")
Back to Archived Journals » Research and Reports in Biochemistry » Volume 5
NUB1 suppression of Huntington toxicity: mechanistic insights
Received 26 February 2015
Accepted for publication 1 April 2015
Published 27 May 2015 Volume 2015:5 Pages 129—136
DOI https://doi.org/10.2147/RRBC.S65546
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Nikolay Dokholyan
Yao Yao, Boxun Lu
Department of Biophysics, School of Life Sciences, Fudan University, Shanghai, People’s Republic of China
Abstract: Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder marked by chorea, dystonia, incoordination, and cognitive and motor disturbance. The major cause of HD is the cytotoxicity of the mutant huntingtin protein (mHTT), encoded by the mutant HTT gene. The mechanism by which mHTT leads to cytotoxicity and neuronal death is unclear, and thus enhancing clearance of the mHTT protein is likely to be an effective approach to treat HD. We have recently identified NUB1 (negative regulator of ubiquitin-like proteins 1) as a modifier of mHTT levels via enhancement of its proteasomal degradation. In this review, we will discuss the mechanism of NUB1-mediated mHTT clearance and potential targeting strategies.
Keywords: drug target discovery, Huntington’s disease, NEDD8, ubiquitination
Huntington’s disease
Huntington’s disease (HD) is an inherited autosomal dominant neurodegenerative disorder caused by an expanded CAG repeat in the huntingtin gene (HTT ) exon 1 which encodes the mutant huntingtin protein (mHTT) with an expanded polyglutamine (polyQ) stretch.1 Despite the loss of normal functions and neuroprotective effects of wild type HTT,2–5 HD is caused mainly by the toxic gain of function of misfolded mHTT.1,6 The mHTT with the expanded polyQ stretch tends to form insoluble aggregates in neurons,7 a common feature shared by most neurodegenerative disorders. Meanwhile, the soluble, intermediate species of mHTT may be the major toxic species.8,9 Cytotoxicity of mHTT leads to loss of GABAergic medium spiny projection neurons in the stratum and atrophy of the cerebral cortex.1,6 The exact mechanism of mHTT toxicity is unclear, and possibly involves dysregulation of gene transcription;10,11 defects in mitochondrial function and cell metabolism;12 elevation of oxidative stress;13 abnormality in calcium signaling;14 alteration of autophagy;15 defects in cellular trafficking;11,16,17 etc. Symptoms of HD include: chorea; dystonia; incoordination; cognitive and motor dysfunction; and premature death.6
Compared to Alzheimer’s disease and Parkinson’s disease, of which most patients are sporadic, HD is a monogenic disorder of which the genetic cause is clear, and thus early diagnosis and establishment of genetic models is relatively easier. As a result, HD is often considered as an important disease model for neurodegenerative disorders. Meanwhile, the conventional targeted-drug-discovery approach does not apply to HD, because the specific mechanism as to how mHTT leads to the disease is unclear, making a direct screening for mHTT “inhibitors” impractical. Thus, we and others consider lowering mHTT level as a promising approach for drug discovery.18 Lowering mHTT should suppress all its downstream toxicity effects, and this has been well validated in a number of different cellular and animal models.19–22 As a result, identification of pathways that can lower mHTT levels is of great therapeutic as well as biological interest.
To achieve this goal, we have recently performed genome-wide screenings for modifiers of mHTT protein levels, and have identified NUB1 as a suppressor of mHTT levels and toxicity via lowering of mHTT protein in the in vivo Drosophila HD models and HD cellular models.20 Here we will discuss the likely mechanism of NUB1-mediated clearance of mHTT and potential therapeutic strategies.
NUB1 protein and its potential linkage with diseases
NUB1, known as negative regulator of ubiquitin-like (UbL) proteins 1, was originally identified as an interactor and regulator of a UbL protein, neural precursor cell expressed, developmentally downregulated 8 (NEDD8).23 NUB1 is a 601-residue-long protein with a calculated 69.1 kDa molecular mass.23 NUB1 and negative regulator of UbL proteins 1 long (NUB1L), a splicing variant of NUB1,24 belong to the UbL–ubiquitin-associated (UBA) family of ubiquitin-binding proteins which play a role in proteasomal degradation.25
NUB1 mRNA expresses ubiquitously in various tissues apart from pancreas, whereas NUB1L mRNA is expressed much less in prostate, leukocyte, liver, and skeletal muscles.24 Notably, although 3.5 kb of NUB1 mRNA was weakly detected in the brain, 69 kDa of NUB1 protein was predominantly expressed in human brain compared with other tissues.26
NUB1 has been reported to be involved in various diseases.26–28 NUB1 was found to bring synphilin-1 to proteasomal degradation and decrease the formation of synphilin-1 positive inclusions in diseases with neurodegenerative α-synucleinopathies.26 NUB1 also induces proteasomal degradation of glycogen synthase kinase 3β, a critical modulator in the pathological hyperphosphorylation of tau, suppressing both tau phosphorylation and aggregation.27 Meanwhile, NUB1/NUB1L causes S-phase transition and apoptosis in renal carcinoma cells by preventing the ubiquitination of cyclin E and P27 via downregulation of NEDD8 conjugation system, suggesting a possible anticancer effect.28
NUB1 in HD
In the previous proteomics study published in 2007, NUB1 was identified as an interactor of the N-terminal fragments of mHTT protein,29 but the biological significance of this interaction is unclear. Six years later, we identified NUB1 as a modulator of mHTT levels and toxicity via unbiased genome-wide screenings.20 This for the first time reveals the potential function of this HTT interactor in HD.
NUB1 downregulates exogenously expressed or endogenous mHTT proteins, while silencing NUB1 leads to mHTT increase in Q68, STHdh cellular models, and human embryonic stem cell-/induced pluripotent stem cell (ESC/iPSC)-derived neurons.20 Meanwhile, NUB1 suppresses mHTT neurotoxicity in in vivo Drosophila models, mammalian-cell models, and iPSC-derived neurons.20 NUB1 leads to significant proteasomal degradation of mHTT and HTT lysine modification is probably involved.20
The above evidence makes NUB1 an appealing drug target for HD, while further validation of phenotypic rescue in the in vivo mammalian HD models is needed. This could be potentially tested by injection of NUB1 overexpressing adeno-associated virus, or crossing the HD models with transgenic models overexpressing NUB1.
Mechanism of NUB1-mediated mHTT clearance
The mechanism of NUB1-mediated mHTT clearance is still not completely clear, yet this information is highly desired because it may provide more targets for potential drug discovery. Our previous study has revealed the major components of the likely pathway, and the mechanism could be further probed from two different angles: revealing domains of NUB1 that are required for the NUB1-mediated clearance, and revealing the other proteins that participate in the pathway.20
NUB1 domains and possible involvement
NUB1 has one UbL domain in N-terminus (Ile85 to Val147) between two coiled coil regions, two UBA domains (UBA1: Asp376 to Asn413; UBA3: Ser477 to His514), a Cys box-like sequence (Lys306 to Glu320), a His box-like sequence (Tyr343 to Tyr362), a bipartite nuclear localization sequence (Arg414 to Arg431), and a PEST sequence (His514 to His568) in C-terminus.23,30,31 NUB1L has an extra UBA domain (UBA2: 432–469 aa) (Figure 1).24
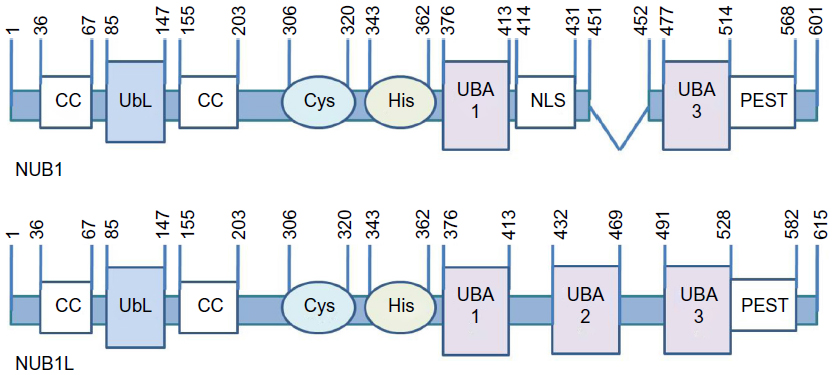 | Figure 1 Domain information of NUB1/NUB1L. |
NUB1/NUB1L are very unique members of UBA–UbL family.25 The UbL domain normally binds to the ubiquitin-interacting motif of the subunit 5a (S5a) of 19s proteasome regulatory particle,25 while in the case of NUB1/NUB1L, it is the C-terminus region between 536 and 584 aa that has the ability to interact with S5a.32 On the other hand, the UBA domain usually binds to ubiquitin.25 As for NUB1/NUB1L, despite interacting with α-peptide bond-linked polyubiquitins via UBA1 in yeast cells,31 they bind to a UbL protein, NEDD8, via C-terminus region between 536 and 584 aa, and NUB1L has an additional binding site between 427 and 474 aa covering UBA2.24
NEDD8 is a highly conserved 81 amino acid protein that shares 60% identity and 80% homology with ubiquitin.24 It is conjugated to its most known substrate cullin family and forms a SCF(Skp-1,cullin,F-box) or CRL(cullin RING ubiquitin ligase) complex as a large subfamily of ubiquitin E3 ligases, controlling the final step of ubiquitination of many substrates, including cyclin E, P27, P21, and P73, and playing an important role in cell cycle progression, apoptosis, tumorigenesis, etc.33,34
NUB1/NUB1L were reported to reduce NEDD8 monomer and conjugated proteins through proteasomal pathway, in which the NEDD8 binding site,24,30 as well as its UbL domain,32 are indispensable. NEDD8 conjugates can bind more effectively with S5a when NUB1 is coexpressed, indicating that interaction of NUB1 with 19s proteasome regulatory particle may play a part in promoting degradation of NEDD8 conjugates.30
NUB1L is also a noncovalent downregulator of another UbL protein, FAT10.35 Notably, the binding site (its three UBA domains) are not required for accelerating degradation of FAT10.36 Instead, NUB1L might bind to the 26s proteasome via UbL domain and induce conformational changes in the 19s regulator to favor binding and/or degradation of FAT10 and conjugated proteins.36
Revealing the domains required for NUB1-mediated mHTT clearance will provide clues for the detailed mechanism. For example, if the C-terminal region is needed, the binding with NEDD8 or S5a is probably involved. If the UbL domain is required, the modulation of NEDD8 or neddylated proteins is involved in the mechanism. In addition, given that NUB1 is an HTT interactor, the interaction between NUB1 and HTT is likely involved in the NUB1 mediated HTT clearance. The NUB1 region that interacts with HTT has been mapped to approximately 442–563 aa by the yeast two-hybrid screens,29 covering the UBA3 and most PEST domains of NUB1. It will be interesting to test whether these domains are required for NUB1-mediated HTT clearance, and this may determine whether the interaction between NUB1 and HTT is required.
NUB1–NEDD8–CUL3 pathway
We have previously shown that the presence of NEDD8 is indispensable for NUB1-mediated mHTT clearance, as is CUL3, a NEDD8 substrate.20 Thus, a possible model is that NUB1 functions as a bridging protein between HTT and neddylated CUL3-based E3 ligases via physical interaction with HTT and NEDD8, promoting the ubiquitin–proteasomal degradation of both mHTT and wtHTT in presence of mHTT.20 To further prove this, in vitro validations that CUL3 is able to ubiquitinate HTT and that NUB1 may enhance the ubiquitination are required.
CUL3 belongs to the cullin family which act as scaffolds for the assembly of multi-subunit ubiquitin E3 ligases; interacting with a RING domain protein ROC1(regulator of cullins 1)/Rbx1(RING-box protein 1) that recruits charged ubiquitin E2s into the complex when activated by neddylation of NEDD8 Gly-76 on its lysine residue;37 and catalyzes the ubiquitination of cullin substrates.34,37 CUL3 itself forms a complex called the BTB–CUL3–Rbx1 ubiquitin ligase, binding to Rbx1 via its C-terminus and to a bric-a-brac-tramtrack broad complex (BTB) domain-containing substrate receptor protein via its N-terminus.38,39 Interestingly, kelch-like family member 22 (KLHL22), a BTB–Kelch adaptor protein of CUL3,40 was also a hit in our previous screen.20 A possible explanation is that KLHL22 may be the substrate receptor protein of CUL3 for HTT (Figure 2). This could be further tested by revealing the potential epistatic effects of KLHL22 on NUB1-mediated HTT clearance, and by reconstituting the complete system in vitro to test if NEDD8–CUL3–KLHL22 is able to ubiquitinate mHTT.
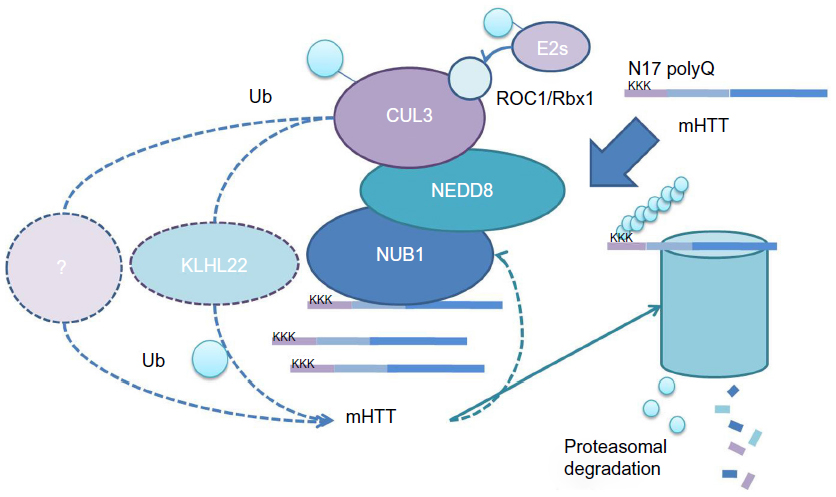 | Figure 2 Possible mechanism models of NUB1-mediated mHTT clearance. |
Alternatively, CUL3 may not directly ubiquitinate HTT, but is indirectly involved. If this is the case, other proteins may mediate the NUB1 effect. Most likely, NUB1 interactors are potential candidates. Proteomics studies revealing NUB1 interactors will be needed. Those proteins could be further tested to see if they show epistatic effects on NUB1-mediated HTT clearance.
The relationship between mHTT and NUB1
While NUB1 modulates mHTT levels, mHTT may play a role in regulating NUB1 levels as well. Based on our previous study, the wild type HTT protein in heterozygous HD cell lines can be regulated by NUB1 silencing or overexpression in the same direction as mHTT protein, but there is no effects in wild type cells.20 Given that NUB1 was reported to have physical interaction with mHTT,29 a possible explanation is that NUB1-induced clearance of HTT is dependent on the activation of NUB1 function by mHTT. Since NUB1 is not an enzyme, the “activation” is likely an enhancement of its interaction with other proteins. As NUB1 has been shown to interact with the proteasome subunit S5a,30,32 one possibility is that the presence of mHTT enhances its interaction with S5a, thus enhancing the proteasomal degradation. To test this, allele-specific knock-down of HTT in heterozygous HD cell lines or overexpression of mHTT in wild type cells can be conducted to see if mHTT specifically regulates NUB1 expression or NUB1 binding with S5a. Meanwhile, we can track NUB1 expression during different stages of HD to detect any potential correlation between expression of mHTT and NUB1. Study of the nature of NUB1–mHTT interaction may help as well.
In addition, endogenous NUB1 expression is lower in the wild type than the HD cells.20 One likely explanation is that the HD cells activate compensatory pathways that elevate NUB1 level to rescue the HD toxicity by reducing mHTT level. This could be further tested by measurement of endogenous NUB1 level in different HD homozygous, heterozygous, and wild type cell lines and in rodent animal models. Similarly, NUB1 was found coexisting with abnormal, proteinase K-resistant α-synuclein and may recognize it from the early stage of protein aggregation in Lewy body disease.41 Detecting NUB1 chaperone activity41 in HD or screening of modulators of NUB1 in mHTT toxicity-induced stress response pathway may help identify the mechanism. One possibility is involvement of interferon-β (IFNβ). The endogenous expression of IFNβ, a NUB1 inducer,23 was found to be upregulated in mice models of spinocerebellar ataxia 7, another polyQ neurodegenerative disorder.42 If IFN is also upregulated in HD cells, the downstream NUB1 is expected to be upregulated.
Targeting strategies for NUB1
NUB1 may not be a directly “druggable” target, because it does not have a receptor or enzymatic function that is usable for compound screening, and its major function is achieved via interaction with other proteins.
One potential approach is to elevate NUB1 expression. NUB1 expression could be induced by IFNβ,23 a US Food and Drug Administration-approved drug for treating multiple sclerosis,43 and thus IFNβ was studied and showed NUB1-dependent HTT lowering and neuronal protection in HD cellular models.20 Further testing of the effect of IFNβ treatment in the in vivo HD animal models using methods such as intraperitoneal injection42 may serve as a proof of concept of potential NUB1-mediated therapy. Given the many side effects of IFNβ,44–46 low-molecular-weight compounds that are easier to deliver or less toxic are more desired compared to IFNβ. Finding such compounds could be achieved by screening for transcriptional enhancers of NUB1 by minigene assays.
Another strategy is enhancing NUB1 functions. Based on the reported adaptor function of NUB1 and our data,20,30 NUB1 is likely functioning via its interaction with HTT, neddylated CUL3, and/or other key proteins in the pathway. As a result, a potential therapeutics strategy is to enhance the interaction between NUB1 and those key proteins required for the modulation. This could be achieved by low-molecular-weight-compound screening using bioluminescence resonance energy transfer technology.47 Similar examples have been successfully applied to identifying Tau–Fyn SH3 interaction inhibitors for Alzheimer’s disease.48
Alternatively, the enzymatic activity or the post-translation modification altering the enzymatic activity could potentially be targeted by small molecules as well. The key enzyme that has been identified is CUL3, of which the function needs to be elevated for mHTT lowering. Given that the activation of cullin E3 ligases is dependent on neddylation,34 the neddylation of CUL3 may provide targets for potential drug discovery. While it is challenging to identify neddylation activators, blocking deneddylation is likely a more feasible approach, which could be achieved by inhibiting the deubiquitinating enzymes (DUB) of NEDD8 (deneddylases). However, one of these enzymes (COP9 signalosome [CSN]), has opposite effects at least in many reported in vivo models,49,50 indicating a much more complicated biochemical process of NEDD8 conjugation system. It is suggested that CSN facilitates cullin–RING ubiquitin ligase complex functions possibly via stabilizing cullin proteins or substrate adaptors.51–54 Therefore, the actual effects in mHTT clearance of CSN and other deneddylases such as deneddylase 1 (DEN1),55 ubiquitin-specific peptidase 21 (USP21),56 ubiquitin C-terminal hydrolase 1 (UCH-L1),57 and ubiquitin C-terminal hydrolase 3 (UCH-L3)58 in in vivo HD models should be studied at first. Using selective DUB inhibitors of the deneddylases above such as 4, 5, 6, 7-Tetrachloroidan-1, 3-dione59 for UCH-L3, or general inhibitors such as ubiquitin aldehyde for UCH family59 and NEDD8 C-terminal vinyl sulfone for DEN1, UCH-L1 and UCH-L3,55,57 will help to identify the role of DUBs in HD and to test potential DUB inhibitor treatment, while finding druggable selective inhibitors for them is of great value, especially for DEN1 which has high selectivity for NEDD8.55 Finally, finding direct activators of CUL3 enzyme activity is also a possible strategy. A summary of different targeting strategies is shown in Table 1.
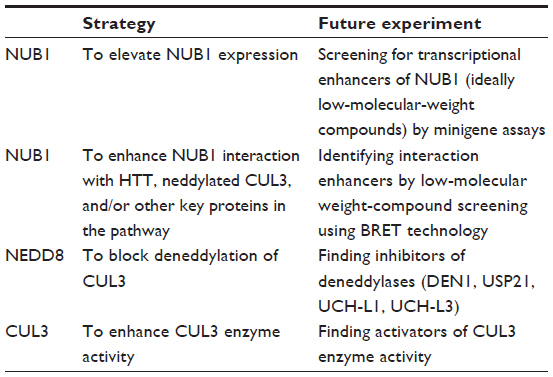 | Table 1 Targeting strategies for NUB1 |
On the other hand, one concern of using NUB1 as an entry point for drug discovery is that NUB1 clears wtHTT as well, at least in some cell lines when mHTT exists.20 When overexpressed by cDNA, NUB1 may reduce wild type HTT protein by a little more than 50%,20 which seems to be within the well-tolerated range based on many studies.4,9,60 In addition, our data have already shown that NUB1 is protective in heterozygous HD-patient iPSC-derived neurons, further confirming the safety of elevating NUB1.20 However, in vivo studies are definitely needed to further confirm the safety (Marian Difiglia, unpublished data, 2015). The preliminary data from our collaborator have shown that NUB1 overexpression by adeno-associated virus injection does not cause any damage within 6 months. Finally, study of the mechanism behind mHTT dependency of NUB1 wtHTT clearance and monitoring wtHTT loss in NUB1 treatment may be required to provide better control of mHTT clearance and the safety issue.
Conclusion
In previous studies, we have already validated NUB1 as a robust suppressor of mHTT toxicity via reducing mHTT levels.20 Here, we further discussed possible mechanisms of NUB1-mediated mHTT clearance via enhanced ubiquitination of mHTT. In addition, we discussed potential explanation of the observed potential feedback relationship between mHTT and NUB1 expression;20 we proposed the hypothesis that HD cells may activate compensatory pathways to elevate NUB1 level to delay disease progression; and we proposed that NUB1-induced clearance of HTT may depend on the activation of NUB1 function by mHTT, possibly via strengthening the interaction with other proteins (Figure 2). Finally, several targeting strategies for NUB1 were also proposed based on the potential mechanism, including elevating NUB1, enhancing NUB1 interaction with other key proteins in the pathway, and enhancing CUL3-based ligases’ function either by direct E3 activators or blocking deneddylation of CUL3 (Figure 3).
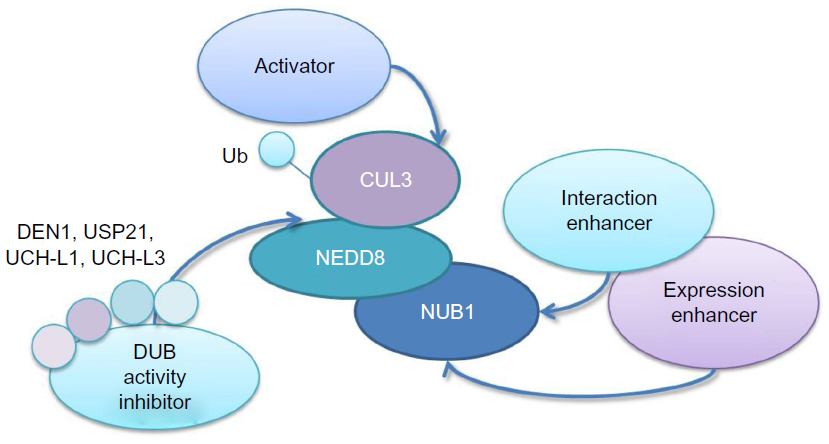 | Figure 3 Targeting strategies for NUB1. |
In summary, the discovery of NUB1 provides new insights into the strategy of lowering mHTT protein, and future experiments are required to provide further understanding of its mechanism and to enhance drug discovery.
Acknowledgments
We would like to thank National Natural Science Foundation of China (31371421, 31422024), Chinese Ministry of Science and Technology (2014AA02502), and Shanghai Pujiang Talent Plan (13PJ1400600) for funding support.
Disclosure
The authors report no conflicts of interest in this work.
References
Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. The Lancet Neurol. 2011;10:83–98. | |
Cattaneo E. Dysfunction of wild-type huntingtin in Huntington disease. News Physiol Sci. 2003;18:34–37. | |
Drouet V, Perrin V, Hassig R, et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann Neurol. 2009;65:276–285. | |
Boudreau RL, McBride JL, Martins I, et al. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther. 2009;17:1053–1063. | |
Auerbach W, Hurlbert MS, Hilditch-Maguire P, et al. The HD mutation causes progressive lethal neurological disease in mice expressing reduced levels of huntingtin. Hum Mol Genet. 2001;10:2515–2523. | |
Walker FO. Huntington’s disease. Lancet. 2007;369:218–228. | |
Vonsattel JP. Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:55–69. | |
Truant R, Atwal RS, Desmond C, Munsie L, Tran T. Huntington’s disease: revisiting the aggregation hypothesis in polyglutamine neurodegenerative diseases. FEBS J. 2008;275:4252–4262. | |
Lu B, Palacino J. A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. 2013;27:1820–1829. | |
Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci. 2005;6:919–930. | |
Kim S, Kim KT. Therapeutic approaches for inhibition of protein aggregation in Huntington’s disease. Exp Neurobiol. 2014;23:36–44. | |
Johri A, Chandra A, Beal MF. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic Biol Med. 2013;62:37–46. | |
Sorolla MA, Reverter-Branchat G, Tamarit J, Ferrer I, Ros J, Cabiscol E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic Biol Med. 2008;45:667–678. | |
Wang JQ, Chen Q, Wang X, et al. Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J Biol Chem. 2013;288:3070–3084. | |
Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015;38:26–35. | |
Gauthier LR, Charrin BC, Borrell-Pagès M, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. | |
Dompierre JP, Godin JD, Charrin BC, et al. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. | |
Yu S, Liang Y, Palacino J, Difiglia M, Lu B. Drugging unconventional targets: insights from Huntington’s disease. Trends Pharmacol Sci. 2014;35:53–62. | |
Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. | |
Lu B, Al-Ramahi I, Valencia A, et al. Identification of NUB1 as a suppressor of mutant Huntington toxicity via enhanced protein clearance. Nat Neurosci. 2013;16:562–570. | |
Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. | |
Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. | |
Kito K, Yeh ET, Kamitani T. NUB1, a NEDD8-interacting protein, is induced by interferon and down-regulates the NEDD8 expression. J Biol Chem. 2001;276:20603–20609. | |
Tanaka T, Kawashima H, Yeh ET, Kamitani T. Regulation of the NEDD8 conjugation system by a splicing variant, NUB1L. J Biol Chem. 2003;278:32905–32913. | |
Su V, Lau AF. Ubiquitin-like and ubiquitin-associated domain proteins: significance in proteasomal degradation. Cell Mol Life Sci. 2009;66:2819–2833. | |
Tanji K, Tanaka T, Mori F, et al. NUB1 suppresses the formation of Lewy body-like inclusions by proteasomal degradation of synphilin-1. Am J Pathol. 2006;169:553–565. | |
Richet E, Pooler AM, Rodriguez T, et al. NUB1 modulation of GSK3beta reduces tau aggregation. Hum Mol Genet. 2012;21:5254–5267. | |
Hosono T, Tanaka T, Tanji K, Nakatani T, Kamitani T. NUB1, an interferon-inducible protein, mediates anti-proliferative actions and apoptosis in renal cell carcinoma cells through cell-cycle regulation. Br J Cancer. 2010;102:873–882. | |
Kaltenbach LS, Romero E, Becklin RR, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007;3:e82. | |
Kamitani T, Kito K, Fukuda-Kamitani T, Yeh ET. Targeting of NEDD8 and its conjugates for proteasomal degradation by NUB1. J Biol Chem. 2001;276:46655–46660. | |
Tanaka T, Yeh ET, Kamitani T. NUB1-mediated targeting of the ubiquitin precursor UbC1 for its C-terminal hydrolysis. Eur J Biochem. 2004;271:972–982. | |
Tanji K, Tanaka T, Kamitani T. Interaction of NUB1 with the proteasome subunit S5a. Biochem Biophys Res Commun. 2005;337:116–120. | |
Tanaka T, Nakatani T, Kamitani T. Inhibition of NEDD8-conjugation pathway by novel molecules: potential approaches to anticancer therapy. Mol Oncol. 2012;6:267–275. | |
Rabut G, Peter M. Function and regulation of protein neddylation. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:969–976. | |
Hipp MS, Raasi S, Groettrup M, Schmidtke G. NEDD8 ultimate buster-1L interacts with the ubiquitin-like protein FAT10 and accelerates its degradation. J Biol Chem. 2004;279:16503–16510. | |
Schmidtke G, Kalveram B, Weber E, et al. The UBA domains of NUB1L are required for binding but not for accelerated degradation of the ubiquitin-like modifier FAT10. J Biol Chem. 2006;281:20045–20054. | |
Pan ZQ, Kentsis A, Dias DC, Yamoah K, Wu K. Nedd8 on cullin: building an expressway to protein destruction. Oncogene. 2004;23:1985–1997. | |
Wimuttisuk W, Singer JD. The Cullin3 ubiquitin ligase functions as a Nedd8-bound heterodimer. Mol Biol Cell. 2007;18:899–909. | |
Choo YY, Hagen T. Mechanism of cullin3 E3 ubiquitin ligase dimerization. PLoS One. 2012;7:e41350. | |
Metzger T, Kleiss C, Sumara I. CUL3 and protein kinases: insights from PLK1/KLHL22 interaction. Cell Cycle. 2013;12:2291–2296. | |
Tanji K, Mori F, Kito K, et al. Synphilin-1-binding protein NUB1 is colocalized with nonfibrillar, proteinase K-resistant α-synuclein in presynapses in Lewy body disease. J Neuropathol Exp Neurol. 2011;70:879–889. | |
Chort A, Alves S, Marinello M, et al. Interferon β induces clearance of mutant ataxin 7 and improves locomotion in SCA7 knock-in mice. Brain. 2013;136:1732–1745. | |
Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet. 2001;357:1576–1582. | |
Goëb J, Cailleau A, Lainé P, et al. Acute delirium, delusion, and depression during IFN-beta-1a therapy for multiple sclerosis: a case report. Clin Neuropharmacol. 2003;26:5–7. | |
Kremenchutzky M, Morrow S, Rush C. The safety and efficacy of IFN-beta products for the treatment of multiple sclerosis. Expert Opin Drug Saf. 2007;6:279–288. | |
Leuschen MP, Filipi M, Healey K. A randomized open label study of pain medications (naproxen, acetaminophen and ibuprofen) for controlling side effects during initiation of IFN beta-1a therapy and during its ongoing use for relapsing-remitting multiple sclerosis. Mult Scler. 2004;10:636–642. | |
Boute N, Jockers R, Issad T. The use of resonance energy transfer in high-throughput screening: BRET versus FRET. Trends Pharmacol Sci. 2002;23:351–354. | |
Cochran JN, Diggs PV, Nebane NM, et al. AlphaScreen HTS and live-cell bioluminescence resonance energy transfer (BRET) assays for identification of Tau-Fyn SH3 interaction inhibitors for Alzheimer disease. J Biomol Screen. 2014;19:1338–1349. | |
Schwechheimer C, Deng XW. COP9 signalosome revisited: a novel mediator of protein degradation. Trends Cell Biol. 2001;11:420–426. | |
Pintard L, Kurz T, Glaser S, Willis JH, Peter M, Bowerman B. Neddylation and deneddylation of CUL-3 is required to target MEI-1/Katanin for degradation at the meiosis-to-mitosis transition in C. elegans. Curr Biol. 2003;13:911–921. | |
Wee S, Geyer RK, Toda T, Wolf DA. CSN facilitates Cullin-RING ubiquitin ligase function by counteracting autocatalytic adapter instability. Nat Cell Biol. 2005;7:387–391. | |
Wu JT, Chan YR, Chien CT. Protection of cullin-RING E3 ligases by CSN-UBP12. Trends Cell Biol. 2006;16:362–369. | |
Wu JT, Lin HC, Hu YC, Chien CT. Neddylation and deneddylation regulate Cul1 and Cul3 protein accumulation. Nat Cell Biol. 2005;7:1014–1020. | |
Enchev RI, Scott DC, da Fonseca PC, et al. Structural basis for a reciprocal regulation between SCF and CSN. Cell Rep. 2012;2:616–627. | |
Gan-Erdene T, Nagamalleswari K, Yin L, Wu K, Pan ZQ, Wilkinson KD. Identification and characterization of DEN1, a deneddylase of the ULP family. J Biol Chem. 2003;278:28892–28900. | |
Gong L, Kamitani T, Millas S, Yeh ET. Identification of a novel isopeptidase with dual specificity for ubiquitin- and NEDD8-conjugated proteins. J Biol Chem. 2000;275:14212–14216. | |
Hemelaar J, Borodovsky A, Kessler BM, et al. Specific and covalent targeting of conjugating and deconjugating enzymes of ubiquitin-like proteins. Mol Cell Biol. 2004;24:84–95. | |
Wada H, Kito K, Caskey LS, Yeh ET, Kamitani T. Cleavage of the C-terminus of NEDD8 by UCH-L3. Biochem Biophys Res Commun. 1998;251:688–692. | |
Mtango NR, Sutovsky M, Vandevoort CA, Latham KE, Sutovsky P. Essential role of ubiquitin C-terminal hydrolases UCHL1 and UCHL3 in mammalian oocyte maturation. J Cell Physiol. 2012;227:2022–2029. | |
Kordasiewicz HB, Stanek LM, Wancewicz EV, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74:1031–1044. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.