")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 6
Obesity – Are we continuing to play the genetic “blame game”?
Authors Venkatesan R, Viswanathan M
Received 2 December 2015
Accepted for publication 29 September 2016
Published 18 November 2016 Volume 2016:6 Pages 11—23
DOI https://doi.org/10.2147/AGG.S52018
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr John Martignetti
Venkatesan Radha,1 Viswanathan Mohan1,2
1Department of Molecular Genetics, Madras Diabetes Research Foundation, 2Dr. Mohan’s Diabetes Specialties Centre, WHO Collaborating Centre for Noncommunicable Diseases, Prevention and Control, IDF Centre for Education, Gopalapuram, Chennai, India
Abstract: Obesity is a heterogeneous disorder, its biological causes being complex. The escalating frequency of obesity during the past few decades is due to environmental factors such as sedentary lifestyle and overnutrition, but who becomes obese at an individual level is determined by genetic susceptibility. Understanding of the molecular cause of obesity is still in its infancy. More research is needed in the area of obesity genetics and epigenetics before formulating public health prescription as presently it would not be cost-effective. This review delineates monogenic form of obesity and the multifactorial nature of common forms of obesity, due to the interaction of genes, environment, and lifestyle.
Keywords: obesity, genes, genomics, monogenic obesity, polygenic obesity
Introduction
Obesity is defined as excess of body weight, and it results from the accumulation of body fat over time because of excess of energy intake or a lack of energy expenditure.1 It is clinically defined as having an increased body mass index (BMI) with the exact definition being ethnic specific. It is also defined using various measures such as waist-to-hip ratio (WHR), waist circumference, and body fat percentage in addition to BMI. It is a complex disorder that is caused by several genetic and nongenetic risk factors2 and is a worldwide epidemic that imposes an enormous burden on individual health as well as public health. According to the World Health Organization (WHO), obesity is one of the most significant contributors to ill health.3 Approximately 500 million people worldwide are estimated to have obesity and 1.4 billion are estimated to be overweight. In less than a generation, the total number of people with obesity has doubled. The prevalence rates of burden of obesity is escalating world over and more so in Asia, Africa, and South America. Increasing childhood obesity is of greater concern throughout the world.
Over the past 100 years, the causes of obesity have been perceived differently. Pituitary/hypothalamic dysfunction was assumed to lead to obesity in the early part of the 20th century.4 Between 1940s and 1970s, it was thought that psychological and psychodynamic aspects were the key factors leading to obesity.5 In 1956, Prader, Labhart, and Willi described the first syndromic form of obesity.6 That genetic factors play a very important role in body weight regulation came from landmark twin and adoption studies in the 1980s and early 1990s.7–9 Subsequently, molecular genetic studies have helped in understanding the genetic underpinning of obesity.10,11
Recently, there is increasing support for the notion that obesity is a neuroendocrine disorder in which both genetic predisposition and environmental risk factors act in concert.12 Indeed, large-scale molecular genetic studies have substantiated that in many cases, genetic predisposition to obesity is due to the combined net effect of polygenic variants. It is estimated that 2.8 million people die each year worldwide due to overweight/obesity. Between 1980 and 2008, the prevalence of obesity worldwide has almost doubled and an increase of overweight individuals from 1.3 to 2 billion is expected by 2030.
Human obesity is a global problem resulting from an excessive accumulation of body fat that can adversely affect health. Global rise in obesity has serious implications resulting in a significant number of diseases including type 2 diabetes mellitus (T2DM), cardiovascular diseases, and even some form of cancer.
Although there is an escalating prevalence of obesity making it a public health problem, its causes and physiological consequences at an individual level still remain elusive. The availability of calorie dense foods, a shift from calorie-poor to calorie-rich environment due to increasing affluence along with increasing sedentary behavior have all played a major role in escalating the obesity epidemic. However, the individual predisposition to becoming obese also has a strong genetic basis. Behind the risk of developing an obese phenotype, genetic factors could be underlying. The “thrifty gene hypothesis”13 states that genes that predispose to obesity would have had a selective advantage in populations that frequently experienced starvation. People possessing these genotypes in today’s obesogenic environment might be those who overreact to it and thus become markedly obese. The fetal programming hypothesis14 says that fetal weight gain tendency is decided by intrauterine environment and mother’s nutritional status and points out that epigenetic regulation of energy regulation pathway genes may be important. The ethnic shift hypothesis15 explains that in some ethnic groups obesity is more prevalent than in the others. This increases the number of obese individuals in the group. The assertive mating hypothesis explains that due to preferences in selection of spouse, there is a significant correlation between BMI of the spouses ultimately resulting in obesity over time.16 In simple terms, obesity could be explained as a consequence of an imbalance in net energy intake and expenditure. The cumulative energy intake and expenditure is balanced and maintained over a period of time, and this phenomenon of regulation is known as energy homeostasis. Energy balance is a conglomerate of traits, such as genes, behavior, diet, and metabolic factors, and each influences the other. Complex genetic factors among these contribute to individual difference in the development of obesity. Gene–environment interactions play an important role in the etiology of obesity. This review examines the role played by genetic and environmental factors in obesity.
Genetic factors
The evidence that obesity has a strong genetic basis comes from different studies. Heritability is the proportion of phenotypic variation among individuals due to genetic contributions. It is difficult to tease out heritability due to family lifestyle like diet pattern and physical activity which also clusters in families. The degree of parental obesity influences the risk, and this is greater if both the parents are obese.17–19 The risk of obesity is 10-fold higher if both the parents are obese and up to 4-fold if either one of the parent is obese.9 The overall familial risk for obesity is estimated to be between 1.5 and 5. Several studies have pointed out that BMI of the offspring is more strongly correlated with maternal BMI,1,18,20,21 suggesting intrauterine influences. Twin, family, and adoption studies suggest that genetic factors play a greater role concerning the predictive value of the parental BMI.22,23 Twin studies have revealed that genetic effects can account for up to 90% of the BMI variance within a population.
A comparison between twins reared apart and raised together proved the heritability of BMI to be same in both the cases. Twin studies have served as model since monozygotic (MZ) twins are genetically identical while dizygotic twins may share only 50% of their genetic material.8 Feinleib et al22 showed that in twin pairs, familial aggregation for obesity results mainly from genetic influence. A 25-year follow-up study using >4,000 MZ and dizygotic twin pairs confirmed these results.23 The heritability of fat mass among MZ twins has been reported to range from 70% to 90% while it is from 35% to 45% in dizygotic twins.
Adoption studies have also shown strong genetic influence on human body weight, correlating more strongly with the BMI of their biological parents than that of their adoptive parents.8 Different racial groups show differing prevalence of obesity, which is one of the evidences for a genetic component of obesity. Obesity prevalence in Asian population is ~35% or less compared to over 50% found in Pima Indians.24 Yet another important factor is the population substructure which could be different in different ethnic groups. This may presuppose that obesity-causing alleles may be present in some specific groups, thus increasing the risk of developing the disease in those populations.
On the basis of genetic and phenotypic characteristics, three types of obesity are seen. Monogenic forms of obesity which can be nonsyndromic or syndromic and polygenic (common) obesity.
Monogenic forms of obesity
Monogenic forms of obesity result from an alteration in a single gene and follow the Mendelian pattern of inheritance, affecting ~5% of the population.25 Two such forms exist – the nonsyndromic and syndromic. Early onset of the disease and an extreme phenotype characterize monogenic obesity.26 Murine models have been useful in understanding the molecular pathogenesis of human obesity.27 Studies of families with extreme obesity have been a source of information on obesity-related mutations.28
Nonsyndromic form of obesity
Both autosomal dominant and recessive forms of obesity have resulted due to several gene mutations. Approximately 200 single-gene mutations are found to cause human obesity, but all these mutations are confined to ~10 genes.29 But these are rare mutations leading to extreme obesity and occur at a very early age.30 Eight gene mutations explain about 10% of such cases of early-onset extreme obesity (Table 1).31 All these genes are in hypothalamic neuronal networks, coding for proteins with main role in leptin–melanocortin signaling pathway affecting food intake and energy expenditure.30
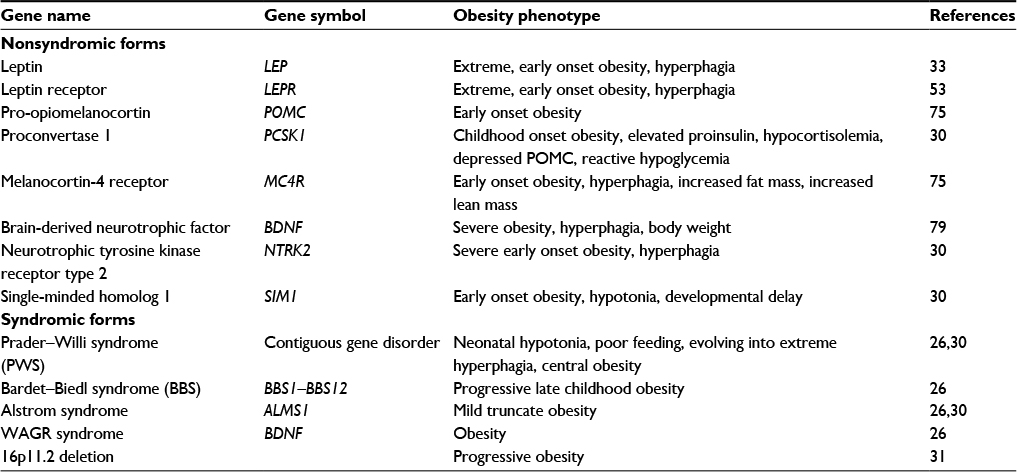 | Table 1 Monogenic forms of obesity Abbreviations: WAGR, Wilms tumor-aniridia-genital anomalies-retardation; POMC, pro-opiomelanocortin. |
Leptin is an adipocyte-derived satiety hormone and is associated with the size of the fat depot. Leptin receptor registers the increased leptin levels. This signal is registered by melanocortin-4 receptor (MC4R). Pro-opiomelanocortin (POMC) and melanocortin system further helps in the regulation of energy homeostasis.
The leptin or LEP gene, located in chr7q31.2, encodes a protein that helps in White body weight regulation.32 The protein plays a role in inhibiting food intake, regulating energy expenditure to maintain adipocyte mass. In 1997, during the course of screening for serum hormone levels in 2 severely obese children of the same family, very low leptin level was noticed.33 Following this finding, studies revealed that leptin deficiency is inherited and commonly seen in extreme early onset obesity.33,34 This was due to a frame shift mutation (delG133), which leads to a truncated protein or a missense mutation Arg105Trp leading to leptin deficiency,35 resulting in loss of sensing of energy need by hypothalamus, thus leading to more consumption of energy and weight gain. On administration of the hormone, energy regulation gets restored in leptin-deficient individuals.
The protein encoded by the LEPR gene (chromosome 1P31.3) is a receptor for leptin and is involved in the regulation of fat metabolism.36 It is reported that a mutation in the splice site of exon 16 is implicated in leptin receptor deficiency, leading to extreme obesity.
The MC4R gene on chromosome 18q21.3 encodes 332 amino acid and is mainly responsible for regulating energy balance.37 It is expressed mainly in the central nervous system contributing to food intake and energy expenditure regulation. A mutation in the MC4R gene leading to receptor nonfunctioning was associated with severe early onset diabetes.38 Impaired MC4R gene activity represents the common cause (1%–6%) of morbid obesity.39,40 More than 150 variants have been described in this gene.41 The defining features of obesity due to MC4R gene mutations is lean body mass that increases hyperinsulinemia.40
POMC deficiency is another cause of obesity. A heterozygous missense mutation, namely Arg236 Gly, in POMC gene resulted abnormal protein which is unable to activate the MC4R. Deficiency of proconvertase 1, due to PC1 gene mutations, is reported as an important cause of obesity. In a child with severe early onset obesity, loss of function mutation in this gene is reported.42
Syndromic forms of obesity
Syndromic forms refer to obesity cases that occur in a distinct set of associated clinical phenotypes, such as mental retardation or organ-specific developmental abnormalities. More than 30 such Mendelian disorders of obesity are recorded, exhibiting extreme genetic heterogeneity. Table 1 presents a list of common forms of early onset syndromic obesity. This includes Wilms tumor-aniridia-genital anomalies-retardation (WAGR) syndrome, Prader–Willi syndrome (PWS), Bardet–Biedl syndrome (BBS), and Alstrom and Cohen syndromes.31 WAGR syndrome is due to a deletion in a region containing Wilm’s tumor (WT1) and PAX6 genes. A deletion in the brain-derived neurotropic factor (BDNF) gene results in an obese phenotype of WAGR syndrome. PWS has many categories characterized by central obesity neonatal hypotonia, hyperphagia, hyperthalamic hypogonadism, and mild mental retardation, with such abnormalities as short stature and peculiar facial features. Loss of expression from potential deletions of 15q11.2 causes BBS is characterized by early onset obesity, associated with progressive cone-rod dystrophy, morphological abnormalities in the fingers, dyslexia, and progressive mental disease. Both Alstrom and Cohen syndromes are associated with childhood truncal obesity and small stature. They are both genetically homogenous and autosomal recessive. Balanced translocations of chromosome 2p13 disrupting the ALMS1 gene or mutations in this gene cause Alstrom syndrome. Mutations in COH1 gene, 8q22, reading a transmembrane protein result in Cohen syndrome.
Another syndrome resulting in morbid obesity and following an autosomal recessive pattern of inheritance is the MO1 syndrome. This is due to a nonsense mutation in the gene encoding a highly conserved ciliary protein, called CEP19.43 The loss of function mutation results in a phenotype characterized by glucose intolerance, dyslipidemia, and insulin resistance. It is interesting to note that the MO1 syndrome, BBS, and Alstrom syndrome are Mendelian disorders, resulting due to mutations in genes encoding ciliary proteins.
Polygenic forms of obesity
A group of alleles responsible for a trait is termed as “polygenic” variants.28 Many such polygenic variants play a role in obesity. Each allele in this case has small effects, and the allelic effects can be additive or nonadditive. Variants of obese genes show variation in frequency between obese subjects making the study of polygenic obesity more complex. Either single-nucleotide polymorphisms (SNPs) or micro satellite markers in the genomic DNA are analyzed in the case of polygenic disorders such as obesity. Unlike in the case of monogenic obesity where a single mutation in a gene is causal in producing the disease phenotype, in polygenic obesity, each polymorphism confers susceptibility to obesity and the presence of an obesogenic environment leads to the phenotype.44
Various molecular genetic approaches are employed in the detection and analysis of obesity genes and their variants. Linkage studies, candidate gene association study, and genome-wide association studies (GWAS) have helped in unraveling the genomics of obesity. Obesity has been measured in terms of body weight (BMI), adiposity, and waist–hip measurements in different studies.
The candidate gene approach
The candidate gene approach is based on a hypothesis. Genes that have been previously implicated in pathways controlling energy intake or expenditure have been chosen as important candidates and analyzed.45 Thus, this approach relies on the current understanding of the biology and pathophysiology of the disease (Table 2). Animal studies, cell-based studies, linkage and positional cloning studies in extreme obese subjects have helped identify candidate genes.46
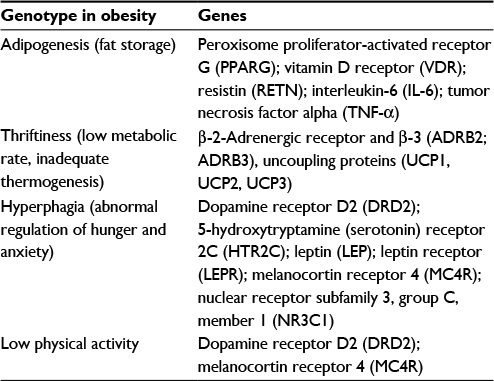 | Table 2 Obesity-related genes |
A total of 127 candidate genes are reported in human obesity gene map.47 Cocaine and amphetamine regulated transcript (CART) gene mutation, Leu34Phe, was identified by this approach in obese children and was found to be associated with less resting energy expenditure resulting in obesity.48 A possible relationship was also pointed out between obesity and mental disorders involving CART as a mediator.49 Large-scale studies suggest that obesity is strongly associated with genetic variants in the MC4R gene, Adrenergic-β-3 receptor (ADRB3) gene, prohormoneconvertase1 (PCSK1) gene, BDNF gene, and endocannabinoid receptor 1 (CNR1)gene. Hypothalamic leptin-melanocortin system is critical for energy balance in humans.32,39–41,50 Long-term therapy with leptin injections is shown to give beneficial effects on lowering fat mass, hyperinsulinemia, and hyperlipidemia in human obese subjects,51–53 but this is not approved as a treatment for common types .
In south Indian population, case–control candidate gene association studies pertaining to obesity were conducted. A research on Adiponectin gene showed interesting results. The 10211T-G polymorphism in the first intron of this gene was associated with hypoadiponectinemia, obesity, and T2DM. Obesity seems to act as an effect modifier, reversing the risk of T2DM in the “G” allele carriers of –11377 C/G polymorphism of the adiponectin gene.54
Another case–control study was also performed to investigate the presence of association of 8 variants of the ADIPOQ gene in a South Indian population.55 The association was tested for obesity, T2DM, and serum adiponectin levels. Of the 8 variants, 4 SNPs, namely +276 G/T (rs1501299), −4522 C/T (rs822393), −11365 C/G (rs266729), and +712 G/A (rs3774261) were significantly associated with T2DM in the SI population. In certain cases, the association of T2DM was mediated through obesity. This study also showed that ADIPOQ gene variants contribute to the genetic risk of T2DM, obesity, and hypoadiponectinemia in the south Indian population.
Several GWAS have shown an association of the fat mass and obesity-associated (FTO) gene variants with obesity and T2DM in various Caucasian56–59 and Asian populations.60–62 Six variants namely rs9940128, rs7193144, and rs8050136 (in intron 1), rs918031 and rs1588413 (in intron 8), and rs11076023 (30 untranslated region) of this gene were investigated to determine the association with obesity and T2DM. It was found that among South Indians, the rs9940128 A/G, rs11076023 A/T, and rs1588413 C/T variants of the FTO gene are associated with T2DM, whereas the rs8050136 C/A variant is associated with obesity.63
However, earlier studies in North Indian Sikhs61 and Asian Indians from the western part of India60 have shown strong association of intron 1 variant rs9939609 T/A of the FTO gene with T2DM independent of BMI. In the case of the rs918031 C/T SNP in intron 8 of the FTO gene, no association was found with T2DM or obesity, which is similar to a study in a Chinese population.64 However, the rs1588413 C/T located in intron 8 showed significant association with T2DM in the study, which was not seen in the Chinese population. The rs1588413 C/T SNP was not associated with obesity in the South Indian population, contrary to the results reported in the Chinese population. This clearly points to ethnic differences in susceptibility to obesity and diabetes between these two Asian populations. Recently, a more comprehensive approach was sought based on the notion that causal variants have high LD value, tag SNPs were used to search for association and determine whether these were in linkage disequilibrium with common variants already reported.65,66
Obesity affects men and women in different ways. There seem to be a higher prevalence of obesity and overweight among women compared to men. This is partly attributable to hormonal changes in a female development particularly in the postmenopausal period. In general, women are comparatively sedentary than men, thus burning lesser calories. The mean waist and hip circumferences between men and women also show significant difference in general. Therefore, to circumvent this confounding factor in all the candidate association studies, the analyses have been carried out by adjusting for age, sex, and also BMI.
Overall, the candidate gene approach resulted in limited success, and this led to the genome-wide approaches.
Genome-wide approaches
Genome-wide linkage studies (GWLS)
GWLS aim to identify chromosomal regions harboring genes for a specific phenotype by making use of the linkage data. Approximately 400 highly polymorphic markers at 10 cM intervals are mapped followed by fine mapping of regions under linkage peaks.67 This study design is applicable to cases with multigenerational pedigree and is not robust to detect phenotypic and genotypic heterogeneities. Candidate gene such as glutamate decarboxylase 2 (GAD2) that regulates food intake,68 ectonucleotide pyrophosphate/phosphodiesterase 1 (ENPP1) that inhibits insulin receptor activity,69 and solute carrier family 6 (SLC6A14) that controls appetite were identified by positional cloning strategy.70 Although many loci have been identified in GWAS, they were not reproducible in subsequent studies. A meta-analysis of 37 genome-wide linkage scans showed no evidence of significant linkage to any of the chromosomal regions studied.71 Till date, PSCK11 is the only region reported by linkage studies to be strongly linked to obesity.
GWAS
HapMap, an international project for the determination of complete range of genetic variations in human genome, is the foundation for GWAS as it gives the information and LD structure of SNPs (“The International HapMap Consortium. A haplotype map of Human Genome.” 2005; The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs.” 2007).
GWAS are a hypothesis-generating, agnostic method in which a large number of genetic markers (SNPs) spanning the entire genome are interrogated for their association with the phenotype of interest. The first gene reported to be associated with obesity was the insulin-induced gene 2 (INSIG2) which was from the Framingham Heart study that involved case–control and family studies of different ethnic groups.72 However, subsequent studies could not replicate these findings.
This was followed by the identification of FTO gene which was unequivocally associated with obesity.56 Frayling et al56 conducted a GWAS to test the correlation between polymorphisms across the entire human genome and T2DM. The rs9939609 polymorphism of FTO gene was strongly associated with T2DM and increased BMI. But after adjustment for BMI, the association was lost, and this was replicated consistently in several populations confirming the association of this SNP with common obesity in several populations such as European, Asian, and African.60–63 Later studies reported other polymorphisms in this region to be consistently associated with early onset childhood and adult obesity (rs142198 and rs17817449).56 Association with other obesity-related traits was also reported.73 It is interesting to note that previously, genome-wide linkage scans in “Framingham Heart Study” and “Family Blood Pressure Program Study” have confirmed BMI linkage with the region where FTO is mapped now.74 Around this region, fine mapping has shown an association with syndrome that includes obesity as one of its features. More than 60 polymorphisms of this gene have been significantly associated with obesity and its related traits.75 The functional mechanism underlying FTO’s role in obesity remains still unknown, although it is believed that the gene could play an important role in food intake control, energy expenditure, and homeostasis. It is however important to recognize that FTO is consistently associated with BMI, waist and hip circumference in all the analyses of heritability.73
Since 2007, obesity and related traits GWAS have been performed in several waves, with larger sample size in each subsequent wave (Table 2).
GWAS for overall adiposity
Much of the focus of GWAS for adiposity has been in finding genes associated with BMI. As of today, there are five waves of discovery for BMI.
The first wave and FTO
Interestingly, the T2DM GWAS of Welcome Trust Case Control Consortium discovered the first locus associated with BMI.56 However, the T2DM association was lost when adjusted for BMI, indicating that the effect of FTO on T2DM was through BMI. Ever since the discovery of FTO as an obesity susceptibility loci, it has been replicated in many populations to investigate its association with BMI and related traits. Human FTO gene is located in chromosome 16, is >400 Kb long, and spans 9 exons. Till date, FTO remains the locus with the largest effect on BMI. Variations in this gene have been associated with risk of overweight and obesity and traits related to them.76
The second wave and MC4R
In the second round of GWAS for BMI, a meta-analysis of 7 studies was done on a large sample size, confirming FTO locus association. Of greater importance is the series of studies convincingly replicating an SNP near the MC4R SNP. 77 The study included 60,352 adults and 5,988 children. The same locus near MC4R has also been associated with BMI in individuals of Indian and European descent.78
The Third wave
At this stage, the Genetic Investigation of Anthropometric Traits (GIANT) consortium brought together GWAS with anthropometric traits. A meta-analysis of 15 cohorts was combined, and replication was followed in these samples.79 Besides confirming FTO and near MC4R, loci in and near TMEM18, GNPDAR, SH2B1, MTCH2, KCTD15, and NEGR1 showed genome-wide significant association with BMI. Subsequently, a meta-analysis was performed.75 Associations were confirmed for genetic variants at various loci as well as the FTO and near MC4R loci. The third wave saw a total of 12 loci associated with BMI.
The fourth wave
To increase the power of the study and to find alleles with low frequency, the GIANT consortium increased the sample size. Eighteen additional novel loci associated with BMI were discovered. At this stage, a total of 32 loci associated with BMI were identified. However, the combined effect of the 32 loci is only 1.45%. Hence, predicting obesity using the risk alleles of these BMI loci is not accurate. These 32 loci together did not show any clinical application to discriminate between obese and nonobese individuals (ROC AUC = 0.575).75
The fifth wave
In the fifth wave, besides confirming all the 32 BMI-associated loci, 7 new loci were identified which explained an additional 0.09% of the variability in the BMI.80
GWAS for abdominal obesity
The distribution of body fat is not accurately reflected by BMI, and hence waist circumference and WHR have been used in GWAS as measures of abdominal fat. Two waves of GWAS for abdominal obesity have been performed.
First wave
A meta-analysis of 16 studies with 38,580 individuals was performed as a part of GIANT consortium. Associations of FTO and near MC4R were confirmed. Furthermore, novel associations of near TFAP2B and MSRA with waist circumference were also observed. These 4 loci were also identified in the GWAS for BMI.81 BMI and waist circumference show strong correlation, and hence the association of these SNPs were also more with overall obesity compared to abdominal obesity.
Second wave
In the second wave, 14 loci were discovered which were more specific to abdominal obesity; 7 of these loci were significantly more pronounced in women given the sex-specific distributions of WHR.82
GWAS for body fat percentage
For adiposity, body fat percentage is a more accurate measurement than BMI which does little to differentiate lean and fat mass. A GWAS combining 15 cohorts and including 36,626 individuals83 was performed. Loci such as FTO, near IRS1, and near SPRY2 were identified. The one near IRS1 locus was found to be associated with a number of metabolic traits, including insulin resistance, T2DM, and cardiovascular disease.
GWAS for extreme obesity
Individuals with extreme obese phenotypes are likely to present more common risk alleles. FTO, near MC4R loci, a locus between MSRA and TNKS were found to be robustly associated with extreme obesity,84 suggesting the importance of specific set of genes susceptible for the phenotype.
GWAS in other populations
Most of the GWAS has been performed in the population of White European descent except for one study which screened a South Asian population at a discovery stage.85 Association studies in non-Europeans can be informative; some loci are likely to be specific to certain ethnic populations. Studies across such loci in different populations might indicate more precisely, where the causal variant or gene might be located.
Of all the gene loci, FTO locus has been replicated consistently across populations of different ethnic backgrounds. Replication studies in populations of East Asian,86–88 South Asian,60,61,63 and African origin89,90 have found convincing associations of FTO locus with BMI and obesity risk. However, risk allele frequency varies substantially between these populations. The near MC4R locus was discovered by GWAS in a population of South Asian descent in the discovery stage. While the risk allele frequency is higher in Asian Indians, the effect size is similar to White Europeans.78 Replication of this locus in other South Asian population is also reported.91
So far the largest GWAS meta-analysis for BMI included results from >100,000 individuals. The BMI meta-analyses resulted in the identification of 97 genome-wide significant loci.92 The common variants identified by GWAS are characterized by modest effect sizes (odds ratio between 1.2 and 1.5 per allele), and the proportion of variability explained by GWAS-identified loci is only ~10%. This could be attributed to a number of reasons, such as lack of coverage of rare variants, genetic heterogeneity across the population, gene–gene and gene–environment interactions, and epigenetic factors. The current list of genetic factors explain only a small proportion of BMI variance, suggesting “missing heritability”93 recognizing the possible contributions of rare variants (MAF, 0.5%). Some of the important loci associated with different measures of obesity are given in Table 3.
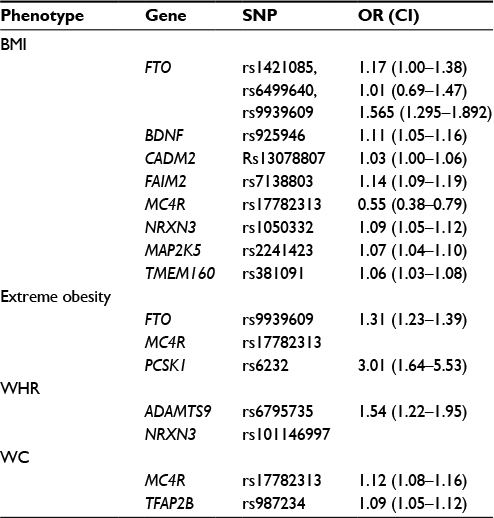 | Table 3 GWAS-identified genes associated with obesity Abbreviations: BMI, body mass index; CI, confidence interval; GWAS, genome-wide association studies; OR, odds ratio; SNP, single-nucleotide polymorphism; WC, waist circumference; WHR, waist-to-hip ratio. |
Although significant developments have been made over the past 10 years in the genetic basis of human obesity, much remains to be done in dissecting out the entire heritability of this disorder. Additional genetic factors need to be identified to explain the greater proportion of the disease.
There exist certain limitations in genetic approaches in unraveling the complexity of obesity. It is recognized that there are several reasons why a certain gene may not show the same disease phenotype. There exists clinical heterogeneity due to different gene dose levels which could also mask clinical changes.
Gene–gene interaction in obesity
The risk of obesity is determined not only by specific genotypes but also by significant gene–gene interactions which could very well be the reason for inconsistency in the replication of single locus association studies. Some interactions of obesity genes have been reported in a Mexican American study. The Pro 12 Ala polymorphisms, PPAR-G2 gene and Try 64 Arg of ADRB 3 gene showed gene–gene interaction, whereby subjects with both the gene variants had significantly higher BMI, insulin, and leptin levels than with only the PPAR 2 gene variant. Similar results were also reported in a Spanish population.94 In a study of a Quebec family, gene–gene interaction was seen in the markers in α-2, β-2, and β-3 adrenergic receptor genes (ADRs) contributing to phenotypic variability in abdominal obesity.95 Women with Gly/Gly genotypes at the β-1 adrenergic receptor gene (ADRB1) and carrying at least one β-3-Arg allele showed an increase in BMI.96
Epigenetics and human obesity
The role of epigenetics in complex diseases such as obesity has initiated a lot of interest in the recent times. Although genetic modifications lead to change in the base sequence of the DNA, epigenetic changes are typically reversible and refer to chemical modification to DNA in the absence of change in the DNA sequence.97 Epigenetic markers are inherited by mitotic cell division, whereas genetic changes are by meiotic division. Epigenetics involves processes such as DNA methylation and posttranslational modifications to histone proteins, such as acetylation. Although the genome is largely stable, the epigenome can get modified reversibly due to nutritional and environmental factors. 98
Epidemiological studies point out that an altered nutritional environment during development, that is maternal nutrition, is associated with a number of chronic diseases such as obesity and T2DM in later life.99,100 Regulation and level of gene expression of individual genes seem to be dependent on epigenetic environment. Generally, DNA methylation at regions such promoters and enhancers is implicated in gene silencing and in the structural genes, in gene expression.
One limitation in such cross-sectional studies is that both methylation levels and the phenotypes are measured at the same time point making it difficult to determine whether the association is a cause or consequence of obesity.
Genes implicated in obesity, appetite control, insulin signaling, and circadian clock regulation have been looked into for methylation markers and lower methylation of some of these genes have been observed in studies.100,101 There are also evidences for epigenetic regulation of specific genes leading to obesity.102 Genome-wide DNA methylation studies have been performed across a large number of genes and CpG sites. Table 4 summarizes some of the findings.
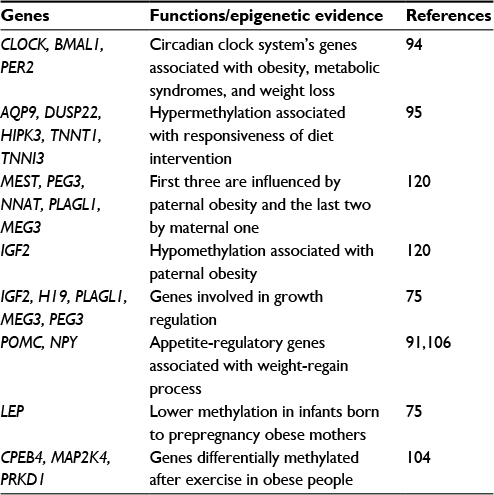 | Table 4 Epigenetic modification and genes associated with obesity125,126 |
Differentially methylated sites were enriched both in obesity candidate genes and in genes with various functions such as adipose tissue functioning. It is now known that methylation profiles do not remain stable throughout life. This was validated by intervention studies.103 Exercise diet and bariatric surgery seem to alter methylation profile in various tissues.94,95,102,104
A small study99 suggested that by reducing fat mass or body weight in obese individuals, alteration in methylation profiles resulted. Some of these methylation marks are results of obese phenotype rather than causal for the condition. These studies are association studies and are yet to establish causal relationship. However, the effect size on phenotypes of differentially methylated sites is likely to be small. As in genetic variations, combinations of multiple differentially methylated sites in several genes could explain variations in phenotype and together will have to be used for developing predictive signatures for obesity.
Higher methylation was observed in appetite-regulatory genes POMC and neuropeptide Y (NPY) genes of weight regainers rather than non-regainers. Lower methylation levels of POMC were associated with weight loss maintenance, while lower total methylation levels in NPY promoter were associated with higher risk of weight regain.106
These are early days for epigenetics and identification of potential biomarkers for obesity is already underway. Unlike genetic markers that are stable, epigenetic markers are modifiable by lifestyle changes in adult and changing the exposure “in utero” making intervention more feasible in the case of unfavorable epigenomic profiles, particularly in the context of complex diseases such as obesity.
Environmental factors
The increasing prevalence of obesity worldwide, the reverse relationship between obesity and socioeconomic class, and the secular trends associated with urbanization provide clear evidence of the environmental influences on weight gain.107,108 According to WHO, reduced physical activity and availability of energy with highly palatable foods represent a nutrition transition which is considered one of the greatest risk for ill health worldwide (http.//www.hsph.harvard.edu).
Gene and environment interactions
During the past few decades, lifestyle changes have had a major role in the obesity epidemic. However, there is convincing evidence that our genome which has remained largely unchanged for generations could still contribute to susceptibility to obesity. Migration studies comparing the risk of disease between populations with different lifestyles provided evidence for gene–environment interactions.109 Compared to Japanese people living in Japan, those migrated to elsewhere are more overweight.109
There is thus an interaction between genetic and environmental factors. This is best illustrated in Pima Indians. Obesity was more in obesogenic environment of Arizona, compared to “restrictive environment” of the Mexican Sierra Mandre Mountains, in the former being 69% are Indians while in the latter only 13%.110 These findings show that despite a similar genetic predisposition, different lifestyles result in differences in prevalence.
A study in 6,000 Danes demonstrated that physical activity brings down the effect of FTO by 30%.111 Recent studies provide evidence that physical activity can bring down the genetic susceptibility to obese phenotype. An interaction between FTO obesity risk genotype and physical activity was repeated by 13 independent studies.112,113 Likewise, a high-level physical activity seems to bring down the combined effect of 12 obesity-associated SNPs by 40%. A few large-scale studies confirmed this interaction between FTO and physical activity while some did not.114
Genes and physical activity interaction
The relationship between excessive TV watching and obesity has been studied in various cross-sectional studies, which have shown a direct association between the two. Gortmaker et al,115observed a 5-fold higher rate in children and adolescents, who watch TV for >5 h a day, compared to those who watch for <2 h. Over a 4-year study period, 60% of obesity incidence was attributed to watching TV.
Physical activity is a determinant of energy and substrate metabolism. A study showed heritability estimates suggest significant genetic effects. For example, duration of exercise improved significantly for those with II and ID genotype of the ACE gene, but not for those with DD genotype.116 The hypoxia inducible factor 1 (HIF1) gene is associated with maximal oxygen uptake after exercise training. 117
Genes and diet interaction
Dietary habits seem to interact with genes by modifying susceptibility to obesity. In 5 independent populations, an interaction between apolipoproteins A polymorphisms and high-saturated fat has been reported.118 Variable responses to diet maybe due to some of the potential susceptibility genes, those that relate to energy homeostasis and thermogenesis, and this includes NPY, melanocortin pathway factors (MC4R), uncoupling proteins (UCPs), and fatty acid binding protein (FABP). Subjects with FABP 54 Thr allele exhibited a better lowering of triglycerides with dietary intervention.119 Consumption of energy-dense, palatable food has been a major culprit in the increase of obesity epidemic.
Obesity risk of FTO genotype was increased when high-fat diet was introduced.114 Diet intervention studies suggest that FTO is probably sensitive to healthy lifestyle in general as it shows similar interaction with diet and physical activity.119
In an obesity intervention study, in a Swedish population, it was shown that after bariatric surgery the FTO obesity allele carriers lost 3 kg less than common allele homozygotes.120 Gastric bypass surgery in 1,001 obese subjects showed that those with risk alleles of 4 obesity genes to be associated with postoperative loss trajectories.121 Thus, obesity genes play a role in therapeutic options, and this seems to be a good approach to form a strategy for personalized treatment in order to achieve higher rates of therapeutic success.
Role of host gut microbiota in obesity
In the last 10 years, genetic and environmental factors dealing with host–microbiota interactions have been investigated by metagenomic approaches. While the human genome is inherited, the human microbiome is acquired from the environment anew every generation. Metagenomic studies have shown differences between microbial gene profiles of obese and nonobese subjects.122 Metagenomic analyses have also shown certain gut microbiota to be predisposing or protective to obesity.123 The gut microbiota contributes to host metabolism by many mechanisms such as fat storage in adipose tissue and modulation of lipid metabolism.
Circadian rhythms and obesity
In humans, there seems to be an intrinsic molecular clock that coordinates and synchronizes the rhythms with the 24-h solar day. This circadian rhythms allow the body to produce peak protein expression once in 24 h. Circadian rhythms are genetically encoded and normally generate internal timing of ~24 h. Circadian clock seems to undergo nutritional programming, and many genes have been identified that disturb the body weight through them.124
One of the greatest health challenges around the globe is the emergence of obesity. Although environmental/lifestyle factors are primary determinants of obesity, the microbial composition of the human gut and epigenetic markers such as DNA methylation pattern changes in genes induced by environmental factors contribute to obesity. However, the genetic susceptibility at the individual level is the basis on which various other factors act to result in obesity. Without the presence of susceptible genetic factors, obesity does not result. Further research in this area is certainly warranted to understand the pathogenesis of the now ubiquitous disorder. With the available knowledge and understanding, we can certainly say that we still play the blame game, the blame being shared between genes, epigenetic factors, and lifestyle, although prevention of obesity would ultimately become a reality only if the environmental and lifestyle factors are modified even in genetically susceptible individuals. The time for action was yesterday!
Disclosure
The authors report no conflicts of interest in this work.
References
Cheung WW, Mao P. Recent advances in obesity: genetics and beyond. ISRN Endocrinol. 2012;2012:536905. | ||
Walley AJ, Asher JE, Froguel P. The genetic contribution to non-syndromic human obesity. Nat Rev Genet. 2009;10:431–442. | ||
Caballero, B. A nutrition paradox – underweight and obesity in developing countries. N Engl J Med. 2005;352:1514–1516. | ||
Fröhlich A. Ein Fall von Tumor der Hypophysis cerebrihne Akromegalie. Wiener Klinische Rundschau. 1901;15(833–836):906–908. | ||
Bruch H. Psychological aspects of overeating and obesity. Psychosomatics. 1964;5:269–274. | ||
Hebebrand J, Hinney A. Environmental and genetic risk factors in obesity. Child Adolesc Psychiatr Clin N Am. 2009;18:83–24. | ||
Bouchard C, Tremblay A, Despres JP, et al. The response to long-term overfeeding in identical twins. N Engl J Med. 1990;322:1477–1482. | ||
Stunkard AJ, Sorensen TI, Hanis C, Teasdale TW, ChakrabortyR, Schull WJ, Schulsinger F. An adoption study of human obesity. N Engl J Med. 1986;314:193–198. | ||
Stunkard AJ, Harris JR, Pedersen NL, McClearn GE. The body-mass index of twins who have been reared apart. N Engl J Med. 1990;322:1483–1487. | ||
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. | ||
Rankinen T, Zuberi A, Chagnon YC, Weisnagel SJ, Argyropoulos G, Walts B, Pérusse L, Bouchard C. The human obesity gene map: the 2005 update. Obesity (Silver Spring). 2006;14:529–644. | ||
Shabana M, Hasnain S. Obesity, more than a cosmetic problem: current knowledge and future prospects of human obesity genetics. Biochem Genet. 2016;54(1):1–28. | ||
Neel JV. Diabetes mellitus: “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet. 1962;14:353–362. | ||
McArdle H, Andersen H, Jones H, Gambling L. Fetal programming: causes and consequences as revealed by studies of dietary manipulation in rats – a review. Placenta. 2006;27:56–60. | ||
Burke MA, Heiland FW, Nadler CM. From “overweight” to “about right”: evidence of a generational shift in body weight norms. Obesity. 2010;18(6):1226–1234. | ||
Hebebrand J, Wulftange H, Goerg T, et al. Epidemic obesity: are genetic factors involved via increased rates of assortative mating? Int J Obes Relat Metab Disord. 2000;24:345–353. | ||
Whitaker RC. Predicting preschooler obesity at birth: the role of maternal obesity in early pregnancy. Pediatrics. 2004;114(1):e29–e36. | ||
Magnusson PK, Rasmussen F. Familial resemblance of body mass index and familial risk of high and low body mass index. A study of young men in Sweden. Int J Obes Relat Metab Disord. 2002;26(9):1225–1231. | ||
Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27(4):325–351. | ||
Lee JH, Reed DR, Price RA. Familial risk ratios for extreme obesity: implications for mapping human obesity genes. Int J Obes Relat Metab Disord. 1997;21(10):935–940. | ||
Xia Q, Grant SF. The genetics of human obesity. Ann N Y Acad Sci. 2013;1281:178–190. | ||
Feinleib M, Garrison RJ, Fabsitz R, et al. The NHLBI twin study of cardiovascular disease risk factors: methodology and summary of results. Am J Epidemiol. 1997;106(4):284–285. | ||
Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. JAMA. 1986;256(1):51–54. | ||
Knowler WC, Pettitt DJ, Saad MF, Bennett PH. Diabetes mellitus in the Pima Indians: incidence, risk factors and pathogenesis. Diabetes Metab Rev. 1990;6(1):1–27. | ||
O’Rahilly S, Farooqi IS, Yeo GS, et al. Minireview: human obesity – lessons from monogenic disorders. Endocrinology. 2003;144:3757–3764. | ||
Farooqi S, O’Rahilly S. Monogenic obesity in humans. Annu Rev Med. 2005;56:443–458. | ||
Lutz T, Woods S. Overview of animal models of obesity. Curr Protoc Pharmacol. 2012;5:61. | ||
Hinney A, Vogel CI, Hebebrand J. From monogenic to polygenic obesity: recent advances. Eur Child Adolesc Psychiatry. 2010;19(3):297–310. | ||
Mutch DM, Clément K. Unravelling the genetics of human obesity. PLoS Genet. 2006;2:e188. | ||
González-Jiménez E, Aguilar Cordero MJ, Padilla López CA, García García I. Monogenic human obesity: role of the leptin melanocortin system in the regulation of food intake and body weight in humans. An Sist Sanit Navar. 2012;35(2):285–293. Spanish. | ||
Albuquerque D, Stice E, Rodriguez-Lopez R, Manco L, Nóbrega C. Current review of genetics of human obesity: from molecular mechanisms to an evolutionary perspective. Mol Genet Genomics. 2015;290:1191–1221. | ||
Dubern B, Clément K. Leptin and leptin receptor-related monogenic obesity. Biochimie. 2012;94:2111–2115. | ||
Montague CT, Farooqi IS, Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature.1997;387:903–908. | ||
Rau H, Reaves BJ, O’Rahilly S, Whitehead JP. Truncated human leptin (delta133) associated with extreme obesity undergoes proteasomal degradation after defective intracellular transport. Endocrinology. 1999;140:1718–1723. | ||
Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat Genet. 1998;18:213–215. | ||
Clément K, Ferré P. Genetics and the pathophysiology of obesity. Pediatr Res. 2003;53:721–725. | ||
Gantz I, Miwa H, Konda Y, et al. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J Biol Chem. 1993;268:15174–15179. | ||
Vaisse C, Clément K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20:113–114. | ||
Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–1095. | ||
Hinney A, Volckmar AL, Knoll N. Melanocortin-4 receptor in energy homeostasis and obesity pathogenesis. Prog Mol Biol Transl Sci.2013;114:147–191. | ||
Challis BG, Pritchard LE, Creemers JW, et al. A missense mutation disrupting a dibasic prohormone processing site in pro-opiomelanocortin (POMC) increases susceptibility to early-onset obesity through a novel molecular mechanism. Hum Mol Genet. 2002;11(17):1997–2004. | ||
Jackson RS, Creemers JW, Farooqi IS, et al. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Investig. 2003;112(10):1550–1560. | ||
Shalata A, Ramirez MC, Desnick RJ, et al. Morbid obesity resulting from inactivation of the ciliary protein CEP19 in humans and mice. Am J Hum Genet. 2013;93(6):1061–1071. | ||
Razquin C, Marti A, Martinez JA. Evidences on three relevant obesogenes: MC4R, FTO and PPARγ. Approaches for personalized nutrition. Mol Nutr Food Res. 2011;55:136–149. | ||
Tabor HK, Risch NJ, Myers RM. Candidate-gene approaches for studying complex genetic traits: practical considerations. Nat Rev Genet. 2002;3(5):391–397. | ||
Bell CG, Walley AJ, Froguel P. The genetics of human obesity. Nat Rev Genet. 2005;6(3):221–234. | ||
Yang W, Kelly T, He J. Genetic epidemiology of obesity. Epidemiol Rev. 2007;29:49–61. | ||
del Giudice EM, Santoro N, Cirillo G, D’Urso L, Di Toro R, Perrone L. Mutational screening of the CART gene in obese children identifying a mutation (Leu34Phe) associated with reduced resting energy expenditure and cosegregating with obesity phenotype in a large family. Diabetes. 2001;50(9):2157–2160. | ||
Mao P. Potential antidepressant role of neurotransmitter CART: implications for mental disorders. Depress Res Treat. 2011:762139. | ||
Hinney A, Volckmar AL, Antel J. Genes and the hypothalamic control of metabolism in humans. Best Pract Res Clin Endocrinol Metab. 2014;28(5):635–647. | ||
Farooqi IS, Jebb SA, Langmack G, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–884. | ||
Farooqi IS, Matarese G, Lord GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103. | ||
Farooqi IS, O’Rahilly S. Leptin: a pivotal regulator of human energy homeostasis. Am J Clin Nutr. 2009;89:980S–984S. | ||
Vimaleswaran KS, Radha V, Ramya K, et al. A novel association of a polymorphism in the first intron of adiponectin gene with type 2 diabetes, obesity and hypoadiponectinemia in Asian Indians. Hum Genet. 2008;123(6):599–605. | ||
Ramya K, Ayyappa KA, Ghosh S, Mohan V, Radha V. Genetic association of ADIPOQ gene variants with type 2 diabetes, obesity and serum adiponectin levels in south Indian population. Gene. 2013;532(2):253–262. | ||
Frayling TM, Timpson NJ, Weedon MN,et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889–894. | ||
Dina C, Meyre D, Gallina S, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39(6):724–726. | ||
Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. | ||
Hinney A, Nguyen TT, Scherag A, et al. Genome wide association (GWA) study for early onset extreme obesity supports the role of fat mass and obesity associated gene (FTO) variants. PLoS One. 2007;2(12):e1361. | ||
Yajnik CS, Janipalli CS, Bhaskar S, et al. FTO gene variants are strongly associated with type 2 diabetes in south Asian Indians. Diabetologia. 2009;52:247–252. | ||
Sanghera DK, Ortega L, Han S, et al. Impact of nine common type 2 diabetes risk polymorphisms in Asian Indian Sikhs: PPARG2 (Pro12Ala), IGF2BP2, TCF7L2 and FTO variants confer a significant risk. BMC Med Genet. 2008;9:59. | ||
Peeters A, Beckers S, Verrijken A, Roevens P, Peeters P, Gaal LV, Hul WV. Variants in the FTO gene are associated with common obesity in the Belgian population. Mol Genet Metab. 2008;93:481–484. | ||
Ramya K, Radha V, Ghosh S, Majumder PP, Mohan V. Genetic variations in the FTO gene are associated with type 2 diabetes and obesity in South Indians (cures-79). Diabetes Technol Ther. 2011;13(1):33–42. | ||
Ogden Cl, Yanovski SZ, Carroll MD, Flegal KM. Epidemiology of Obesity. Gastroenterology. 2007;132:2087–2102. | ||
Vimaleswaran KS, Loos RJ. Progress in the genetics of common obesity and type 2 diabetes. Expert Rev Mol Med. 2010;12:e7. | ||
Tascan M, Musso G, Hao T, Vidal M, MacRae CA, Roth FP. Selecting causal genes from genomewide association studies via functionally coherent subnetworks. Nat Methods. 2015;12(2):154–159TF. | ||
Bell CG, Benzinou M, Siddiq A et al. Genome-wide linkage analysis for severe obesity in French caucasians finds significant susceptibility locus on chromosome 19q. Diabetes. 2004;53(7):1857–1865. | ||
Boutin P, Dina C, Vasseur F,. GAD2 on chromosome 10p12 is a candidate gene for human obesity. PLoS Biol. 2003;1(3):e68. | ||
Suviolahti E, Oksanen LJ, Öhman M, et al. The SLC6A14 gene shows evidence of association with obesity. J Clin Invest. 2003;112(11):1762–1772. | ||
Meyre D, Bouatia-Naji N, Tounian A, et al. Variants of ENPP1 are associated with childhood and adult obesity and increase the risk of glucose intolerance and type 2 diabetes. Nat Genet. 2005;37(8):863–867. | ||
Saunders CL, Chiodini BD, Sham P, et al. Meta-analysis of genome-wide linkage studies in BMI and obesity. Obesity. 2007;15(9):2263–2275. | ||
Herbert A, Gerry NP, McQueen MB, et al. A common genetic variant is associated with adult and childhood obesity. Science. 2006;312:279–283. | ||
Wardle J, Llewellyn C, Sanderson S, Plomin R. The FTO gene and measured food intake in children. Int J Obes. 2008;33(1):42–45. | ||
Wu X, Cooper RS, Borecki I, et al. A combined analysis of genomewide linkage scans for body mass index, from the National Heart, Lung, and Blood Institute family blood pressure program. Am J Hum Genet. 2002;70(5):1247–1256. | ||
Speliotes EK, Willer CJ, Berndt SI, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–948. | ||
Scuteri A, Sanna S, Chen W-M, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3:e115. | ||
Loos RJF, Lindgren CM, Li S, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40:768–775. | ||
Chambers JC, Elliott P, Zabaneh D, et al. Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nat Genet. 2008;40:716–718. | ||
Thorleifsson G, Walters GB, Gudbjartsson DF, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;41:18–24. | ||
Day FR, Loos RJ. Developments in obesity genetics in the era of genome-wide association studies. J Nutrigenet Nutrigenomics. 2011;4:222–238. | ||
Willer CJ, Speliotes EK, Loos RJ, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. | ||
Heid IM, Jackson AU, Randall JC, et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010;42:949–960. | ||
Kilpelainen TO, Zillikens MC, Stancakova A, et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat Genet. 2011;43:753–760. | ||
Scherag A, Dina C, Hinney A, et al. Two new loci for body-weight regulation identified in a joint analysis of genomewide association studies for early-onset extreme obesity in French and German study groups. PLoS Genet. 2010;6:e1000916. | ||
Taylor AE, Sandeep MN, Janipalli CS, et al. Associations of FTO and MC4R variants with obesity traits in Indians and the role of rural/urban environment as a possible effect modifier. J Obes. 2011;2011:307542. | ||
Shi J, Long J, Gao Y-T, et al. Evaluation of genetic susceptibility loci for obesity in Chinese women. Am J Epidemiol. 2010;172:244–254. | ||
Dorajoo R, Blakemore AIF, Sim X, et al. Replication of 13 obesity loci among Singaporean Chinese, Malay and Asian-Indian populations. Int J Obes (Lond). 2012;36(1):159–163. | ||
Croteau-Chonka DC, Marvelle AF, Lange EM, et al. Genome-wide association study of anthropometric traits and evidence of interactions with age and study year in Filipino women. Obesity. 2011;19:1019–1027. | ||
Grant SFA, Li M, Bradfield JP, et al. Association analysis of the FTO gene with obesity in children of Caucasian and African ancestry reveals a common tagging SNP. PLoS One. 2008;3:e1746. | ||
Hennig B, Fulford A, Sirugo G, et al. FTO gene variation and measures of body mass in an African population. BMC Med Genet. 2009;10:21. | ||
Been LF, Nath SK, Ralhan SK, et al. Replication of association between a common variant near melanocortin-4 receptor gene and obesity-related traits in Asian Sikhs. Obesity. 2009;18:425–429. | ||
den Hoed M, Ekelund U, Brage S, et al. Genetic susceptibility to obesity and related traits in childhood and adolescence. Diabetes. 2010;59:2980–2988. | ||
Llewellyn CH, Trzaskowski M, Plomin R, Wardle J. Finding the missing heritability in pediatric obesity: the contribution of genome-wide complex trait analysis. Int J Obes (Lond). 2013;37(11):1506–1509. | ||
Milagro FI, Campión J, Cordero P, et al. A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss. FASEB J. 2011;25:1378–1389. | ||
Moleres A, Campión J, Milagro FI, et al. Differential DNA methylation patterns between high and low responders to a weight loss intervention in overweight or obese adolescents: the EVASYON study. FASEB J. 2013;27:2504–2512. | ||
Ochoa MC, Marto A, Azcona C, et al. Gene-gene interaction between PPAR G 2 and ADR B3 increases obesity risk in children and adolescents. Int J Obes Relat Metab Disord. 2004;28(3):37–41. | ||
Lavebratt C, Almgren M, Ekström TJ. Epigenetic regulation in obesity. Int J Obes (Lond). 2012;36:757–765. | ||
Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 2007;27:363–388. | ||
Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. 1989;298:564–567. | ||
Schellong K, Schulz S, Harder T, Plagemann A. Birth weight and long-term overweight risk: systematic review and a meta-analysis including 643,902 persons from 66 studies and 26 countries globally. PLoS One. 2012;7:e47776. | ||
van Dijk SJ, Molloy PL, Varinli H, Morrison JL, Muhlhausler BS; members of EpiSCOPE. Epigenetics and human obesity. Int J Obes. 2015;39:85–97. | ||
Cordero P, Campion J, Milagro FI, et al. Leptin and TNF-alpha promoter methylation levels measured by MSP could predict the response to a low-calorie diet. J Physiol Biochem. 2011;67:463–470. | ||
Barres R, Kirchner H, Rasmussen M, et al. Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep. 2013;3:1020–1027. | ||
Rönn T, Volkov P, Davegårdh C, et al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue. PLoS Genet. 2013;9:e1003572. | ||
Bouchard L, Rabasa-Lhoret R, Faraj M, et al. Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction. Am J Clin Nutr. 2010;91:309–321. | ||
van der Klaauw AA, Farooqi IS. The hunger genes: pathways to obesity. Cell. 2015;161(1):119–132. | ||
Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA. 2014;311:806–814. | ||
Popkin BM. Global nutrition dynamics: the world is shifting rapidly toward a diet linked with noncommunicable diseases. Am J Clin Nutr. 2006;84:289–298. | ||
Ravussin E, Valencia ME, Esparza J, Bennett PH, Schulz O. Effects of a traditional lifestyle on obesity in Pima Indians. Diabetes Care. 1994;17:1067–1074. | ||
Andreasen CH, Stender-Petersen KL, Mogensen MS, et al. Low physical activity accentuates the effect of the FTO rs9939609 polymorphism on body fat accumulation. Diabetes. 2008;57:95–101. | ||
Vimaleswaran KS, Li S, Zhao JH, et al. Physical activity attenuates the body mass index-increasing influence of genetic variation in the FTO gene. Am J Clin Nutr. 2009;90:425–428. | ||
Cauchi S, Stutzmann F, Cavalcanti-Proença C, et al. Combined effects of MC4R and FTO common genetic variants on obesity in European general populations. J Mol Med. 2009;87:537–546. | ||
Rampersaud E, Mitchell BD, Pollin TI, et al. Physical activity and the association of common FTO gene variants with body mass index and obesity. Arch Intern Med. 2008;168:1791–1797. | ||
Jonsson A, Renström F, Lyssenko V, et al. Assessing the effect of interaction between an FTO; variant (rs9939609) and physical activity on obesity in 15,925 Swedish and 2,511 Finnish adults. Diabetologia. 2009;52:1334–1338. | ||
Gortmaker SL, Must A, Sobol AM, et al. Television viewing as a cause of increasing obesity amongchildren in the United States, 1986–1990. Arch Pediatr Adolesc Med. 1996;150(4):356–62. | ||
Keogh JW, Palmer BR, Taylor D, et al. ACE and UCP2 gene polymorphisms and their association with baseline and exercise-related changes in the functional performance of older adults. Peer J. 2015;3:e980. | ||
Lindholm ME, Rundqvist H. Skeletal muscle hypoxia-inducible factor-1 and exercise. Exp Physiol. 2016;101(1):28–32. | ||
Smith CE, Tucke KL, Arnett DK, et al. Apolipoprotein A2 Polymorphism Interacts with Intakes of Dairy Foods to Influence Body Weight in 2 U.S. Populations. J Nutr. 2013;143(12):1865–1871. | ||
Burgio E, Lopomo A, Migliore L. Obesity and diabetes: from genetics to epigenetics. Mol Biol Rep. 2015;42:799–818. | ||
Sarzynski MA, Jacobson P, Rankinen T, Carlsson B, Sjostrom L, Bouchard C, Carlsson LM. Associations of markers in 11 obesity candidate genes with maximal weight loss and weight regain in the SOS bariatric surgery cases. Int J Obes (Lond). 2011;35(5):676–683. | ||
Still, CD, Wood GC, Chu X, et al. High allelic burden of four obesity SNPs is associated with poorer weight loss outcomes. Obesity (Silver Spring). 2011;19(8):1676–1683. | ||
Ussar S, Griffin NW, Bezy O, et al. Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab. 2015;22(3):516–530. | ||
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1131. | ||
Wang D, Chen S, Liu M, Liu C. Maternal obesity disrupts circadian rhythms of clock and metabolic genes in the offspring heart and liver. Chronobiol Int. 2015;32(5):1–12. | ||
Vidal AC, Murphy SK, Murtha AP, et al. Associations between antibiotic exposure during pregnancy, birth weight and aberrant methylation at imprinted genes among offspring. Int J Obes (Lond). 2013;37:907–913. | ||
Lesseur C, Armstrong DA, Paquette AG, Koestler DC, Padbury JF, Marsit CJ. Tissue-specific Leptin promoter DNA methylation is associated with maternal and infant perinatal factors. Mol Cell Endocrinol. 2013;381:160–167. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.