")
Back to Journals » Hepatic Medicine: Evidence and Research » Volume 8
Obeticholic acid for the treatment of primary biliary cholangitis in adult patients: clinical utility and patient selection
Authors Bowlus C
Received 14 June 2016
Accepted for publication 22 July 2016
Published 1 September 2016 Volume 2016:8 Pages 89—95
DOI https://doi.org/10.2147/HMER.S91709
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Gerry Lake-Bakaar
Christopher L Bowlus
Division of Gastroenterology and Hepatology, University of California Davis, Davis, CA, USA
Abstract: Primary biliary cholangitis (PBC), previously known as primary biliary “cirrhosis”, is a rare autoimmune liver disease characterized by the hallmark autoantibodies to mitochondrial antigens and immune-mediated destruction of small bile duct epithelial cells leading to cholestasis and cirrhosis. Surprisingly, while immune modulators have not been effective in the treatment of PBC, supplementation with the hydrophilic bile acid (BA) ursodeoxycholic acid (UDCA) has been demonstrated to slow the disease progression. However, a significant minority of PBC patients do not have a complete response to UDCA and remain at risk of continued disease progression. Although the mechanisms of action are not well understood, UDCA provided proof of concept for BA therapy in PBC. Obeticholic acid (OCA), a novel derivative of the human BA chenodeoxycholic acid, is a potent agonist of the nuclear hormone receptor farnesoid X receptor, which regulates BA synthesis and transport. A series of clinical trials of OCA in PBC, primarily in combination with UDCA, have established that OCA leads to significant reductions in serum alkaline phosphatase that are predicted to lead to improved clinical outcomes, while dose-dependent pruritus has been the most common adverse effect. On the basis of these studies, OCA was given conditional approval by the US Food and Drug Administration with plans to establish the long-term clinical efficacy of OCA in patients with advanced PBC.
Keywords: primary biliary cholangitis, nuclear receptors, farnesoid X receptor, bile acid, obeticholic acid, ursodeoxycholic acid
Primary biliary cholangitis
Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis, is a chronic cholestatic liver disease with an autoimmune basis, affecting mostly middle-aged women.1 The inflammatory response leads to slow destruction of small intrahepatic bile ducts and progression at varying rates to cirrhosis leading to liver transplantation or death.2 PBC is typically suspected in asymptomatic patients when cholestatic liver biochemistries are encountered, as part of the evaluation of symptoms related to cholestasis such as pruritus, or during the evaluation of cirrhosis. The diagnosis of PBC requires fulfillment of at least two of three criteria including an elevated serum alkaline phosphatase (ALP), the presence of antimitochondrial antibodies (AMA), and liver histology compatible with PBC. Because AMA is detected in 90%–95% of patients with PBC and in less than 1% of normal controls, liver biopsy is rarely required for the diagnosis of PBC.3 In addition to diagnosis, liver biopsy was historically used for staging of fibrosis but has been replaced by noninvasive methods such as transient elastography.
The immunopathology of PBC is well understood.4 The major targets of the AMA have been identified and include the E2 subunits of the pyruvate dehydrogenase complex and related proteins.5,6 In fact, the target is restricted to lipoic acid modification of a specific lysine residue. Moreover, T-cell responses from both CD4+ and CD8+ cells are enhanced in both peripheral blood and liver of patients with PBC and are directed to the same epitopes. Genome-wide association studies have implicated a number of immune-related genes, including IL-12, in susceptibility to PBC.7 Both epidemiologic and experimental studies support the idea that environmental exposures also play an important role in breaking tolerance to the self-antigens.6 Nevertheless, attempts at altering the natural history of PBC through immune modulation have to date been unsuccessful.8
Until recently, ursodeoxycholic acid (UDCA) was the only drug approved by the US Food and Drug Administration (FDA) for the treatment of PBC.9 UDCA was first shown to have favorable effects in patients with PBC in the 1980s,2 and at a dose of 13–15 mg/kg/d, improves liver biochemistries, slows histological progression, and improves liver transplantation-free and overall survival, at least in those treated in the early stages of the disease.2,9,10 Because PBC is a rare and slowly progressive disease, individual clinical trials have lacked the power to demonstrate clinically meaningful differences in outcomes in most cases. This limitation has led to comparisons of the rates of actual clinical outcomes in patients treated with UDCA to predicted rates of outcomes based upon natural history models applied to the same population assuming that they had not been treated with UDCA. The updated Mayo natural history model for PBC is a validated prognostic model of liver transplant-free survival, and when applied to a cohort of PBC patients who had received 13–15 mg/kg UDCA daily for a mean of 8 years (range 1–22 years), a Markov model predicted a significantly better liver transplant-free survival with UDCA compared to the spontaneous survival rate predicted by the Mayo model.11 While the overall survival rates without liver transplantation were 84% and 66% at 10 and 20 years, respectively, in early-stage patients, only 6% and 22% were predicted to progress to liver transplantation or death after 10 and 20 years, respectively. These survival rates were similar to that of an age- and sex-matched control population. In contrast, the probability of death or liver transplantation was significantly increased in patients treated in late stages of the disease.10
Other indicators of poor outcome despite UDCA treatment include an incomplete biochemical response to UDCA.12–14 Several studies have proposed various criteria as predictors of treatment success with UDCA.12,13,15–19 Studies of large patient cohorts from France and the United Kingdom demonstrated that reduction of the ALP and aspartate transaminase (AST) to <1.5 × the upper limit of normal (ULN) and a normal total bilirubin after 1 year of UDCA therapy (Paris II criteria) was the best criterion for distinguishing high- and low-risk groups of patients.16,20 More recent studies of an international cohort of over 4,500 PBC patients further characterized the relationship between levels of serum ALP and total bilirubin at 1-year of follow-up and the risk of liver transplantation or death21 leading to the PBC GLOBE score, which incorporates age at initiation of UDCA therapy, levels of serum ALP, total bilirubin, albumin, and platelets (http://www.globalpbc.com/globe).22 These studies have been critical to our understanding of the natural history of PBC in the era of UDCA and for the appropriate selection of patients for additional therapies, namely, obeticholic acid (OCA).
Obeticholic acid
In addition to their roles in the solubilization of lipids for digestion, bile acids (BAs) have more recently been found to act as signaling molecules, notably through the farnesoid X receptor (FXR), a nuclear receptor expressed in the liver, intestine, adrenal glands, and kidneys that has a central role in the synthesis and enterohepatic circulation of BAs.23 OCA is a derivative of chenodeoxycholic acid, the primary human BA, and is the natural ligand for FXR. OCA is approximately 100 times more potent as an FXR ligand than chenodeoxycholic acid24 and demonstrates high selectivity with minimal activity to the another described BA receptor, G protein-coupled bile acid receptor 1 (GPBAR1 or TGR5). Activation of FXR in the liver reduces conversion of cholesterol to BAs by downregulating the primary enzymes involved in BA synthesis, including cytochrome P450 7A1 (CYP7A1) and CYP8B1. BA toxicity in the liver is also reduced by increasing other BA-modifying enzymes and promoting the transport of BAs. By activating FXR in the ileum, OCA decreases BA reabsorption through downregulation of the apical sodium-dependent BA transporter and increased expression of fibroblast growth factor 19, which in the liver also decreases BA synthesis through CYP7A1.25 Preclinical studies have also suggested that OCA may have antifibrotic properties26,27 and the potential to improve portal hypertension.28
Two Phase II studies and a pivotal Phase III study have been completed that have investigated the safety and efficacy of OCA in patients with PBC (Table 1). An international, Phase II, double-blind, placebo-controlled, dose–response study evaluated the effects of OCA on ALP, other liver enzymes, and safety in patients with PBC with persistently elevated ALP levels >1.5–10× the ULN while on a stable dose of UDCA for at least 6 months.29 A total of 165 patients were randomized to placebo, OCA 10, 25, or 50 mg once daily for 12 weeks while continuing UDCA at the preenrollment dose. Compared with the placebo group, the groups taking 10, 25, or 50 mg OCA had a greater mean percent decrease in their ALP (–23.7%±17.8%, –24.7%±17.9%, –21.0%±27.6%, respectively, vs 2.6%±12.5%, P<0.0001). Significant decreases in alanine transaminase (ALT) and gamma-glutamyl transpeptidase were also seen in all treatment groups compared with placebo.
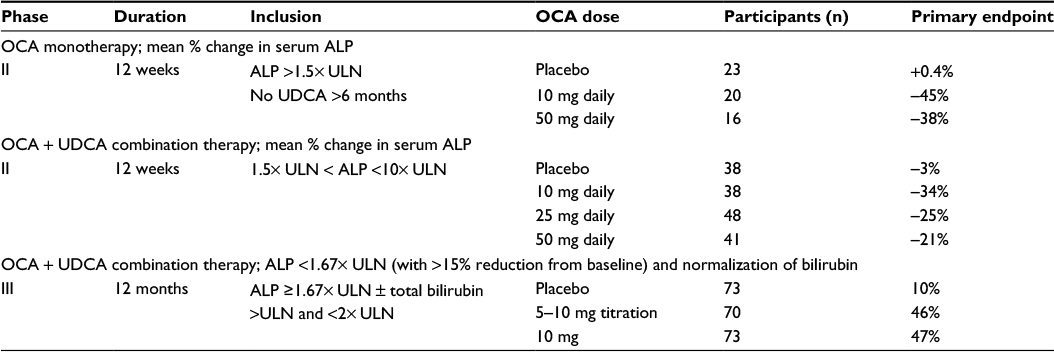 | Table 1 Randomized controlled trials of OCA for the treatment of PBS Abbreviations: PBS, primary biliary cholangitis; OCA, obeticholic acid; ALP, alkaline phosphatase; ULN, upper limit of normal; UDCA, ursodeoxycholic acid. |
In a long-term safety extension (LTSE) of this study, 78 PBC subjects were started on 10 mg daily and titrated every 8 weeks to a maximum dose of 50 mg.30 Safety and efficacy assessments occurred at baseline, pre- and posttitration, and every 3 months. At 10 mg daily OCA, mean ALP levels decreased by 71 U/L (19%), and a further decrease of 23 U/L (9%) occurred after titration from 10 to 25 mg. Mean ALP values increased by 8 U/L (2%) with titration from 25 to 50 mg. Pruritus worsened, or was reported as a new adverse effect (AE), following titration to 25 mg in 46% of patients and in 17% of patients after titration to 50 mg, suggesting that a dose at or below 25 mg might provide the best balance between efficacy and tolerability.
The efficacy of OCA as a monotherapy was also evaluated in a Phase II, international, double-blind, placebo-controlled study of OCA in patients with PBC and persistently elevated ALP levels (≥1.5–10× ULN) who had not been taking UDCA for at least 6 months.31 Fifty-nine patients received placebo, OCA 10 mg or 50 mg once daily for 12 weeks. The OCA 10 mg group showed the greatest ALP decrease, from 3.9× ULN pretreatment to 1.9× ULN, at the end of the study.
Interim results of the only Phase III study of OCA, PBC OCA International Study of Efficacy (POISE; NCT01473524), were the basis for the recent FDA’s conditional approval of OCA for the treatment of PBC with an incomplete response to UDCA (Figure 1).32 The POISE trial is an international, multicenter, placebo-controlled, randomized clinical trial designed to study the safety and efficacy of once-daily OCA in PBC patients with an incomplete response to or unable to tolerate UDCA. The primary end point of the 12-month double-blind portion of the trial was a composite of an ALP level <1.67× ULN with a ≥15% reduction from baseline and a normal bilirubin level compared to placebo. Two hundred and seventeen patients were randomized to daily doses of placebo, 10 mg OCA, or 5 mg OCA titrated to 10 mg OCA based on clinical response at 6 months. The median UDCA dose was 15.3 mg/kg, and 7% of patients were UDCA intolerant. All three treatment groups were well matched. The primary end point was met in an intention to treat analysis with response rates of 10% in the placebo group compared to 47% in the 10 mg OCA group and 46% in the 5–10 mg OCA group (both OCA groups P<0.0001 vs placebo). The placebo group experienced a mean decrease in ALP from baseline of 5%, compared to a mean decrease of 39% in the 10 mg OCA dose group and 33% in the 5–10 mg OCA titration group (both OCA groups P<0.0001 vs placebo). In addition to the reduction in ALP, patients treated with either dose of OCA had a greater decrease in total bilirubin compared to placebo (–0.02 and –0.05, respectively, vs 0.12; P<0.001), suggesting that there was improvement in liver function. Furthermore, both OCA groups met prespecified secondary end points of clinical biomarkers, including gamma-glutamyl transpeptidase, ALT, and AST as well as markers of inflammation. More than 95% of subjects completing the double-blind portion of the study were enrolled into an optional open-label, long term safety extension, which is ongoing
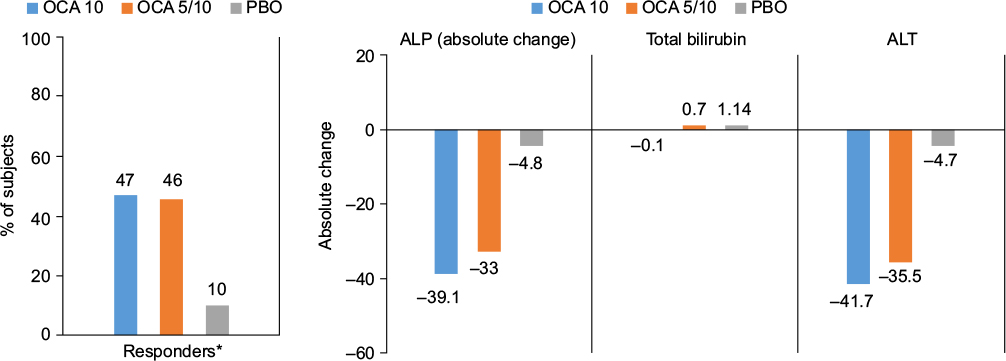 | Figure 1 Primary and secondary end points of POISE trial comparing obeticholic acid 10 mg daily, 5 mg daily titrating to 10 mg daily, and placebo. Abbreviations: POISE, PBC OCA International Study of Efficacy; OCA, obeticholic acid; ALP, alkaline phosphatase; ALT, alanine aminotransferase; PBO, placebo. |
The most common AE of OCA in PBC is dose-related pruritus. Although pruritus is prevalent in PBC, there was a clear increase in frequency and severity of pruritus in patients treated with OCA compared to placebo. For example, in the Phase II study by Mason et al,33 pruritus was reported by 50% of subjects taking placebo compared to 47%, 85%, and 80% of subjects taking OCA 10, 25, and 50 mg, respectively. Both severity and discontinuation rates were dose dependent. No subjects in the placebo group discontinued compared to discontinuation rates due to pruritus of 8%, 8%, and 24% in the 10, 25, and 50 mg OCA groups, respectively. In the long-term extension of this study30 in which 78 participants were started on 10 mg OCA and OCA dose was increased every 8 weeks, pruritus occurred in almost all patients (n=71, 91%) ranging from mild in 35% to moderate in 42% and severe in 23%. All other AEs occurred in <20% of patients. In the Phase II study of OCA as monotherapy,31 pruritus was again the most common AE with similar rates. However, in the LTSE phase of this trial, with mean exposure time to OCA of 3.2 years, pruritus improved in severity with continued treatment despite the number of days of exposure being similar each year.34
In the POISE trial where the dose of OCA was further reduced, pruritus was again the most frequently reported AE associated with OCA treatment, but was mostly mild-moderate: (placebo: 38%, OCA 10 mg: 68%, OCA 5–10 mg titration: 56%) with only 1% discontinuation due to pruritus in the OCA 5–10 mg arm vs 10% in OCA 10 mg arm. As noted, the response rate at 12 months was not significantly different in the OCA 5–10 mg arm compared to the 10 mg OCA arm. Thus, the recommended starting dose is 5 mg oral daily, with the consideration of increasing the dose to 10 mg daily after 3 months depending upon tolerability and biochemical response.
In addition to pruritus, liver-related adverse events and reductions in high-density lipoprotein -cholesterol (HDL-C) have been observed. In a pooled analysis of the three placebo-controlled trials in PBC, the exposure-adjusted incidence rates for all serious and otherwise clinically significant liver-related adverse reactions, and isolated elevations in liver biochemical tests, per 100 patient exposure years were 5.2 in the OCA 10 mg group, 19.8 in the OCA 25 mg group, and 54.5 in the OCA 50 mg group compared to 2.4 in the placebo group. In the POISE trial, dose-dependent 20% and 9% reductions from baseline mean HDL-C levels were observed at 2 weeks in patients in the 10 and 5–10 mg titration arms, respectively, compared to 2% in the placebo arm. No significant further reductions were seen at month 12, with 19% and 12% reductions from baseline mean HDL-C level in the 10 and 5–10 mg titration arms, respectively, compared to 2% in the placebo arm.
Clinical utility
OCA has clearly demonstrated the ability to induce changes in liver biochemistries that are associated with a lower risk of liver transplantation or death, but has not yet demonstrated a direct impact on clinical outcomes or quality of life. In addition, the changes in ALP and bilirubin have not been validated as surrogates to clinical end points in any PBC study. Nevertheless, there is a preponderance of evidence establishing their association with clinical outcomes and biologic plausibility, especially with bilirubin, to anticipate that reductions in ALP and total bilirubin will translate into improved clinical outcomes. Results of the ongoing COBALT trial35 will establish whether OCA impacts clinical outcomes and to what degree.
In the clinic, most PBC patients will still be adequately treated with UDCA monotherapy and will not require the addition of OCA. However, the 20%–40% of PBC patients who do not have an adequate biochemical response to UDCA will be candidates for OCA. In addition, OCA is a new alternative for the small number of PBC patients who cannot tolerate UDCA.
Patient selection
According to the FDA, OCA is indicated for the treatment of PBC in combination with UDCA in adults with an “inadequate” response to UDCA, or as monotherapy in adults unable to tolerate UDCA. Although the POISE study had strict enrollment criteria and definition of an inadequate response, the FDA approval allows for clinical judgment by the physician in determining whether an individual patient has responded adequately to UDCA. In most cases, an inadequate response to UDCA may be defined by one that leaves a patient at greater risk of liver transplantation or death compared to an age- and sex-matched control population. Several criteria, including those already discussed, have been developed and validated for this purpose (Table 2). The Global PBC Score has the advantage of being based upon the largest cohort and provides a direct comparison to the survival rates of an age- and sex-matched population. In contrast, the Paris I criteria provides a simple 1–2–3 score based upon total bilirubin <1× ULN; AST <2 × ULN; and ALP <3 × ULN. However, in clinical practice, several factors in addition to biochemical response to UDCA should be kept in mind when considering OCA to PBC patients. These include factors that might lower the threshold for treatment, such as advanced fibrosis or younger age, and factors that might increase the threshold for treatment, including severe pruritus and cardiovascular risk factors.
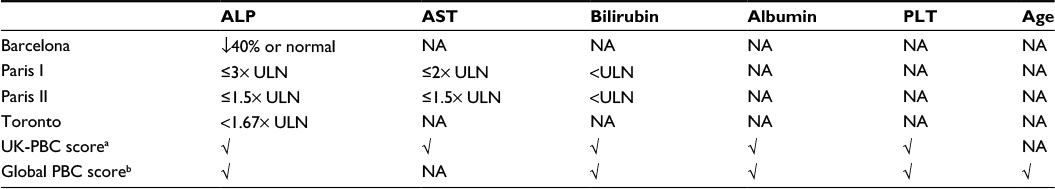 | Table 2 Biochemical response criteria for PBS treated with ursodeoxycholic acid Notes: aUK-PBC risk scores = 1 − baseline survival functionexp(0.0287854 × (ALP12 × ULN – 1.722136304) – 0.0422873 × (((TA12 × ULN/10)–1) – 8.675729006) + 1.4199 × (ln(BIL12 × ULN/10) + 2.709607778) − 1.960303 × (ALB12 × LLN – 1.17673001) – 0.4161954 × (PLT12 × LLN – 1.873564875)), where baseline survivor function = 0.982 (at 5 years); 0.941 (at 10 years); 0.893 (at 15 years). Calculator available at http://www.uk-pbc.com/resources/tools/riskcalculator/. bGLOBE score = 0.044378 × age at start of UDCA therapy + 0.93982 × ln(BIL12 × ULN) + 0.335648 × ln(ALP12 × ULN) − 2.266708 × ALB12 × LLN − 0.002581 × PLT12 per 109/L + 1.216865. Calculator available at http://www.globalpbc.com/globe. Abbreviations: PBS, primary biliary cholangitis; ALP, alkaline phosphatase; AST, aspartate aminotransferase; PLT, platelet count; NA, not applicable; ALP12, ALP after 12 months treatment; ALT, alanine aminotransferase; TA12, AST or ALT after 12 months treatment; BIL12, total bilirubin after 12 months treatment; ALB12, albumin after 12 months treatment; PLT12, platelet count after 12 months treatment; ULN, upper limit of normal; LLN lower limit of normal; UDCA, ursodeoxycholic acid. |
In the POISE study, patients were required to have an ALP >1.67 × ULN (approximately 200 U) or a bilirubin >ULN and <2 × ULN either despite UDCA or intolerance to UDCA. However, the Global PBC Study Group analysis demonstrated that the risk of liver transplantation or death begins increasing even at an ALP > ULN. In addition, the impact on risk is even greater with total bilirubin elevations and starts at levels that are >0.5 × ULN, ie, even within the normal range. Thus, in a patient with an ALP > ULN but <1.67 × ULN, treatment with OCA might be considered particularly in a younger patient or a patient with advanced fibrosis or cirrhosis. In addition, treatment should be considered in any patient with a bilirubin above ULN.
Patients with the most advanced disease, ie, those with a total bilirubin >2 × ULN, portal hypertension, or a Model for End-stage Liver Disease score ≥15 were excluded from the POISE trial but may be those most likely to benefit from therapy. However, data on the use of OCA in this population do not exist. Although the COBALT study35 will include PBC patients with more advanced disease, patients with more advanced disease, that is, Child–Pugh Class B and C cirrhosis, will not likely qualify. In the absence of data, the current recommendation is to start OCA at 5 mg once “weekly” and increase the dosage as tolerated to a maximum dose of 10 mg twice weekly at least 3 days apart (OCALIVA36 package insert).
The most challenging patients to manage with OCA are likely to be those with baseline moderate-to-severe pruritus. Importantly, PBC patients with severe pruritus requiring current or prior systemic treatments, such as BA sequestrants or rifampicin, were excluded from the POISE trial32 but may represent those at greatest needed. Aggressive and appropriate management of this symptom will be critical to ensuring that OCA reaches its full potential in the treatment of PBC patients. The mechanisms of pruritus in cholestasis and the pruritus induced by OCA are not well understood. Despite the common use of sequestrants to reduce serum BA levels, there is little empiric evidence that these treatments are effective, and in fact, there is no correlation between serum BA levels and the severity of pruritus. Similarly, it is perplexing that while OCA appears to improve cholestasis, it worsens pruritus. According to the OCA package insert, the management of intolerable pruritus with OCA should include antihistamines or BA-binding resins, dose reductions of OCA, or interruptions in OCA for up to 2 weeks. However, the use of antihistamines in cholestatic pruritus is not supported by any evidence and, typically, is only effective in sedating patients. BA-binding resins are poorly tolerated and must be separated from OCA and UDCA by at least 4 hours, often leading to reduced adherence. Dose reductions including alternate day dosing and “drug holidays” are likely to be the most effective and practical approach to dealing with pruritus. The use of rifampicin, sertraline, or naltrexone for the management of pruritus induced or worsened by OCA was not allowed in the POISE trial,32 and the safety and efficacy of these medications in this setting has not been established.
The impact of the effects of OCA on HDL-C and cardiovascular risk is not known, and neither is it known how to optimally manage these changes. Hypercholesterolemia in PBC is common and typically is due to an increase in lipoprotein X, which is identified as HDL-C by standard clinical labs, but there does not appear to be an increased risk of cardiovascular disease in PBC patients. A 7-year prospective study of 312 PBC patients found no statistically significant difference in atherosclerotic death rates between PBC patients and an age- and sex-matched control group.37 Similarly, a study of 400 PBC patients followed up over 6.2 years found the incidence of cardiovascular events in PBC patients was similar to that of the general population.38 These findings are despite the fact that PBC is associated with a higher frequency of smoking compared to healthy controls. Small clinical trials in PBC patients have demonstrated that HMG-CoA reductase inhibitors (statins) are effective in lowering low-density lipoprotein-cholesterol without any significant impact on HDL-C, and these in general have been well tolerated. In PBC patients with advanced disease and without cardiovascular risk factors, the reductions in HDL-C are unlikely to be clinically significant. In PBC patients with higher cardiovascular risk factors or a history of cardiovascular disease, the risk–benefit analysis becomes much more difficult, especially in those with early-stage disease.
Conclusion
In April 2016, the US FDA granted accelerated approval of OCA for the treatment of PBC in combination with UDCA in adults who fail to respond adequately to UDCA, or as monotherapy in adults who cannot tolerate UDCA. In these patients, OCA has demonstrated that it improves ALP and other liver biochemistries, which are associated with improvements in long-term clinical outcomes. The long-term efficacy of OCA still needs to be delineated, and its clinical benefit on overall or transplant-free survival may be difficult to establish, given the slow progressive nature of the disease, rarity of the condition, and ethical concerns of carrying out a placebo-controlled trial now that OCA is approved for use in these patients. Tolerability in PBC patients who have baseline severe pruritus might limit its use in these patients unless appropriate management steps are taken, most likely with dose adjustments. Nevertheless, OCA offers the first new treatment for PBC in decades and the potential to slow the progression in the group of patients at greatest need. Patient selection will require individual assessment of the risk of disease progression to liver transplant and death, which can be predicted accurately with the GLOBE score, response to UDCA, cardiovascular risk factors, and baseline pruritus.
Disclosure
The author has received grant support from and is a member of the advisory board and speaker bureau for Intercept Pharmaceuticals. The author reports no other conflicts of interests in this work.
References
Selmi C, Bowlus CL, Gershwin ME, Coppel RL. Primary biliary cirrhosis. Lancet. 2011;377(9777):1600–1609. | ||
Momah N, Lindor KD. Primary biliary cirrhosis in adults. Expert Rev Gastroenterol Hepatol. 2014;8(4):427–433. | ||
Bowlus CL, Gershwin ME. The diagnosis of primary biliary cirrhosis. Autoimmun Rev. 2014;13(4–5):441–444. | ||
Kawata K, Kobayashi Y, Gershwin ME, Bowlus CL. The immunophysiology and apoptosis of biliary epithelial cells: primary biliary cirrhosis and primary sclerosing cholangitis. Clin Rev Allergy Immunol. 2012;43(3):230–241. | ||
Wang J, Budamagunta MS, Voss JC, et al. Antimitochondrial antibody recognition and structural integrity of the inner lipoyl domain of the E2 subunit of pyruvate dehydrogenase complex. J Immunol. 2013;191(5):2126–2133. | ||
Chen RC, Naiyanetr P, Shu SA, et al. Antimitochondrial antibody heterogeneity and the xenobiotic etiology of primary biliary cirrhosis. Hepatology. 2013;57(4):1498–1508. | ||
Hirschfield GM, Liu X, Xu C, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med. 2009;360(24):2544–2555. | ||
Tsuda M, Moritoki Y, Lian ZX, et al. Biochemical and immunologic effects of rituximab in patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Hepatology. 2012;55(2):512–521. | ||
Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ. Primary biliary cirrhosis. Hepatology. 2009;50(1):291–308. | ||
Corpechot C, Carrat F, Bahr A, Chrétien Y, Poupon R-E, Poupon R. The effect of ursodeoxycholic acid therapy on the natural course of primary biliary cirrhosis. Gastroenterology. 2005;128(2):297–303. | ||
Kim WR, Lindor KD, Locke GR, et al. Epidemiology and natural history of primary biliary cirrhosis in a U.S. community. Gastroenterology. 2000;119(6):1631–1636. | ||
Kuiper EM, Hansen BE, de Vries RA, et al. Improved prognosis of patients with primary biliary cirrhosis that have a biochemical response to ursodeoxycholic acid. Gastroenterology. 2009;136(4):1281–1287. | ||
Pares A, Caballeria L, Rodes J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology. 2006;130(3):715–720. | ||
Zein CO, Angulo P, Lindor KD. When is liver biopsy needed in the diagnosis of primary biliary cirrhosis? Clin Gastroenterol Hepatol. 2003;1(2):89–95. | ||
Corpechot C, Abenavoli L, Rabahi N, et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology. 2008;48(3):871–877. | ||
Corpechot C, Chazouilleres O, Poupon R. Early primary biliary cirrhosis: biochemical response to treatment and prediction of long-term outcome. J Hepatol. 2011;55(6):1361–1367. | ||
Kumagi T, Guindi M, Fischer SE, et al. Baseline ductopenia and treatment response predict long-term histological progression in primary biliary cirrhosis. Am J Gastroenterol. 2010;105(10):2186–2194. | ||
Azemoto N, Kumagi T, Abe M, et al. Biochemical response to ursodeoxycholic acid predicts long-term outcome in Japanese patients with primary biliary cirrhosis. Hepatol Res. 2011;41(4):310–317. | ||
Zhang LN, Shi TY, Shi XH, et al. Early biochemical response to ursodeoxycholic acid and long-term prognosis of primary biliary cirrhosis: results of a 14-year cohort study. Hepatology. 2013;58(1):264–272. | ||
Carbone M, Mells GF, Pells G, et al. Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid. Gastroenterology. 2013;144(3):560–569. | ||
Lammers WJ, van Buuren HR, Hirschfield GM, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study. Gastroenterology. 2014;147(6):1338–1349 e1335; quiz e1315. | ||
Lammers WJ, Hirschfield GM, Corpechot C, et al. Development and validation of a scoring system to predict outcomes of patients with primary biliary cirrhosis receiving ursodeoxycholic acid therapy. Gastroenterology. 2015;149(7):1804–1812 e1804. | ||
Lindor KD. Farnesoid X receptor agonists for primary biliary cirrhosis. Curr Opin Gastroenterol. 2011;27(3):285–288. | ||
Pellicciari R, Fiorucci S, Camaioni E, et al. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45(17):3569–3572. | ||
Flores A, Mayo MJ. Primary biliary cirrhosis in 2014. Curr Opin Gastroenterol. 2014;30(3):245–252. | ||
Fiorucci S, Antonelli E, Rizzo G, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004;127(5):1497–1512. | ||
Fiorucci S, Rizzo G, Antonelli E, et al. Cross-talk between farnesoid-X-receptor (FXR) and peroxisome proliferator-activated receptor gamma contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J Pharmacol Exp Ther. 2005;315(1):58–68. | ||
Verbeke L, Farre R, Trebicka J, et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology. 2014;59(6):2286–2298. | ||
Hirschfield GM, Mason A, Luketic V, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. 2015;148(4):751–761 e758. | ||
Hirschfield GM, Mason AL, Gordon SC, et al. A long term safety extension trial of the farnesoid X receptor (FXR) agonist obeticholic acid (OCA) and UDCA in primary biliary cirrhosis (PBC). Hepatology. 2011;54:429A. | ||
Kowdley KV, Jones D, Luketic V. The OCA PBC Study Group. An international study evaluating the farnesoid X receptor agonist obeticholic acid as monotherapy in PBC. J Hepatol. 2012;54:S13. | ||
Nevens F, Andreone P, Mazzella G, et al. The first primary biliary cirrhosis (PBC) Phase III trial in two decades-an international study of the FXR agonist obeticholic acid in PBC patients. J Hepatol. 2014;60:S525. | ||
Mason A, Luketic V, Lindor K, et al. Farnesoid-X receptor agonists: a new class of drugs for the treatment of PBC? An international study evaluating the addition of obeticholic acid (INT-747) to ursodeoxycholic acid. Hepatology. 2010;52:357A. | ||
Adorin L. Long-term treatment of primary biliary cirrhosis with the FXR agonist obeticholic acid shows durable efficacy. Poster presented at: EASL mono-conference on PBC; May 23–24, 2014; Milan, Italy: 14. | ||
Intercept. Phase 3 Study of Obeticholic Acid Evaluating Clinical Outcomes in Patients with Primary Biliary Cirrhosis (COBALT). Available from: https://clinicaltrials.gov/ct2/show/NCT02308111. NLM identifier: NCT02308111. Accessed August 15, 2016. | ||
OCALIVA(R) (obeticholic acid). New York, NY: Intercept Pharmaceuticals; 2016. | ||
Crippin JS, Lindor KD, Jorgensen R, et al. Hypercholesterolemia and atherosclerosis in primary biliary cirrhosis: what is the risk? Hepatology. 1992;15(5):858–862. | ||
Longo M, Crosignani A, Battezzati PM, et al. Hyperlipidaemic state and cardiovascular risk in primary biliary cirrhosis. Gut. 2002;51(2):265–269. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.