’ healthcare in the treatment of cancer and rare and degenerative diseases. Genomic data are more important when data are structured and the genes and variants are linked with clinical data, including the type and treatments implemented.73
AI Genomics and the Phenotype Prediction Problem
A ful understanding of patient’s genomics is mandatory in order to design robust and efficient therapeutics. Phenotype prediction problems are highly undetermined because the number of monitored genetic probes are much higher than the number of observed samples.74 A robust prediction of phenotypes (diseases) is carried out through a robust sampling of the uncertainty space of the problem. Let us consider a binary classifier 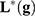 , that is, an application between the set of genes that serve to discriminate a given phenotype, for instance, disease and healthy controls, or cancer metastasis and the absence of it. The discriminatory signatures are those that minimize the prediction error
, that is, an application between the set of genes that serve to discriminate a given phenotype, for instance, disease and healthy controls, or cancer metastasis and the absence of it. The discriminatory signatures are those that minimize the prediction error 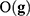 , which is the difference in a given norm between the observed and the predicted classes, or equivalently, maximize the predictive accuracy, that is, the number of samples of the training set that are correctly predicted. As in any inverse problem, the uncertainty space of the phenotype prediction problem,
, which is the difference in a given norm between the observed and the predicted classes, or equivalently, maximize the predictive accuracy, that is, the number of samples of the training set that are correctly predicted. As in any inverse problem, the uncertainty space of the phenotype prediction problem, 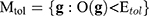 , is composed by the sets of high predictive networks with similar predictive accuracy; that is, those sets of genes
, is composed by the sets of high predictive networks with similar predictive accuracy; that is, those sets of genes  that classify the samples with a prediction error
that classify the samples with a prediction error 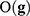 lower than
lower than 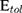 .6–8
.6–8
As in any other inverse problem, the cost function topography in phenotype prediction problems is composed of several flat curvilinear valleys20,21 where the genetic signatures are located, all of them with a similar predictive accuracy of the training set. Nevertheless, in the phenotype prediction problem, the size of the high discriminatory genetic signatures varies, that is, high discriminatory genetic networks of different complexity exist, and the optimization of 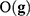 is not always performed in the same space dimension. This fact complicates the sampling approach to understand the altered genetic pathways involved in the disease progression. Besides, due to the high underdetermined character of the phenotype prediction problems, their associated uncertainty space has a very high dimension, and the characterization of the involved biological pathways is very ambiguous because there exist many equivalent genetic networks that predict the phenotype with similar accuracies.75–77 The important working hypothesis is that by sampling the uncertainty space of the phenotype prediction problems, we are able to understand the altered genetic pathways of the disease in order to use this knowledge in precision medicine for diagnosis, prognosis, and treatment optimization.
is not always performed in the same space dimension. This fact complicates the sampling approach to understand the altered genetic pathways involved in the disease progression. Besides, due to the high underdetermined character of the phenotype prediction problems, their associated uncertainty space has a very high dimension, and the characterization of the involved biological pathways is very ambiguous because there exist many equivalent genetic networks that predict the phenotype with similar accuracies.75–77 The important working hypothesis is that by sampling the uncertainty space of the phenotype prediction problems, we are able to understand the altered genetic pathways of the disease in order to use this knowledge in precision medicine for diagnosis, prognosis, and treatment optimization.
Different interesting methods were proposed by Cernea et al8 and successfully applied in the analysis of Triple Negative Breast Cancer metastasis, comparing the results obtained with Bayesian networks.78 Bayesian networks are utilized to model the genetic signatures' distribution related to the phenotype prediction, 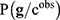 , according to Bayes’ rule:79–81
, according to Bayes’ rule:79–81
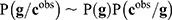
In this expression 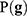 is the prior distribution used to model the genetic signatures and
is the prior distribution used to model the genetic signatures and 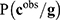 is the probability of the genetic signature
is the probability of the genetic signature  , that depends on its predictive accuracy
, that depends on its predictive accuracy 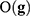 .82–84 Bayesian networks are computationally much more expensive than the Fisher, Holdout and Random samplers.6 Besides, the probabilistic parameterization of the uncertainty space is not unique. Considering all the plausible networks have to provide similar results in sampling the altered genetic pathways to other samplers. Different types of algorithms could be used in tackling precision medicine problems, such as k-Nearest-Neighbour classifier,85 Extreme Learning Machines,86 Random Forest,87 Support Vector Machines88 or Deep Neural Networks.89 Despite the wide range of classifiers, poor results are generally observed due to the impact of noise in data, mainly in the observed class of the samples of the training set, and the use of irrelevant features in the discrimination.5,64,77
.82–84 Bayesian networks are computationally much more expensive than the Fisher, Holdout and Random samplers.6 Besides, the probabilistic parameterization of the uncertainty space is not unique. Considering all the plausible networks have to provide similar results in sampling the altered genetic pathways to other samplers. Different types of algorithms could be used in tackling precision medicine problems, such as k-Nearest-Neighbour classifier,85 Extreme Learning Machines,86 Random Forest,87 Support Vector Machines88 or Deep Neural Networks.89 Despite the wide range of classifiers, poor results are generally observed due to the impact of noise in data, mainly in the observed class of the samples of the training set, and the use of irrelevant features in the discrimination.5,64,77
Once the altered genetic pathways are robustly sampled, the next step consists of performing an optimum selection of the therapeutics tailored according to the patient’s genetic profile. This procedure aims to dramatically increase the success rate and is particularly important to understand the disease mechanisms in a given individual.
Case Study: Analysis of Metastasis and Survival in TNBC
Introduction and Methods
We illustrated the importance of genomic robust sampling in precision medicine and uncertainty analysis with the analysis of the Triple Negative Breast Cancers (TNBC) phenotype. The algorithms utilized have been successfully utilized in the analysis of breast cancer and lymph node metastasis,8,90 in Sarcopenia,91 Multiple Sclerosis,92 Multiple Myeloma93 and Inclusion Body Myositis.94 More specifically, we performed a robust sampling in order to find out the altered genetic pathways by the metastasis events. To do so, we used the microarray analyzed by Jézéquel et al,95 which is deposited in the Gene Expression Omnibus (GEO) under the acronym GSE58812. The dataset covers data from 107 patients identified with TNBC and controlled for metastasis (44 degenerated and 63 were cured after a monitoring period of 7 years).
The comparison of different algorithms across different studies is an objective but challenging way of evaluating the performance of different methodologies. However, it is rather difficult to make comparisons, since each algorithm suffers different bias in the dataset, due to inaccuracies, lack of data or inconsistencies. Furthermore, it is more important to use the same independent set of data in order to perform a fair evaluation.
To make algorithms more robust and less sensitive to bias, the Fisher’s Ratio Sampler, the Holdout Sampler and the Random Sampler are trained in different data bags and, afterwards LOOCV is carried out by consensus. That is, given a set of decision makers, the decision taken by the majority tends to be accurate when the set of decision makers tends to be infinite. That implies that during the training stage, only the best performing data bags are selected to make the blind validation.
In this case, we have used the following samplers to find the set of genes that are involved in the metastasis and survival in TNBC and compared it to the BNs (Figure 3).
- Fisher’s Ratio Sampler: this algorithm considers the discriminatory strength of the differentially expressed genes according to its Fisher’s Ratio in order to induce a prior sampling distribution of the genetic networks. The sampled networks are established using this prior distribution and their likelihood is established via an LOOCV via a k-NN classifier.
- Holdout Sampler: a set of random 75/25 data bag holdouts were generated from the database, where 75% of the data is used for training and 25% for validation. In our case study, we have generated 1000 holdouts. In each holdout, the genes are ranked according to the Fisher’s ratio, maximizing the distance between the centers of the gene expression in each class and minimizing the intra-variance. The Fisher’s ratio was combined with a previous fold-change analysis to avoid the genes with high FR and low fold-change due to small dispersions within each class.
- Random Sampler: this scheme selects genes and builds gene signatures of variable lengths. The algorithm shares similarities with the Fisher’s Ratio Sampler, but, in this case, the prior sampling distribution is uniform within the differentially expressed genes, instead of being proportional to the Fisher’s Ratio. The sampled networks' likelihood is established via an LOOCV via a k-NN classifier.
- Bayesian Networks: the Bayesian Network selects genes by building genetic signatures prior to distribution by using an acyclic graph. This algorithm is used to sample the posterior distribution of the genetic signatures,
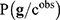 , according to Bayes rule:
, according to Bayes rule: 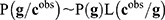 , where
, where 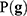 is the prior distribution to sample the genetic signatures and
is the prior distribution to sample the genetic signatures and 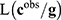 is the likelihood of the genetic signature
is the likelihood of the genetic signature  , that depends on the its predictive accuracy
, that depends on the its predictive accuracy  .
.
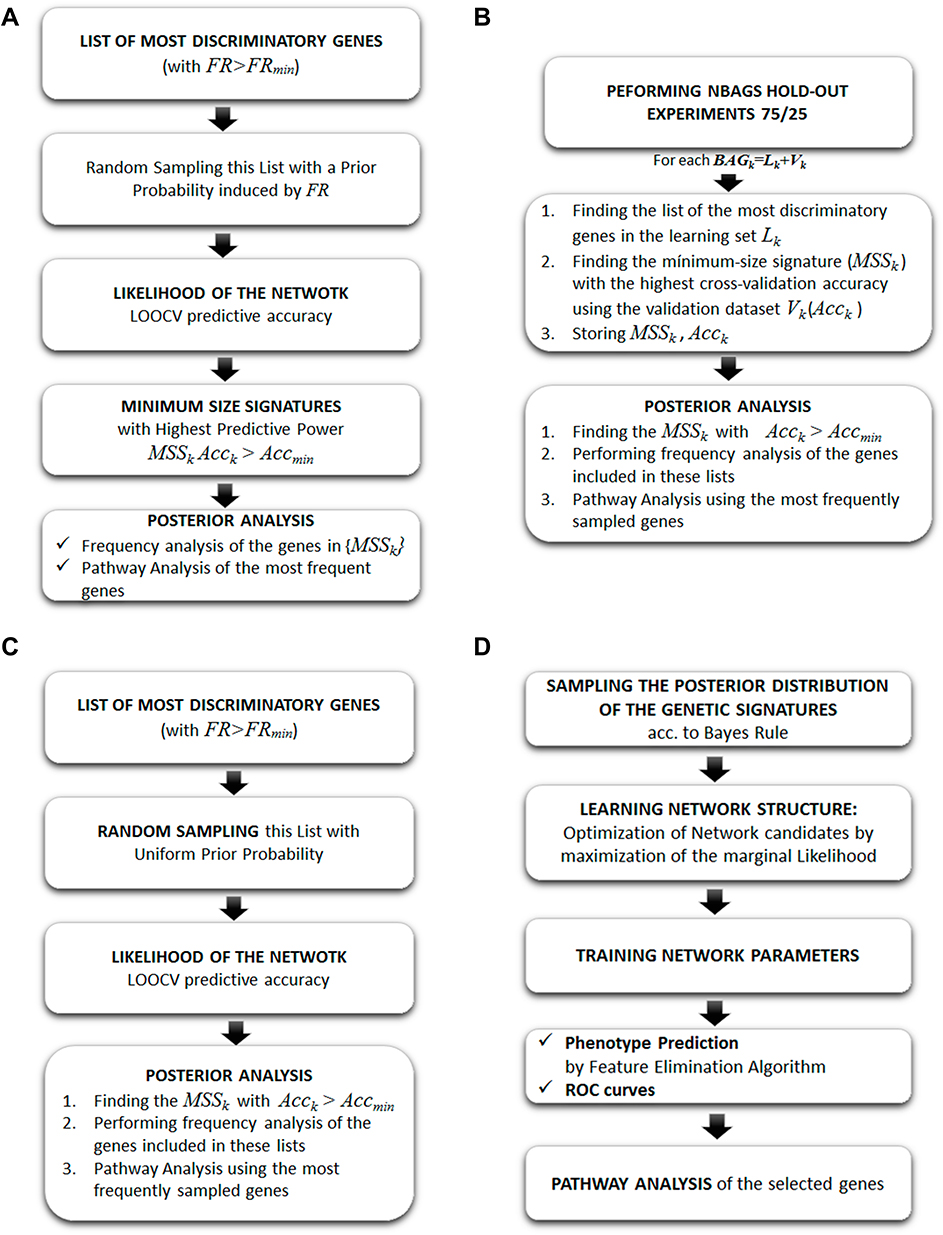 |
Figure 3 Algorithm workflow of (A) Fisher’s ratio sampler; (B) Holdout sampler; (C) Random sampler; (D) Bayesian network. Reproduced from: Cernea et al. Robust pathway sampling in phenotype prediction. Application to triple nagtive cancer. BMC Bioinformatics. In press.96 |
Results and Discussion
Tables 1 and 2 show the 15 most frequently sampled genes by each of these algorithms. The most frequent gene, LINC00630, appears in both Fisher’s ratio sampler and Random sampler algorithms, and Holdout sampler models it with a lower frequency. Other common genes appraised by all algorithms were STC1, LOC100506272, BAIAP2-AS1, LOC646482.
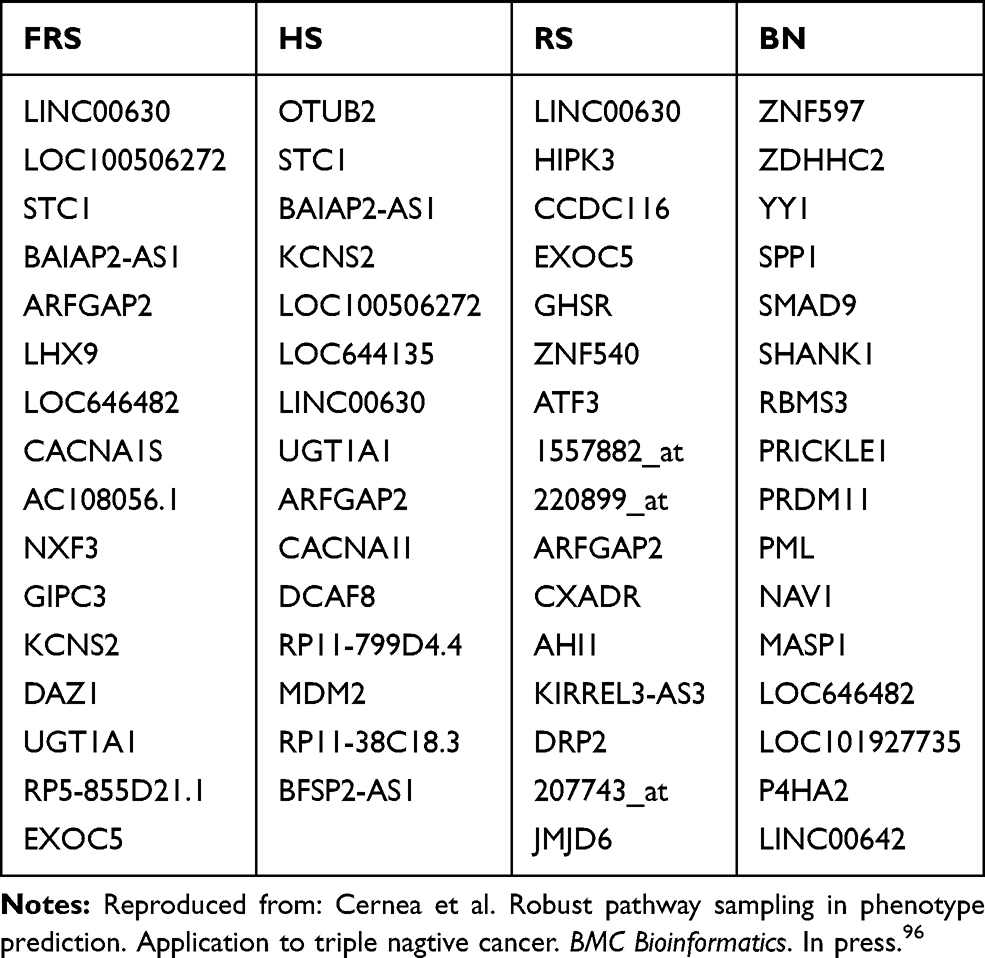 |
Table 1 Metastasis Prediction: List of Most-Frequently Sampled Genes by the Different Algorithms: Fisher’s Ratio Sampler, Holdout Sampler, Random Sampler and Bayesian Network |
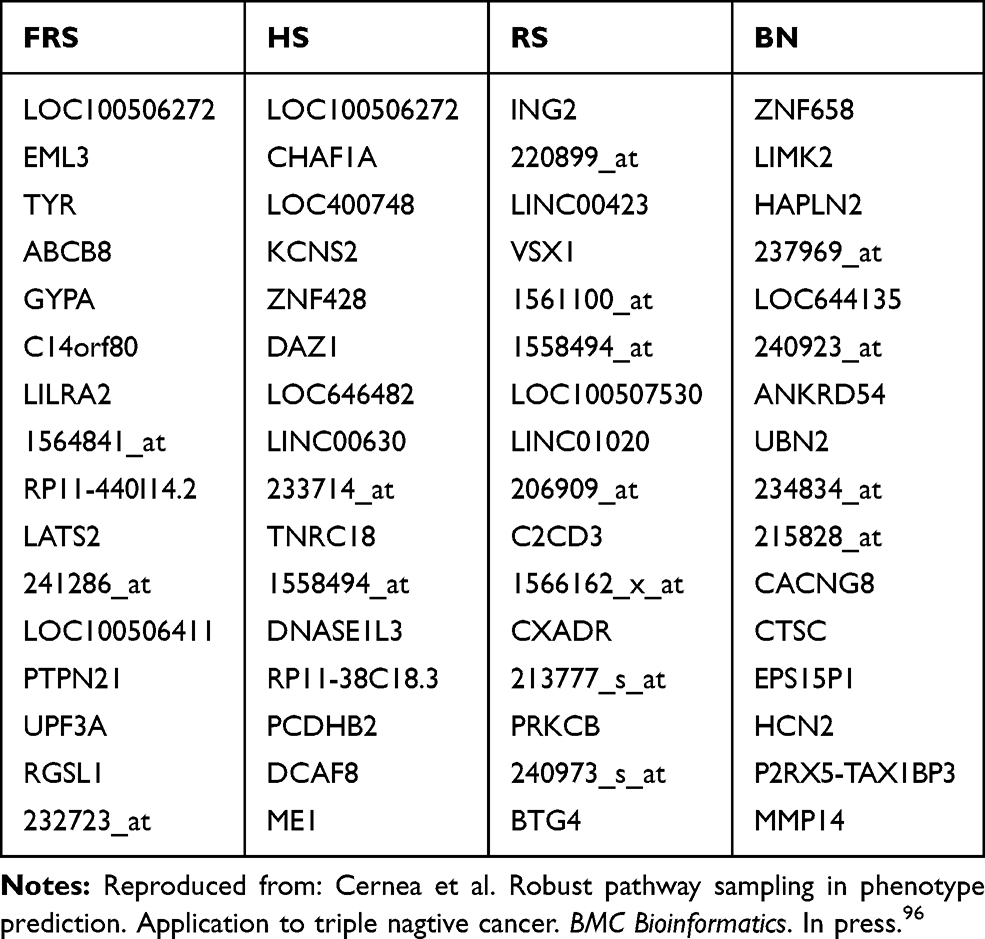 |
Table 2 Survival Prediction: List of the Most-Frequently Sampled Genes by the Different Algorithms |
We also showed the phenotype centered network obtained from Bayesian Network algorithm in Figure 4, which illustrates the directed graph formed by the 68 genes most associated with the survival phenotype. It should be noted that this graph is not unique, since there exist other plausible probabilistic parameterizations of the uncertainty space related to this phenotype prediction problem, as we have proven by using other sampling algorithms. Therefore, the conclusions for the pathway analysis and drug design obtained with just one of these parameterizations, might be biased.
The most important genes provided by the random sampler were: 1) HIPK3, which codes a serine/threonine-protein kinase, involved in transcription regulation and apoptosis. 2) CCDC116 (coiled-coil domain containing 116), mainly found in the testis and associated with risk in multiple kinds of cancers.97 3) EXOC5 which is linked to peptide hormone metabolism. 4) GHSR, Growth Hormone Secretagogue Receptor which is linked to the CAMP signaling pathway.
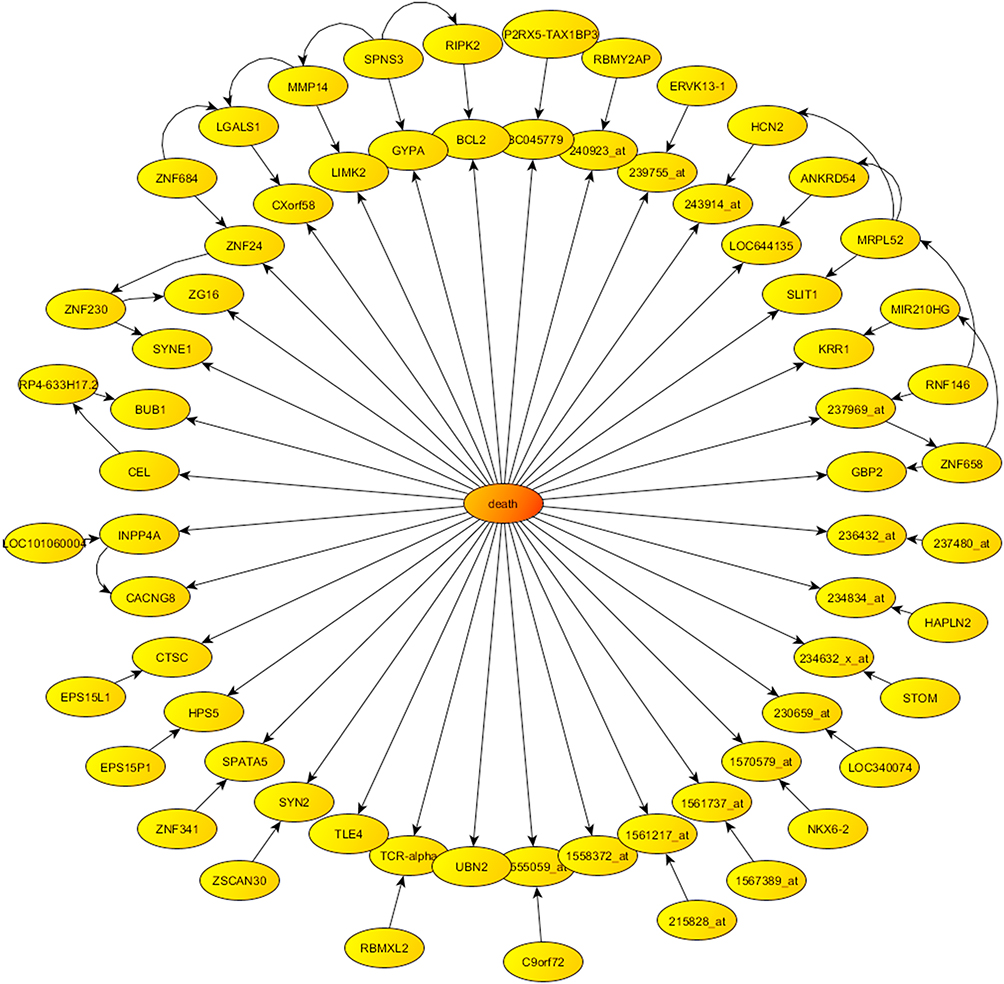 |
Figure 4 Phenotype centered network provided by Bayesian networks in the case of survival. |
Bayesian networks sampled genes are mainly related to the TGF-beta Receptor Signaling and MTOR Signaling Pathway: 1) ZNF597 encodes a zinc finger protein, involved in expression and transcription. 2) ZDHHC2 is called Palmitoyltransferase or Reduced expression associated with metastasis protein in liver.98 3) YY1 is highly expressed in various types of cancers and regulates tumorigenesis through multiple pathways, such as in breast cancer.99
Tables 3 and 4 show the top genetic pathways inferred with the most frequently sampled genes by each algorithm.
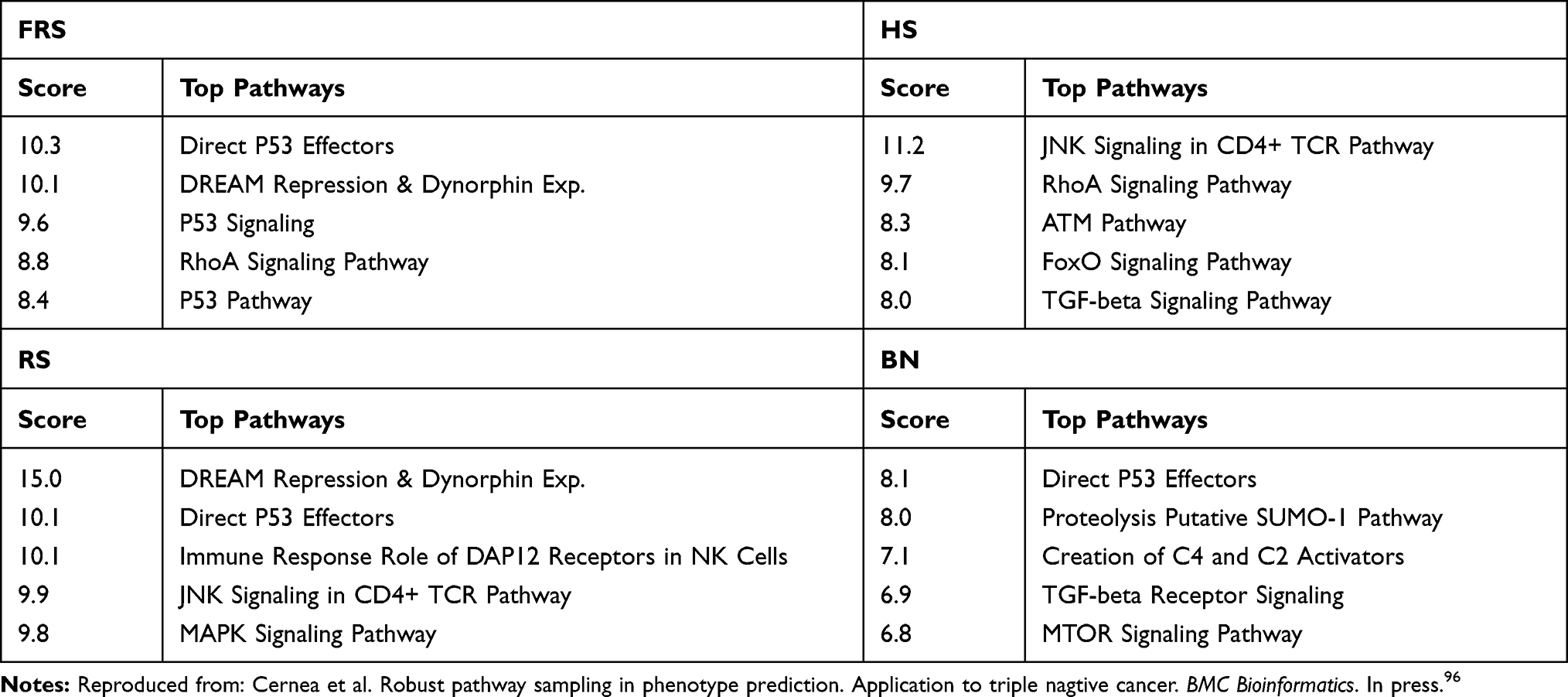 |
Table 3 Metastasis Prediction: Pathways Sampled by Different Algorithms Ranked According to the Scoring Provided by GeneAnalytics |
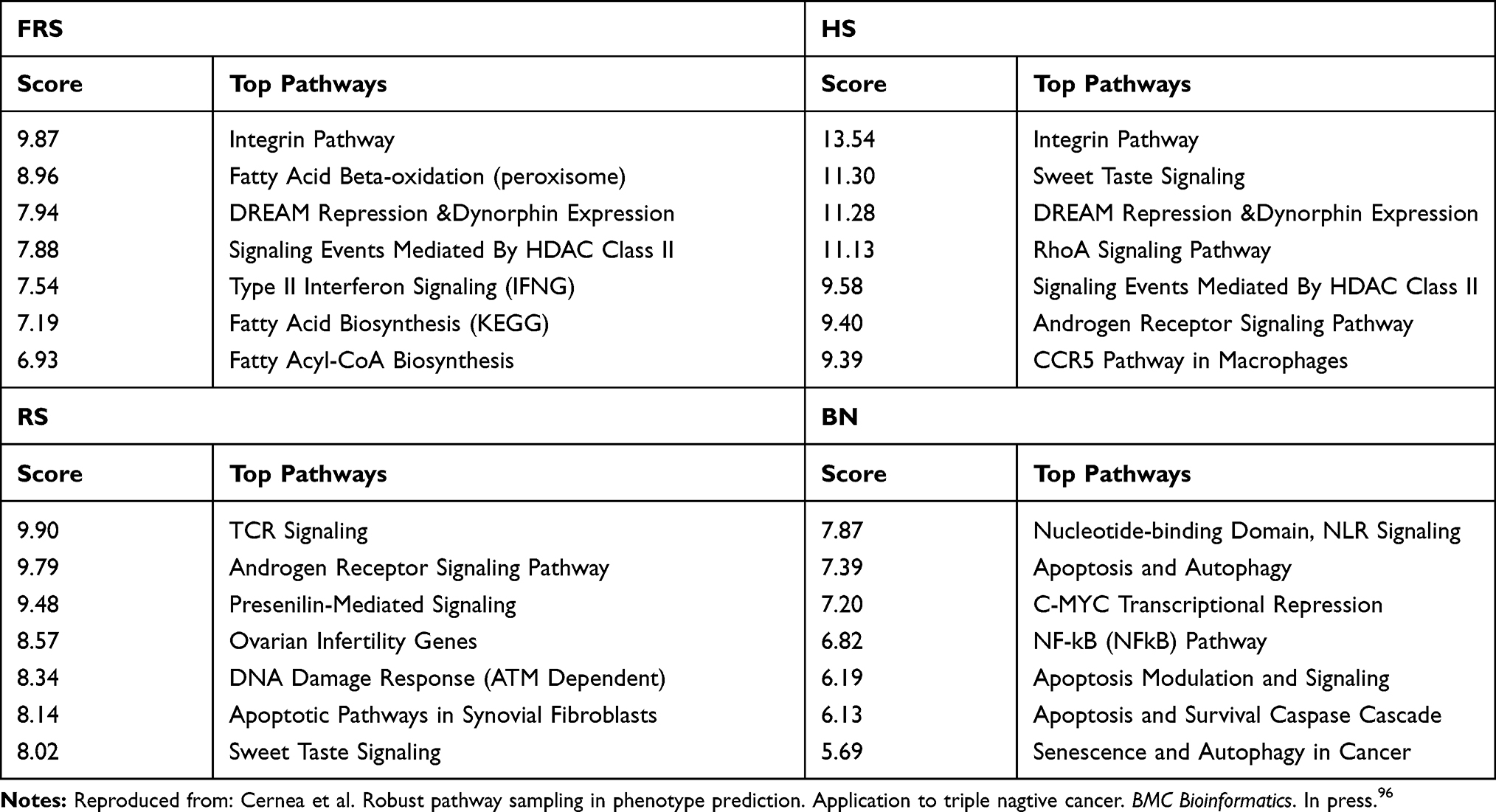 |
Table 4 Survival Prediction: Pathways Sampled by Different Algorithms Ranked According to the Scoring Provided by GeneAnalytics |
The main conclusions obtained from this analysis are the following:
- in the Metastasis prediction most of the samplers identified the Direct P53 Effectors as the main altered pathway. Other common pathways found were the DREAM Repression and Dynorphin Expression and TGF-beta Signaling Pathway. Besides phagocytosis, a major mechanism in the immune system defense, seems to play a crucial role.
- In the survival prediction the integrin pathway appears to be crucial. The role of integrins in metastasis has been highlighted by Ganguly et al,100 among others.
- A robust understanding of diseases from a genomic point of view is required in order to carry out Precision Medicine.
- In all the cases, the pathways are involved in cancer and in immune response. Consequently, this study confirms the vision that the altered pathways should be independent from the sampling approach and the classifier utilized.
Application to Drug Design
The previous analysis can be used to perform drug selection and repositioning via CMAP methodologies.101–103
The main drugs found that can revert the genes that are deregulated in TNBC metastasis:
- Geldanamycin HL60 (1e-06), which is an antitumor antibiotic that inhibits the function of Heat Shock Protein 90 (HSP90), which plays important roles in the regulation of cell cycle, cell growth, cell survival, apoptosis, angiogenesis and oncogenesis. This drug has been repositioned in leukaemia cell lines (HL60).
- LY-294002 HL60 and MCF7 (1e-05) are potent inhibitors of Phosphatidylinositol 3-Kinases. PI3K inhibitors are effective in inhibiting tumor progression (Yang et al, 2019). LY294002 is also a BET inhibitor having both anti-inflammatories and anti-cancer properties. It has been found that the expression of the growth promoting transcription factor Myc is blocked by BET inhibitors (eg, Alderton, 2011). This drug has been repositioned in Leukaemia (HL60) and Breast Cancer cell lines.104
- Trichostatin A (TSA) is a potent and specific inhibitor of HDAC class I/II. Histone deacetylase inhibitors (HDAC inhibitors, HDACi, HDIs) are chemical compounds that inhibit histone deacetylases. By removing acetyl groups, HDACs reverse chromatin acetylation and alter transcription of oncogenes and tumor suppressor genes.105,106
Therefore, the use of deep sampling approaches combined with Connectivity Map technologies (CMAP) allows fast finding of possible compounds and the design of new drugs that maximize the mechanisms of action needed to counteract the progression of the disease. The results obtained in this case study back the introduction of AI in Precision Medicine, since the drugs obtained are also currently being tested in TNBC.107
Expert Opinion
Precision medicine is on the forefront of new paradigms in medical care. Since understanding a patient’s health mechanisms is an arduous task, the use of Artificial Intelligence has become a crucial tool in understanding patients’ genomics in order to generate tailored therapeutics. In this sense, Biological Invariance has arisen as a new paradigm in genomics, proteomics, metabolomics or precision medicine, which states that the analysis of genomics and potential therapeutics should be independent of the sampling methodology and the classifier utilized for their inference. Predominantly, phenotype prediction and the analysis of altered pathways have become an important discipline in drug discovery and precision medicine, that is, finding a set of genes that prospectively differentiates a specified phenotype disease with respect to a control sample. The problem is highly undetermined since the number of monitored genetic probes exceeds the number of samples; therefore, this creates ambiguity in the identification that must be solved with the aid of Artificial Intelligence and Deep sampling techniques.
Independently of the methods, robust AI schemes are necessary to integrate information from different sources and to reduce the inherent uncertainty spaces existing in this kind of analysis. Furthermore, Artificial Intelligence shall be employed to elucidate complex genetic data and all the research information available on the current and future health status of patients in order to improve patients’ quality of life and reduce medical costs. This process is supposed to be, such as in the sampling of altered pathways, independent from the methodology.
We believe that any AI protocol to boost Precision Medicine should be iterative and learn from experience. Algorithms and methodologies should be kept simple and fast, since they would yield to statistically the same results and statistically with the same accuracy.
However, the utilization of AI in Precision Medicine, despite being promising, still has some limitations and challenges. Precision medicine, such as Drug Design, Palliative Medicine, Healthcare monitoring, and Preventive medicine could be considered problems of high variability. This means that ML approaches have a wide range of learning and boundary conditions in order to be trained properly. As ML is extremely data-hungry, the requirement algorithms are very complex, and its training is extremely demanding. We require the development of hybrid machine learning techniques that are capable of sampling different classifiers, boundary conditions, attributes in the training stage, while optimization of the ML architecture is carried out. In this sense, it would be interesting to develop new hybrid optimization-ML techniques, such as Stochastic Gradient Descent with Momentum108 or Adam optimizer.109
Disclosure
The authors report no conflicts of interest in this work.
References
1. Becker A. Artificial intelligence in medicine: what is it doing for us today? Health Policy Technol. 2019;8:198–205. doi:10.1016/j.hlpt.2019.03.004
2. Mesko B. The role of artificial intelligence in precision medicine. Expert Rev Precis Med Drug Develop. 2017;2:239–241. doi:10.1080/23808993.2017.1380516
3. Elenko E, Underwood L, Zohar D. Defining digital medicine. Nat Biotechnol. 2015;33:456–461. doi:10.1038/nbt.3222
4. Jiang F, Jiang Y, Zhi H, et al. Artificial intelligence in healthcare: past, present and future. Stroke Vascular Neurol. 2017;2:e000101. doi:10.1136/svn-2017-000101
5. deAndrés-Galiana EJ, Fernández-Martínez JL, Saligan LN, Sonis ST. Impact of microarray preprocessing techniques in unraveling biological pathways. J Comp Biol. 2016;23:957–968. doi:10.1089/cmb.2016.0042
6. Williams AM, Liu Y, Regner KR, Jotterand F, Liu P, Liang M. Artificial intelligence, physiological genomics, and precision medicine. Physiol Genomics. 2018;50:237–243. doi:10.1152/physiolgenomics.00119.2017
7. Mesko B, Drobni Z, Benyei E, Gergely B, Gyorffy Z. Digital health is a cultural transformation of traditional healthcare. mHealth. 2017;3:1–8. doi:10.21037/mhealth.2017.08.07
8. Cernea A, Fernández-Martínez JL, deAndrés-Galiana EJ, et al.. Comparison of different sampling algorithms for phenotype prediction.
9. Cernea A, Fernández-Martínez JL, deAndrés-Galiana EJ, et al. Sampling Defective Pathways in Phenotype Prediction Problems via the Fisher’s Ratio Sampler.
10. Cernea A, Fernández-Martínez JL, deAndrés-Galiana EJ, et al. Sampling defective pathways in phenotype prediction problems via the Holdout sampler.
11. de Dombal FT, Leaper DJ, Staniland JR, McCann AP, Horrocks JC. Computer-aided diagnosis of acute abdominal pain. Br Med J. 1972;2:9–13. doi:10.1136/bmj.2.5804.9
12. Adams ID, Chan M, Clifford PC, et al. Computer aided diagnosis of acute abdominal pain: a multicentre study. Br Med J. 1986;293:800–804. doi:10.1136/bmj.293.6550.800
13. Farrar JT. Use of a digital computer in the analysis of intestinal motility records. IRE Trans Med Electron. 1960;ME-7:259–263. doi:10.1109/IRET-ME.1960.5008074
14. Brodman K, van Woerkom AJ. Computer-aided diagnostic screening for 100 common diseases. JAMA. 1966;197:901–905. doi:10.1001/jama.1966.03110110125029
15. Heaf PJ. Automation in medicine. Proc R Soc Med. 1964;1148-1149:57.
16. Fujita H, Doe K, Pencil LE, Chua KG. Image feature analysis and computer-aided diagnosis in digital radiography. 2. Computerized determination of vessel sizes in digital subtraction angiography. Med Phys. 1987;14:549–556. doi:10.1118/1.596066
17. Geivers H, Schaper W, Serves J, Xhonneux R. Cardiac excitability determined by electronic computer. Pflugers Arch Gesamte Physiol Menschen Tiere. 1967;298:185–190. doi:10.1007/BF00364699
18. Chan HP, Doi K, Galhotra S, Vyborny CJ, MacMahon H, Jokich PM. Image feature analysis and computer-aided diagnosis in digital radiography. 1. Automated detection of microcalcifications in mammography. Med Phys. 1987;14:538–548.
19. Debray JR, Chrétien J, Gueniot M, et al. A new concept of preventative medicine using automatic data processing by computer. II. Application to diseases and risks of the digestive apparatus and to cardiovascular diseases and risks. Ann Med Interne (Paris). 1969;120:589–596.
20. Fernández-Martínez JL, Fernández-Muñiz Z, Tompkins MJ. On the topography of the cost functional in linear and nonlinear inverse problems. Geophysics. 2012;77:W1–W5. doi:10.1190/geo2011-0341.1
21. Fernández-Martínez JL, Pallero JLG, Fernández-Muñiz Z, Pedruelo-González LM. From Bayes to Tarantola: new insights to understand uncertainty in inverse problems. J App Geophys. 2013;98:62–72. doi:10.1016/j.jappgeo.2013.07.005
22. Li H, Luo M, Zheng J, et al. An artificial neural network prediction model of congenital heart disease based on risk factors: a hospital-based case-control study. Medicine. 2017;96:e6090. doi:10.1097/MD.0000000000006090
23. Kind Y, Akiva P, Choman E, et al. Performance analysis of a machine learning flagging system used to identify a group of individuals at a high risk for colorectal cancer. PLoS One. 2017;12:e0171759. doi:10.1371/journal.pone.0171759
24. Mai NV, Krauthammer M. Controlling testing volume for respiratory viruses using machine learning and text mining. AMIA Annu Symp Proc. 2017;2016:1910–1919.
25. Fuller C, Cellura AP, Hibler BP, Burris K. Computer-aided diagnosis of melanoma. Semin Cutan Med Surg. 2016;35:25–30. doi:10.12788/j.sder.2016.004
26. González G, Ash SY, Vegas Sanchez-Ferrero G, et al. Disease staging and prognosis in smokers using deep learning in chest computed tomography. Am J Respir Crit Care Med. 2018;197:193–203. doi:10.1164/rccm.201705-0860OC
27. Young SD, You W, Wang W. Toward automating HIV identification: machine learning for rapid identification of HIV-related social media data. J Acquir Immune Defic Syndr. 2017;74:S128–S131. doi:10.1097/QAI.0000000000001240
28. Dente CJ, Bradley M, Schobel S, et al. Towards precision medicine: accurate predictive modeling of infectious complications in combat casualties. J Trauma Acute Care Surg. 2017;83:609–616. doi:10.1097/TA.0000000000001596
29. Dagliati A, Marini S, Sacchi L, et al. Machine learning methods to predict diabetes complications. J Diabetes Sci Technol. 2017;12:295–302. doi:10.1177/1932296817706375
30. Sinha N, Dauwels J, Kaiser M, et al. Predicting neurosurgical outcomes in focal epilepsy patients using computational modelling. Brain. 2017;140:319–322. doi:10.1093/brain/aww299
31. Li Z, Liu H, Du X, et al. Integrated machine learning approaches for predicting ischemic stroke and thromboembolism in atrial fibrillation. AMIA Annu Symp Proc. 2016;2016:799–807.
32. Fernández-Ovies FJ, Alférez-Baquero ES, de Andrés-galiana EJ, Cernea A, Fernández-Muñiz Z, Fernández-Martínez JL. Detection of breast cancer using infrared thermography and deep neural networks. In: Rojas I, Valenzuela O, Rojas F, Ortuño F, editors. Bioinformatics and Biomedical Engineering. Vol. 11466. IWBBIO 2019. Lecture Notes in Computer Science; 2019:514–523.
33. Sunwoo L, Kim YJ, Choi SH, et al. Computer-aided detection of brain metastasis on 3D MR imaging: observer performance study. PLoS One. 2017;12:e0178265. doi:10.1371/journal.pone.0178265
34. Lee H, Troschel FM, Tajmir S, et al. Pixel-level deep segmentation: artificial intelligence quantifies muscle on computed tomography for body morphometric analysis. J Digit Imaging. 2017;30:487–498. doi:10.1007/s10278-017-9988-z
35. Lee H, Tajmir S, Lee J, et al. Fully automated deep learning system for bone age assessment. J Digit Imaging. 2017;30:427–441. doi:10.1007/s10278-017-9955-8
36. Suzuki K, Shiraishi J, Abe H, MacMahon H, Doi K. False-positive reduction in computer-aided diagnostic scheme for detecting nodules in chest radiographs by means of massive training artificial neural network. Acad Radiol. 2005;12:191–201. doi:10.1016/j.acra.2004.11.017
37. Park E, Chang HJ, Nam HS. Use of machine learning classifiers and sensor data to detect neurological deficit in stroke patients. J Med Internet Res. 2017;19:e120. doi:10.2196/jmir.7092
38. Tsipouras MG, Giannakeas N, Tzallas AT, et al. A methodology for automated CPA extraction using liver biopsy image analysis and machine learning techniques. Comput Methods Programs Biomed. 2017;140:61–68. doi:10.1016/j.cmpb.2016.11.012
39. Niel O, Boussard C, Bastard P. Artificial intelligence can predict GFR decline during the course of ADPKD. Am J Kidney Dis. 2018;71:911–912. doi:10.1053/j.ajkd.2018.01.051
40. Zhang J, Song Y, Xia F, et al. Rapid and accurate intraoperative pathological diagnosis by artificial intelligence with deep learning technology. Med Hypotheses. 2017;107:98–99. doi:10.1016/j.mehy.2017.08.021
41. Alvarez-Machancoes O, Fernández-Martínez JL. Using artificial intelligence methods to speed up drug discovery. Exp Opin Drug Discov. 2019;14:769–777. doi:10.1080/17460441.2019.1621284
42. Scannel JW, Blanckley A, Boldon H, et al. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11:191–200. doi:10.1038/nrd3681
43. Speck-Planche A, Cordeiro MNDS. Enabling virtual screening of potent and safer antimicrobial agents against noma: mtk-QSBER model for simultaneous prediction of antibacterial activities and ADMET properties. Mini Rev Med Chem. 2015;15(15):194–202. doi:10.2174/138955751503150312120519
44. Tenorio-Borroto E, Ramirez FR, Speck-Planche A, et al. QSPR and flow cytometry analysis (QSPR-FCA): review and new findings on parallel study of multiple interactions of chemical compounds with immune cellular and molecular targets. Curr Drug Metab. 2014;15:414–428. doi:10.2174/1389200215666140908101152
45. Martínez-Arzate SG, Tenorio-Borroto E, Barbabosa Pliego A, et al. PTML model for proteome mining of B-cell epitopes and theoretical-experimental study of Bm86 protein sequences from Colima, Mexico. J Proteome Res. 2017;16:4093–4103. doi:10.1021/acs.jproteome.7b00477
46. Tenorio-Borroto E, Ramírez FR, Speck-Planche A, et al. QSPR and flow cytometry analysis (QSPR-FCA): review and new findings on parallel study of multiple interactions of chemical compounds with immune cellular and molecular targets. Curr Drug Metab. 2014;15:414–428. doi:10.2174/1389200215666140908101152
47. Ferreira Da Costa J, Silva D, Caamaño O, et al. Perturbation theory/machine learning model of ChEMBL data for dopamine targets: docking, synthesis, and assay of New l-Prolyl-l-leucyl-glycinamide Peptidomimetics. ACS Chem Neurosci. 2018;9:2572–2587. doi:10.1021/acschemneuro.8b00083
48. Romero-Duran FJ, Alonso N, Yanez M, et al. Brain-inspired cheminformatics of drug-target brain interactome, synthesis, and assay of TVP1022 derivatives. Neuropharmacology. 2016;103:270–278. doi:10.1016/j.neuropharm.2015.12.019
49. Speck-Planche A. Multicellular target QSAR model for simultaneous prediction and design of anti-pancreatic cancer agents. ACS Omega. 2009;4:3122–3132. doi:10.1021/acsomega.8b03693
50. Speck-Planche A, Cordeiro MNDS. De novo computational design of compounds virtually displaying potent antibacterial activity and desirable in vitro ADMET profiles. Med Chem Res. 2017;26:2345–2356. doi:10.1007/s00044-017-1936-4
51. Kleandrova VV, Ruso JM, Speck-Planche A, et al. Enabling the discovery and virtual screening of potent and safe antimicrobial peptides simultaneous prediction of antibacterial activity and cytotoxicity. ACS Comb Sci. 2016;18:490–498. doi:10.1021/acscombsci.6b00063
52. Swinney DC, Jason TI. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507–519. doi:10.1038/nrd3480
53. Xie L, Ge X, Tan H, et al. Towards structural systems pharmacology to study complex diseases and personalized medicine. PLOS Comp Bio. 2009;10:e1003554. doi:10.1371/journal.pcbi.1003554
54. Arrell DK, Terzic A. Network systems biology for drug discovery. Clin Pharmacol Ther. 2010;88:120–125. doi:10.1038/clpt.2010.91
55. Gulshan V, Peng L, Coram M, et al. Development and validation of a deep learning algorithm for detection of diabetic retinopathy in retinal fundus photographs. JAMA. 2016;316:2402–2410. doi:10.1001/jama.2016.17216
56. Halevy A, Norvig P, Pereira F. The unreasonable effectiveness of data. IEEE Intell Syst. 2009;24:8–12. doi:10.1109/MIS.2009.36
57. Fernández-Muñiz Z, Fernández-Martínez JL, Srinivasan S, Mukerji T. Comparative analysis of the solution of linear continuous inverse problems using different basis expansions. J App Geophys. 2015;113:92–102. doi:10.1016/j.jappgeo.2014.12.010
58. Fernández-Martínez JL, Cernea A. Exploring the uncertainty space of ensemble classifiers in face recognition. Int J Pattern Recognit Artif Intell. 2015;29:1556002. doi:10.1142/S0218001415560029
59. Hicks JL, Uchida TK, Seth A, Rajagopal A, Delp SL. Is my model good enough? Best practices for verification and validation of musculoskeletal models and simulations of movement. J Biomech Eng. 2015;137:0209051–02090524. doi:10.1115/1.4029304
60. van Hulse V, Khoshgoftaar TM, Napolitano A. Experimental perspectives on learning from imbalanced data
61. Van Hulse J. Data Quality in Data Mining and Machine Learning. Boca Raton (FL): s.n.; 2007.
62. Fernández-Martínez. JL. High-dimensional data analysis. US20140222749A1 US. March 06, 2011.
63. Fernández-Martínez. JL. Model reduction and uncertainty analysis in inverse problems. Leading Edge. 2015;34:1006–1016. doi:10.1190/tle34091006.1
64. Fernández-Martínez JL, Fernández-Muñiz Z. The curse of dimensionality in inverse problems. J Comp App Math. 2019;369:112571.
65. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–795. doi:10.1056/NEJMp1500523
66. Libbrecht MW, Noble WS. Machine learning applications in genetics and genomics. Nature Rev Genet. 2015;16:321–332. doi:10.1038/nrg3920
67. Weng SF, Reps J, Kai J, Garibaldi JM, Qureshi N. Can machine-learning improve cardiovascular risk prediction using routine clinical data? PLoS One. 2017;12:e0174944. doi:10.1371/journal.pone.0174944
68. Berger MF, Mardis ER. The emerging clinical relevance of genomics in cancer medicine. Nat Rev Clin Oncol. 2018;15:353–365. doi:10.1038/s41571-018-0002-6
69. Do H, Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin Chem. 2015;61:64–71. doi:10.1373/clinchem.2014.223040
70. Network, Cancer Genome Atlas Research. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi:10.1038/nature07385
71. Van Allen EM, Wagle N, Stojanov P, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014;20:682–688. doi:10.1038/nm.3559
72. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi:10.1038/nbt.2696
73. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi:10.1158/2159-8290.CD-12-0095
74. deAndrés-Galiana EJ, Fernández-Martínez JL, Sonis ST. Design of biomedical robots for phenotype prediction problems. J Comp Biol. 2016;23:678–692. doi:10.1089/cmb.2016.0008
75. Fernández-Martínez JL, Pallero J, Fernández-Muñiz Z. The effect of noise and Tikhonov’s regularization in inverse problems. Part I: the linear case. J Appl Geophys. 2014;108:176–185. doi:10.1016/j.jappgeo.2014.05.006
76. Fernández-Martínez JL, Pallero J, Fernández-Muñiz Z. The effect of noise and Tikhonov’s regularization in inverse problems. Part II: the nonlinear case. J Appl Geophys. 2014;108:186–193. doi:10.1016/j.jappgeo.2014.05.005
77. deAndrés-Galiana EJ, Fernández-Martínez JL, Sonis ST. Sensitivity analysis of gene ranking methods in phenotype prediction. J Biomed Inf. 2016;64:255–264. doi:10.1016/j.jbi.2016.10.012
78. Cernea A, Fernández-Martínez JL, deAndrés-Galiana EJ, Galván JA, Pravia CG. Analysis of clinical prognostic variables for triple negative breast cancer histological grading and lymph node metastasis. J Med Inf Decis Making. 2018;1:14. doi:10.14302/issn.2641-5526.jmid-18-2488
79. Jiang X, Barmada MM, Visweswaran S. Identifying genetic interactions in genome-wide data using Bayesian networks. Genet Epidemiol. 2010;34:575–581. doi:10.1002/gepi.20514
80. Hageman RS, Leduc MS, Korstanje R, Paigen B, Churchill GA. A Bayesian framework for inference of the genotype-phenotype map for segregating populations. Genetics. 2011;187:1163–1170. doi:10.1534/genetics.110.123273
81. McGeachi MJ, Chang HH, Weiss ST. CGBayesNets: conditional gaussian Bayesian network learning and inference with mixed discrete and continuous data. PLoS Comput Biol. 2014;10:e1003676. doi:10.1371/journal.pcbi.1003676
82. Josan A. Conditional reasoning with subjective logic. J Multiple Valued Logic Soft Comput. 2008;15:5–38.
83. Cassells W, Schoenberger A, Graboys TB. Interpretation by physicians of clinical laboratory results. N Engl J Med. 1978;299:999–1001. doi:10.1056/NEJM197811022991808
84. Su C, Andrew A, Karagas MR, Borsuk ME. Using Bayesian networks to discover relations between genes, environment and disease. BioData Min. 2013;6:6. doi:10.1186/1756-0381-6-6
85. Altman NS. An Introduction to Kernel and Nearest-Neighbor Nonparametric Regression. Vol. 46. The American Statistician; 1992:175–185.
86. Huang GB, Zhu QY, Siew CK. Extreme learning machines: theory and applications. Neurocomputing. 2006;70:489–501. doi:10.1016/j.neucom.2005.12.126
87. Breiman L. Random Forests. Mach Learn. 2001;45:5–32. doi:10.1023/A:1010933404324
88. Cortes C, Vapnik VN. Support-vector networks. Mach Learn. 1995;20:273–297. doi:10.1007/BF00994018
89. Zaccone G, Karim MR, Menshawy A. Deep Learning with TensorFlow. Birmingham (UK): Packt Publishing Ltd; 2017.
90. Cernea A, Fernández-Martínez JL, deAndrés-Galiana EJ, Galván JA, García Pravia C. Analysis of clinical prognostic variables for triple negative breast cancer histological grading and lymph node metastasis. J Med Inf Decis Making. 2018;1:14. doi:10.14302/issn.2641-5526.jmid-18-2488
91. Cernea A, Fernández-Martínez JL, deAndrés-Galiana EJ, et al. Prognostic networks for unraveling the biological mechanisms of Sarcopenia. Mech Ageing Dev. 2019;182:111129. doi:10.1016/j.mad.2019.111129
92. deAndrés-Galiana EJ, Bea G, Fernández-Martínez JL, Saligan LN. Analysis of defective pathways and drug repositioning in multiple sclerosis via machine learning approaches. Comput Biol Med. 2019;115:103492. doi:10.1016/j.compbiomed.2019.103492
93. Fernández-Martínez JL, deAndrés-Galiana EJ, Fernández-Ovies FJ, Cernea A, Kloczkowski A. Robust sampling of defective pathways in multiple myeloma. Int J Mol Sci. 2019;20:4681. doi:10.3390/ijms20194681
94. Fernández-Martínez JL, Álvarez O, deAndrés EJ, de la Viña JFS, Huergo L. Robust sampling of altered pathways for drug repositioning reveals promising novel therapeutics for inclusion body myositis. J Rare Dis Res Treat. 2019;4:7–15. doi:10.29245/2572-9411
95. Jezequel P, Loussouarn D, Guerin-Charbonnel C, et al. Gene-expression molecular subtyping of triple-negative breast cancer tumours: importance of immune response. Breast Cancer Res. 2015;20:43. doi:10.1186/s13058-015-0550-y
96. Cernea A, Fernandez-Martinez JL, deAndres-Galiana EJ, Fernandez-Ovies FJ, Alvarez-Machancoses O, Fernandez-Muñiz Z, Saligan LN, Sonis ST, Robust pathway sampling in phenotype prediction. Application to triple negative Cancer. BMC Bioinformatics. 2020;21(2):89. doi:10.1186/s12859-020-3356-6.
97. Qin N, Wang C, Lu Q, et al. A cis-eQTL genetic variant of the cancer-testis gene CCDC116 is associated with risk of multiple cancers. Hum Genet. 2017;136:987. doi:10.1007/s00439-017-1827-2
98. Oyama T, Miyoshi Y, Koyama K, et al. Isolation of a novel gene on 8p21. 3-22 whose expression is reduced significantly in human colorectal cancers with liver metastasis. Cancer. 2000;29:9–15.
99. Wan M, Huang W, Kute TE, et al. Yin Yang 1 plays an essential role in breast cancer and negatively regulates p27. Am J Pathol. 2012;180:2120–2133. doi:10.1016/j.ajpath.2012.01.037
100. Ganguly KK, Pal S, Moulik S, Chatterjee A. Integrins and metastasis. Cell Adh Migr. 2013;7:251–261. doi:10.4161/cam.23840
101. Alvarez O, Fernández-Martínez JL. The importance of biological invariance in drug design. J Sci Tech Res. 2019;18:13211–13212.
102. Subramanian A, Narayan R, Corsello SM, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171:1437–1452. doi:10.1016/j.cell.2017.10.049
103. Chan J, Wang X, Turner JA, Baldwin NE, Gu J, Stegle O. Breaking the paradigm: dr Insight empowers signature-free, enhanced drug repurposing. Bioinformatics. 2019;35:2818–2826. doi:10.1093/bioinformatics/btz006
104. Alderton GK. Targeting MYC? You BET. Nature Rev Drug Discov. 2011;10:732–733. doi:10.1038/nrd3569
105. Zucchetti B, Shimada AK, Katz A, Curigliano G. The role of histone deacetylase inhibitors in metastatic breast cancer. Breast. 2019;43:130–134. doi:10.1016/j.breast.2018.12.001
106. Yang J, Nie J, Ma X, et al. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. 2019;18:26. doi:10.1186/s12943-019-0954-x
107. Crown J, O’Shaughnessy J, Gullo G. Emerging targeted therapies in triple-negative breast cancer. Ann Oncol. 2012;23:vi56–vi65. doi:10.1093/annonc/mds196
108. Roux NL, Schmidt M, Bach FR. A stochastic gradient method with an exponential convergence _rate for finite training sets. Adv Neural Inf Process Syst. 2012;2663–2671.
109. Zhang Z 2018, Improved Adam optimizer for deep neural networks.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.