")
Back to Journals » OncoTargets and Therapy » Volume 10
NVP-AUY922, a novel HSP90 inhibitor, inhibits the progression of malignant pheochromocytoma in vitro and in vivo
Authors Lian JP, Lin D, Xie X, Xu Y, Xu L, Meng L, Zhu Y
Received 14 December 2016
Accepted for publication 13 March 2017
Published 19 April 2017 Volume 2017:10 Pages 2219—2226
DOI https://doi.org/10.2147/OTT.S130236
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Jianpo Lian,1,* Dengqiang Lin,1,* Xing Xie,1,* Yunze Xu,2 Lieyu Xu,1 Li Meng,1 Yu Zhu1
1Department of Urology, Ruijin Hospital, 2Department of Urology, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Purpose: Malignant pheochromocytoma (PCC) is a rare tumor with a very poor prognosis and no effective treatments. The aim of this study was to assess the efficacy of a novel second-generation synthetic heat-shock protein 90 (HSP90) inhibitor, NVP-AUY922, to treat malignant PCC in vitro and in vivo.
Materials and methods: Cell Counting Kit-8 (CCK-8) and Transwell assays were used to assess the effects of NVP-AUY922 on the proliferation and migration of the PCC cell line PC12. Flow cytometry was used to determine the effects of NVP-AUY922 on apoptosis and cell-cycle progression. Activation of phosphatidylinositol-3-kinase (PI3K)/protein kinase B (PKB/AKT) and mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling was measured using a Western blot analysis. In vivo, a mouse xenograft model was used to test the effects of intraperitoneal injection of NVP-AUY922 on tumor growth.
Results: NVP-AUY922 was found to be cytotoxic in PC12 cells at lower concentrations compared with 17-allylamino-17-demethoxygeldanamcyin (17-AAG). NVP-AUY922 inhibited the proliferation of PC12 cells in a time- and concentration-dependent manner and decreased the rate of migration of PC12 cells. Furthermore, we found that HSP90 inhibition induced cell-cycle arrest and apoptosis. In vivo, administration of NVP-AUY922 reduced PCC tumor growth without significant weight loss. Finally, we observed the modulation of MEK/ERK and PI3K/AKT signaling in response to NVP-AUY922 exposure.
Conclusion: NVP-AUY922 exhibits potent anti-PCC activities in vitro and in vivo and represents a promising therapeutic small molecule for treating malignant PCC.
Keywords: pheochromocytoma, HSP90, NVP-AUY922, xenograft model, therapy
Introduction
Pheochromocytoma (PCC) is a rare catecholamine-secreting tumor derived from chromaffin cells. PCC tumors are mainly found in the adrenal medulla, while ~10%–20% arise in the ganglia of the sympathetic nervous system and are referred to as paragangliomas.1 Although malignant PCC accounts for ~10%, it has a considerably high mortality rate, with the 5-year survival rate of patients as low as 44%, due to difficulties in diagnosing and treating PCC tumors.1,2 Surgery, radiotherapy (iodine-131 metaiodobenzylguanidine [I-131 MIBG]), and conventional cytotoxic chemotherapy aid to temporarily delay disease progression and prevent the recurrence of PCC, but their impact on long-term survival has not yet been determined.3,4 Therefore, there exists a need for more effective adjuvant or neoadjuvant therapies.
Targeted antitumor therapies have been extensively studied as promising novel therapeutic strategies for malignant PCC.4–6 Previous studies have shown that heat-shock protein 90 (HSP90), a highly conserved adenosine triphosphate (ATP)-dependent multi-chaperone protein required for the correct conformation, activity, function, and stability of >200 client proteins, is highly expressed in malignant PCC.7–11 The major client proteins stabilized by HSP90 are involved in growth, differentiation, cell-cycle progression, apoptosis, tumor invasion, metastasis, and angiogenesis. In addition, HSP90 has been shown to activate mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) and phosphatidylinositol-3-kinase (PI3K)/protein kinase B (PKB/AKT) signaling. Therefore, targeting HSP90 represents a beneficial therapeutic strategy that might result in the inhibition of several key oncogenic pathways.
Geldanamycin and its derivative, 17-allylamino-17-demethoxygeldanamcyin (17-AAG), are the most commonly used HSP90 inhibitors. However, severe side effects and chemoresistance in response to 17-AAG treatment have been reported.12,13 N-Vinylpyrrolidone (NVP)-AUY922 is a purine-scaffold derivative and non-geldanamycin analog of 17-AAG. We have previously shown that NVP-AUY922 exerts its effects through mimicking the ATP/adenosine diphosphate (ADP)-binding interface of the N-terminus of HSP90 and potently inhibiting the naive activity of multi-chaperone and complexed client proteins, without the same degree of hepatotoxicity as its geldanamycin counterparts.14 The aim of this study was to evaluate the efficacy of NVP-AUY922 using in vivo and in vitro models of malignant PCC.
Materials and methods
Cell line and reagents
Rat PCC PC12 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% horse serum, 5% fetal bovine serum (FBS; Sigma-Aldrich, St Louis, MO, USA), and antibiotic/antimycotic at 37°C with 5% CO2. The HSP90 inhibitor, NVP-AUY922, and 17-AAG were purchased from Selleck Chemicals (Houston, TX, USA) and solubilized in dimethyl sulfoxide (DMSO) to a concentration of 1 mM/mL before use. Cell Counting Kit-8 (CCK-8) was purchased from Dojindo (Tokyo, Japan). All primary antibodies (anti-β-actin, anti-cyclin B1, anti-Bax, anti-heat-shock protein 70 (HSP70), anti-phospho-AKT (S473), anti-AKT, anti-phospho-ERK1/2 (T202/Y204), anti-ERK1/2, anti-phospho-MEK, and anti-MEK) were acquired from Cell Signaling Technology (Boston, MA, USA).
Cell proliferation assay
The effect of NVP-AUY922 and 17-AAG on cell proliferation was determined by the CCK-8 assay. Tumor cells were cultured in 96-well plates at a density of 3×103/well in 200 μL complete culture medium for 24 h. Varying concentrations of NVP-AUY922 or 17-AAG were then added to each well and incubated for 24, 48, or 72 h. After each time point, 100 μL of the culture medium containing 10% CCK-8 was added to each well and incubated for 2 h before measuring the absorbance at 450 nm.
Cell migration assay
Cell migration was assessed using a modified Boyden chamber (BD Biosciences), as per the manufacturer’s protocol. PC12 cells were seeded at 200,000 cells per chamber, and cell migration was stimulated for 8 h with 10% serum in the absence (control [CTR]) or presence of NVP-AUY922 at a final concentration of 75 or 150 nM/mL.
Western blot
PC12 cells, treated with the indicated concentrations of NVP-AUY922 for certain times, were collected. PC12 cells were pelleted, washed once with phosphate-buffered saline (PBS), and then incubated in ice-cold lysis buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1% Triton X-100, 100 μg/mL phenylmethylsulfonyl fluoride, and 1 mM DL-Dithiothreitol) for 15 min. The lysates were centrifuged at 12,000 rpm for 30 min at 4°C, and the protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). For each group, 30 μg of protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using 10% polyacrylamide gels and transferred to polypropylene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% nonfat milk and then incubated with primary antibodies (anti-AKT, antiphospho-AKT, anti-MEK, antiphospho-MEK, anti-ERK, antiphospho-ERK, anti-HSP70, and β-actin). The membranes were then washed three times for 10 min with Tris-buffered saline (50 mM Tris, pH 7.4, 0.9% NaCl, 0.05% Tween 20) and incubated with horseradish peroxidase-conjugated secondary antibodies (Abcam, Cambridge, MA, USA). Membranes were again washed three times for 10 min with Tris-buffered saline with Tween 20 (TBST). The bands were visualized using Pierce enhanced chemiluminescence (Thermo Fisher Scientific).
Flow cytometry
Following treatment for 24 and 48 h with NVP-AUY922, PC12 cells were stained with propidium iodide (PI) to monitor cell-cycle progression. The percentage of cells undergoing apoptosis was measured using the Annexin V-FITC Apoptosis Detection Kit (Becton Dickinson, NJ, USA) per manufacturer’s instructions.
PCC mouse xenograft model
All animal studies were conducted in accordance with the principles and procedures outlined in the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee in Teaching and Research of Shanghai Jiao Tong University.
Briefly, PC12 cells (1×106 cells/100 μL) were implanted subcutaneously into the left hind flank of 4–5 weeks old female athymic nude mice. Tumor volume was measured using Vernier calipers, and mice were administered intraperitoneal injections of saline or NVP-AUY922 (35 mg/kg) once the tumor volume was found to be measurable (80–150 mm3, ~5–8 days post injection). Mice were treated every other day for 2 weeks, and the changes were checked once every 2 days. Post experimentation, the mice were sacrificed and the tumors were excised and weighed.
Detection of apoptosis in excised tumors
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL; KeyGen Biotech, Nanjing, China) was carried out per manufacturer’s instructions. Briefly, paraffin-embedded tumor samples were cut into 5-mm sections and mounted on glass slides. After TUNEL, five randomly selected high-powered visual fields were evaluated for each section, and apoptotic cells were identified based on intense brown nuclear staining. Here, the percentage of apoptotic cells is expressed as the average number of apoptotic cells per sample.
Statistical analysis
Tumor volume and weight are presented as mean ± standard deviation. All statistical analyses and graphical presentations were performed using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA). In all experiments, P<0.05 was considered to be statistically significant. For all in vitro assays, each individual experiment was performed at least three times.
Ethical approval
All studies were conducted in accordance with the guidelines of the National Institutes of Health for the Care and Use of Laboratory Animals and approved by the Animal Care and Use Committee in Teaching and Research, Shanghai Jiao Tong University.
Results
Inhibition of HSP90 correlates with increased HSP70 expression and suppression of cell proliferation and migration
As increased HSP70 expression is a hallmark of HSP90 inhibition, we determined HSP70 protein levels after exposure to NVP-AUY922 (100 nM) or 17-AAG (100 nM) after 24, 48, or 72 h. As expected, we observed elevated HSP70 protein levels in PC12 cells post treatment with both HSP90 inhibitors (Figure 1A).
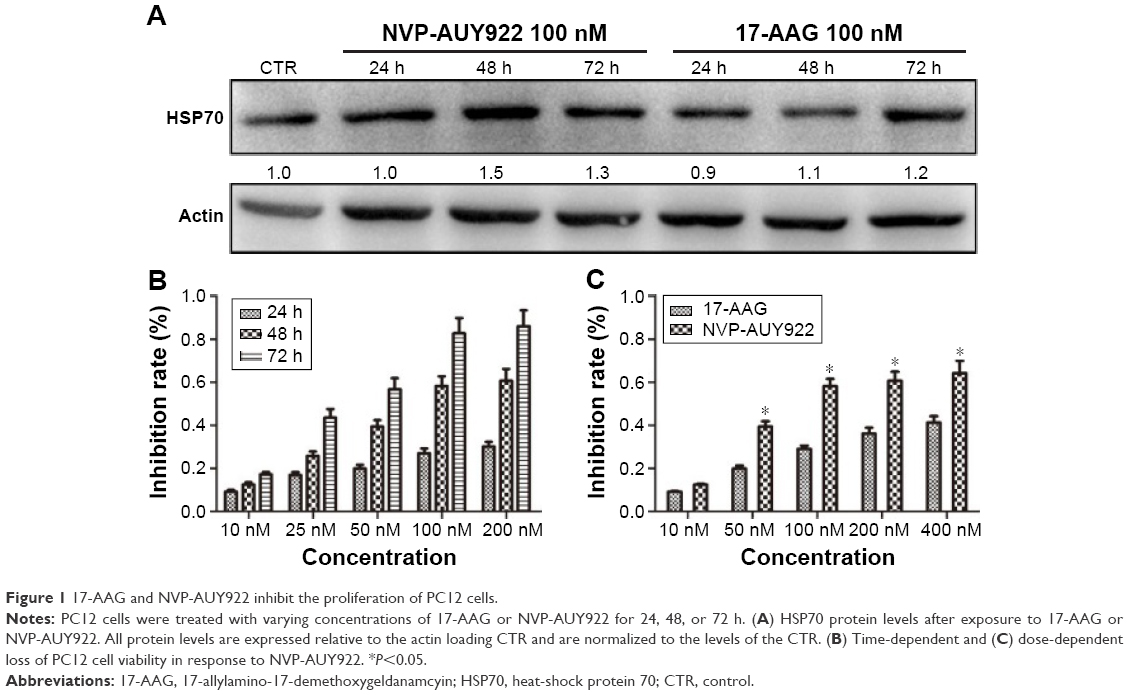 | Figure 1 17-AAG and NVP-AUY922 inhibit the proliferation of PC12 cells. |
We next evaluated the effects of NVP-AUY922 on cell proliferation. NVP-AUY922 inhibited PC12 cell proliferation in a dose- and time-dependent manner. The half maximal inhibitory concentrations (IC50s) at 48 and 72 h were found to be 75 and 30 nM, respectively (Figure 1B). We observed that lower concentrations of NVP-AUY922 could induce a higher anti-proliferative effect on PC12 cells compared to 17-AAG (Figure 1C). As a model to test the effect of NVP-AUY922 on tumor metastasis, we measured cellular migration using Transwell assays. We found a significant decrease in the number of migrating PC12 cells in response to treatment with NVP-AUY922 in a dose-dependent manner (P<0.05; Figure 2).
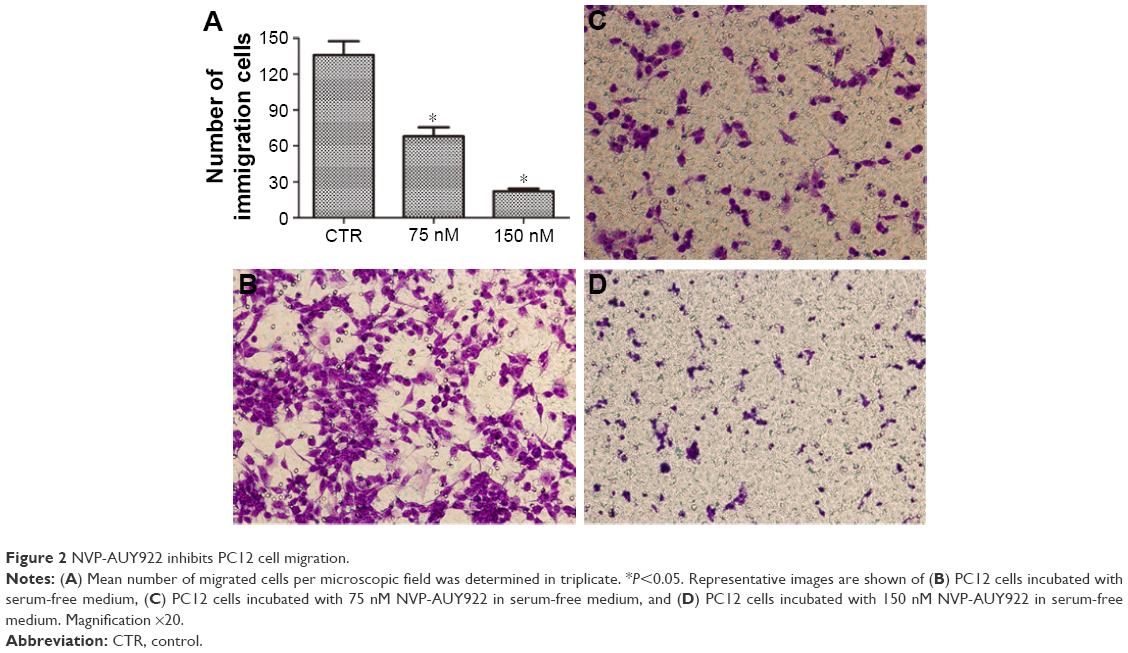 | Figure 2 NVP-AUY922 inhibits PC12 cell migration. |
NVP-AUY922 induces cell-cycle arrest and apoptosis in PC12 cells
To explore the possible mechanism(s) underlying the inhibition of proliferation by NVP-AUY922, cell-cycle analysis was performed. Treatment with NVP-AUY922 for 24 h significantly decreased the percentage of cells in the G1 phase and increased the percentage of cells in the G2 phase (P<0.05; Figure 3A and B). We also found that NVP-AUY922 blocked protein expression of cyclin B1, a cell cycle-associated protein (Figure 3C).
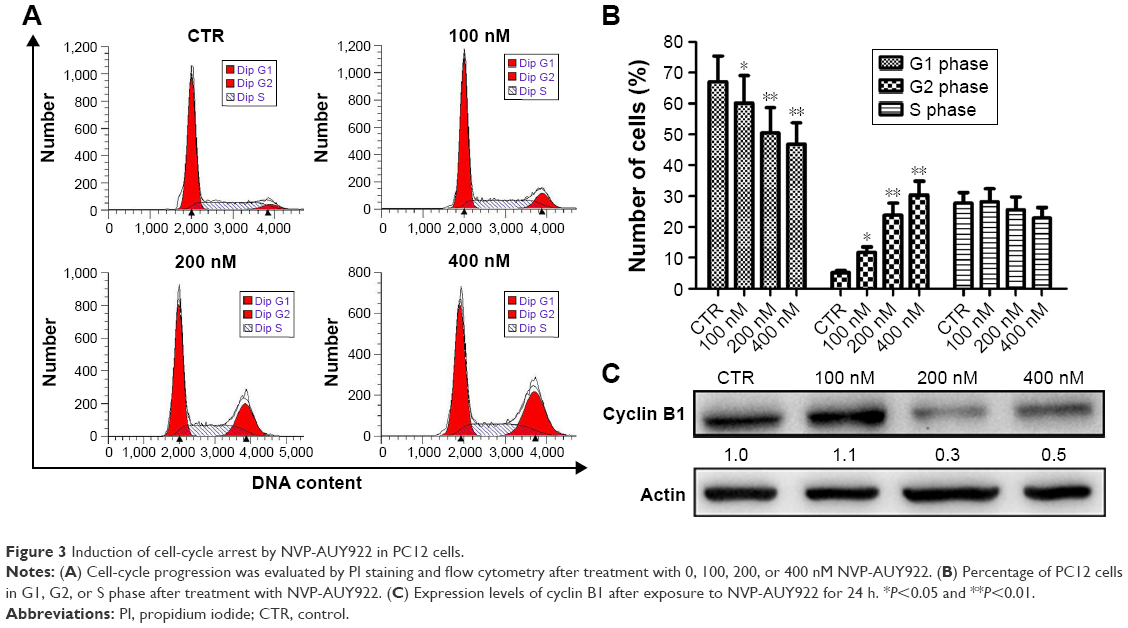 | Figure 3 Induction of cell-cycle arrest by NVP-AUY922 in PC12 cells. |
We also characterized the effect of NVP-AUY-922 on apoptosis in PC12 cells using Annexing V-FITC/PI staining. We observed a significant increase in the percentage of apoptotic PC12 cells after exposure to NVP-AUY922 for 48 h (Figure 4A and B). Consistent with our flow cytometric analysis results, we found that the protein expression of Bax increased in response to NVP-AUY922 after 48 h (Figure 4C).
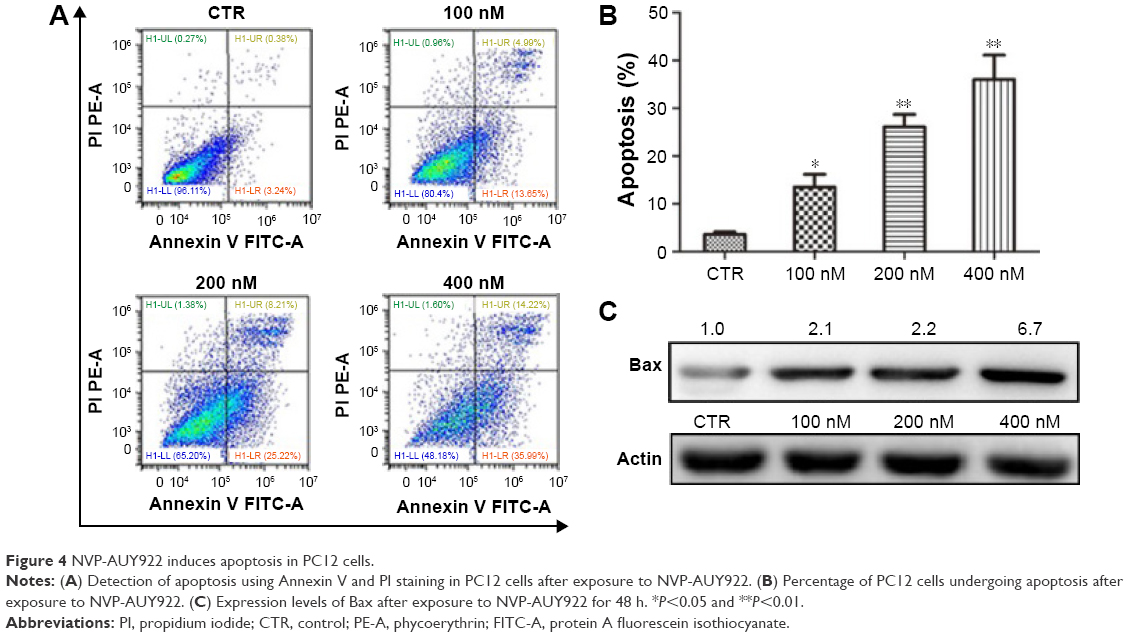 | Figure 4 NVP-AUY922 induces apoptosis in PC12 cells. |
NVP-AUY922 reduces the growth of PCC tumors in a PC12 xenograft model
Given our observation that NVP-AUY922 exhibits potent anti-cancer activity in vitro, we hypothesized that NVP-AUY922 could reduce the growth of PCC tumors in vivo. We therefore established a PC12 cell xenograft model in athymic nude mice. The PC12 tumors grew at a steady rate in mice treated with vehicle only, whereas NVP-AUY922 significantly reduced tumor volume after 15 days (P<0.05; Figure 5A and B). Furthermore, NVP-AUY922 reduced the mean tumor weight compared to that in mice administered vehicle only, without affecting the overall body weight (P<0.01; Figure 5C and D). Additionally, NVP-AUY922 markedly increased apoptosis (18.0%±1.08%) in PCC tumors compared with the CTR group (2.4%±0.15%; P<0.01; Figure 5E).
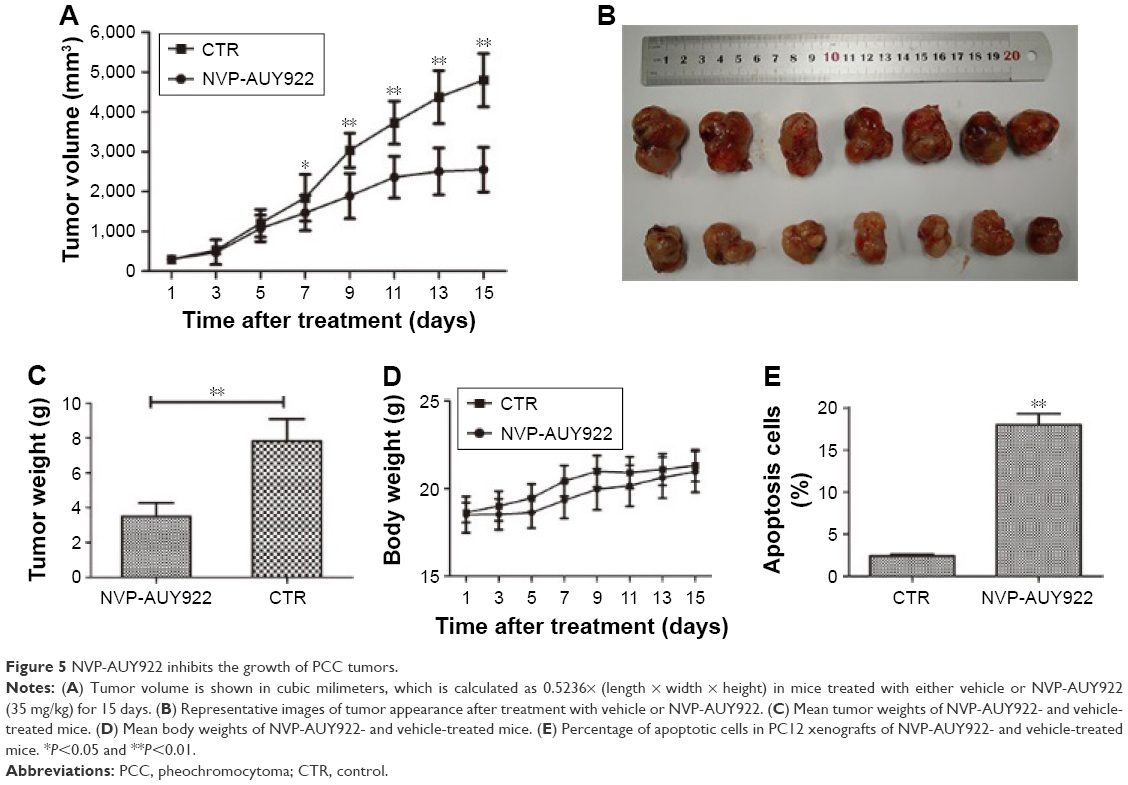 | Figure 5 NVP-AUY922 inhibits the growth of PCC tumors. |
NVP-AUY922 modulates PI3K/AKT and MEK/ERK signaling in PC12 cells
The PI3K/AKT MEK/ERK pathways play important roles in PCC tumorigenesis and chemoresistance. We therefore assessed the effects of HSP90 inhibition on the activation of AKT, MEK, and ERK in response to treatment with increasing concentrations of NVP-AUY922 for 24 h. NVP-AUY922 significantly inhibited phosphorylation of AKT (S472), MEK (S217/221), and ERK (T202/Y204). Interestingly, exposure to NVP-AUY922 slightly decreased the protein expression levels of AKT, MEK, and ERK (Figure 6).
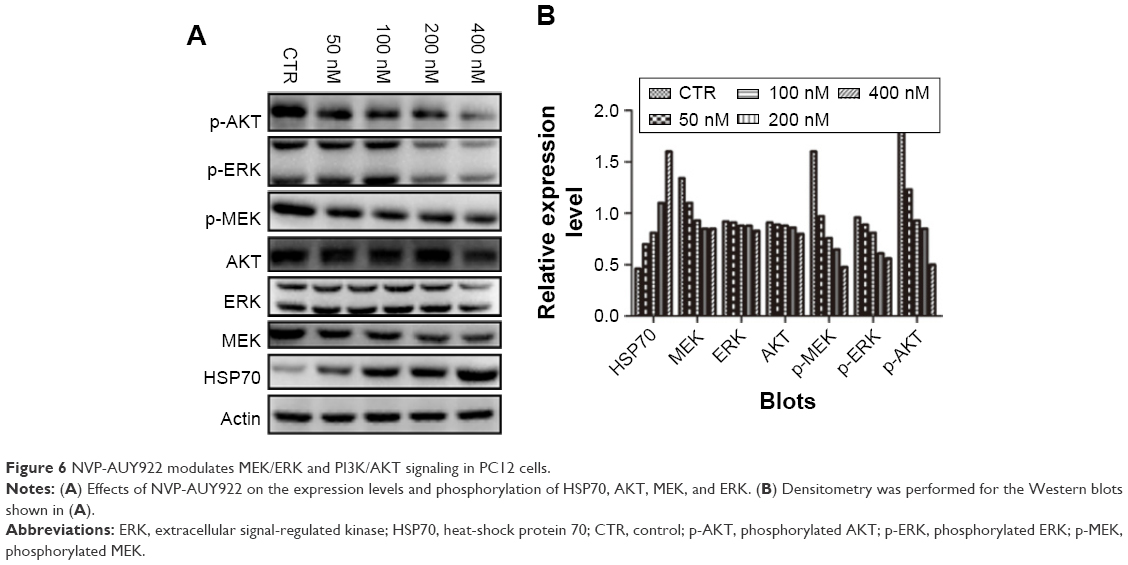 | Figure 6 NVP-AUY922 modulates MEK/ERK and PI3K/AKT signaling in PC12 cells. |
Discussion
The array of treatment options for malignant PCC is extremely limited due to the low incidence of PCC tumors and their complex pathogenesis. Accordingly, there are few effective cytotoxic drugs used to treat malignant PCC. As HSP90 has been shown to regulate the maturation and stability of proteins that are essential for tumor growth and metastasis, it has emerged as a promising therapeutic target.8,13 Until recently, geldanamycin (17-AAG) has been the most extensively studied HSP90 inhibitor in cancer therapy. However, its use has been associated with severe hepatotoxicity and chemoresistance.12,13,15 We hypothesized that NVP-AUY922, a novel small molecule HSP90 inhibitor, could overcome 17-AAG resistance with less pronounced side effects.15 In this study, we showed that treatment with NVP-AUY922 effectively reduced tumor growth and induced apoptosis in malignant PCC in vitro and in vivo. We therefore consider NVP-AUY922 as a valid alternative therapy for malignant PCC, to be used as a single agent or in combination therapy.
Here, we showed that NVP-AUY922 could significantly inhibit the proliferation and the migration of PC12 cells in a time- and dose-dependent manner, with a lower IC50 compared to 17-AAG.
We also found a dose-dependent induction of apoptosis and cell-cycle arrest in response to NVP-AUY922 treatment. Finally, in a mouse model of malignant PCC, administration of NVP-AUY922 significantly inhibited tumor growth without any indication of toxicity to vital organs.
We also hypothesized that NVP-AUY922 treatment could lead to the inactivation of various HSP90-specific oncogenic “client” proteins. We therefore assessed the levels of activity of the PI3K/AKT and MEK/ERK pathways, both known to be involved in PCC progression.16 Initially, we determined HSP70 protein levels, a hallmark of HSP90 inhibition, and found a dose-dependent upregulation of HSP70 expression after 24 h in PC12 cells treated with NVP-AUY922. HSP70 upregulation contrasted with reductions in the phosphorylation of AKT, MEK, and ERK, but with no obvious change in the total levels of these three signaling proteins. AKT and MAPK/ERK have been shown to exhibit cross-talk with respect to their roles, which results in protumorigenic signals.16,17 In this instance, inhibition of both AKT and MEK/ERK signaling by NVP-AUY922 could explain its potent effects on apoptosis and cell-cycle arrest, as simultaneous inactivation of multiple oncogenic signaling pathways has been shown to be beneficial with regard to overcoming chemoresistance.
Conclusion
Our results collectively show that NVP-AUY922 effectively inhibits proliferation of malignant PCC and leads to reduction in the growth of PCC tumors in a xenograft mouse model. This effect appears to be mediated by downregulation and deactivation of proteins involved in survival and carcinogenesis. We propose that NVP-AUY922 is a promising novel chemotherapeutic small molecule for the treatment of malignant PCC.
Acknowledgments
Lian JP, Lin D, and Xie X shared the first authorship. This study was supported by grants from the National Natural Science Foundation of China (no 81272936) and the Shanghai Municipal Natural Science Foundation (nos 134119a2700 and 17ZR1417300).
Disclosure
The authors report no conflicts of interest in this work.
References
Klingler HC, Klingler PJ, Martin JK Jr, Smallridge RC, Smith SL, Hinder RA. Pheochromocytoma. Urology. 2001;57(6):1025–1032. | ||
Zarnegar R, Kebebew E, Duh QY, Clark OH. Malignant pheochromocytoma. Surg Oncol Clin N Am. 2006;15(3):555–571. | ||
Carrasquillo JA, Pandit-Taskar N, Chen CC. I-131 metaiodobenzylguanidine therapy of pheochromocytoma and paraganglioma. Semin Nucl Med. 2016;46(3):203–214. | ||
Fishbein L. Pheochromocytoma and paraganglioma: genetics, diagnosis, and treatment. Hematol Oncol Clin North Am. 2016;30(1):135–150. | ||
Lowery AJ, Walsh S, McDermott EW, Prichard RS. Molecular and therapeutic advances in the diagnosis and management of malignant pheochromocytomas and paragangliomas. Oncologist. 2013;18(4):391–407. | ||
Mohammed AA, El-Shentenawy AM, Sherisher MA, El-Khatib HM. Target therapy in metastatic pheochromocytoma: current perspectives and controversies. Oncol Rev. 2014;8(2):249. | ||
Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006;66(18):9153–9161. | ||
Banerji U. Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res. 2009;15(1):9–14. | ||
Montemurro M, Bauer S. Treatment of gastrointestinal stromal tumor after imatinib and sunitinib. Curr Opin Oncol. 2011;23(4):367–372. | ||
Xu Y, Qi Y, Rui W, et al. Expression and diagnostic relevance of heat shock protein 90 and signal transducer and activator of transcription 3 in malignant pheochromocytoma. J Clin Pathol. 2013;66(4):286–290. | ||
Boltze C, Mundschenk J, Unger N, et al. Expression profile of the telomeric complex discriminates between benign and malignant pheochromocytoma. J Clin Endocrinol Metab. 2003;88(9):4280–4286. | ||
Piper PW, Millson SH. Mechanisms of resistance to HSP90 inhibitor drugs: a complex mosaic emerges. Pharmaceuticals (Basel). 2011;4(11):1400–1422. | ||
Jarosz D. HSP90: a global regulator of the genotype-to-phenotype map in cancers. Adv Cancer Res. 2016;129:225–247. | ||
Lee KH, Lee JH, Han SW, et al. Antitumor activity of NVP-AUY922, a novel heat shock protein 90 inhibitor, in human gastric cancer cells is mediated through proteasomal degradation of client proteins. Cancer Sci. 2011;102(7):1388–1395. | ||
Gaspar N, Sharp SY, Pacey S, et al. Acquired resistance to 17-allylamino-17-demethoxygeldanamycin (17-AAG, tanespimycin) in glioblastoma cells. Cancer Res. 2009;69(5):1966–1975. | ||
Nölting S, Garcia E, Alusi G, et al. Combined blockade of signalling pathways shows marked anti-tumour potential in pheochromocytoma cell lines. J Mol Endocrinol. 2012;49(2):79–96. | ||
Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–3074. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.