")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 14
Novel Treatments from Inhibition of the Intestinal Sodium–Hydrogen Exchanger 3
Authors Kovesdy CP, Adebiyi A , Rosenbaum D, Jacobs JW, Quarles LD
Received 20 August 2021
Accepted for publication 11 November 2021
Published 1 December 2021 Volume 2021:14 Pages 411—420
DOI https://doi.org/10.2147/IJNRD.S334024
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Csaba P Kovesdy,1 Adebowale Adebiyi,2 David Rosenbaum,3 Jeffrey W Jacobs,3 L Darryl Quarles1
1Division of Nephrology, University of Tennessee Health Science Center, Memphis, TN, USA; 2Department of Physiology, University of Tennessee Health Science Center, Memphis, TN, USA; 3Medical Affairs, Ardelyx, Inc., Boston, MA, USA
Correspondence: Csaba P Kovesdy
University of Tennessee Health Science Center, Memphis VA Medical Center, 956 Court Avenue, Room B222, Memphis, TN, 38163, USA
Tel +901 448-2985
Email [email protected]
Abstract: Plasma membrane sodium–hydrogen exchangers (NHE) transport Na+ into cells in exchange for H+. While there are nine isoforms of NHE in humans, this review focuses on the NHE3 isoform, which is abundantly expressed in the gastrointestinal tract, where it plays a key role in acid–base balance and water homeostasis. NHE3 inhibition in the small intestine results in luminal sodium and water retention, leading to a general decrease in paracellular water flux and diffusional driving force, reduced intestinal sodium absorption, and increased stool sodium excretion. The resulting softer and more frequent stools are the rationale for the development of tenapanor as a novel, first-in-class NHE3 inhibitor to treat irritable bowel syndrome with constipation. NHE3 also has additional therapeutic implications in nephrology. Inhibition of intestinal NHE3 also lowers blood pressure by reducing intestinal sodium absorption. Perhaps, the most novel effect is its ability to decrease intestinal phosphate absorption by inhibiting the paracellular phosphate absorption pathway. Therefore, selective pharmacological inhibition of NHE3 could be a potential therapeutic strategy to treat not only heart failure and hypertension but also hyperphosphatemia. This review presents an overview of the molecular and physiological functions of NHE3 and discusses how these functions translate to potential clinical applications in nephrology.
Keywords: sodium–hydrogen exchanger 3, sodium–hydrogen exchanger 3 inhibitors, paracellular phosphate absorption pathway, hyperphosphatemia, chronic kidney disease, heart failure
Introduction: The Role of NHE3
Sodium–hydrogen antiporters or sodium–proton exchangers (NHE), of which there are nine total isoforms in humans, are a class of both plasma and organellar membrane proteins that transport Na+ into the cell and H+ out of the cell, as well as regulate luminal pH and cation composition of the intracellular compartments.1 Isoforms of NHE have different tissue and subcellular distributions: some NHEs function primarily on the plasma membrane, and others are present on intracellular organelles.1 NHE1 is expressed in the plasma membrane of virtually all tissues2 and plays an important role in cardiac health.3 NHE2 is expressed in the gastrointestinal (GI) tract and contributes to the maintenance of stomach pH and homeostasis, whereas NHE3 predominates in the GI tract and kidneys.4,5 NHE4 and NHE5 are abundant in the stomach and brain, respectively.6,7 NHE6-9 are expressed in the intracellular compartments of many organ system membranes, but of these NHE8 is the exception and expressed apically.1,8–10
NHE3 differs from other isoforms as it continuously recycles between the apical membrane of the epithelial and endosomal compartment, playing a key role in water and sodium homeostasis.11 The physiological roles of NHE3 make it an attractive target for developing pharmacological inhibitors, which may be effective novel treatments for hypertension, constipation, diabetes mellitus, electrolyte disturbances (hyperphosphatemia), and congestive heart failure.
This review aims to present the molecular and physiological functions of NHE3 and how this information translates to clinical practice. The discussion will focus on the significance of NHE3 as a target for disease modification and recent advancements in the understanding of hyperphosphatemia and phosphate management in chronic kidney disease (CKD) patients.
Structure of the NHE3 Protein
All human NHE isoforms have similar structures, consisting of two functionally distinct domains.12 However, the amino acid make-up of both domains varies among the NHE isoforms. An N-terminal of NHE3 is made up of 454 amino acids.13,14 It is a 12 pass transmembrane domain that mediates ion exchange and the cytosolic C-terminus of 376 amino acids, regulates the transport rate, and interacts with the cytoskeleton and other ancillary molecules.13,14 Separate regions of the C-terminal have been described as the “switch domain” for their role in setting NHE3 activity.15,16 The C-terminal domain of NHE3 also binds to multiple proteins involved in NHE3 regulation.16 Studies suggest that these regulatory proteins may form organized complexes that interact with NHE3 and with each other, thus stimulating or inhibiting the NHE3 activity.17,18
Evolution of NHE3 Across Species
NHE originated as intracellular exchangers first seen in yeast, slime mold, and plant species.19 Of the two NHE subgroups, plasma membrane and intracellular, NHE3 genes belong to the plasma membrane subgroup.19 Human NHE3 genes have their origins in the worm NHX2 gene, known to be the first evolved plasma membrane NHE.19 Studies in the C. elegans have shown that the ortholog of mammalian NHE3, CeNHX2, is recycled on and off the apical membrane of the gut epithelial cells.20 This suggests that the emergence of CeNHX2 was necessary to maintain the proton gradient required for nutrient absorption.20
Genetics and Gain/Loss of Functions
NHE3, which is encoded by the SLC9A3 gene, plays a critical role in the absorption of sodium and fluids and regulates acid–base homeostasis.21 Studies of NHE3 knockout mice showed that lack of NHE3 results in reduced intestinal structural integrity, causing the animals to develop alkaline diarrhea, hyponatremia, and metabolic acidosis.21 A study of individuals with congenital sodium diarrhea found that nine participants from unrelated families had an absent or mutated NHE3 protein, as demonstrated by SLC9A3 mutations.22 Congenital sodium diarrhea is characterized by watery diarrhea after birth, high fecal loss of sodium, and metabolic acidosis.23 Thus, loss or marked reduction of NHE3 function in humans was observed to have similar effects as those observed in NHE3 knockout mice.21,22 To our knowledge, there are no models for a gain of function.
Mechanisms of NHE3 Regulation
NHE3 regulation falls into two forms: acute and chronic.24–27 Acute regulation is a rapid, reversible process that occurs within minutes or hours.28,29 NHE3 contains multiple phosphorylation sites,27,30 and activity is modulated by protein kinases, including protein kinase A (PKA)27 and serum- and glucocorticoid-inducible kinase 1 (SGK1).30 Trafficking is another method of acute regulation. NHE3 differs from other NHE isoforms in that it can travel between the plasma membrane and intracellular compartments, and trafficking is the temporary compartmentalization of transport activity.11 NHE3 is also capable of dynamic interaction with numerous proteins, and its activity can be modulated by protein-to-protein association.17,31 Chronic NHE3 regulation, accounted for by transcriptional regulation, is an ongoing process that often involves multiple overlapping pathways.32,33 Glucocorticoids, aldosterone, metabolic acidosis, serotonin, and proinflammatory cytokines have been shown to modify NHE3 expression.32,34–36 There may also be a metabolic role in NHE3 activity. One example is the stimulation of renal NHE3 by fructose, which increases sodium reabsorption in proximal tubules.37 Fructose can also sensitize the proximal tubule to angiotensin II.38 These data may partially explain the mechanism by which a fructose diet induces renal injury and hypertension.37,38 Additionally, chronic metabolic acidosis has been shown to increase NHE3 protein abundance in an animal model.39
Physiological Properties of NHE3
In addition to the kidney, which plays a major role in maintaining water and sodium balances, the importance of the GI tract in the regulation of sodium balance is increasingly recognized.40,41 Expressed in the intestinal tract, kidney, and gallbladder, NHE3 is key to intestinal sodium and water absorption.4,5,42 Present most abundantly in the GI tract and the kidneys, NHE3 helps the kidneys in their primary function of maintaining water and sodium homeostasis in the body through regulation of sodium absorption.5 Key physiological functions of the NHE3 pathway include absorption of sodium, HCO3, NH4+, and water, and modulating the absorption of other nutrients (eg, dipeptides and amino acids) through the H+ gradient.24,43–46 The contribution of NHE3 to maintaining sodium homeostasis and acid–base balance is supported by recent results from an NHE3 knockout mouse model.21
However, NHE3 may be more than a sodium transporter – some studies have suggested an ability to modify other genes. In humans, NHE3 expression is decreased in patients with inflammatory bowel disease.47 NHE3 knockout mice overexpress genes that induce the proinflammatory cytokines that target NHE3 as part of a homeostatic response to impaired sodium absorption.48 In animal models, NHE3 deficiency compromises immune response and may be linked to changes to genes related to stress and inflammation.48,49 Microarray analysis of NHE3 knockout mice showed that genes involved in response to stress, inflammation, and chemotaxis were altered, in addition to genes of ion transporters and ion channels.49 NHE3 knockout mice have also been shown to have a compromised innate immune response.48 This ability is consistent with studies showing changes in the expression level of genes in various brain regions in NHE1 knockout mice.50
Physiology of NHE3 Inhibition
NHE3 inhibition results in luminal sodium and water retention, leading to a general decrease in paracellular water flux and diffusional driving force.51 Reduced intestinal sodium absorption and increased stool sodium excretion lead to modest intracellular proton retention that is proposed to induce a pH-sensitive conformational change in claudin proteins, directly reducing paracellular permeability specific to phosphate through the tight junction.51 These physiological effects have been shown to translate to softer and more frequent stools, decreased blood pressure, and reduced phosphate concentrations.52
The role of NHE3 in maintaining sodium and water homeostasis is confirmed by pharmacological data from NHE3 inhibitor trials. The importance of NHE3 for intestinal sodium absorption is supported by reduced intestinal sodium absorption and increased renal sodium reabsorption in animal studies of a systemic NHE3 inhibitor for the treatment of hypertension.53 These effects were consistent with reductions in urinary sodium and increases in fecal sodium seen in both animal and human trials of another NHE3 inhibitor.54 Studies of intestinal epithelial cellular models demonstrated that an NHE3 inhibitor blocks NHE3-mediated proton efflux, resulting in a rapid (<1 minute) reduction in intracellular pH and a correspondingly rapid increase in transepithelial electrical resistance, which inhibits proton secretion coupled to sodium absorption by NHE3.51 There are, to the best of our knowledge, no disputes on the differing roles of NHE3 in intestinal sodium absorption in animals and humans.
In contrast to sodium, the impact of genetic deletion of NHE3 on phosphate absorption in mice have been variable. Pan and coworkers reported that NHE knockout in mice results in a large and significant decrease in urinary phosphate excretion,55 which would be consistent with reduced intestinal absorption. In contrast, Xue and coworkers did not report changes in phosphate absorption or excretion in their NHE3 knockout mouse model,21 but this does not necessarily have serious implications for the understanding of NHE3 in humans; results from animal models of NaPi2b did not correspond to those seen in human trials,56,57 and it is possible that NHE3 inhibitors are interacting with other pathways not present in animals.
Disease Targets for NHE3 Inhibition
The importance of sodium and water homeostasis for the proper functioning of multiple biological systems58 makes NHE3 a potential therapeutic target for numerous disease states important to nephrologists,53,59,60 such as hypertension, heart failure, diabetes mellitus, constipation, and hyperphosphatemia (Table 1).
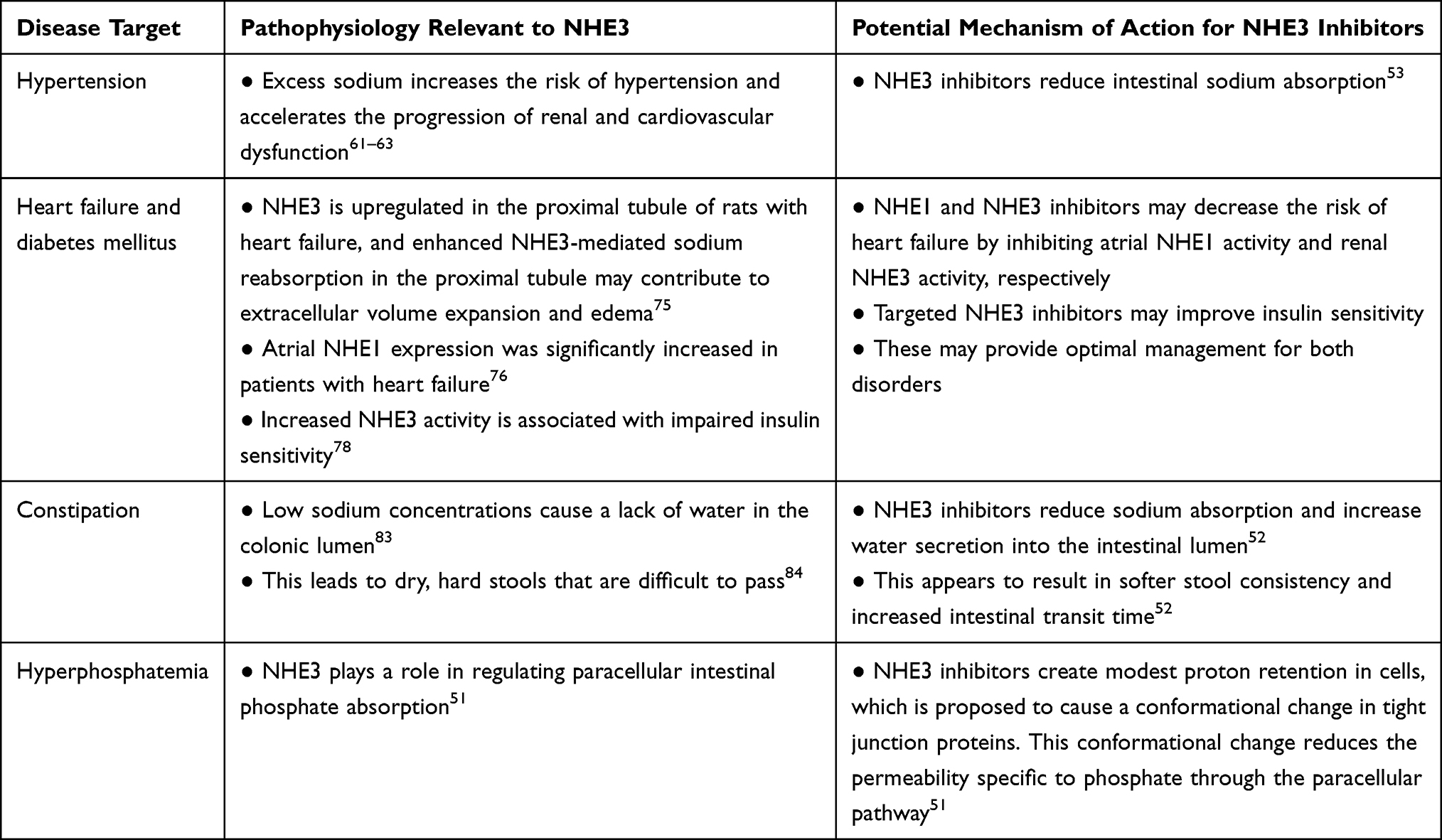 |
Table 1 Disease Targets for Sodium–Hydrogen Exchanger 3 (NHE3) |
Hypertension: Excess sodium increases the risk of hypertension and accelerates the progression of renal and cardiovascular dysfunction.61–63 Reduction of GI sodium absorption through NHE3 inhibition may be an effective method of combating hypertension and associated negative clinical outcomes.53,60 The natriuretic effects of the proximal tubule SGLT2 inhibitor empagliflozin, which lowers blood pressure, may be determined by NHE3.64 A study of the NHE3 inhibitor SAR218034 found that the investigational drug decreased sodium absorption in the gut and substantially reduced systolic blood pressure in rats.60 Tenapanor, a novel targeted NHE3 inhibitor, has been shown to reduce blood pressure, fluid volume, and left ventricular hypertrophy in salt-fed nephrectomized rats.54 Although human studies have been conducted,54 the impact on hypertension has not been published yet as of the time of this paper.
Diabetes: Diabetic patients may also benefit from NHE3 inhibitors. In vitro study of both human and mouse jejunums found that NHE3 knockdown suppressed glucose uptake via sodium-glucose co-transporter 1 (SGLT1), implying an essential role of NHE3 in small intestinal glucose absorption.65 Therefore, a potential clinical application of NHE3 inhibition is the treatment of type 2 diabetes by reducing intestinal glucose uptake.65 Given that type 2 diabetes is almost 2.5 times as likely to develop in patients with hypertension compared to those with normal blood pressure,66 diabetic patients may also benefit from the ability of NHE3 inhibitors to combat high blood pressure.
Heart failure and diabetes mellitus: Heart failure and diabetes mellitus are common, intertwined comorbidities.67,68 Diabetes is an independent risk factor for heart failure,69 and insulin resistance is associated with risk of heart failure.70 The insulin insensitivity and adipokine abnormalities that characterize diabetes contribute to heart failure,70,71 and neurohormonal systems activated in heart failure (eg, norepinephrine, angiotensin II) negatively impact insulin sensitivity and microvascular disease in diabetes.72,73 It is thought that a mechanism connecting these physiological activities is increased activities of NHE3 and NHE1.74 Inoue et al found that NHE3 is upregulated in the proximal tubule of rats with heart failure by transcriptional, translational, and posttranslational mechanisms.75 The study also suggested that enhanced NHE3-mediated sodium reabsorption in the proximal tubule may contribute to extracellular volume expansion and edema (hallmarks of heart failure).75 Compared with patients without heart failure, atrial NHE1 expression was significantly increased in patients with heart failure with preserved ejection fraction and atrial fibrillation.76 Impaired insulin sensitivity (linked to the progression of diabetes)77 is associated with increased NHE3 activity.78 Therapies that disrupt both NHE1 and NHE3 had a substantially greater impact on heart failure in diabetes patients in comparison to other factors like myocardial infarction or stroke.76,79,80 A potential logical explanation for this effect is that such therapies are particularly effective when both NHE isoforms are upregulated. Empagliflozin, an SGLT2 inhibitor, inhibits NHE in human cardiomyocytes.76 In patients with type 2 diabetes and high cardiovascular risk, empagliflozin reduced heart failure hospitalization and cardiovascular death, with a consistent benefit in patients with and without baseline heart failure.81 Sodium glucose cotransporter 2 may functionally interact with NHE3 in the proximal tubule, suggesting that SGLT2 inhibition could be an effective treatment for blood glucose effect-dependent and independent nephroprotective actions and reduction and cardiovascular mortality.82 Therefore, it is logical to further explore the potential for systemic NHE inhibitors to decrease the risk of heart failure and improve glucose tolerance.74
Constipation: The mechanisms that lead to constipation make this complication another target for treatment with NHE3 inhibitors. Lack of water in the colonic lumen as a result of low sodium concentrations leads to dry, hard stools that are difficult to pass.83–85 NHE3 inhibitors reduce sodium absorption in the gut, leading to increased water secretion into the intestinal lumen.52,86 This may accelerate intestinal transit time and result in softer stool consistency.52 An NHE3 inhibitor has been shown to increase stool sodium excretion and soften stool consistency in human trials; the effects of irritable bowel syndrome with constipation (IBS-C), a common GI condition characterized by abdominal pain and prolonged GI transit, were alleviated in trial participants.87,88 The NHE3 inhibitor was well-tolerated, and diarrhea of mild-to-moderate severity was the most common adverse event (13.3%).88 This therapy was approved by the FDA in September of 2019.89
Hyperphosphatemia: Over 80% of the patients with CKD on dialysis require treatment for hyperphosphatemia,90 a condition associated with negative clinical outcomes.91 Kestenbaum et al found that serum phosphate concentrations >3.5 mg/dL were associated with a significantly increased risk of death in CKD patients, and mortality risk increased linearly with each subsequent 0.5 mg/dL increase in serum phosphate concentrations.92 Phosphate retention also drives increases in FGF23,93 which is independently associated with all-cause mortality and heart failure,94 and increases in PTH,93 which is predictive of cardiovascular mortality.95 Tenapanor is a first-in-class, minimally absorbed, small-molecule NHE3 inhibitor with a unique mechanism of action that effectively reduces phosphate levels.51 Inhibition of NHE3 by this therapy reduced proton secretion coupled with sodium absorption, resulting in reduced sodium absorption and modest proton retention in the cells.51 This modest intracellular proton retention generated is proposed to modulate tight junction proteins (claudins) resulting in increased transepithelial electrical resistance (TEER) and reducing permeability specific to phosphate, thereby decreasing phosphate absorption through the paracellular pathway.51 Models of NHE3 knockout cells demonstrated that the effect of tenapanor on transepithelial electrical resistance and the paracellular pathway is due exclusively to on-target inhibition of NHE3 and is likely due to decreased intracellular pH.51 Onset of increased TEER and reduced paracellular phosphate permeability is near-immediate.51 (Figure 1) When administered orally to rats, tenapanor acted exclusively in the GI tract. The systemic availability of tenapanor was well below its in vitro potency of 5 nM when tested through plasma pharmacokinetic studies, as well as autoradiography and mass balance studies performed with (14) C-tenapanor.54 The plasma tenapanor levels were below the limit of quantification in >99% of the patients on dialysis receiving tenapanor.96 Thus, tenapanor likely does not inhibit NHE3 in the kidney. Tenapanor acts specifically on sodium and phosphate and does not affect the overall balance of other ions such as potassium, calcium, and magnesium.54 The in vivo specificity of tenapanor is highlighted by its lack of impact on chloride, an anion that likely has similar paracellular permeability characteristic to phosphate.51
 |
Figure 1 An NHE3 blocker reduces intestinal phosphate absorption through the paracellular pathway. (A) The intestinal paracellular phosphate absorption pathway: The sodium–hydrogen exchanger isoform 3 (NHE3) promotes the exchange of one sodium ion for one proton across the cell membrane.5 The paracellular pathway, which is favored by electrical and chemical gradients, mediates the majority of intestinal phosphate absorption in humans.51,109 Phosphate ions move down their electrical and chemical concentration gradients and are absorbed through the tight junction (TJ) complexes.51,109. (B) Effect of an NHE3 blocker on the intestinal paracellular phosphate absorption pathway: Treatment with an NHE3 blocker inhibits sodium–proton exchange (1), leading to a modest intracellular proton accumulation.51 This is proposed to cause conformational changes in tight junction proteins (2) and a consequent increase in transepithelial electrical resistance (TEER) that reduces paracellular permeability specific to phosphate (3)51. |
Tenapanor effectively reduced phosphate levels in multiple clinical trials.97–100 At 12 weeks, tenapanor administration lowered serum phosphorus from baseline concentrations of 8.1 mg/dL to 5.5 mg/dL in the efficacy analysis set.97 In a long-term Phase 3 study, tenapanor administration lowered serum phosphorus in subjects from baseline concentrations of 7.7 mg/dL to 5.1 mg/dL at 26 weeks in the efficacy analysis set.98 A recent trial that compared the effectiveness of a combination of tenapanor and binder vs placebo and binder showed that tenapanor plus binder resulted in a 0.65 mg/dL larger mean serum phosphate reduction from baseline compared to placebo plus binder.100 The study included 236 patients undergoing maintenance dialysis with hyperphosphatemia (defined in this trial as serum phosphorus 5.5–10 mg/dL inclusive) despite receiving phosphate binder therapy (sevelamer, nonsevelamer, sevelamer plus nonsevelamer, or multiple nonsevelamer binders).100 Almost twice as many patients treated with tenapanor and binder achieved phosphate <5.5 mg/dL compared to patients treated with placebo and binder (37–50% vs 18–24%, p<0.05).100 This dual-mechanism approach may be particularly relevant for patients with persistent hyperphosphatemia.100 Adverse events were largely limited to softened stool and a modest increase in bowel movement frequency, consistent with tenapanor’s mechanism of action that increases stool sodium and water content.97
Phosphate homeostasis is intricately connected to the regulation of hormones and minerals affected in CKD mineral and bone disorder, such as vitamin D, calcium, fibroblast growth factor-23, and parathyroid hormone.101,102 Thus, phosphate control via NHE3 inhibition may also have the potential to improve morbidity and mortality related to CKD-mineral and bone disorder.103 Reduction of phosphate levels via NHE3 inhibition in the GI tract could also protect against vascular calcification,104 a common complication of CKD associated with significant morbidity and mortality.105,106 The effects of NHE3 inhibition and of phosphate lowering therapies in general on clinical outcomes will need to be examined in randomized controlled trials.
Summary and Future Directions
There is a gap between the latest biological understanding of the effects of NHE3 and clinical practice/therapy development.51,54,107,108 Leveraging the latest understanding of the importance of NHE3 for multiple disease states may help researchers and clinicians achieve better patient outcomes. Inhibition of GI NHE3 results in increased sodium and water excretion as well as reduced paracellular permeability to phosphate. The effects of NHE3 inhibition translate to a range of systemic changes, including softer and more frequent stools, decreased blood pressure, and reduced phosphate concentrations. Reduced sodium absorption via NHE3 inhibition may be an effective treatment for hypertension, constipation, diabetes mellitus, and congestive heart failure. The clinical utility of NHE3 inhibition in these disease states will need to be tested in clinical trials. Targeted NHE3 inhibition has the potential to be particularly impactful for the treatment of hypertension, constipation, and achieving and maintaining normal phosphate homeostasis.
More broadly, the role of the GI tract in maintaining systemic homeostasis, alongside the kidney, should be considered in clinical practice and research as it is established that the GI tract is a site of absorption for solutes such as phosphate and sodium as well as water and acid–base balance. The connection between the GI tract and kidney and consideration of the GI tract as a site of focus to offload some of the kidneys’ efforts certainly warrant further exploration.
Statement of Ethics
In this review article, no new research study was conducted that prospectively assigns human participants or groups of humans to one or more health-related interventions, and therefore, no patients were enrolled or subjected to therapies. Thus, there are no requirements for any ethical approval or informed consent. The process of developing this article complies with internationally accepted standards for research practice and reporting.
Acknowledgments
Writing support was provided by Xelay Acumen Group.
Funding
Writing support was funded by Ardelyx, Inc.
Disclosure
Dr Csaba P Kovesdy is a consultant for Abbott, Akebia, Amgen, Ardelyx, Astra-Zeneca, Bayer, Cara Therapeutics, Rockwell, Takeda, Tricida and Vifor. Dr Jeffery W Jacobs and Dr David Rosenbaum are employees of Ardelyx, Inc. In addition, Dr Jeffrey W Jacobs has a patent US8541448B2 issued to Ardelyx. The authors report no other conflicts of interest in this work.
References
1. Nakamura N, Tanaka S, Teko Y, Mitsui K, Kanazawa H. Four Na+/ H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J Biol Chem. 2005;280(2):1561–1572. doi:10.1074/jbc.M410041200
2. SLC9A1. The human protein atlas. Available from: https://www.proteinatlas.org/ENSG00000090020-SLC9A1/cell.
3. Engelhardt S, Hein L, Keller U, Klämbt K, Lohse MJ. Inhibition of Na(+)-H(+) exchange prevents hypertrophy, fibrosis, and heart failure in beta(1)-adrenergic receptor transgenic mice. Circ Res. 2002;90(7):814–819. doi:10.1161/01.RES.0000014966.97486.C0
4. Hoogerwerf WA, Tsao SC, Devuyst O, et al. NHE2 and NHE3 are human and rabbit intestinal brush-border proteins. Am J Physiol. 1996;270(1 Pt 1):G29–41. doi:10.1152/ajpgi.1996.270.1.G29
5. Schultheis PJ, Clarke LL, Meneton P, et al. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat Genet. 1998;19(3):282–285. doi:10.1038/969
6. Pizzonia JH, Biemesderfer D, Abu-Alfa AK, et al. Immunochemical characterization of Na+/H+exchanger isoform NHE4. Am J Physiol Renal Physiol. 1998;275(4):F510–F517. doi:10.1152/ajprenal.1998.275.4.F510
7. Baird NR, Orlowski J, Szabó EZ, et al. Molecular cloning, genomic organization, and functional expression of Na+/H+ exchanger isoform 5 (NHE5) from human brain. J Biol Chem. 1999;274(7):4377–4382. doi:10.1074/jbc.274.7.4377
8. Numata M, Orlowski J. Molecular cloning and characterization of a novel (Na+, K+)/H+ exchanger localized to the trans-Golgi network. J Biol Chem. 2001;276(20):17387–17394. doi:10.1074/jbc.M101319200
9. Brett CL, Wei Y, Donowitz M, Rao R. Human Na(+)/H(+) exchanger isoform 6 is found in recycling endosomes of cells, not in mitochondria. Am J Physiol Cell Physiol. 2002;282(5):C1031–1041. doi:10.1152/ajpcell.00420.2001
10. Zhang J, Bobulescu IA, Goyal S, Aronson PS, Baum MG, Moe OW. Characterization of Na+/H+ exchanger NHE8 in cultured renal epithelial cells. Am J Physiol Renal Physiol. 2007;293(3):F761–F766. doi:10.1152/ajprenal.00117.2007
11. D’Souza S, Garcia-Cabado A, Yu F, et al. The epithelial sodium-hydrogen antiporter Na+/H+ exchanger 3 accumulates and is functional in recycling endosomes. J Biol Chem. 1998;273(4):2035–2043. doi:10.1074/jbc.273.4.2035
12. Wakabayashi S, Shigekawa M, Pouyssegur J. Molecular physiology of vertebrate Na+/H+ exchangers. Physiol Rev. 1997;77(1):51–74. doi:10.1152/physrev.1997.77.1.51
13. Alexander RT, Grinstein S. Tethering, recycling and activation of the epithelial sodium-proton exchanger, NHE3. J Exp Biol. 2009;212(Pt 11):1630–1637. doi:10.1242/jeb.027375
14. Zizak M, Cavet ME, Bayle D, et al. Na(+)/H(+) exchanger NHE3 has 11 membrane spanning domains and a cleaved signal peptide: topology analysis using in vitro transcription/translation. Biochemistry. 2000;39(27):8102–8112. doi:10.1021/bi000870t
15. Donowitz M, Mohan S, Zhu CX, et al. NHE3 regulatory complexes. J Exp Biol. 2009;212(Pt 11):1638–1646. doi:10.1242/jeb.028605
16. Levine SA, Nath SK, Yun CHC, et al. Separate C-terminal domains of the epithelial specific brush border Na+//H+ exchanger isoform NHE3 are involved in stimulation and inhibition by protein kinases/growth factors. J Biol Chem. 1995;270(23):13716–13725. doi:10.1074/jbc.270.23.13716
17. Li X, Zhang H, Cheong A, et al. Carbachol regulation of rabbit ileal brush border Na+ -H + exchanger 3 (NHE3) occurs through changes in NHE3 trafficking and complex formation and is Src dependent. J Physiol. 2004;556(Pt 3):791–804. doi:10.1113/jphysiol.2004.060921
18. Cha B, Kenworthy A, Murtazina R, Donowitz M. The lateral mobility of NHE3 on the apical membrane of renal epithelial OK cells is limited by the PDZ domain proteins NHERF1/2, but is dependent on an intact actin cytoskeleton as determined by FRAP. J Cell Sci. 2004;117(Pt 15):3353–3365. doi:10.1242/jcs.01180
19. Brett CL, Donowitz M, Rao R. Evolutionary origins of eukaryotic sodium/proton exchangers. Am J Physiol Cell Physiol. 2005;288(2):C223–239. doi:10.1152/ajpcell.00360.2004
20. Nehrke K. A reduction in intestinal cell pHi due to loss of the Caenorhabditis elegans Na+/H+ exchanger NHX-2 increases life span. J Biol Chem. 2003;278(45):44657–44666. doi:10.1074/jbc.M307351200
21. Xue J, Thomas L, Tahmasbi M, et al. An inducible intestinal epithelial cell-specific NHE3 knockout mouse model mimicking congenital sodium diarrhea. Clin Sci (Lond). 2020;134(8):941–953. doi:10.1042/CS20200065
22. Janecke AR, Heinz-Erian P, Yin J, et al. Reduced sodium/proton exchanger NHE3 activity causes congenital sodium diarrhea. Hum Mol Genet. 2015;24(23):6614–6623. doi:10.1093/hmg/ddv367
23. Holmberg C, Perheentupa J. Congenital Na+ diarrhea: a new type of secretory diarrhea. J Pediatr. 1985;106(1):56–61. doi:10.1016/S0022-3476(85)80465-0
24. He P, Yun CC. Mechanisms of the regulation of the intestinal Na+/H+ exchanger NHE3. J Biomed Biotechnol. 2010;2010:238080. doi:10.1155/2010/238080
25. Ikuma M, Kashgarian M, Binder HJ, Rajendran VM. Differential regulation of NHE isoforms by sodium depletion in proximal and distal segments of rat colon. Am J Physiol Gastrointest Liver Physiol. 1999;276(2):G539–G549. doi:10.1152/ajpgi.1999.276.2.G539
26. Lamprecht G, Weinman EJ, Yun CHC. The Role of NHERF and E3KARP in the cAMP-mediated Inhibition of NHE3 *. J Biol Chem. 1998;273(45):29972–29978. doi:10.1074/jbc.273.45.29972
27. Zhao H, Wiederkehr MR, Fan L, Collazo RL, Crowder LA, Moe OW. Acute inhibition of Na/H exchanger NHE-3 by cAMP. Role of protein kinase a and NHE-3 phosphoserines 552 and 605. J Biol Chem. 1999;274(7):3978–3987. doi:10.1074/jbc.274.7.3978
28. Janecki AJ, Montrose MH, Zimniak P, et al. Subcellular redistribution is involved in acute regulation of the brush border Na+/H+ exchanger isoform 3 in human colon adenocarcinoma cell line caco-2: PROTEIN KINASE C-MEDIATED INHIBITION OF THE EXCHANGER *. J Biol Chem. 1998;273(15):8790–8798. doi:10.1074/jbc.273.15.8790
29. Zhang Y, Magyar CE, Norian JM, Holstein-Rathlou N-H, Mircheff AK, McDonough AA. Reversible effects of acute hypertension on proximal tubule sodium transporters. Am J Physiol-Cell Physiol. 1998;274(4):C1090–C1100. doi:10.1152/ajpcell.1998.274.4.C1090
30. Wang D, Sun H, Lang F, Yun CC. Activation of NHE3 by dexamethasone requires phosphorylation of NHE3 at Ser663 by SGK1. Am J Physiol Cell Physiol. 2005;289(4):C802–810. doi:10.1152/ajpcell.00597.2004
31. Yun CH, Oh S, Zizak M, et al. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proc Natl Acad Sci U S A. 1997;94(7):3010–3015. doi:10.1073/pnas.94.7.3010
32. Laghmani K, Borensztein P, Ambühl P, et al. Chronic metabolic acidosis enhances NHE-3 protein abundance and transport activity in the rat thick ascending limb by increasing NHE-3 mRNA. J Clin Invest. 1997;99(1):24–30. doi:10.1172/JCI119128
33. Ambühl P, Amemiya M, Preisig PA, Moe OW, Alpern RJ. Chronic hyperosmolality increases NHE3 activity in OKP cells. J Clin Invest. 1998;101(1):170–177.
34. Yun CH, Gurubhagavatula S, Levine SA, et al. Glucocorticoid stimulation of ileal Na+ absorptive cell brush border Na+/H+ exchange and association with an increase in message for NHE-3, an epithelial Na+/H+ exchanger isoform. J Biol Chem. 1993;268(1):206–211. doi:10.1016/S0021-9258(18)54135-1
35. Cho JH, Musch MW, Bookstein CM, McSwine RL, Rabenau K, Chang EB. Aldosterone stimulates intestinal Na+ absorption in rats by increasing NHE3 expression of the proximal colon. Am J Physiol. 1998;274(3):C586–594.
36. Amin MR, Malakooti J, Sandoval R, Dudeja PK, Ramaswamy K. IFN-γ and TNF-α regulate human NHE3 gene expression by modulating the Sp family transcription factors in human intestinal epithelial cell line C2BBe1. Am J Physiol-Cell Physiol. 2006;291(5):C887–C896. doi:10.1152/ajpcell.00630.2005
37. Queiroz-Leite GD, Crajoinas RO, Neri EA, et al. Fructose acutely stimulates NHE3 activity in kidney proximal tubule. Kidney Blood Press Res. 2012;36(1):320–334. doi:10.1159/000343390
38. Cabral PD, Hong NJ, Hye Khan Ma, et al. Fructose stimulates Na/H exchange activity and sensitizes the proximal tubule to angiotensin II. Hypertension. 2014;63(3):e68–73. doi:10.1161/HYPERTENSIONAHA.113.02564
39. Ambühl PM, Amemiya M, Danczkay M, et al. Chronic metabolic acidosis increases NHE3 protein abundance in rat kidney. Am J Physiol. 1996;271(4 Pt 2):F917–925. doi:10.1152/ajprenal.1996.271.4.F917
40. Chen Y, Asico LD, Zheng S, et al. Gastrin and D1 dopamine receptor interact to induce natriuresis and diuresis. Hypertension. 2013;62(5):927–933. doi:10.1161/HYPERTENSIONAHA.113.01094
41. Lennane RJ, Peart WS, Carey RM, Shaw J. A comparison on natriuresis after oral and intravenous sodium loading in sodium-depleted rabbits: evidence for a gastrointestinal or portal monitor of sodium intake. Clin Sci Mol Med. 1975;49(5):433–436. doi:10.1042/cs0490433
42. Abedin MZ, Giurgiu DI, Abedin ZR, Peck EA, Su X, Smith PR. Characterization of NA+/H+ exchanger isoform (NHE1, NH32 and NHE3) expression in prairie dog gallbladder. J Membr Biol. 2001;182(2):123–134. doi:10.1007/s00232-001-0038-9
43. Watanabe C, Kato Y, Ito S, Kubo Y, Sai Y, Tsuji A. Na+/H+ exchanger 3 affects transport property of H+/oligopeptide transporter 1. Drug Metab Pharmacokinet. 2005;20(6):443–451. doi:10.2133/dmpk.20.443
44. Li HC, Du Z, Barone S, et al. Proximal tubule specific knockout of the Na+/H+ exchanger NHE3: effects on bicarbonate absorption and ammonium excretion. J Mol Med (Berl). 2013;91(8):951–963. doi:10.1007/s00109-013-1015-3
45. Shi H, Zhao X, Ding Z, et al. Na+/H+ exchanger regulates amino acid-mediated autophagy in intestinal epithelial cells. Cell Physiol Biochem. 2017;42(6):2418–2429. doi:10.1159/000480184
46. Gawenis LR, Stien X, Shull GE, et al. Intestinal NaCl transport in NHE2 and NHE3 knockout mice. Am J Physiol Gastrointest Liver Physiol. 2002;282(5):G776–784. doi:10.1152/ajpgi.00297.2001
47. Sullivan S, Alex P, Dassopoulos T, et al. Downregulation of sodium transporters and NHERF proteins in IBD patients and mouse colitis models: potential contributors to IBD-associated diarrhea. Inflamm Bowel Dis. 2009;15(2):261–274. doi:10.1002/ibd.20743
48. Kiela PR, Laubitz D, Larmonier CB, et al. Changes in mucosal homeostasis predispose NHE3 knockout mice to increased susceptibility to DSS-induced epithelial injury. Gastroenterology. 2009;137(3):
49. Laubitz D, Larmonier C, Bai A, et al. Colonic gene expression profile in NHE3-deficient mice: evidence for spontaneous distal colitis. Am J Physiol Gastrointest Liver Physiol. 2008;295:G63–G77. doi:10.1152/ajpgi.90207.2008
50. Zhou D, Xue J, Gavrialov O, Haddad G. Na+/H+ exchanger 1 deficiency alters gene expression in mouse brain. Physiol Genomics. 2004;18:331–339. doi:10.1152/physiolgenomics.00076.2004
51. King AJ, Siegel M, He Y, et al. Inhibition of sodium/hydrogen exchanger 3 in the gastrointestinal tract by tenapanor reduces paracellular phosphate permeability. Sci Transl Med. 2018;10:456. doi:10.1126/scitranslmed.aam6474
52. Rosenbaum DP, Yan A, Jacobs JW. Pharmacodynamics, safety, and tolerability of the NHE3 inhibitor tenapanor: two trials in healthy volunteers. Clin Drug Investig. 2018;38(4):341–351. doi:10.1007/s40261-017-0614-0
53. Linz D, Wirth K, Linz W, et al. Antihypertensive and laxative effects by pharmacological inhibition of sodium-proton-exchanger subtype 3-mediated sodium absorption in the gut. Hypertension. 2012;60(6):1560–1567. doi:10.1161/HYPERTENSIONAHA.112.201590
54. Spencer AG, Labonte ED, Rosenbaum DP, et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci Transl Med. 2014;6(227):227ra236. doi:10.1126/scitranslmed.3007790
55. Pan W, Borovac J, Spicer Z, et al. The epithelial sodium/proton exchanger, NHE3, is necessary for renal and intestinal calcium (re) absorption. Am J Physiol Renal Physiol. 2012;302(8):F943–F956. doi:10.1152/ajprenal.00504.2010
56. Schiavi SC, Tang W, Bracken C, et al. Npt2b deletion attenuates hyperphosphatemia associated with CKD. J Am Soc Nephrol. 2012;23(10):1691–1700. doi:10.1681/ASN.2011121213
57. Larsson TE, Kameoka C, Nakajo I, et al. NPT-IIb inhibition does not improve hyperphosphatemia in CKD. Kidney Int Rep. 2018;3(1):73–80. doi:10.1016/j.ekir.2017.08.003
58. Molnar C, Gair J. Concepts of biology: 1st Canadian edition; 2015.
59. Bradford EM, Sartor MA, Gawenis LR, Clarke LL, Shull GE. Reduced NHE3-mediated Na+ absorption increases survival and decreases the incidence of intestinal obstructions in cystic fibrosis mice. Am J Physiol Gastrointest Liver Physiol. 2009;296(4):G886–898. doi:10.1152/ajpgi.90520.2008
60. Linz B, Hohl M, Reil JC, Böhm M, Linz D. Inhibition of NHE3-mediated sodium absorption in the gut reduced cardiac end-organ damage without deteriorating renal function in obese spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2016;67(3):225–231. doi:10.1097/FJC.0000000000000336
61. Mente A, O’Donnell MJ, Rangarajan S, et al. Association of urinary sodium and potassium excretion with blood pressure. N Engl J Med. 2014;371(7):601–611. doi:10.1056/NEJMoa1311989
62. Ohta Y, Tsuchihashi T, Kiyohara K, Oniki H. High salt intake promotes a decline in renal function in hypertensive patients: a 10-year observational study. Hypertens Res. 2013;36(2):172–176. doi:10.1038/hr.2012.155
63. Zhao X, Yang X, Zhang X, et al. Dietary salt intake and coronary atherosclerosis in patients with prehypertension. J Clin Hypertens. 2014;16(8):575–580. doi:10.1111/jch.12362
64. Onishi A, Fu Y, Patel R, et al. A role for tubular Na(+)/H(+) exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am J Physiol Renal Physiol. 2020;319(4):F712–f728. doi:10.1152/ajprenal.00264.2020
65. Chan LKY, Wang Y, Ng EKW, Leung PS. Na(+) /H(+) exchanger 3 blockade ameliorates type 2 diabetes mellitus via inhibition of sodium-glucose co-transporter 1-mediated glucose absorption in the small intestine. Diabetes Obes Metab. 2018;20(3):709–717. doi:10.1111/dom.13151
66. Gress TW, Nieto FJ, Shahar E, Wofford MR, Brancati FL. Hypertension and antihypertensive therapy as risk factors for type 2 diabetes mellitus. Atherosclerosis Risk in Communities Study. N Engl J Med. 2000;342(13):905–912. doi:10.1056/NEJM200003303421301
67. Greenberg BH, Abraham WT, Albert NM, et al. Influence of diabetes on characteristics and outcomes in patients hospitalized with heart failure: a report from the organized program to initiate lifesaving treatment in hospitalized patients with heart failure (OPTIMIZE-HF). Am Heart J. 2007;154(2):
68. Tenenbaum A, Motro M, Fisman EZ, et al. Functional class in patients with heart failure is associated with the development of diabetes. Am J Med. 2003;114(4):271–275. doi:10.1016/S0002-9343(02)01530-9
69. van Melle JP, Bot M, de Jonge P, de Boer RA, van Veldhuisen DJ, Whooley MA. Diabetes, glycemic control, and new-onset heart failure in patients with stable coronary artery disease: data from the heart and soul study. Diabetes Care. 2010;33(9):2084–2089. doi:10.2337/dc10-0286
70. Paolillo S, Rengo G, Pellegrino T, et al. Insulin resistance is associated with impaired cardiac sympathetic innervation in patients with heart failure. Eur Heart J Cardiovasc Imaging. 2015;16(10):1148–1153.
71. Ai M, Otokozawa S, Asztalos BF, et al. Adiponectin: an independent risk factor for coronary heart disease in men in the Framingham offspring Study. Atherosclerosis. 2011;217(2):543–548. doi:10.1016/j.atherosclerosis.2011.05.035
72. Marangou AG, Alford FP, Ward G, et al. Hormonal effects of norepinephrine on acute glucose disposal in humans: a minimal model analysis. Metabolism. 1988;37(9):885–891. doi:10.1016/0026-0495(88)90124-2
73. Hollenberg NK, Price DA, Fisher ND, et al. Glomerular hemodynamics and the renin-angiotensin system in patients with type 1 diabetes mellitus. Kidney Int. 2003;63(1):172–178. doi:10.1046/j.1523-1755.2003.00701.x
74. Packer M. Activation and inhibition of sodium-hydrogen exchanger is a mechanism that links the pathophysiology and treatment of diabetes mellitus with that of heart failure. Circulation. 2017;136(16):1548–1559. doi:10.1161/CIRCULATIONAHA.117.030418
75. Inoue BH, Santos L, Pessoa TD, et al. Increased NHE3 abundance and transport activity in renal proximal tubule of rats with heart failure. Am J Physiol Regul Integr Comp Physiol. 2012;302(1):R166–R174. doi:10.1152/ajpregu.00127.2011
76. Trum M, Riechel J, Lebek S, et al. Empagliflozin inhibits Na+/H+ exchanger activity in human atrial cardiomyocytes. ESC Heart Failure. 2020;7(6):4429–4437. doi:10.1002/ehf2.13024
77. Wu W-C, Wei J-N, Chen S-C, et al. Progression of insulin resistance: a link between risk factors and the incidence of diabetes. Diabetes Res Clin Pract. 2020;161:108050.
78. Klisic J, Hu MC, Nief V, et al. Insulin activates Na+/H+ exchanger 3: biphasic response and glucocorticoid dependence. Am J Physiol Renal Physiol. 2002;283(3):F532–F539. doi:10.1152/ajprenal.00365.2001
79. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. New Engl J Med. 2015;373(22):2117–2128. doi:10.1056/NEJMoa1504720
80. Borges-Júnior FA, Dos Santos DS, Benetti A, et al. Empagliflozin inhibits proximal tubule NHE3 activity, preserves GFR and restores euvolemia in nondiabetic rats with induced heart failure. bioRxiv. 2020. doi:10.1101/2020.07.16.207118
81. Fitchett D, Zinman B, Wanner C, et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA-REG OUTCOME® trial. Eur Heart J. 2016;37(19):1526–1534. doi:10.1093/eurheartj/ehv728
82. Novikov A, Vallon V. Sodium glucose cotransporter 2 inhibition in the diabetic kidney: an update. Curr Opin Nephrol Hypertens. 2016;25(1):50–58. doi:10.1097/MNH.0000000000000187
83. Devroede GJ, Phillips SF. Conservation of sodium, chloride, and water by the human colon. Gastroenterology. 1969;56(1):101–109. doi:10.1016/S0016-5085(69)80071-5
84. McRorie J, Pepple S, Rudolph C. Effects of fiber laxatives and calcium docusate on regional water content and viscosity of digesta in the large intestine of the pig. Dig Dis Sci. 1998;43(4):738–745. doi:10.1023/A:1018805812321
85. Bharucha AE, Dorn SD, Lembo A, Pressman A. American gastroenterological association medical position statement on constipation. Gastroenterology. 2013;144(1):211–217. doi:10.1053/j.gastro.2012.10.029
86. Guignard J-P, Sulyok E. Chapter 81 - renal morphogenesis and development of renal function. In: Gleason CA, Devaskar SU, editors. Avery’s Diseases of the Newborn (Ninth Edition). Philadelpia: W.B. Saunders; 2012:1165–1175.
87. Chey WD, Lembo AJ, Rosenbaum DP. Tenapanor treatment of patients with constipation-predominant irritable bowel syndrome: a phase 2, randomized, placebo-controlled efficacy and safety trial. Am J Gastroenterol. 2017;112(5):763–774. doi:10.1038/ajg.2017.41
88. Chey WD, Lembo AJ, Rosenbaum DP. Efficacy of tenapanor in treating patients with irritable bowel syndrome with constipation: a 12-week, placebo-controlled phase 3 trial (T3MPO-1). Am J Gastroenterol. 2020;115(2):281–293. doi:10.14309/ajg.0000000000000516
89. IBSRELA® (tenapanor) tablets, for oral use [prescribing information]. Ardelyx; 2019.
90. Phosphate binder use, last 3 months. DOPPS practice monitor; 2020. Available from: https://www.dopps.org/DPM-HD/Files/maxPBINDER_use_c_overallTAB.htm.
91. Slinin Y, Foley RN, Collins AJ. Calcium, phosphorus, parathyroid hormone, and cardiovascular disease in hemodialysis patients: the USRDS waves 1, 3, and 4 study. J Am Soc Nephrol. 2005;16(6):1788–1793. doi:10.1681/ASN.2004040275
92. Kestenbaum B, Sampson JN, Rudser KD, et al. Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol. 2005;16(2):520–528. doi:10.1681/ASN.2004070602
93. Qadeer HA, Bashir K. Physiology, phosphate. In: StatPearls [Internet]. Treasure Island, FL: StatPearls Publishing; 2020.
94. Ix JH, Katz R, Kestenbaum BR, et al. Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study). J Am Coll Cardiol. 2012;60(3):200–207. doi:10.1016/j.jacc.2012.03.040
95. Hagström E, Hellman P, Larsson TE, et al. Plasma parathyroid hormone and the risk of cardiovascular mortality in the community. Circulation. 2009;119(21):2765–2771. doi:10.1161/CIRCULATIONAHA.108.808733
96. Block GA, Rosenbaum DP, Leonsson-Zachrisson M, et al. Effect of tenapanor on interdialytic weight gain in patients on hemodialysis. Clin J Am Soc Nephrol. 2016;11(9):1597–1605. doi:10.2215/CJN.09050815
97. Block GA, Rosenbaum DP, Yan A, Chertow GM. Efficacy and safety of tenapanor in patients with hyperphosphatemia receiving maintenance hemodialysis: a randomized phase 3 trial. J Am Soc Nephrol. 2019;30(4):641–652.
98. Chertow GM, Yang Y, Rosenbaum DP. Long-term safety and efficacy of tenapanor for the control of serum phosphorus in patients with chronic kidney disease on dialysis.
99. Rosenbaum DP, Sprague SM. Tolerability of tenapanor, an investigational, first-in-class, nonbinder therapy for the control of serum phosphorus in patients with chronic kidney disease on dialysis.
100. Pergola PE, Rosenbaum DP, Yang Y, Chertow GM. A randomized trial of tenapanor and phosphate binders as a dual-mechanism treatment for hyperphosphatemia in patients on maintenance dialysis (AMPLIFY). J Am Soc Nephrol. 2021;32(6):1465–1473. doi:10.1681/ASN.2020101398
101. Nussey S, Whitehead S. The parathyroid glands and vitamin D. In: Endocrinology: An Integrated Approach. Oxford: BIOS Scientific Publishers; 2001.
102. Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79(12):1370–1378. doi:10.1038/ki.2011.47
103. Martin KJ, González EA. Prevention and control of phosphate retention/hyperphosphatemia in CKD-MBD: what is normal, when to start, and how to treat? Clin J Am Soc Nephrol. 2011;6(2):440–446. doi:10.2215/CJN.05130610
104. Labonté ED, Carreras CW, Leadbetter MR, et al. Gastrointestinal inhibition of sodium-hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J Am Soc Nephrol. 2015;26(5):1138–1149. doi:10.1681/ASN.2014030317
105. Kraus MA, Kalra PA, Hunter J, Menoyo J, Stankus N. The prevalence of vascular calcification in patients with end-stage renal disease on hemodialysis: a cross-sectional observational study. Ther Adv Chronic Dis. 2015;6(3):84–96. doi:10.1177/2040622315578654
106. Cano-Megías M, Guisado-Vasco P, Bouarich H, et al. Coronary calcification as a predictor of cardiovascular mortality in advanced chronic kidney disease: a prospective long-term follow-up study. BMC Nephrol. 2019;20(1):188. doi:10.1186/s12882-019-1367-1
107. Tonelli M, Pannu N, Manns B. Oral phosphate binders in patients with kidney failure. N Engl J Med. 2010;362(14):1312–1324. doi:10.1056/NEJMra0912522
108. Hutchison AJ, Smith CP, Brenchley PE. Pharmacology, efficacy and safety of oral phosphate binders. Nat Rev Nephrol. 2011;7(10):578–589. doi:10.1038/nrneph.2011.112
109. Saurette M, Alexander RT. Intestinal phosphate absorption: the paracellular pathway predominates? Exp Biol Med. 2019;244(8):646–654. doi:10.1177/1535370219831220
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.