")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 7
Novel treatment strategies in triple-negative breast cancer: specific role of poly(adenosine diphosphate-ribose) polymerase inhibition
Authors Audeh MW
Received 5 May 2014
Accepted for publication 10 July 2014
Published 3 October 2014 Volume 2014:7 Pages 307—316
DOI https://doi.org/10.2147/PGPM.S39765
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
M William Audeh
Division of Medical Oncology, Samuel Oschin Cancer Institute, Cedars-Sinai Medical Center, Los Angeles, CA, USA
Abstract: Inhibitors of the poly(adenosine triphosphate-ribose) polymerase (PARP)-1 enzyme induce synthetic lethality in cancers with ineffective DNA (DNA) repair or homologous repair deficiency, and have shown promising clinical activity in cancers deficient in DNA repair due to germ-line mutation in BRCA1 and BRCA2. The majority of breast cancers arising in carriers of BRCA1 germ-line mutations, as well as half of those in BRCA2 carriers, are classified as triple-negative breast cancer (TNBC). TNBC is a biologically heterogeneous group of breast cancers characterized by the lack of immunohistochemical expression of the ER, PR, or HER2 proteins, and for which the current standard of care in systemic therapy is cytotoxic chemotherapy. Many “sporadic” cases of TNBC appear to have indicators of DNA repair dysfunction similar to those in BRCA-mutation carriers, suggesting the possible utility of PARP inhibitors in a subset of TNBC. Significant genetic heterogeneity has been observed within the TNBC cohort, creating challenges for interpretation of prior clinical trial data, and for the design of future clinical trials. Several PARP inhibitors are currently in clinical development in BRCA-mutated breast cancer. The use of PARP inhibitors in TNBC without BRCA mutation will require biomarkers that identify cancers with homologous repair deficiency in order to select patients likely to respond. Beyond mutations in the BRCA genes, dysfunction in other genes that interact with the homologous repair pathway may offer opportunities to induce synthetic lethality when combined with PARP inhibition.
Keywords: PARP, triple negative breast cancer, PARP inhibitors
Introduction
Breast cancers defined by the lack of demonstrable expression of the ER, PR, or HER2 proteins represent approximately 15%–20% of all breast cancers, and have been labeled “triple-negative” breast cancers (TNBCs), although they do not truly represent a homogeneous group with biologically uniform behavior. Classic morphologic descriptions by light microscopy and simple immunohistochemistry have led to the identification of this subset as TNBC for clinical utility, facilitating the assumption that they represent a discrete clinical entity. While clinical case series and reviews have indeed identified a generally poor prognosis for these cancers in comparison to ER-positive and HER2-positive cancers,1–4 owing in large part to the absence of effective targeted therapies with broad activity in breast cancers defined only as TNBC. However, significant biological diversity exists within this subgroup, as revealed by molecular analysis on a variety of platforms.5 It is this biological diversity within the TNBC subset that now directs the design of clinical trials, as targets identified within subsets of TNBC have led to trials utilizing targeted therapies in sensitive subsets defined by the presence of predictive biomarkers. One such targeted therapy of interest in the treatment of TNBC is the family of compounds capable of inhibiting the poly(adenosine diphosphate [ADP]-ribose) polymerase (PARP) enzyme. By inhibiting primarily PARP-1 and -2, PARP inhibitors currently undergoing investigation induce a number of biological effects, most importantly the inhibition of the deoxyribonucleic acid (DNA)-repair pathway involved in base-excision repair (BER). The basis for considering PARP inhibitors as potential therapies for TNBC and the challenges encountered and expected in the application of PARP inhibitors in the TNBC population are the subject of this review.
Redefining TNBC based on meaningful biology
TNBCs have been identified and defined in routine clinical practice by the absence of immunohistochemical staining for three proteins: ER, PR, and HER2. The further definition of breast cancer by gene-expression patterns based on messenger ribonucleic acid (mRNA) rather than protein yielded the intrinsic molecular subtypes luminal A, luminal B, HER2-enriched, and basal type.6,7 TNBC was assumed to fall within the basal subtype, as they share the absence of immunohistochemical markers; however, considerable evidence has shown that not all TNBCs are truly basal by gene expression.8–10 Molecular analysis of TNBC cohorts by a variety of platforms, including microarray, DNA copy-number variation, and DNA sequencing,5,10,11 reveal that approximately 20%–25% of TNBCs do not display basal breast cancer genomic markers, and conversely between 25% and 30% of basal breast cancers defined genomically do not show TNBC immunohistochemistry. Therefore, not all TNBCs defined by immunohistochemistry will be basal breast cancers, as defined by genomic profiling. More importantly, these findings call into question the validity of utilizing targeted therapies in cohorts of breast cancer defined simply as TNBC12 without further molecular stratification, and should perhaps lead to reconsideration of therapies that have produced negative results in such unselected trial populations.
Within the TNBC cohort, significant biological diversity exists as well, resulting in variability of clinical outcomes in response to chemotherapy.13,14 Lehmann et al11 identified six defined subtypes within a cohort of TNBC based on gene expression, identified as basal-like (BL)-1, BL2, mesenchymal, mesenchymal stem-like (MSL), immunomodulatory, and luminal androgen receptor (LAR). Genomic cluster analysis identified specific genes and pathways characterizing these subsets, such as proliferative and DNA-repair pathways in BL1; EGF, MET and Wnt in BL2; immune signaling in immunomodulatory; cell motility in both mesenchymal and mesenchymal stem-like; and hormonal signaling in the luminal androgen receptor. Significant clinical diversity between these robustly defined molecular subtypes was observed, as evidenced by markedly different responsiveness to neoadjuvant chemotherapy: 52% pathologic complete remission (pCR) in BL1, and 0 pCR in BL2, for example.15 A meta-analysis of gene expression in 190 TNBCs identified by immunohistochemistry and also identified as BL by the intrinsic gene set, also revealed considerable clinical diversity in prognosis, with signatures of an activated stroma showing a poorer prognosis, while signatures reflecting BRCA1 loss, reduced mTOR signaling, and an activated Src pathway were associated with a better prognosis.
The most common aspect of biology within the TNBC cohort, identified by the Cancer Genome Atlas Network,5 appears to be mutation or deletion of TP53, observed in 71%. (The prevalence of mutations in TP53 or related pathway members rose to nearly 100% when limited to BL cancers.) The high frequency of p53 dysfunction in TNBC does unite this group as having significant genomic instability, likely due to some defect in DNA-repair proficiency.
The best-known such defect in DNA repair in TNBC is due to mutation in BRCA1 or BRCA2, and is associated with germ-line inheritance. Over 2,000 different mutations have been identified in BRCA1 and BRCA2, with most being deletions or insertions, as well as single-nucleotide substitutions, in both coding and noncoding sequences.16 Most mutations result in frameshift, truncation, and splice-site changes leading to protein dysfunction, and large genomic rearrangements, particularly in BRCA1, are also frequent. While copy-number variation is not typical in germ-line BRCA1 and BRCA2 mutations, somatic loss of heterozygosity is a frequent consequence of loss of BRCA function.
The prevalence of BRCA1 or BRCA2 germ-line mutation in breast cancer in general is estimated to be 5%–10%, while in TNBC the prevalence has been assessed as between 10.6% and 19.8%.17,18 However, 75%–80% of cancers arising in BRCA1 carriers and approximately 50% of those in BRCA2 carriers are TNBC.19 As will be discussed in this paper, the DNA-repair defect in BRCA-mutated cancers, primarily affecting double-strand break repair through homologous recombination, places increased dependence on the proficiency of base-excision repair, and therefore the function of PARP, in these cancers. Inhibition of PARP function in BRCA-mutated cancers leads to cell death,20,21 and has led to numerous clinical trials with PARP inhibitors in this population. The characteristic genomic instability common to both BRCA1-mutated cancers and TNBC in general has been termed “BRCAness”,22–24 and forms the basis for the interest in TNBC as a potential target for PARP inhibitors.
BRCAness has been characterized in a number of ways, beginning with extreme genomic instability, a high frequency of TP53 mutations, loss of RAD51-focus formation, and sensitivity to DNA cross-linking agents,25 as well as BRCA-like array comparative genomic hybridization,26 and presumed markers of DNA-repair proficiency, such as RAD51 functionality, PARP-1 expression levels, and other BER-pathway members.24 To the extent that homologous repair deficiency (HRD) is considered essential for BRCAness, HRD assays have also been investigated27,28 in TNBC.
Somatic inactivation of BRCA1 by promoter hypermethylation occurs in approximately 15% of all sporadic breast cancers, and in TNBC the frequency is as high as 27%–37%.26,29 The majority of BRCA1-methylated cancers display a BRCAness profile defined by a BRCA1-like or BRCA2-like comparative genomic hybridization assay. However, the biological effects of somatic inactivation of BRCA1 are not entirely equivalent to the effects of a germ-line BRCA1 mutation, as evidenced by different responsiveness to standard neoadjuvant chemotherapy, with a 63% pCR observed in germ-line BRCA1 and 35% pCR in cancers with somatic inactivation of BRCA1.30 Nevertheless, cancers lacking a germ-line BRCA mutation but displaying BRCAness do have similar clinical features, including younger age of onset, apparent dysfunction of homologous DNA repair, and possibly increased sensitivity to platinum chemotherapy.31 Therefore, a substantial proportion of TNBCs possess some form of BRCA dysfunction by either germ-line or somatic inactivation of BRCA1 or BRCA2, and those that do not may still harbor other defects in DNA repair leading to HRD, and producing characteristic genomic instability, high frequency of p53-pathway dysfunction, and as a result possible sensitivity to PARP inhibition.28 This direction of clinical research is however complicated by the imprecision of defining clinical cohorts by TNBC status alone, and by the challenges of defining BRCAness in cancers lacking a known BRCA germ-line mutation, a cohort that makes up the majority of TNBC.
Targeting poly(ADP-ribose) polymerase with PARP inhibitors
The PARP family of enzymes, first described in 1963,32 encompasses 17 enzymes, six of which utilize nicotinamide adenine dinucleotide (NAD+) as the substrate to synthesize polymeric chains of ADP-ribose for the purpose of posttranslational modification of target proteins.33,34 Of these, PARP-1 is the primary enzyme involved in the regulation of DNA repair, although PARP-2 and PARP-3 are also involved to a lesser extent. PARP-1 is a nuclear protein with two zinc-finger domains that bind to areas of single-strand DNA breaks within seconds of the damage, and initiates the formation of a poly-ADP scaffold that recruits other members of the BER pathway, such as XRCC1.35 The BER pathway is not essential for cellular survival and the ultimate repair of stalled replication forks and double-strand breaks in HR-proficient cells. However, in the absence of BRCA1 or BRCA2 function, double-strand break repair through homologous recombination is impaired; the loss of BER through PARP inhibition in such cells produces catastrophic genomic instability and cell death.20,21 This synergy between the intrinsic HRD caused by the loss of BRCA function and the induced loss of BER function through PARP inhibition has been termed “synthetic lethality”.36 The intense recent interest in PARP inhibition as a form of cancer therapy was promoted by the observation of significant cell death in BRCA-deficient cell lines treated only with a PARP inhibitor in the absence of any other DNA-damaging agents. The intrinsic sensitivity of BRCA-mutated cancer to PARP inhibition was thus established, and led to the clinical development of several compounds believed to be PARP inhibitors. (See Table 1 for a timeline of PARP-inhibitor development.)
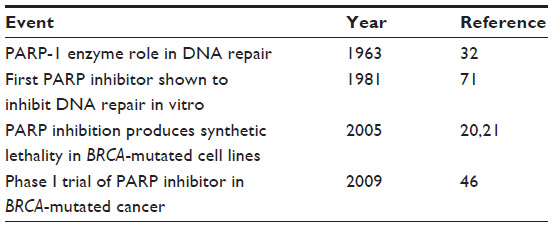 | Table 1 Timeline of PARP-inhibitor development |
Inhibitors of PARP-1 were initially discovered over 30 years ago,34 and the original molecular structure formed the basis for the development of PARP inhibitors currently in clinical trials, utilizing nicotinamide-like scaffolds with modified side-chain groups.37 Inhibition of the PARP-1 and PARP-2 enzymes is thought to be achieved primarily through small-molecule binding to the catalytic domain that binds NAD+. However, the effect of the PARP-inhibitor side chain extending outside the NAD catalytic site may also be to “trap” the PARP enzyme at the site of DNA damage, preventing its usual release from the DNA molecule.38–40
“PARP trapping” may therefore be an additional action of some PARP inhibitors that may contribute to their potential cytotoxicity. In response to the presence of single-strand DNA breaks, PARP-1 binds to the damaged site through its zinc-finger domains. This binding activates the catalytic activity of PARP-1, and the initiation of poly(ADP) polymerization. This produces not only a poly(ADP) scaffold that recruits and binds other DNA-repair proteins but also adds poly(ADP) moieties to PARP-1 itself, leading under normal cell physiology to an increasingly negative charge on the enzyme, and eventual dissociation from the DNA.
While the preclinical data suggested the requirement for HRD as mediated through loss of BRCA function for sensitivity to PARP inhibition, interest was also generated in utilizing PARP inhibition in association with the induction of DNA damage by chemotherapy in the more general cohort of TNBC, regardless of BRCA-mutation status.41 Unfortunately, this initial approach utilized the agent BSI-201, or iniparib, a molecule thought to be a PARP inhibitor, but which was ultimately found to be lacking PARP-1- and -2-inhibitor activity.42 Prior to the reporting of a lack of PARP-inhibitory activity, however, two large and highly publicized trials with iniparib in TNBC were undertaken, and nearly put an end to the development of PARP inhibitors in cancer therapy.43
Clinical trials with iniparib in triple-negative breast cancer – a false start for PARP inhibitors
Iniparib was initially developed as a PARP inhibitor, and showed evidence of PARP inhibition in peripheral blood mononuclear cells by over 50% after a single dose in a Phase I trial.42 However, no other biomarkers of PARP inhibition, such as induction of γ-H2AX foci, were reported, and the chemical structure of the compound was not revealed to consultants knowledgeable in the chemistry of PARP inhibitors.43 A randomized, open-label Phase II trial combining iniparib with chemotherapy, carboplatinum, and gemcitabine, versus chemotherapy alone, in 123 women with metastatic TNBC was reported at the 2009 American Society of Clinical Oncology Meeting, and subsequently published.41 The BRCA status of the trial participants was not reported; however, the response rate rose from 32% to 52% with the addition of iniparib, and improvements in both progression-free (3.6 months versus 5.9 months) and overall survival (7.7 months to 12.3 months) were reported. Based on the significantly positive results, a Phase III trial with the same design was immediately conducted. Within 1 month of the publication of the Phase II results, the sponsors of the trial reported that the Phase III trial had failed to confirm the Phase II results, with no difference between the control arm and the iniparib arm.42
The failure of the iniparib Phase III trial to meet its end point was initially believed to indicate a lack of efficacy of PARP-inhibitor therapy in TNBC, until definitive in vitro cellular assays for functional PARP inhibition revealed that iniparib did not significantly inhibit PARP-1 or PARP-2 catalytic activity, and could not be classified as a PARP inhibitor.44 While initial preclinical observations of PARP-1 enzyme inhibition by metabolites of iniparib were made in cell-free systems, three functional cell-based assays were used to compare iniparib to two known PARP inhibitors with chemical structures quite different from iniparib: olaparib and veliparib.37 The assays compared 1) selective induction of apoptosis or inhibition of colony formation in HR-deficient cell lines with BRCA2 or ATM mutations, 2) selective sensitization of HR-proficient cells to topoisomerase I poisons, and 3) inhibition of the formation of poly(ADP-ribose) polymer (pADPr) in intact cells. In contrast to the distinct activity of olaparib and veliparib in these assays, iniparib exhibited little or no ability to selectively kill HR-deficient cells, sensitize cells to topoisomerase I poisons, or inhibit pADPr formation in intact cells. Although iniparib did display cytotoxicity in normal and neoplastic cells at high (>40 μmol/L) concentrations, its mechanism of action appeared unlikely to be via PARP inhibition. While these and similar findings42,45 put to rest the development of iniparib as a PARP inhibitor, they also led to renewed interest in true PARP inhibitors as active therapeutic single agents, or in combination with chemotherapy, in tumors with identifiable HRD, such as germ-line BRCA mutations.46 PARP inhibitors have been studied in clinical trials in ovarian cancer and breast cancer47,48 and a number of other tumor types,49 with activity mainly in individuals with known germ-line BRCA mutations,46 although there is a rationale for use in cancers assumed to have somatically acquired HRD, such as TNBC without germ-line BRCA mutation.
Clinical trials with PARP inhibitors in breast cancer and the rationale for use in TNBC
A number of early phase clinical trials have assessed the activity of PARP inhibitors in cohorts of patients that included or were limited to breast cancer. These trials utilized PARP inhibitors either as single agents, attempting to elicit synthetic lethality in sensitive cohorts, or in combination with chemotherapy, where the action of PARP inhibition as a chemosensitizer cannot be readily separated from any element of synthetic lethality. While most responses have been seen in patients with BRCA germ-line mutations, with often significant and long-lasting clinical benefit, the broad early clinical experience with a variety of PARP inhibitors has led to intense clinical research interest in expanding the population of patients potentially sensitive to PARP inhibition, including TNBC. The available data on PARP inhibitors in breast cancer is summarized in the following sections.
Olaparib (AZD 2811)
Olaparib, formerly known as AZD 2811, has been the PARP inhibitor most extensively studied in breast cancer to date (if one excludes iniparib for the reasons described earlier). A Phase I study in human subjects was conducted with olaparib beginning in 2005, and enriched for patients with BRCA germ-line mutations.46 Of the 60 patients in the trial, objective responses were only seen among the 22 who were known BRCA carriers. Of the nine breast cancer patients (molecular subtype not specified) in this trial, three had BRCA2 mutations, two of whom had objective responses, including a complete remission lasting over 60 weeks.
The Phase I trial was followed by one of the two largest trials thus far of a PARP inhibitor in breast cancer.48 A Phase II trial of olaparib as a single agent enrolled 54 patients with germ-line BRCA mutations and metastatic breast cancer previously treated with a median of three prior chemotherapy regimens, as reported by Tutt et al.48 Two dose cohorts were studied, and 29 of those enrolled had TNBC. Of the 13 TNBC patients in the higher-dose cohort, seven of 13 (54%) showed a partial response, and four of 13 (31%) had stable disease; however, even in the lower-dose cohort, four of 16 (25%) TNBC patients showed a partial response, and seven of 16 (44%) had stable disease. Responses were not limited to TNBC, however, and the objective response rate for the entire higher-dose cohort, which included ER-positive and HER2-positive patients was 41%, with a median progression-free survival of nearly 6 months. The conclusion of this trial was that responsiveness to PARP inhibition in breast cancer was not a feature of TNBC but of BRCA-mutated breast cancer.
A more recent trial of olaparib in nearly 300 patents with a variety of BRCA-mutated cancers was reported at the American Society of Clinical Oncology Meeting in 2013 by Kaufman et al, and included 62 patients with breast cancer.49 Eight of 62, or 12.9%, had an objective response, with disease stabilization in 47%. The lower response rate than the prior trial may be explained by the fact that this cohort was more heavily pretreated than in the Tutt et al trial, with the mean number of prior chemotherapies for metastatic disease being 4.6. More importantly, 47.6% (20 of 42) of patients had received prior platinum (including 26% cisplatin and 45% carboplatin), a factor that may predict resistance to PARP-inhibitor therapy.50
In contrast to the response rates seen in the Tutt et al and Kaufman et al trials, an earlier Phase II trial of single-agent olaparib in a cohort of eight BRCA-mutated breast cancer patients reported by Gelmon et al51 showed no objective responses, although minor tumor regression and disease stabilization were seen. In addition, no objective responses were seen in 15 TNBC patients lacking BRCA mutations. The apparent discrepancy in response rates between these trials has not been adequately explained, although the heterogeneity of TNBC and BRCA mutations may be a factor, and the lack of clinical response in the TNBC cohort is in contrast to in vitro data with BRCA wild-type TNBC cell lines, in which olaparib displayed significant antiproliferative and cytotoxic activity.45,52 Clinical trials are planned with olaparib in metastatic BRCA-mutated breast cancer as a single agent in comparison to physician choice, without specificity for the TNBC subtype. However, trials with olaparib in early stage breast cancer in both the adjuvant and neoadjuvant settings, given concurrently with or following chemotherapy, have specified not only BRCA mutation but also TNBC as entry criteria. In this trial design, both the potential for synthetic lethality as well as chemosensitization are being studied.
Veliparib
Veliparib, also known as ABT-888, has had extensive clinical testing in breast cancer, although almost entirely in combination with cytotoxic chemotherapy, and thus acting as a chemosensitizer rather than seeking to induce synthetic lethality in the setting of BRCA mutation. Veliparib has been well tolerated when combined with cytotoxic agents at full dose, unlike most other PARP inhibitors, and this is thought to be due the lack of significant PARP trapping,39 with probable lesser potency as a single agent. Data in cell lines derived from TNBC indicated diminished potency of veliparib in combination with cisplatin, in comparison to olaparib and rucaparib.52 However, the combination of veliparib with the alkylating agent temozolomide in a tumor xenograft model utilizing a variety of tumors (including breast cancer) showed considerable antimetastatic effect, even in tumors resistant to temozolomide alone.53 Other chemotherapy partners for veliparib, such as metronomic cyclophosphamide, also appear to be promising, based on Phase I data.54 The veliparib/temozolomide regimen was taken into a Phase II clinical trial in metastatic breast cancer,55 with results presenting data in abstract form on 41 patients, including 15 with TNBC. Responses were seen, although primarily in BRCA-mutation carriers. A larger Phase II trial in metastatic breast cancer, limited to BRCA carriers only, was then undertaken with this regimen as one arm of a three-arm trial, with the other arms being veliparib in combination with carboplatin and paclitaxel, and carboplatin/paclitaxel alone. The results of this trial are not yet available, but will not answer the question of the utility of veliparib in TNBC without BRCA mutation. However, a 71-patient neoadjuvant trial, adding the combination of veliparib plus carboplatin to standard neoadjuvant chemotherapy, included 38 women with TNBC and was presented at the 2013 San Antonio Breast Cancer Symposium.2 A pCR rate of 52% was seen with the veliparib/platinum combination versus 26% in the control arm in this exploratory study in the I-SPY network. A 300-patient neoadjuvant trial in TNBC, regardless of BRCA-mutation status, is underway with veliparib, carboplatin, and poclitaxel.
Niraparib
Niraparib, also known as MK4827, is a catalytic site inhibitor of PARP-1 and -2 activity, and has PARP-trapping activity as well, exceeding that of olaparib and veliparib.39 Niraparib showed significant single-agent activity in a Phase I trial in a cohort of 100 patients with various tumor types, including 29 BRCA-mutation carriers.56 Twelve breast cancer patients were included in this trial, and two of four BRCA carriers with breast cancer showed responses, in addition to two non-BRCA carriers with stable disease. (The breast cancer molecular subtype was not reported). The reported response rate as a single agent in BRCA-mutated cancers was between 40% and 50%. An international Phase III trial in metastatic BRCA-mutated breast cancer, not limited to TNBC, is under way with single-agent niraparib versus physician choice of single-agent standard chemotherapy.
Rucaparib
Rucaparib, also known as AG-014699 and PF-01367338, has similar PARP catalytic inhibitory activity to olaparib and niraparib, as well as similar PARP-trapping activity.40 Rucaparib displayed in vitro cytotoxicity to TNBC cell lines superior to olaparib and veliparib,52 and in a separate study57 showed preferential in vitro cytotoxicity to cell lines homozygous for BRCA1 or BRCA2 mutation, either by germ-line loss or somatic methylation. A Phase II study in BRCA-mutated cancers, including 17 breast cancers, was reported in abstract form in 2011,58 with an overall clinical benefit rate of 32% and an overall response rate of 5%. (The frequency of TNBC was not reported.) Rucaparib is now under development in BRCA-mutated cancers.
BMN-673
BMN-673 also has both catalytic PARP-inhibition and PARP-trapping activity; however, the PARP-trapping potency has been reported to be 100-fold greater than that of olaparib.40 In vitro and in a human mammary xenograft model, BMN 673 targeted tumor cells with mutations in BRCA1, BRCA2 (and PTEN) with greater potency than other PARP inhibitors.59 Initial clinical results with this compound as a single agent in Phase I were reported in 2013.60 In a cohort of 39 patients, eight breast cancer patients were included (six with BRCA mutations), with responses seen in two of the mutation carriers. A Phase III trial is underway in metastatic BRCA-mutated breast cancer. It is unclear whether the increased PARP-trapping activity and in vitro potency relative to existing PARP inhibitors will translate into differing clinical activity and/or toxicity. Table 2 summarizes the results of single-agent PARP-inhibitor therapy in metastatic breast cancer.
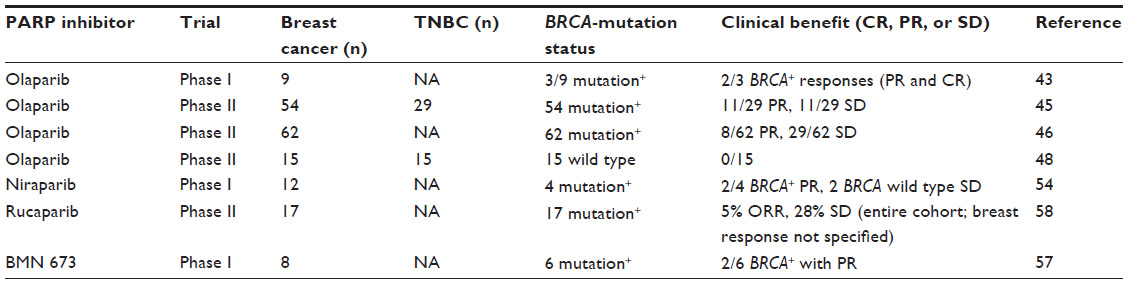 | Table 2 Clinical trials with single-agent PARP inhibitors in metastatic breast cancer (including TNBC) |
Targeting TNBC with PARP inhibitors: biomarkers of HRD and beyond
It is clear that the majority of responses seen in TNBC treated with PARP inhibitors have been in association with BRCA germ-line mutations, with the assumption that a somatic event, such as loss of heterozygosity, methylation silencing, or inactivating mutation, in the wild-type allele has rendered the malignant cells homozygous deficient for either BRCA1 or BRCA2, with the resulting profound defect in HR. Therefore, clinical trials are under way in the adjuvant, neoadjuvant, and metastatic setting for BRCA-mutated TNBC. In this patient cohort, the presence of a germ-line BRCA mutation is assumed to be the primary predictive biomarker of response to PARP inhibition, and the TNBC subset has been selected, in large part, due to the absence of targeted therapies with evidence of efficacy in this cohort of breast cancer. However, with only a minority of TNBC harboring germ-line BRCA mutations, efforts to identify other biomarkers of potential sensitivity to PARP inhibition in the remaining, broader TNBC population have been undertaken. Of these, the most significant has been the search for other biomarkers of HRD in TNBC.
The high degree of genomic instability observed in TNBC without germ-line BRCA mutation has suggested the presence of other germ-line or somatically acquired defects in DNA-repair proficiency, and has encouraged the search for biomarkers indicative of HRD. The loss of the highly accurate HR process for double-strand breaks, and its replacement by error-prone pathways, such as nonhomologous end joining, is thought to leave a specific array of genomic defects, a characteristic “genomic scar” for which a biomarker assay may be found. A biomarker of HRD could then be used as a predictive biomarker for responsiveness to chemotherapy known to produce double-stranded DNA breaks, such as platinum agents and alkylating agents, or more importantly to PARP inhibitors.
One approach to the detection of HRD in tumors has been to study the expression levels of DNA-repair genes and pathways, either by mRNA or proteomics61 and from such studies, specific genes, such as RECQL4,62 as well as RAD51 and Ku80,63 have been used to predict sensitivity to DNA-damaging chemotherapy and PARP inhibitors. Beyond specific repair genes, a genome-wide transcriptome of HR has recently been reported.64 However, such expression-based biomarkers do not include an assessment of the extent of the characteristic genomic scar, the functional consequence of HRD. An HRD score, derived from determining the extent of loss of heterozygosity of DNA regions spanning lengths between 15 Mb and whole chromosomes, using single-nucleotide polymorphism analysis, was applied to a cohort of 55 primarily TNBC patients treated with platinum-based neoadjuvant therapy, 13 of whom carried germ-line BRCA mutations.27 The HRD score correlated with the likelihood of pathologic response more accurately than the presence of a BRCA mutation, and a number of non-germ-line TNBCs showed high HRD scores and pCR. A biomarker that combines both HRD-associated gene-expression levels, as well as assessment of HRD-specific DNA damage, would likely be beneficial in identifying TNBC (and other tumors) lacking germ-line BRCA mutation, but harboring HRD due to other defects. Such a biomarker may then be predictive of sensitivity to PARP inhibition and synthetic lethality.
Beyond BRCA and related DNA-repair genes, a number of other genes have been identified that may affect sensitivity to PARP inhibition in tumors with wild-type BRCA. PTEN loss has been identified as enhancing sensitivity to PARP inhibition, possibly through the effects on BRCA expression, and PARP inhibitors have had clinical activity in tumors with wild-type BRCA and loss of PTEN.65 Mutations in PTEN, PI3K, AKT, INPP4B, and other members of the PI3K pathway appear to occur in a substantial proportion of TNBCs.5 Importantly, specific inhibition of PI3K has been shown to impair BRCA1 and BRCA2 expression, and sensitize BRCA-proficient TNBC in a patient-derived xenograft model.66 Such studies have led to a Phase I trial utilizing a PI3K inhibitor in combination with olaparib in TNBC, both BRCA-mutated and wild type, with preliminary results in ten TNBC patients reported in June 2014.67 While an overall response rate of 30% was seen (three of ten TNBC patients), nearly all carried germ-line BRCA mutations, raising the question as to whether the responses may have been due to PARP inhibition alone. If this combined approach proves successful in non-BRCA-mutated TNBC, another mechanism of inducing synthetic lethality, by combining PARP inhibition and PI3K-pathway blockade, may be identified.
Other instances of mutations causing synthetic lethality with PARP inhibition in BRCA-proficient tumors have been identified through in vitro studies, including loss of CDK1, required for the phosphorylation of BRCA1 in the damage response68; loss of the cohesin complex, responsible for the stability of replication forks and necessary for HR in G2;69 and, in the case of activating EGFR mutations, dual inhibition of EGFR and PARP.70 It will be important to determine whether these varied genetic lesions will ultimately result in some form of HRD, and will therefore be detected by a more general HRD biomarker, or whether individual biomarkers for each mutation will be needed to confirm their potential for synergy with PARP inhibition.
Summary
PARP inhibitors have shown significant clinical activity, particularly as single agents, in tumors harboring defects in double-strand DNA-break repair, the best studied being ovarian and breast cancers with BRCA germ-line mutations, in which they have induced synthetic lethality. Several PARP inhibitors are in Phase III clinical trials, and it is likely that a PARP inhibitor will be approved for clinical use in BRCA-mutated ovarian cancer in 2014. Although in vitro measures of potency show wide variation in PARP catalytic inhibition and PARP trapping at the DNA-binding site, such differences have yet to show clinical relevance.
In TNBC, a significant minority are due to BRCA germ-line mutations, and this cohort will likely benefit from therapy with a PARP inhibitor in the metastatic setting, as well as ultimately in adjuvant and neoadjuvant therapy. Several large, international trials are under way in 2014 in TNBC with BRCA germ-line mutations. The majority of TNBCs, however, have wild-type BRCA1 and BRCA2, yet within this group, a subset may have intrinsic HRD due to other causes, and strong evidence exists to suggest this is the case. Predictive biomarkers for the detection of clinically significant HRD are under development and nearing clinical application, and will be essential for identifying the broader TNBC cohort that may benefit from PARP-inhibitor therapy. In addition, a number of related pathways, ultimately converging on DNA repair, appear promising as additional opportunities for PARP inhibition, either by mutation-based or targeted therapy-induced synthetic lethality with PARP inhibition. It is clear that the classification of this subset of breast cancer as simply “triple negative” is entirely inadequate for the appropriate application of this promising new class of therapeutic molecules, and will soon be replaced by a more biologically accurate set of biomarkers, particularly those that define defects in DNA repair.
Disclosure
The author has served as a consultant to Tesaro and Myriad Genetics.
References
Dent R, Trudeau M, Pritchard KI, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007; 13(15 Pt 1):4429–4434. | |
[No authors listed]. Positive results for drug combo in I-SPY 2 trial. Cancer Discov. 2014;4(2):OF2. | |
Haffty BG, Yang Q, Reiss M, et al. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J Clin Oncol. 2006;24(36):5652–5657. | |
Guarneri V, Dieci MV, Conte P. Relapsed triple-negative breast cancer: challenges and treatment strategies. Drugs. 2013;73(12):1257–1265. | |
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. | |
Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. | |
Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. | |
Han B, Audeh W, Jin Y, Bagaria S, Cui X. Biology and treatment of basal-like breast cancer. In: Schatten H, editor. Cell and Molecular Biology of Breast Cancer. New York: Springer; 2013. | |
Bertucci F, Finetti P, Cervera N, et al. How basal are triple-negative breast cancers? Int J Cancer. 2008;123(1):236–240. | |
Prat A, Adamo B, Cheang MC, Anders CK, Carey LA, Perou CM. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist. 2013;18(2):123–133. | |
Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. | |
Oakman C, Moretti E, Pacini G, Santarpia L, Di Leo A. Triple negative breast cancer: a heterogeneous subgroup defined by what it is not. Eur J Cancer. 2011;47 Suppl 3:S370–S372. | |
Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. 2014;232(2):142–150. | |
Ignatiadis M, et al. A meta-analysis of gene expression profiling studies identifies clinically relevant oncogenic pathways in basal-like breast cancer. Cancer Res. 2009;69(24 Suppl 3):106. | |
Masuda H, Bedard P, Haibe-Kains, et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res. 2013;19(19):5533–5540. | |
Karami F, Mehdipour P. A comprehensive focus on global spectrum of BRCA1 and BRCA2 mutations in breast cancer. Biomed Res Int. 2013;2013:928562. | |
Hartman AR, Kaldate RR, Sailer LM, et al. Prevalence of BRCA mutations in an unselected population of triple-negative breast cancer. Cancer. 2012;118(11):2787–2795. | |
Gonzalez-Angulo AM, Timms KM, Liu S, et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res. 2011;17(5):1082–1089. | |
Bayraktar S, Glück S. Systemic therapy options in BRCA mutation-associated breast cancer. Breast Cancer Res Treat. 2012;135(2):355–366. | |
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. | |
Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. | |
Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–819. | |
Chalasani P, Livingston R. Differential chemotherapeutic sensitivity for breast tumors with “BRCAness”: a review. Oncologist. 2013;18(8):909–916. | |
De Summa S, Pinto R, Sambiasi D, et al. BRCAness: a deeper insight into basal-like breast tumors. Ann Oncol. 2013;24 Suppl 8: viii13–viii21. | |
Turner N, Moretti E, Siclari O, et al. Targeting triple negative breast cancer: is p53 the answer? Cancer Treat Rev. 2013;39(5):541–550. | |
Lips EH, Mulder L, Oonk A, et al. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer. 2013;108(10):2172–2177. | |
Telli M, Jensen KC, Abkevich V, et al. Homologous recombination deficiency (HRD) score predicts pathologic response following neoadjuvant platinum-based therapy in triple negative and BRCA1/2 mutation associated breast cancer. Cancer Res. 2012;72(24 Suppl 3):PD09-04. | |
Timms K, Abkevich V, Neff C, Morris B, et al. Frequency of homologous repair defects across breast cancer subtypes. 2013: Poster presented at: 104th Annual Meeting of the American Association of Cancer Research; April 6–10, 2013; Washington. | |
Rice JC, Ozcelik H, Maxeiner P, Andrulis I, Futscher BW. Methylation of the BRCA1 promoter is associated with decreased BRCA1 mRNA levels in clinical breast cancer specimens. Carcinogenesis. 2000;21(9):1761–1765. | |
Lips EH, Mulder L, Hannemann J, et al. Indicators of homologous recombination deficiency in breast cancer and association with response to neoadjuvant chemotherapy. Ann Oncol. 2011;22(4):870–876. | |
Oonk AM, van Rijn C, Smits MM, et al. Clinical correlates of ‘BRCAness’ in triple-negative breast cancer of patients receiving adjuvant chemotherapy. Ann Oncol. 2012;23(9):2301–2305. | |
Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963;11(1):39–43. | |
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8(3):193–204. | |
Steffen JD, Brody JR, Armen RS, Pascal JM. Structural implications for selective targeting of PARPs. Front Oncol. 2013;3:301. | |
Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10(4):293–301. | |
Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26(22):3785–3790. | |
Giannini G, Battistuzzi G, Vesci L, et al. Novel PARP-1 inhibitors based on a 2-propanoyl-3H-quinazolin-4-one scaffold. Bioorg Med Chem Lett. 2014;24(2):462–466. | |
De Lorenzo SB, Patel AG, Hurley RM, Kaufmann SH. The elephant and the blind men: making sense of PARP inhibitors in homologous recombination deficient tumor cells. Front Oncol. 2013;3:228. | |
Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599. | |
Murai J, Huang SY, Renaud A, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13(2):433–443. | |
O’Shaughnessy J, Osborne C, Pippen JE, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med. 2011;364(3):205–214. | |
Mateo J, Ong M, Tan DS, Gonzalez MA, de Bono JS. Appraising iniparib, the PARP inhibitor that never was – what must we learn? Nat Rev Clin Oncol. 2013;10(12):688–696. | |
Sinha G. Downfall of iniparib: a PARP inhibitor that doesn’t inhibit PARP after all. J Natl Cancer Inst. 2014;106(1):djt447. | |
Patel AG, De Lorenzo SB, Flatten KS, Poirier GG, Kaufmann SH. Failure of iniparib to inhibit poly(ADP-ribose) polymerase in vitro. Clin Cancer Res. 2012;18(6):1655–1662. | |
Pierce A, McGowan PM, Cotter M, et al. Comparative antiproliferative effects of iniparib and olaparib on a panel of triple-negative and non-triple-negative breast cancer cell lines. Cancer Biol Ther. 2013;14(6):537–545. | |
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. | |
Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. | |
Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–244. | |
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germ-line BRCA1/2 mutation: an open-label phase II study. J Clin Oncol. 2013;31 Suppl:11024. | |
Chan SL, Mok T. PARP inhibition in BRCA-mutated breast and ovarian cancers. Lancet. 2010;376(9737):211–213. | |
Gelmon KA, Hirte HW, Robidoux A, et al. Can we define tumors that will respond to PARP inhibitors? A phase II correlative study of olaparib in advanced serous ovarian cancer and triple-negative breast cancer. J Clin Oncol. 2010;28(Suppl 15):3002. | |
Chuang HC, Kapuriya N, Kulp SK, Chen CS, Shapiro CL. Differential anti-proliferative activities of poly(ADP-ribose) polymerase (PARP) inhibitors in triple-negative breast cancer cells. Breast Cancer Res Treat. 2012;134(2):649–659. | |
Palma JP, Wang YC, Rodriguez LE, et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. 2009;15(23):7277–7290. | |
Kummar S, Ji J, Morgan R, et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res. 2012;18(6):1726–1734. | |
Isakoff SJ, Overmoyer B, Tung NM, et al. A phase II trial of the PARP inhibitor veliparib (ABT888) and temozolomide for metastatic breast cancer. J Clin Oncol. 2010;28(Suppl 15):1019. | |
Sandhu SK, Schelman WR, Wilding G, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14(9):882–892. | |
Drew Y, Mulligan EA, Vong WT, et al. Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst. 2011;103(4):334–346. | |
Drew Y, Ledermann A, Jones A, et al. Phase II trial of the poly(ADP-ribose) polymerase (PARP) inhibitor AG-014699 in BRCA 1 and 2-mutated, advanced ovarian and/or locally advanced or metastatic breast cancer. J Clin Oncol. 2011;29(Suppl 15):3104. | |
Shen Y, Rehman FL, Feng Y, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19(18):5003–5015. | |
De Bono JS, Mina LA, Gonzalez M, et al. First-in-human trial of novel oral PARP inhibitor BMN 673 in patients with solid tumors. J Clin Oncol. 2013;31(Suppl 15):2580. | |
Cardnell RJ, Byers LA. Proteomic markers of DNA repair and Pl3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res. 2013;19(22):6322–6328. | |
Santarpia L, Iwamoto T, Di Leo A, et al. DNA repair gene patterns as prognostic and predictive factors in molecular breast cancer subtypes. Oncologist. 2013;18(10):1063–1073. | |
Pitroda SP, Pashtan IM, Logan HL, et al. DNA repair pathway gene expression score correlates with repair proficiency and tumor sensitivity to chemotherapy. Sci Transl Med. 2014;6(229):229ra42. | |
Peng G, Lin CJ, Mo W, et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat Commun. 2014;5:3361. | |
Mendes-Pereira AM, Martin SA, Brough R, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1(6–7):315–322. | |
Ibrahim YH, García-García C, Serra V, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2(11):1036–1047. | |
Matulonis UW, Wolf GM, Birrer MJ, et al. Phase I study of oral BKM120 and oral olaparib for high-grade serous ovarian cancer (HGSC) or triple-negative breast cancer (TNBC). J Clin Oncol. 2014;32(Suppl 5):2510. | |
Johnson N, Li YC, Walton ZE, et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med. 2011; 17(7):875–882. | |
O’Neil NJ, van Pel DM, Hieter P. Synthetic lethality and cancer: cohesin and PARP at the replication fork. Trends Genet. 2013;29(5):290–297. | |
Nowsheen S, Cooper T, Stanley JA, Yang ES. Synthetic lethal interactions between EGFR and PARP inhibition in human triple negative breast cancer cells. PLoS One. 2012;7(10):e46614. | |
Durkacz BW, Irwin J, Shall S. Inhibition of (ADP-ribose) biosynthesis retards DNA repair but does not inhibit DNA repair synthesis. Biochem Biophys Res Commun. 1981;101:1433–1441. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.