")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 16
Obstacles to Early Diagnosis and Treatment of Alpha-1 Antitrypsin Deficiency: Current Perspectives
Authors Quinn M, Ellis P, Pye A , Turner AM
Received 6 October 2020
Accepted for publication 30 November 2020
Published 16 December 2020 Volume 2020:16 Pages 1243—1255
DOI https://doi.org/10.2147/TCRM.S234377
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Garry Walsh
Mark Quinn,1 Paul Ellis,1 Anita Pye,1 Alice M Turner1,2
1Institute of Applied Health Research, University of Birmingham, Birmingham, UK; 2University Hospitals Birmingham, Birmingham, UK
Correspondence: Alice M Turner
Institute of Applied Health Research, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK
Tel +44 121 371 3885
Email [email protected]
Abstract: This review summarizes the current research and outlooks regarding the obstacles to diagnosing and treating early alpha-1-antitrypsin deficiency (AATD). It draws on prior systematic reviews and expert surveys to discover precisely what difficulties exist in early diagnosis and treatment of AATD and elucidate potential solutions to ease these difficulties. The perceived rarity of AATD may translate to a condition poorly understood by primary care physicians, and even many respiratory physicians, which results in opportunities for diagnosis being missed, especially in mild or asymptomatic patients. There are diagnostic techniques involving biomarkers and home testing methods which could improve the rate of early diagnosis. With respect to treatment, AATD involves treating two separate pathologies, lung disease and liver disease. The only specific AATD treatment, augmentation therapy, has proven ability in treating lung disease but not liver disease. Alpha-1-antitrypsin (AAT) synthesized in the liver can form damaging polymers that also result in reduced circulating AAT levels and, whilst liver transplantation is used to effectively treat AATD, it is inappropriate in early disease. Novel therapeutic areas such as gene editing and increasing autophagy are therefore being researched as future treatments. Ultimately, diagnosis and treatment are intrinsically linked in AATD, with earlier diagnosis leading to better treatment options and thus better patient outcomes.
Keywords: emphysema, cirrhosis, diagnostic screening programs, chronic obstructive pulmonary disease
Introduction
Alpha-1-antitrypsin deficiency (AATD) is a hereditary, metabolic disorder that is defined by low serum concentration of the protease inhibitor, alpha-1-antitrypsin (AAT).1 In patients with AATD, the lack of AAT causes an imbalance in protease-antiprotease activity, this imbalance is the main factor in the pathogenesis of the disease.2 AAT normally acts to counteract serine proteases, such as neutrophil elastase, from causing proteolytic damage to the lungs. In deficient individuals this proteolysis continues unchecked and thus degrades elastin, compromising alveolar structure.2 The continued progression of this mechanism of action will eventually lead to the clinical presentation of symptoms of chronic obstructive pulmonary disease (COPD).2
AAT is encoded by the SERPINA1 gene and it is mutations on this gene that cause deficiency. AATD is a codominant condition and the severity of deficiency is dependent on the inherited genotype.3 There have been over 100 mutations described in the literature, each with varying levels of deficiency and consequently pathogenicity.3 This includes null variants, which are mutations that can lead to a stop codon being transcribed so no functional protein is synthesized. The normal allele is designated M and the normal genotype designated PiMM, with Pi representing protease inhibitor. This genotype codes for normal AAT serum concentrations, which are commonly accepted to be 1.0–2.2 g/L or 20–53 µM.4 The most common deficient alleles are S and Z, which code for roughly 60% and 15%, respectively, of normal AAT serum concentrations.4 AATD is widely known in Europe as a disease of the Caucasian population but a comprehensive review of the prevalence of the S and Z alleles showed that the spread by migrating populations means they are found in many countries around the world.5 Prevalence varies within a given geographical region, and neighboring countries can often have widely different incidences. High incidence of the Z allele was seen in North Africa (28.6 per 1,000 population in Egypt) and North/Central Asia (15.0 per 1,000 population in Saudi Arabia) but none detected in Central/South Africa. The prevalence of AATD in the Brazilian population, which is known to be racially diverse and includes European immigrants, has been shown to be 2.8%, which is similar to other countries, with the Z allele found in 0.8%.6 The S allele has been found in high numbers in at least nine South American countries, where numbers of greater than 30 per 1,000 population were observed.
PiSZ and PiZZ are the most commonly occurring genotypes that are linked with the highest pathogenicity, although those heterozygous with M and Z/null can carry higher risk if they also have certain exposures, ie, smoking.7 Increasing evidence suggests that the SZ variant may also only lead to significant disease in the presence of such exposures.8,9
Those who are homozygous for PiZZ can also present with liver disease. This is because the Z allele leads to severe misfolding of the protein, and as AAT is secreted from hepatocytes the misfolded protein polymerizes causing hepatocyte damage. The pathophysiology for this damage is believed to originate primarily from stress in the endoplasmic reticulum due to accumulation of Z-AAT. Continuous hepatocyte destruction eventually manifests as chronic liver disease, initially fibrosis and eventually cirrhosis.10
There is increasing interest in both the pulmonary and hepatology literature in early diagnosis of COPD11 and liver fibrosis,12 respectively, with the hope that early diagnosis, lifestyle modifications, and aggressive treatment could reduce progression to more symptomatic disease, with consequent morbidity and reduced life expectancy. Since AATD may cause both of these problems, and onset of disease tends to be at a younger age than in non-AATD related disease, it is logical that the healthcare community would wish to prioritize early diagnosis in these individuals. The aim of this review is to collate and condense from the current literature factors seen as the main obstacles to early diagnosis and management of AATD. In constructing this review, we used the 2017 European Respiratory Society (ERS) working statement on the diagnosis and treatment of AATD as a starting point.13 This was an update on the international statement published by ERS, in conjunction with the American Thoracic Society (ATS) some years previously,14 and focused on the diagnosis and treatment of AATD and associated lung disease, using systematic review methodology to identify literature up to that point. Subsequent targeted literature searches focused on systematic reviews, expert surveys, and randomized controlled trials (RCTs) that might contain data on early diagnosis or treatment that were published after this review.
Diagnosis
When is AATD Diagnosed?
There are three main approaches adopted in the diagnosis of AATD; testing individuals who are symptomatic for AATD related pathology, ie, lung or liver disease, testing of individuals who may be genetically predisposed to AATD, and screening.13 Guidelines for testing have been laid out by the World Health Organization (WHO) via their 2014 Global Initiative for COPD which recommend routine testing for all COPD patients in areas with high AATD prevalence.15 The American Thoracic Society/European Respiratory Society (ATS/ERS)14 went further, stating testing for AATD should be conducted for all COPD patients, all patients with unresponsive adolescent asthma, and all patients with either cryptogenic liver disease, granulomatosis with polyangiitis, bronchiectasis, or panniculitis. Traditionally, however, testing has only been carried out on individuals presenting with early onset emphysema. This diagnostic pathway has resulted in AATD being a condition which is likely both underdiagnosed and diagnosed too late for some therapeutic interventions to have maximum effect.
Underdiagnosis of AATD
A recent update to a paper calculating the prevalence of PiZZ AATD in Europe, through secondary analysis of cohort studies, found prevalence to be approximately 0.02–0.05%, which equates to a potential cohort of 119,594 individuals.16 Data was collected from studies considered to be representative of the general population, with cohort studies of patients with AATD-related diseases such as COPD and liver cirrhosis excluded. In this work, the authors noted the substantial gap in epidemiological data surrounding AATD which highlights the underdiagnosed nature of the condition. There have been several recent papers, with wide scope, that have reported underdiagnosis of AATD.17,18 Several different factors have been hypothesized to try and explain this, ranging from the wide variability in AATD pathogenicity which makes definitive diagnosis difficult, to the fact that the usual clinical presentation of AATD is very similar to COPD and therefore may be misdiagnosed. A Europe-wide survey of experts in AATD found that underdiagnosis is a key problem in the management of AATD; most clinicians (77%) believed that only about 15% of cases of AATD were being diagnosed, with the remainder surveyed estimating diagnosis was roughly 30%.19 Underdiagnosis was further demonstrated when comparing the clinicians’ estimates for confirmed cases of AATD diagnosis to published prevalence: on average the clinicians’ estimates were 90% lower than the figures ascertained from epidemiological studies. When queried as to why this underdiagnosis exists, a large majority (76.9%) of clinicians cited a lack of awareness of AATD among primary care as the biggest obstacle. A significant number (15.4%) also stated no availability of treatment as another obstacle for diagnosis, presumably because in the absence of treatment some clinicians feel making a diagnosis is not worthwhile. Notably guidance on treatment varies worldwide.18,20
A potential factor worth considering as an obstacle to diagnosis is the availability of a nationwide AATD screening program; of the 13 countries represented in the survey only four had a screening program in place.19 The use of a screening program would theoretically cast a wide net for potential cases, especially those who are heterozygous for deficient alleles and may be asymptomatic or present with mild COPD symptoms. Table 1 considers whether AATD meets the classical criteria for screening, and more recent proposed adaptations to this following the advent of genetic technologies.21 Although population wide screening has drawbacks related to cost effectiveness, targeted screening of at-risk groups and family screening both have the potential to be powerful tools in improving the diagnostic rate of AATD, recognized in the newer criteria, which appear to advocate a more targeted approach.
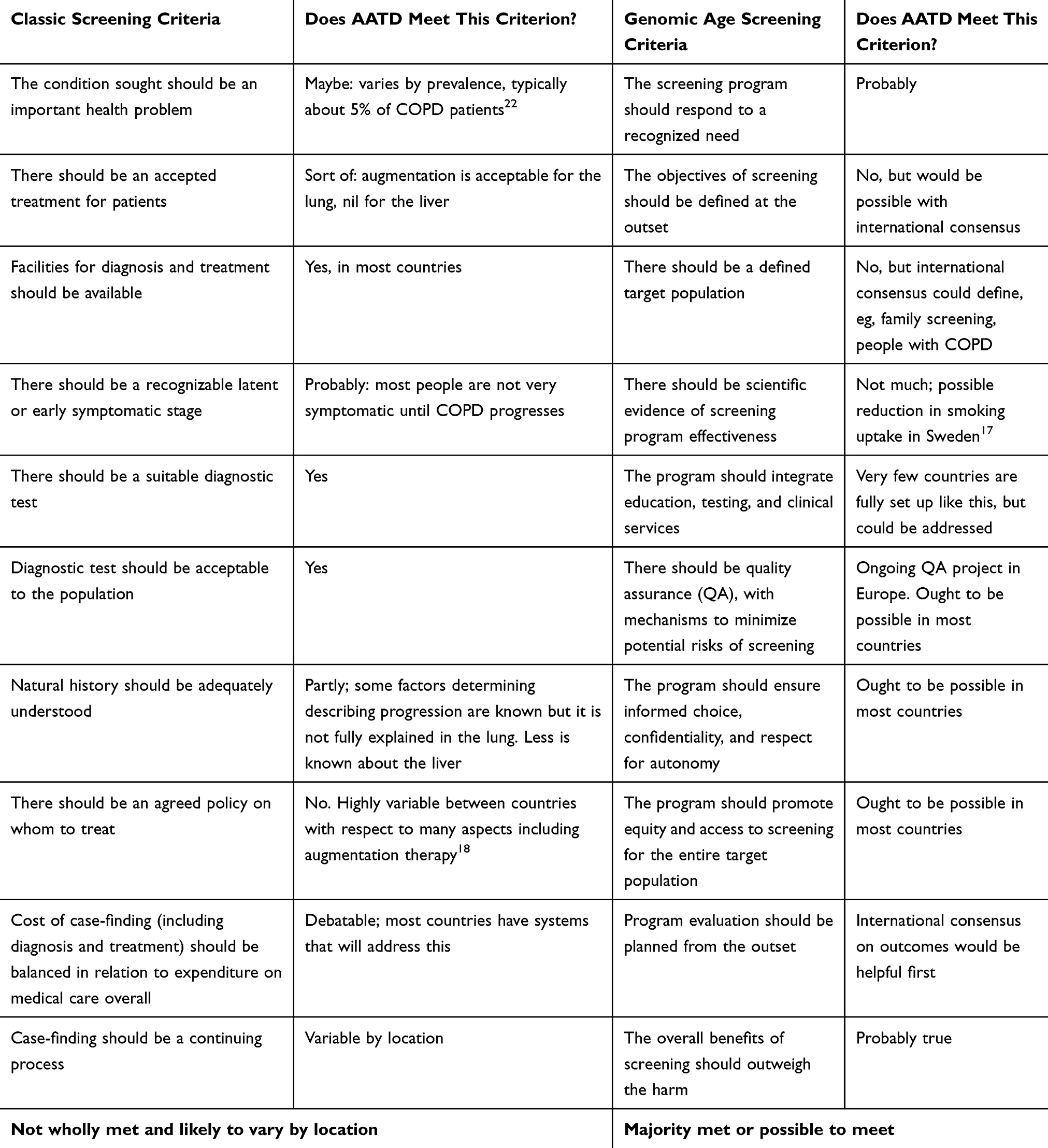 |
Table 1 Screening Criteria in Relation to AATD |
Table 1 shows that whilst some criteria for screening are met, not all are reached, regardless of which system is used.
All the clinicians surveyed in Europe had access to AAT serum concentration measuring and isoelectric focusing for diagnosis, such that a screening programme could be set up. However, only 73% have access to gene sequencing. Opening up this technology could have a dramatic impact on the incidence of AATD, especially in detecting heterozygous cases or those with very rare genetic variants, as shown in a study from Canada.23 Whilst over 100 mutations of the SERPINA1 gene have been described in the literature, diagnosis has been mostly restricted to the most pathogenic, PiSZ and PiZZ genotypes by many laboratories. Removing this obstacle by gene sequencing all potential cases could significantly address some aspects of the underdiagnosis problem, in that rarer alleles would be identified routinely, and the technology could be incorporated in some countries alongside other genetic testing programs. Sequencing would, however, add cost to screening, and this would need to be balanced against the potential value for a country of conducting such an intervention, as alluded to in Table 1. In Sweden, screening was introduced for a short time, but the only impact at age 34 was on smoking uptake; no difference was seen in other pulmonary parameters or symptoms.24 Cost-effectiveness modeling using lifetime horizon parameters has been done in COPD case finding,25 and similar methods might need to be employed to convince healthcare funders that a screening approach for AATD is affordable. Given the variable prevalence of AATD worldwide such studies would almost certainly need to be country or region specific.
Delays in Diagnosis: The Role of Public Health, Laboratory Methods, and Education
Another factor to consider as an obstacle to diagnosis is the time taken for diagnoses to be confirmed, with the majority of clinicians (60%) estimating diagnosis took longer than 5 years.19 One might speculate that this diagnostic odyssey means potential cases are lost due to patients losing faith and not attending follow-up clinic visits. It is also fair to assume in that time span some patients’ health deteriorates to the point where they die before diagnosis is confirmed. These types of delays in diagnosis are seen to be common, as reported in Canada, where average diagnosis time was 7 years.26 There have been positive strides taken to address this obstacle in many locations. In Germany a public health campaign raising awareness for AATD testing and offering test kits significantly improved case detection, albeit performing better in a selected rather than unselected population.27 In Spain a form of test kit which utilizes dried blood spots, hence is possible for patients to perform themselves, was also examined and found feasible.28 Lateral flow test kits (similar to a pregnancy test) have also been used internationally with success.29 Opportunistic testing at hospital admission for COPD has also shown promise in a small study.30
A review addressing areas for research within AATD also underlined the underdiagnosis problem; approximately 90% of cases were undetected.31 The authors indicated that since AATD clinical presentation was so similar to other pulmonary pathologies, patients were often misdiagnosed. This would ring true when looking at recent epidemiological studies that estimated, globally, there are over 1.0 million individuals with the most severely deficient genotypes, ie, PiSZ and PiZZ,5 yet clinicians still view it as a rare disease. The authors argue that, due to the wide variability in the severity of symptoms of AATD patients, a new diagnostic approach is required. As there is no universally accepted diagnostic algorithm, AAT serum concentration in COPD patients is widely used. Accounting for the effects of inflammation on AAT level (because AAT is an acute phase protein) by concurrent measurement of CRP is also advocated.32 However, even with the addition of CRP, use of levels to determine advanced testing could easily miss heterozygous individuals who exhibit greater variability in AAT serum concentration and yet could still be at risk of developing COPD. Use of genetic technologies as described above could overcome this limitation, albeit at considerable cost. Another approach using measurement of α1-globulin alongside liver function and inflammatory markers, as a means of determining whether genotyping is worthwhile has also been proposed.33
Although AATD has been thought of primarily as a pulmonary disease and most diagnoses are detected through symptoms of lung pathology, it is important to also understand the importance of liver disease as a diagnostic pathway; especially within neonates as AATD is their most common cause of liver disease.34,35 It is possible that screening at the neonatal level is unnecessary, but targeted testing of individuals who have exclusively liver disease (no COPD) may be even more valuable than testing COPD patients; this approach may yield higher detection rates of AATD than testing COPD patients,31 albeit with varying prevalence depending on age. A review focused on improving the management of AATD in children concluded that there should be greater awareness of the condition within pediatric medicine, especially in cases of liver disease of unknown etiology.34 The authors reviewed a variety of clinical presentations that could indicate PiZZ AATD including increased bilirubin, hepatomegaly, hepatosplenomegaly, chronic hepatitis, and failure to thrive, and found neonatal cholestasis to be the most associated feature, with a screening study of 200,000 neonates finding 17% of those with PiZZ genotype presenting with the pathology. The authors concede that, because a wide range of neonatal disorders cause liver disease, an AATD diagnosis is not always obvious. They suggest that to remove this obstacle to diagnosis, pediatricians should be made more aware of the hepatic pathology of AATD and understand that a correct and early diagnosis can put patients on the appropriate treatment path to help prevent developing severe lung disease. They also note the added benefit a neonatal diagnosis can provide in allowing parents to avail themselves of the services of genetic counselors. Table 2 summarizes the strategies tested to date to raise awareness and reduce delay in diagnosis.
 |
Table 2 Methods to Reduce Under-Diagnosis or Delayed Diagnosis of AATD |
Case-Finding Instead of Screening
As there is no defining clinical sign for AATD diagnosis, the use of targeted detection across a range of phenotypes has to be considered; this would be consistent with the more nuanced approach to screening now advocated (Table 1). It is conceivable that AATD could be identified through screening for other lung pathologies; for example, if small airways disease (SAD) or emphysema were seen on a CT scan done for lung cancer screening it might trigger tests for COPD38 and AATD. Typically cancer screening focuses on heavy smokers though, so there would be risks to this approach, mainly missing cases who have never smoked or have low smoke exposure, and in some countries questions have been raised about the cost-effectiveness of such CT based programs.39 Case finding for AATD in individuals with COPD diagnosed at a young age, with low smoke exposure, or with a family history of COPD would also be an obvious method, consistent with most current international guidance, but inadequately implemented in most countries at present. For example in the UK only 2.2% of people with COPD below the age of 60 had been tested for AATD in one primary care group.40 Importantly case finding for COPD has been found to be feasible, beneficial, and cost-effective in primary care, suggesting that adding an extra step to test for AATD in a subset of people is something that might be practical and affordable.25,41
The parallel process for liver disease would be to screen people who appear to have liver fibrosis. Serum concentration of gamma glutamyl transferase (GGT) was seen to be raised in children with PiZZ genotype,42 although it is not a perfect marker in adults since it may also relate to lung disease,43 and classically rises with alcohol consumption as well. The possibility of using liver pathology as a signpost for AATD via elastography as an imaging biomarker has also been investigated; a comparative study concluded that increased shear wave speed, as determined by a variety of elastography techniques, correlated significantly with AATD diagnosis (P<0.009).44 A threshold of 7.9 kPa seemed to relate well to diagnosis and histological fibrosis in cohort studies.45–47 However the utility of widespread AATD case findings based on elastography or liver function tests in the blood has not been conducted.
One novel way of detecting subgroups within COPD or liver fibrosis who might be more at risk of having AATD could be use of biomarkers that detect disease progression in either the lung or the liver. Desmosine and isodesmosine, both products of elastin degradation, are raised in AATD COPD patients48 and could hold promise as a biomarker for measuring progression of AATD-caused COPD.49 Results from the RAPID/RAPID Extension trial showed a correlation between desmosine/isodesmosine levels and CT lung density used as a clinical marker of progression of emphysema.50 Ma et al50 suggested that desmosine/isodesmosine levels may therefore be useful in evaluation of therapeutic agents that can decrease elastin degradation and lung matrix injury. Z-AAT polymers could also be a potential marker to confirm at-risk patients with AATD,51 although further research is needed because little is known about how Z-AAT polymers are synthesized and secreted, in comparison to M-AAT polymers, nor which risk groups altered levels mark out most clearly. The use of imaging biomarkers has also been investigated for identifying and evaluating lung disease in AATD. A review conducted into the role computed tomography played in the management of AATD confirmed that CT image assessment could be used to identify AATD-related emphysema in the lungs and could map disease progression, thereby allowing predictions to be made on the appropriateness of therapy.52 Clearly use of CT needs to be balanced against risks of radiation exposure and also of cost; secondary analysis of scans being done for another reason (eg, cancer screening) would be a more sensible approach than using CT specifically for emphysema detection.
The consensus in the literature is that patients presenting with either liver or lung disease come into contact with many healthcare professionals, over long periods of time, so opportunities for diagnosis exist. Figure 1 illustrates a possible ideal health system maximizing detection and onward referral of patients at risk of deterioration. After a diagnosis has been made patients should be followed-up 3–12 monthly with lung function and other clinical assessments, as described by the European Respiratory Society.13
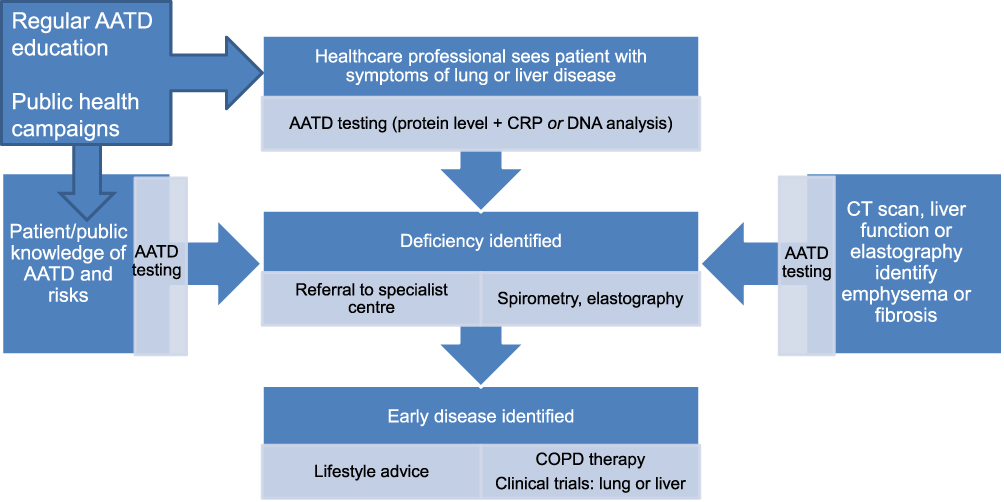 |
Figure 1 Optimized detection of AATD, In the above system patients or healthcare professionals could access AATD testing, and it would also be automatically triggered by identification of emphysema or liver fibrosis on tests done for other reasons. Educated healthcare professionals would always test for AATD in relevant respiratory and hepatic presentations, and patients aware of (for example) a family history of AATD or COPD would know that they should get tested and would access it themselves either before or after a healthcare professional saw them. Referral to specialist centers would occur early after a positive laboratory test. |
Treatment
Determining the prognosis can be difficult in AATD, because of the variability of presentation and subsequent progression, and it is conceivable that clinicians only feel there is benefit to early diagnosis if they can then prognosticate, thus know who to treat. The use of multifaceted tools or scores to evaluate COPD patients has been explored, in which the BODE score (BMI, airflow obstruction, dyspnoea, exercise capacity) was better at differentiating survival than forced expiratory volume 1 (FEV1) alone or the GOLD classification (Global Initiative for COPD),53 and shows a similar pattern in AATD.54 However scores like this generally perform better at population than individual level. Once prognosis has been established then efforts to treat AATD can be explored. Treatment of AATD has traditionally been lung-centric and has aimed to delay the onset of emphysema, reduce the frequency of pulmonary exacerbations, and improve quality-of-life. This section will not just encompass those therapies targeting lung disease, but also future therapies to alleviate AATD-related liver disease.
Lung Disease
The treatment options available to AATD patients at present are broadly similar to patients who have COPD unrelated to AATD, with symptoms being managed by bronchodilators, inhaled steroids, pulmonary rehabilitation, possibly lung volume reduction treatment, and lung transplantation.55 The only specific AATD treatment licensed worldwide has been plasma-purified AAT, given intravenously; this treatment has been coined augmentation therapy as it augments the patient’s deficient AAT serum concentration. Of these, the interventions that might be applicable to patients diagnosed early at present are inhaled therapies, rehabilitation, and augmentation.
Inhaled Therapies
In the case of an AATD patient diagnosed early, we would hope that lung function will be less impaired (if at all) hence augmentation therapy might not be necessary, and certainly is not recommended in most international guidance.13,20 The main goal is to prevent the development of lung disease, and risk factors for emphysema progression should be assessed as early as possible. There are limited studies looking at the benefits of inhaled therapies in AATD, but those patients with COPD are treated following COPD guidelines so we would expect evidence in usual COPD to apply. Inhaled therapies are prescribed according to the degree of breathlessness and inhaled corticosteroids have been shown to reduce FEV1 decline in PiZZ AATD patients with persistent eosinophilia.56
Augmentation
Augmentation therapy has had significant beneficial effects for patients in trials; the RAPID trial found lung density decline, by quantitative computed tomography (CT), in treated patients was significantly slower than in untreated patients, and earlier trials showed trends in this direction.57 However, it is important to note that this was only observed for quantitative CT measurements performed at total lung capacity (TLC) alone rather than functional residual capacity (FRC) alone or TLC and FRC combined. A systematic review and meta-analysis also found noteworthy results related to augmentation therapy;55 in pooled data from three RCTs for a total of 320 participants where lung density was measured via CT scan, annual deterioration in lung density was significantly less on augmentation than on placebo with a difference of 0.79 g/L/year. However, this was the only significant result, with no effect demonstrated on secondary outcomes including lung function and quality-of-life. Nevertheless it is important as change in lung density by quantitative CT is related to mortality in AATD.58 Another meta-analysis of clinical studies found that decline in FEV1 was 23% slower in patients receiving augmentation.59 However this decline was only significant in the subset of patients with predicted FEV1 of 30–65%, those with <30% or >65% predicted were not significant. This suggested that augmentation therapy was only appropriate for those AATD patients with moderate lung disease, although it may also be argued that it is the ongoing rate of deterioration rather than a single baseline value that is of relevance for augmentation, since it only modifies ongoing decline.
Whilst most international guidelines set a threshold FEV1 for receipt of augmentation therapy,20 these vary and having any threshold at all may be inappropriate given that response to augmentation appears to be independent of baseline FEV1.60,61 However there is little evidence that augmentation therapy can improve patient outcomes in early disease, since the majority of RCTs excluded patients with higher levels of lung function, which are likely to occur in those with early disease (summarized in Table 3). Theoretically similar effects might be expected in any patient with emphysema, but evidence to support this is lacking.
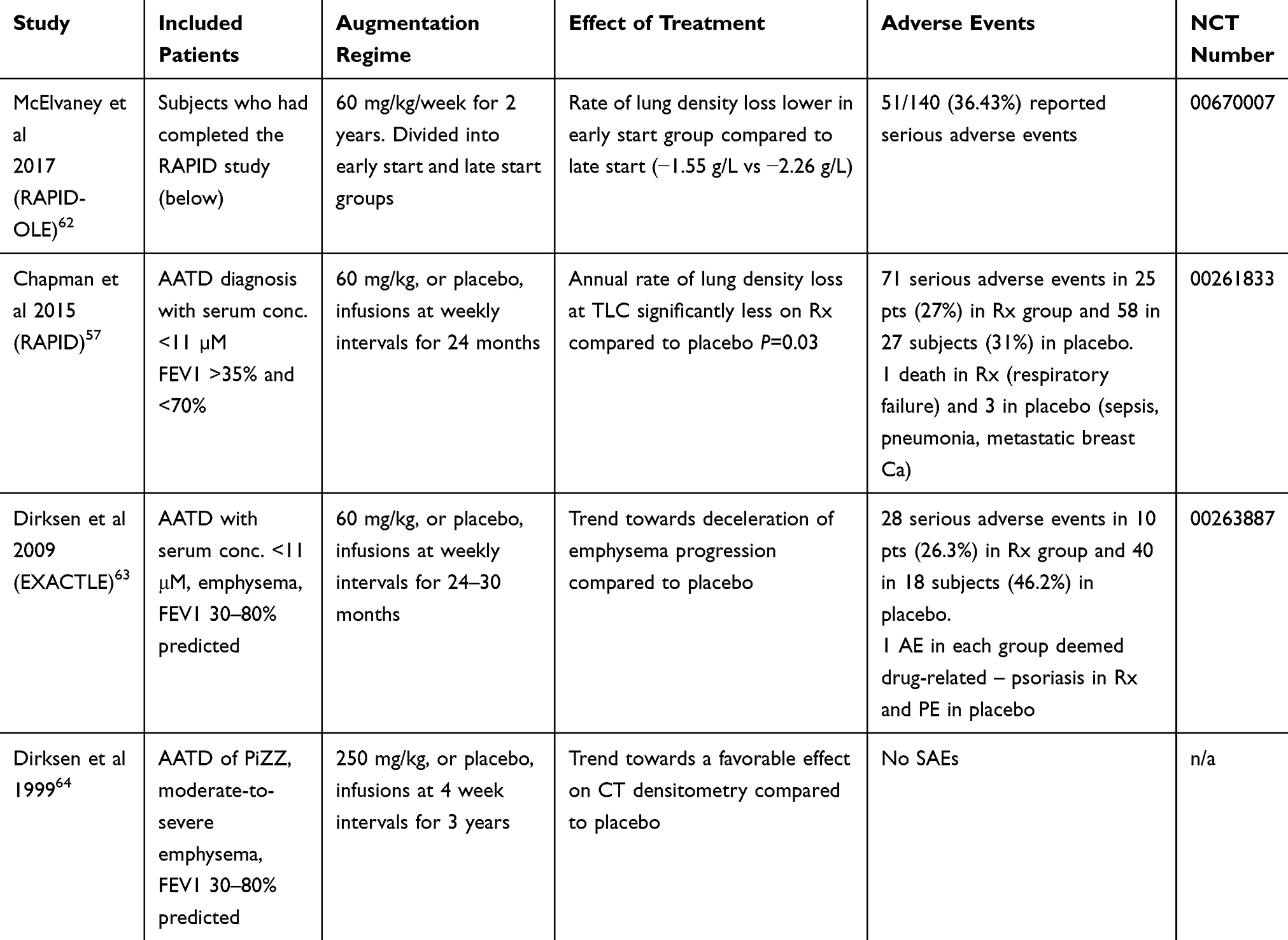 |
Table 3 Studies of Augmentation Therapy |
Table 3 shows that inclusion criteria excluded most patients likely to have early disease, with upper limits on FEV1 of 70–80% predicted. Patients with FEV1 ≤70% in the context of emphysema would likely have moderate severity (or worse) COPD; those ≥80% would be mild COPD. Whilst it is possible someone could be newly diagnosed with this level of lung function impairment this may not represent early disease, given that delay in diagnosis is common. Thus there is little evidence from RCTs of efficacy of augmentation in early disease.
One approach to clinical decision-making about augmentation might be to use the treatment in any patient who has lung disease which appears to be progressing;65 this would of course require a period of observation, which many patients and clinicians would be reluctant to do in the presence of symptomatic disease. However in cases diagnosed early, where there might be few symptoms, a period of observation might be more acceptable; this requires exploration with the patient and physician community, particularly in countries where augmentation is already widely available. Selection of the optimal method to pick up decline would also be helpful; whilst CT densitometry has proven best at this in trials regular use of CT would have logistical issues in many centers, and potential harms from regular radiation exposure. Identification of physiological or blood markers would be more desirable; in our own work gas transfer appeared to be much better than FEV1 at detecting densitometry defined emphysema progression, for instance.58
When reviewing the results from a European expert survey the provision of augmentation therapy for AATD patients is inconsistent; Germany and France each have approximately 60% of their patients receiving the treatment while the UK and the Netherlands have 0% and 5%, respectively.19 Reasons for these disparities include differences in regulations around augmentation therapy; countries which granted approval to a specific named brand drug (Prolastin) had higher uptake of augmentation therapy than countries without approval. Other commonly cited obstacles to augmentation were infusion time (33.3%), and frequency of infusions (16.7%), and in many health systems cost will also be a factor, which will be greater if treatment is extended to early stages.19 This would suggest that augmentation therapy has enough downsides that a search for alternatives is required. Indeed many studies of alternative strategies are under way, which have been reviewed elsewhere.66 Many of the trials of alternative lung directed therapies are recruiting patients in earlier stages of disease than augmentation is typically used; specific subgroup analyses by disease stage may be of interest in order to determine which treatments should/could be used at each stage. Table 4 summarizes currently recruiting studies and the severity stages applicable.
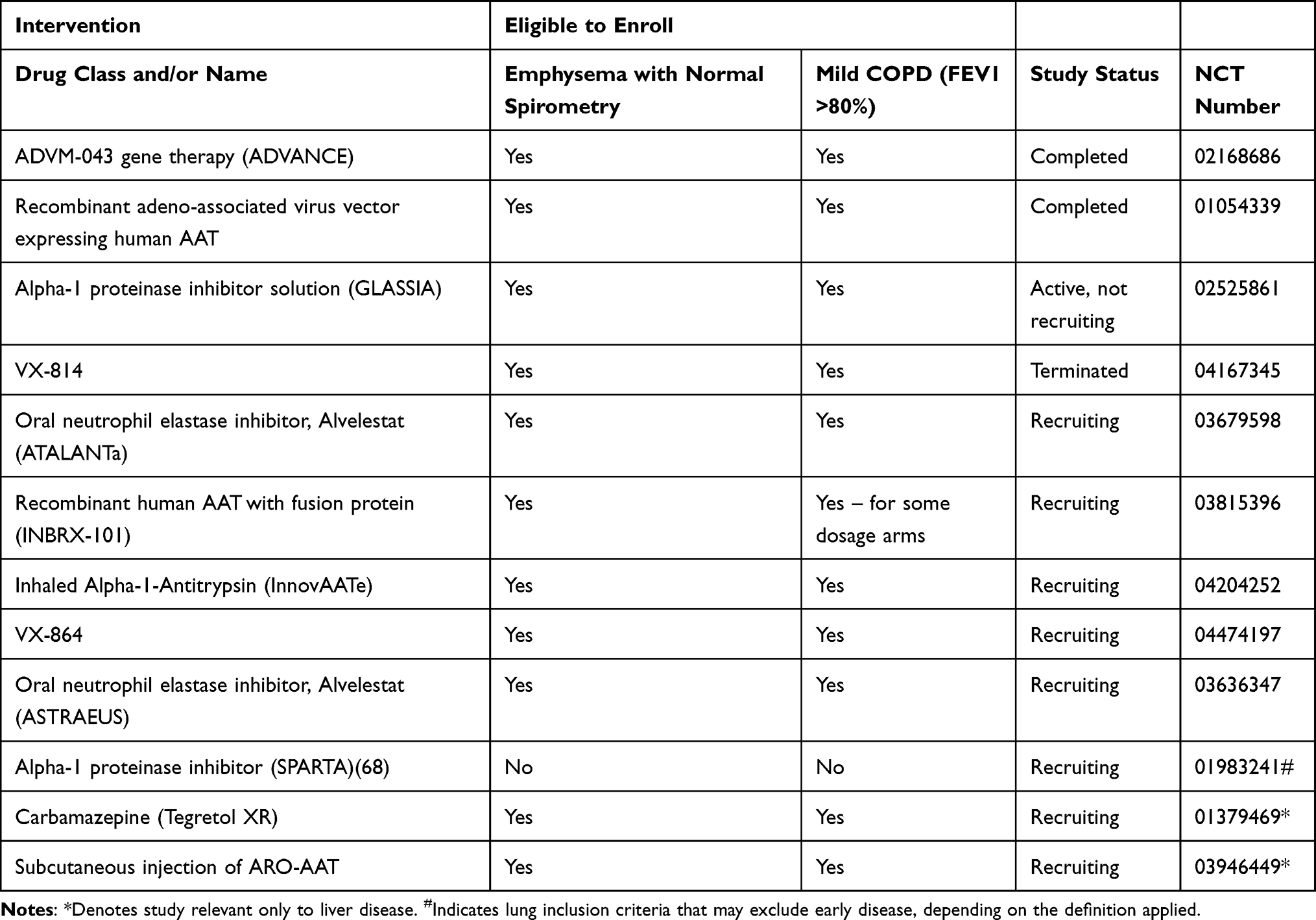 |
Table 4 RCTs of Treatments Ongoing and Potentially Applicable to Early Disease |
Whilst these new treatments have been reviewed extensively,66 it is worth considering them briefly here for their potential in early disease. A synthetic recombinant AAT (rAAT) fused with another molecule to reduce any immune reactions with non-human proteins is being investigated in a dose-escalating Phase I trial; this would have similar limitations to augmentation in that it requires intravenous infusions, which might not be popular with relatively asymptomatic patients, although the frequency is likely to be less, which may be helpful. In order to tackle the obstacles associated with intravenous augmentation, inhaled augmentation may have promise, and is in Phase 3 testing. Another avenue to consider in AATD treatment is that of addressing antiprotease-protease balance – instead of increasing antiprotease activity (ie, AAT) this decreases protease activity (ie, neutrophil elastase). Clinical trials are ongoing that use a synthetic neutrophil elastase inhibitor and will measure neutrophil elastase activity through measuring desmosine/isodesmosine concentration as a marker of neutrophil elastase action. This mechanism would potentially be applicable and modifiable in all disease stages. Treatments which address raising native AAT levels, such as small molecule approaches that prevent polymerization, would also be attractive in early lung disease for similar reasons. Notably the current trials targeting these two mechanisms are oral, so may be more acceptable to patients with few symptoms, such as those diagnosed at an early stage.
Liver Disease
Whilst augmentation therapy has proven effective at decelerating severe lung disease there are many manifestations of AATD that require their own specialist treatment, the most pertinent of these is liver disease. A systematic review has reported the prevalence and natural history of AATD associated liver disease, being around 15% on average with a bimodal distribution peaking in childhood and later life.68 This indicates two different tracks to development of significant disease, one rapid in childhood and the other much more indolent. This has implications for what we would consider “early diagnosis” in the liver – for children this would presumably have to be neonatal screening, whilst for adults it might mean those with abnormal LFTs or elastography (as described in the case finding section). The authors noted that when carrying out their search the only available treatment option was liver transplantation, which had good outcomes in both children and adults with AATD,68 though this is of course a treatment reserved for advanced disease. It should be noted however that the wider issues of relative shortage of pediatric organs for transplantation69 might preclude much in the way of benefit from early diagnosis for the minority of patients who will develop clinically significant childhood liver disease. Early detection of more indolent adult liver disease could have more potential for benefit, particularly with the newer drugs that are becoming available in this field.
There are four novel therapeutic areas that could be appropriate for treating early onset liver disease, which function by interfering in accumulation of Z-AAT polymers in the liver. One of these therapeutic areas is gene editing technologies, namely small interfering RNA (siRNA) and clustered regularly interspaced short palindromic repeats (CRISPR/Cas9). The siRNA molecules target RNA that encodes AAT protein and thus reduce its production and therefore accumulation. CRISPR/Cas9 is being used to edit the SERPINA1 gene to stop producing Z-AAT; both of these methods have proven effective in mouse models and clinical trials have been started. The second therapeutic area is autophagic enhancers which increase the autophagy and disposal of Z-AAT polymers; they have also been moved to the clinical trial stage. Chaperone molecules were another therapeutic area trialed; these work by inhibiting enzymes related to histones that then increase Z-AAT secretion from the liver. After the success of 4-phenylbuterate (4-PBA) in mouse models where a two-fold increase in circulating AAT levels was seen70 it was hypothesized that this may be a promising therapy to prevent emphysema and reduce accumulation of Z protein within the liver.71 However, preliminary trials failed to show an increase in AAT blood levels in AATD71 and also resulted in significant adverse events,72 indicating that further research is needed in this area. Finally there is a monoclonal antibody that has a structure designed to fit into the reactive loop of Z-AAT and thus prevent aggregation on the polymers and thus liver accumulation.73 Some such molecules have been tested in mammalian cell lines.74 Each of these drugs has potential to slow down, or prevent onset of liver disease, and those which also elevate AAT levels in the blood have some potential to treat or prevent lung disease. However, the rate of progression of adult liver disease, and the optimal non-invasive way of monitoring this to select patients at risk of rapid decline, and in whom newer treatments would be expected to have greater benefit requires further research in large cohorts.
Currently, therefore, it would be harder to target drugs to early liver disease as we are less certain which patients have disease with potential to progress, and how to pick up progression early. Similar issues to treatment of early disease, such as cost and willingness of patients to take drugs by non-oral routes if asymptomatic would also apply to management of early liver disease.
Conclusion
The obstacles to early diagnosis and treatment could be seen as similar in nature to one another. A lack of awareness of the condition, naturally arising from its rarity, has led to a paucity of treatment research and development until recently; meaning there is only one disease-specific therapy, augmentation therapy, available at present. Even this treatment has had trouble convincing regulators of its efficacy, thus erecting barriers for patients trying to access it. The nature of the disease, presenting with two different pathologies, has made developing a universal treatment difficult. Detecting all cases of AATD and showing its true prevalence in our population would not only give clinical researchers the chance to conduct large RCTs, but also sound the alarm to the research community at large that there is action needed to be taken in helping remove the significant disease burden AATD places on patients. To address this is likely to require public health programs, investment in validated test strategies accessible to all clinicians, as well as widespread education of the physician and patient community, especially in areas where the disease is most likely to present – pulmonology, hepatology, and pediatrics, alongside primary care clinicians who conduct COPD reviews in most countries.
Disclosure
MQ, PE, and AP report no competing interests. AMT reports grants and personal fees from CSL Behring and Vertex and grants from Grifols and Arrowhead Inc, outside the submitted work, has had grants or honoraria from CSL Behring, Vertex, Arrowhead and Grifols within the last 5 years, and grant funding from Alpha 1 Foundation, ATS Foundation and Chest Foundation for work in AATD, and reports no other potential conflicts of interest for this work.
References
1. Strnad P, et al. Alpha1-antitrypsin deficiency. N Engl J Med. 2020;382:1443–1455. doi:10.1056/NEJMra1910234
2. Abboud RT, Tanya N, Jung B, et al. Alpha1-antitrypsin deficiency: a clinical-genetic overview. Appl Clin Genet. 2011;4:55–65. doi:10.2147/TACG.S10604
3. Haq I, Irving JA, Saleh AD, et al. Deficiency mutations of alpha-1 antitrypsin. effects on folding, function, and polymerization. Am J Respir Cell Mol Biol 2016;54:71–80. doi:10.1165/rcmb.2015-0154OC
4. Brantly ML, Wittes JT, Vogelmeier CF, et al. Use of a highly purified α1-antitrypsin standard to establish ranges for the common normal and deficient α1-antitrypsin phenotypes. Chest. 1991;100:703–708. 3 doi:10.1378/chest.100.3.703
5. de Serres FJ, Blanco I Prevalence of α1-antitrypsin deficiency alleles PI*S and PI*Z worldwide and effective screening for each of the five phenotypic classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: a comprehensive review Ther Adv Respir Dis. 2012;6(5):277–295. doi:10.1177/1753465812457113
6. Russo R, et al. Prevalence of alpha-1 antitrypsin deficiency and allele frequency in patients with COPD in Brazil. J Bras Pneumol. 2016;42(5):311–316 doi:10.1590/S1806-37562015000000180
7. Molloy K, et al. Clarification of the risk of chronic obstructive pulmonary disease in alpha1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med. 2014;189:419–427. doi:10.1164/rccm.201311-1984OC
8. Franciosi AN, et al. Clarifying the risk of lung disease in sz alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2020;202:73–82. doi:10.1164/rccm.202002-0262OC
9. Franciosi AN, et al. SZ alpha-1 antitrypsin deficiency and pulmonary disease: more like MZ, not like ZZ. Thorax. 2020. doi:10.1136/thoraxjnl-2020-215250
10. Mahadeva R, et al. Heteropolymerization of S, I, and Z alpha1-antitrypsin and liver cirrhosis. J Clin Invest. 1999;103:999–1006. doi:10.1172/JCI4874
11. Fazleen A, Wilkinson T Early COPD: current evidence for diagnosis and management. Ther Adv Respir Dis. 2020;14:1753466620942128. doi:10.1177/1753466620942128
12. Harris R, Harman DJ, Card TR, et al. Prevalence of clinically significant liver disease within the general population, as defined by non-invasive markers of liver fibrosis: a systematic review. Lancet Gastroenterol Hepatol. 2017;2:288–297. doi:10.1016/S2468-1253(16)30205-9
13. Miravitlles M, et al. European respiratory society statement: diagnosis and treatment of pulmonary disease in alpha1-antitrypsin deficiency. Eur Respir J. 2017;50.
14. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168:818–900.
15. Global initiative for obstructive lung disease. Available from: www.goldcopd.com.
16. Blanco I, Bueno P, Diego I, et al. Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chron Obstruct Pulmon Dis. 2017;12:561–569. 10.2147/COPD.S125389
17. Casas F, et al. Indications for active case searches and intravenous alpha-1 antitrypsin treatment for patients with alpha-1 antitrypsin deficiency chronic pulmonary obstructive disease: an update. Arch Bronconeumol. 2015;51:185–192. 10.1016/j.arbres.2014.05.008
18. Greulich T, et al. European screening for alpha1 -antitrypsin deficiency in subjects with lung disease. Clin Respir J. 2017;11:90–97. 10.1111/crj.12310
19. Horvath I, et al. Diagnosis and management of alpha1-antitrypsin deficiency in Europe: an expert survey. ERJ Open Res. 2019;5.
20. Attaway A, Majumdar U, Sandhaus RA, et al. An analysis of the degree of concordance among international guidelines regarding alpha-1 antitrypsin deficiency. Int J Chron Obstruct Pulmon Dis. 2019;14:2089–2101. 10.2147/COPD.S208591
21. Andermann A, et al. Revisiting Wilson and Jungner in the genomic age: a review of screening criteria over the past 40 years. Bull World Health Organ. 2008;86:317–319. 10.2471/BLT.07.050112
22. Brode SK, Ling SC, Chapman KR. Alpha-1 antitrypsin deficiency: a commonly overlooked cause of lung disease. CMAJ. 2012;184:1365–1371. doi:10.1503/cmaj.111749
23. Gupta N, Gaudreault N, Thériault S, et al. Granularity of, SERPINA1 alleles by DNA sequencing in CanCOLD. Eur Respir J. 2020. 56 2000958 10.1183/13993003.00958-2020
24. Tanash HA, Nystedt-Düzakin M, Montero LC, et al. The Swedish α 1 -antitrypsin screening study: health status and lung and liver function at age 34. Ann Am Thorac Soc. 2015;12:807–812. 10.1513/AnnalsATS.201410-452OC
25. Lambe T, Adab P, Jordan RE, et al. Model-based evaluation of the long-term cost-effectiveness of systematic case-finding for COPD in primary care. Thorax. 2019;74:730–739. 10.1136/thoraxjnl-2018-212148
26. Bradi AC, Audisho N, Casey DK, et al. Alpha-1 antitrypsin deficiency in Canada: regional disparities in diagnosis and management. COPD. 2015;12 Suppl 1:15–21. 10.3109/15412555.2015.1021908
27. Greulich T, Nell C, Herr C, et al. Results from a large targeted screening program for alpha-1-antitrypsin deficiency: 2003–2015. Orphanet J Rare Dis. 2016;11:75. 10.1186/s13023-016-0453-8
28. Garcia-Palenzuela R, et al. [Detection of alpha-1 antitrypsin deficiency: A study on patients diagnosed with chronic obstructive pulmonary disease in primary health care]. Semergen. 2017;43:289–294. Spanish.
29. Greulich T, et al. Real world evaluation of a novel lateral flow assay (AlphaKit (R) QuickScreen) for the detection of alpha-1-antitrypsin deficiency. Respir Res. 2017;Volume 12:151. 10.1186/s12931-018-0826-8
30. Tasch JJ, et al. A novel approach to screening for alpha-1 antitrypsin deficiency: inpatient testing at a teaching institution. Chronic Obstr Pulm Dis. 2018;5:106–110.
31. Torres-Duran M, Lopez-Campos JL, Barrecheguren M, et al. Alpha-1 antitrypsin deficiency: outstanding questions and future directions. Orphanet J Rare Dis. 2018;13:114. 10.1186/s13023-018-0856-9
32. Sanders CL, Ponte A, Kueppers F, et al. The effects of inflammation on alpha 1 antitrypsin levels in a national screening cohort. COPD. 2018;15:10–16. 10.1080/15412555.2017.1401600
33. Scarlata S, et al. Electrophoretic alpha1-globulin for screening of alpha1-antitrypsin deficient variants. Clinical Chemistry Laboratory Med. 2020.
34. De Tommaso AM, et al. Diagnosis of alpha-1-antitrypsin deficiency by DNA analysis of children with liver disease. Arq Gastroenterol. 2001;38:63–68. 10.1590/S0004-28032001000100012
35. Sharp HL History of the first description of childhood liver disease in AATD. COPD. 2013;10 Suppl 1:13–16. 10.3109/15412555.2013.766165
36. de Miguel-diez J, Jiménez-García R, López de Andrés A, et al. Effectiveness of an intervention to improve management of COPD using the AUDIT methodology: results of the neumo-advance study. Clin Drug Investig. 2019;39:653–664. 10.1007/s40261-019-00787-4
37. Nolte JL, Ataya A, Merrill H, et al. Alpha1-antitrypsin deficiency—increased knowledge and diagnostic testing after viewing short instructional video. COPD. 2017;14:52–55. 10.1080/15412555.2016.1245280
38. Ruparel M, Quaife SL, Dickson JL, et al. Prevalence, symptom burden, and underdiagnosis of chronic obstructive pulmonary disease in a lung cancer screening cohort. Ann Am Thorac Soc. 2020;17:869–878. 10.1513/AnnalsATS.201911-857OC
39. Black WC, et al. Cost-effectiveness of CT screening in the national lung screening trial. N Engl J Med. 2014;371:1793–1802. 10.1056/NEJMoa1312547
40. Soriano JB, et al. Trends of testing for and diagnosis of alpha1-antitrypsin deficiency in the UK: more testing is needed. Eur Respir J. 2018;52.
41. Jordan RE, et al. Targeted case finding for chronic obstructive pulmonary disease versus routine practice in primary care (TargetCOPD): a cluster-randomised controlled trial. Lancet Respir Med. 2016;4:720–730. 10.1016/S2213-2600(16)30149-7
42. Lin HC, et al. Alpha1-antitrypsin deficiency: transition of care for the child with AAT deficiency into adulthood. Curr Pediatr Rev. 2019;15:53–61. 10.2174/1573396314666181113094517
43. Holme J, et al. Studies of gamma-glutamyl transferase in alpha-1 antitrypsin deficiency. COPD. 2010;7:126–132. 10.3109/15412551003631733
44. Reiter R, et al. Comparison of non-invasive assessment of liver fibrosis in patients with alpha1-antitrypsin deficiency using magnetic resonance elastography (MRE), acoustic radiation force impulse (ARFI) Quantification, and 2D-shear wave elastography (2D-SWE). PLoS One. 2018;13:e0196486. 10.1371/journal.pone.0196486
45. Clark VC, et al. Clinical and histologic features of adults with alpha-1 antitrypsin deficiency in a non-cirrhotic cohort. J Hepatol. 2018. 69 1357–1364 10.1016/j.jhep.2018.08.005
46. Kumpers J, et al. Assessment of liver phenotype in adults with severe alpha-1 antitrypsin deficiency (Pi*ZZ genotype). J Hepatol. 2019;71:1272–1274. 10.1016/j.jhep.2019.08.011
47. Hamesch K, et al. Liver fibrosis and metabolic alterations in adults with alpha-1-antitrypsin deficiency caused by the Pi*ZZ Mutation. Gastroenterology. 2019;157:705–19 e18. 10.1053/j.gastro.2019.05.013
48. Viglio S, et al. MEKC of desmosine and isodesmosine in urine of chronic destructive lung disease patients. Eur Respir J. 2000;15:1039–1045. 10.1034/j.1399-3003.2000.01511.x
49. Fregonese L, et al. Long-term variability of desmosine/isodesmosine as biomarker in alpha-1-antritrypsin deficiency–related COPD. COPD. 2011;8:329–333 10.3109/15412555.2011.589871
50. Ma S, et al. The effect of alpha-1 proteinase inhibitor on biomarkers of elastin degradation in alpha-1 antitrypsin deficiency: an analysis of the RAPID/RAPID Extension trials. Chronic Obstr Pulm Dis. 2017;4(1):34–44.
51. Mahadeva R, et al. Polymers of Z alpha1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am J Pathol. 2005;166:377–386. 10.1016/S0002-9440(10)62261-4
52. Campos MA, Diaz AA The role of computed tomography for the evaluation of lung disease in alpha-1 antitrypsin deficiency. Chest. 2018;153:1240–1248. 5 10.1016/j.chest.2017.11.017
53. Celli BR, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:1005–1012. 10.1056/NEJMoa021322
54. Thabut G, et al. Performance of the BODE index in patients with alpha1-antitrypsin deficiency-related COPD. Eur Respir J. 2014;44:78–86. 10.1183/09031936.00168113
55. Edgar RG, et al. Treatment of lung disease in alpha-1 antitrypsin deficiency: a systematic review. Int J Chron Obstruct Pulmon Dis. 2017;12:1295–1308. 10.2147/COPD.S130440
56. Low EV, et al. ICS use may modify FEV1 decline in α1-antitrypsin deficiency patients with relatively high blood eosinophils. Respiration 2018; 95: 114–121 10.1159/000481867
57. Chapman KR, et al. Intravenous augmentation treatment and lung density in severe alpha1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386:360–368. 10.1016/S0140-6736(15)60860-1
58. Green CE, et al. Lung density associates with survival in alpha 1 antitrypsin deficient patients. Respir Med. 2016;112:81–87. 10.1016/j.rmed.2016.01.007
59. Chapman KR, et al. Augmentation therapy for alpha1 antitrypsin deficiency: a meta-analysis. COPD. 2009;6:177–184. 10.1080/15412550902905961
60. Highly specialised technology evaluation human alpha1-proteinase inhibitor for treating emphysema [ID856]: evaluation Report. NICE, 2019.
61. Ficker JH, et al. Alpha-1 antitrypsin (A1-PI) treatment slows emphysema progression independent of baseline FEV1. Eur Respir J. 2017;50:OA3416.
62. McElvaney NG, et al. Long-term efficacy and safety of alpha1 proteinase inhibitor treatment for emphysema caused by severe alpha1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE). Lancet Respir Med. 2017;5:51–60. 10.1016/S2213-2600(16)30430-1
63. Dirksen A, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J. 2009;33:1345–1353. 10.1183/09031936.00159408
64. Dirksen A, et al. A randomized clinical trial of alpha (1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med. 1999;160:1468–1472. 10.1164/ajrccm.160.5.9901055
65. Stockley RA, et al. Individualized lung function trends in alpha-1-antitrypsin deficiency: a need for patience in order to provide patient centered management? Int J Chron Obstruct Pulmon Dis. 2016;11:1745–1756. 10.2147/COPD.S111508
66. Pye A, Turner AM Experimental and investigational drugs for the treatment of alpha-1 antitrypsin deficiency. Expert Opin Investig Drugs. 2019;28:891–902. 10.1080/13543784.2019.1672656
67. Sorrells S, et al. SPARTA clinical trial design: exploring the efficacy and safety of two dose regimens of alpha1-proteinase inhibitor augmentation therapy in alpha1-antitrypsin deficiency. Respir Med. 2015;109:490–499. 10.1016/j.rmed.2015.01.022
68. Townsend S, et al. Presentation and prognosis of liver disease in alpha-1 antitrypsin deficiency. Expert Rev Gastroenterol Hepatol. 2018;12:745–747. 10.1080/17474124.2018.1477589
69. Sasaki K, et al. Should we be utilizing more liver grafts from pediatric donation after circulatory death donors? A national analysis of the SRTR from 2002–2017. Transplantation. 2020. Publish Ahead of Print 10.1097/TP.0000000000003458
70. Burrows JA, et al. Chemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiency. Proc Natl Acad Sci U S A. 2000;97:1796–1801. 10.1073/pnas.97.4.1796
71. Teckman JH Lack of effect of oral 4-phenylbutyrate on serum alpha-1-antitrypsin in patients with alpha-1-antitrypsin deficiency: a preliminary study. J Pediatr Gastroenterol Nutr. 2004;39:34–37.
72. Gonzalez-Peralta RP, et al. 4-Phenyl butyrate mediated secretion rescue in patients with alpha 1-antitrypsin (aat) deficiency: A pilot study. Hepatology. 2006;44:211A.
73. Chang Y-P, Mahadeva R, Chang W-SW, et al. Identification of a 4-mer peptide inhibitor that effectively blocks the polymerization of pathogenic Z α 1-Antitrypsin. Am J Respir Cell Mol Biol. 2006;35:540–548. 5 10.1165/rcmb.2005-0207OC
74. Ordonez A, et al. A single-chain variable fragment intrabody prevents intracellular polymerization of Z alpha1-antitrypsin while allowing its antiproteinase activity. FASEB J. 2015;29:2667–2678. 10.1096/fj.14-267351
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.