")
Back to Journals » OncoTargets and Therapy » Volume 13
Novel Small Molecular Compound AE-848 Potently Induces Human Multiple Myeloma Cell Apoptosis by Modulating the NF-κB and PI3K/Akt/mTOR Signaling Pathways
Authors Xu Y, Feng X, Zhou Q, Jiang W, Dai Y, Jiang Y, Liu X, Li S, Wang Y, Wang F, Li A, Zheng C
Received 7 July 2020
Accepted for publication 4 December 2020
Published 22 December 2020 Volume 2020:13 Pages 13063—13075
DOI https://doi.org/10.2147/OTT.S270090
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Gaetano Romano
Yaqi Xu,1– 3 Xiaoli Feng,4 Qian Zhou,1,2,5 Wen Jiang,1,2,6 Yibo Dai,1– 3 Yang Jiang,1– 3 Xiaoli Liu,1– 3 Shuo Li,1,2,7 Yongjing Wang,1– 3 Fang Wang,6 Ai Li,1– 3 Chengyun Zheng1– 3
1Department of Hematology, The Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, People’s Republic of China; 2Institute of Biotherapy for Hematological Malignancies, Shandong University, Jinan, Shandong, People’s Republic of China; 3Shandong University-Karolinska Institute Collaboration Laboratory for Stem Cell Research, Jinan, Shandong, People’s Republic of China; 4Clinical Laboratory, The Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, People’s Republic of China; 5Department of Hematology, Linyi Central Hospital, Linyi, Shandong, People’s Republic of China; 6Institute of Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, People’s Republic of China; 7Binzhou Medical University Hospital, Binzhou, Shandong, People’s Republic of China
Correspondence: Chengyun Zheng; Ai Li
Hematology Department of the Second Hospital of Shandong University, 247th of Beiyuan Road, Jinan, Shandong, People’s Republic of China
Tel +86 13675319282; +86 176 6008 0688
Email [email protected]; [email protected]
Background: We aimed to investigate the anti-multiple myeloma (MM) activity of the new small molecular compound AE-848 (5-bromo-2-hydroxyisophthalaldehyde bis[(1-methyl-1H-benzimidazol-2-yl)hydrazone]) and its underlying anti-MM mechanism.
Methods: Cell viability and apoptosis were detected and quantified by using MTT and flow cytometry, respectively. JC-1 dye-related techniques were used to assess mitochondrial membrane potential (MMP). Western blotting was applied to detect the expression of NF-κB and PI3K/Akt/mTOR pathway-associated proteins. The in vivo activity of AE-848 against MM was evaluated in a MM mouse model.
Results: Application of AE-848 into the in vitro cell culture system significantly reduced the viability and induced apoptosis of the MM cell lines, RPMI-8226 and U266, in a dose- and time-dependent manner, respectively. JC-1 dye and Western blotting analysis revealed that AE-848 induced the cleavage of caspase-8, caspase-3, and poly ADP-ribose polymerase (PARP), resulting in loss of mitochondrial membrane potential (MMP). Both the NF-κB and PI3K/AKT/mTOR signaling pathways were involved in AE-848-induced apoptosis of U266 and RPMI8226 cells. Moreover, AE-848 leads to cell cycle arrest of MM cells. Its anti-MM efficacy was further confirmed in a xenograft model of MM. AE-848 administration significantly inhibited MM tumor progression and prolonged the survival of MM-bearing mice. More importantly, our results demonstrated that AE-848 markedly induced primary MM cell apoptosis.
Conclusion: Our results for the first time showed that the small compound AE-848 had potent in vitro and in vivo anti-myeloma activity, indicating that AE-848 may have great potential to be developed as a drug for MM treatment.
Keywords: small molecular compound, AE-848, multiple myeloma, apoptosis, NF-κB and PI3K/Akt/mTOR pathway
Introduction
Multiple myeloma (MM) is a malignant tumor characterized by abnormal hyperplasia of plasma cells in the bone marrow. It accounts for approximately 10% of hematopoietic tumors worldwide, and its incidence is increasing globally.1 The widespread filling of malignant plasma cells in bone marrow leads to multiple osteolytic lesions, repeated infections, anemia, hypercalcemia, hyperviscosity syndrome and kidney damage, which can eventually result in adverse consequences.2
The pathogenesis of MM is extremely complex, involving a variety of cytokines, adhesion molecules, signal transduction pathways, cellular genetic abnormalities, and the bone marrow microenvironment.3 Among these, nuclear factor κB (NF-κB) is a key factor that selectively binds to the enhancer of B cell kappa-light chain to regulate the expression of many genes. Hyperactivated NF-κB signaling thus serves as an important prognostic biomarker and therapeutic target for MM.4,5 The PI3K/Akt/mTOR signaling pathway controls a number of biological processes critical to tumorigenesis, including apoptosis, transcription, translation, and cell cycle.6 A growing number of studies have shown that inhibition of the PI3K/Akt/mTOR signaling pathway triggers apoptosis in MM cells.7,8
In the recent years advances in treatments, including immunomodulatory drugs (eg, thalidomide and lenalidomide),9 proteasome inhibitors (eg, bortezomib and carfilzomib),10 and autologous transplantation,11 and so on, has changed outcome of MM patients tremendously. However, MM remains incurable, and there is a demand for novel therapeutic compounds. In the present study, we evaluated anti-MM activity of the small molecule 5-bromo-2-hydroxyisophthalaldehyde bis[(1-methyl-1H-benzimidazol-2-yl)hydrazone] (AE-848), which was cytotoxic to both MM-derived cell lines and primary MM cells. And administration of AE-848 significantly inhibited myeloma growth and prolonged survival of myeloma bearing mice. These findings together suggest a therapeutic potential of AE-848 for the treatment of MM.
Materials and Methods
General
1H NMR spectra were recorded on a Bruker DRX spectrometer at 600 MHz using DMSO-d6 as the solvent. Melting points (Mp) were determined using a Stuart melting point apparatus. All reagents and solvents were purchased from commercial sources and used without further purification.
Synthesis
Synthesis of 2-Hydrazino-1-Methyl-1H-Benzimidazole (1)
Ethanol (10 mL) was added to 2-chloro-1-methyl-1H-benzimidazole (1 g, 6.4 mmol), followed by addition of hydrazine (1 mL, 32 mmol) under stirring at room temperature. The solution was heated to 70 °C and allowed to stirred for 1 h, and then the reaction mixture was cooled to room temperature. After filtration and elimination of the solvent, the product was obtained as a white solid ([2-hydrazino-1-methyl-1H-benzimidazole, Mp: 148–150 °C. 1H NMR (600 MHz, CDCl3): 1.25–1.29 (s, 2H), 3.82–3.85 (s, 3H), 7.29–7.36 (m, 2H), 7.38–7.42 (d, 1H),7.81–7.84 (d, 1H), 7.86–7.89 (d, 1H)]).
Synthesis of 5-Bromo-2-Hydroxy-1,3-Benzenedicarboxaldehyde (2)
Trifluoroacetic acid (15 mL) was added to 4-bromophenol, followed by the addition of hexamethylenetetramine (1.62 g, 11.5 mmol) at room temperature under stirring. The solution was heated to 120 °C and refluxed for 12 h under argon. HCl (4N, 30 mL) was added to the reaction mixture, which was then stirred for another 1 h. The reaction mixture was cooled to room temperature and the crude product was obtained by filtration and elimination of the solvent. The pure product was obtained by column separation (eluent: petroleum ether/ethyl acetate = 3:1) as a yellow solid (5-bromo-2-hydroxy-1,3-benzenedicarboxaldehyde, Mp: 126–128 °C. 1H NMR (600 MHz, DMSO-d6): 8.12–8.16 (s, 2H), 10.19–10.21 (s, 2H), 11.55–11.70 (s, 1H)).
Synthesis of AE-848 (3)
Ethanol (10 mL) was added to a mixture of 2-hydrazino-1-methyl-1H-benzimidazole (1 mmol) and 5-bromo-2-hydroxy-1,3-benzenedicarboxaldehyde (1 mmol), and 2 drops of acetic acid were added. The precipitate formed after stirring for 30 min. The reaction mixture was refluxed for 1–2 h and filtrated when it became hot. The crude product was obtained by washing with hot methanol, and the pure product was purified by crystallizing with tert-butyl alcohol and DMF (2:1 (v/v)) [AE-848, Mp: 156–158 °C. 1H NMR (600 MHz, DMSO-d6): 3.88–3.92 (s, 6H), 6.85–7.30 (m, 8H), 7.44–7.49 (s, 2H), 7.86–7.93 (s, 2H), 8.40–8.46 (s, 2H), 11.10–11.45 (s, 1H)].
Reagents
AE-848 was dissolved in dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO, USA) at a concentration of 50 mM and stored at 37°C until use. Human CD38-PE monoclonal antibody was obtained from Miltenyi (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Annexin V-FITC/propidium iodide (PI) detection kit and mitochondrial membrane potential (MMP) detection kit were purchased from KeyGEN BioTECH (Jiangsu, China). MTT was obtained from Solarbio (Beijing, China). Antibodies against Caspase-8, Caspase-3, cleaved poly ADP-ribose polymerase (PARP), P65, phosphatidylinositol 3 kinase (PI3K), Akt, mammalian target of rapamycin (mTOR), GAPDH and β-actin were purchased from Cell Signaling Technology (Danvers, MA, USA). And antibodies against NF-κB2 P100/P52, NF-κB1 P105/P50, Rel-B, c-Rel and Histone-3 were purchased from Abcam (Cambridge, UK). Pan-caspase inhibitor, Z-VAD-FMK (zVAD), was purchased from R&D Systems (Minneapolis, MN, USA).
Cell Culture
Human MM cell lines (U266 and RPMI8226) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) at 37 °C in a humidified incubator with 95% air and 5% CO2. Cells in the logarithmic growth phase were selected for subsequent experiments.
Primary MM Cells
The human sample study was approved by the Human Ethics Research Committee of the Second Hospital of Shandong University (approval no. KYLL-2019(KJ)P-0205). Bone marrow specimens were obtained from newly diagnosed or relapsed/refractory patients with MM. Bone marrow mononuclear cells (BMMCs) by Ficoll-Hypaque (Tianjin HY Bioscience Co., Ltd., Tianjin, China) were isolated via density gradient centrifugation and further cultured in RPMI-1640 medium with 10% FBS. Blood samples from healthy volunteers were collected and treated with Ficoll-Hypaque density gradient centrifugation to isolate peripheral blood mononuclear cells (PBMCs) following the reagent instructions. Each patient and healthy volunteer provided informed consent. The study was approved by the Human Ethics Research Committee of the Second Hospital of Shandong University in accordance with the Declaration of Helsinki.
Cell Viability Analysis
MTT assay was conducted to detect cell viability.12 U266 and RPMI8226 cells were seeded at 2×104 cells/well and treated with 2.5, 5, 10, or 20 µM AE-848 for 12 h, or with 5 µM AE-848 for 24 and 48 h, respectively. The cells were incubated with MTT at room temperature for 4 h. Subsequently, an appropriate amount of DMSO was then added and oscillated on the oscillator for 15 min. The optical density at 570 nm was measured with a multifunctional microplate reader (Synergy Neo, BioTek, Winooski, VT, USA), and the values were expressed as absorbance.
Apoptosis Analysis
U266 and RPMI8226 cells (2×105 cells/well) were incubated with AE-848 (2.5, 5, and 10 µM) for 12 h or incubated with 5 µM AE-848 for 24 h and 48 h. The cells were then incubated with Annexin V in the dark for 15 min. Subsequently, 2 µL PI was added to the cell suspension, and the apoptosis rate was measured using a flow cytometer (BD Biosciences, San Jose, CA, USA).
For eight primary MM samples, the cells (2×105 cells/well) were inoculated into 24-well plates with 5 μM AE-848 for 12 h. The cultured cells were then collected, washed, and stained with anti-human CD38-PE monoclonal antibody in PBS for 20 min at room temperature in the dark. Afterwards, the cells were washed with PBS twice and resuspended in 100 μL binding buffer containing 2 μL Annexin-V. The percentage of apoptotic cells was measured using FACS.
Analysis of MMP
JC-1 dye was used to detect MMPs.13 The images were taken with an inverted fluorescent microscope. Briefly, U266 and RPMI8226 cells (2×105 cells/well) were treated with AE-848 (5µM) for 12 h. After incubation, the MM cells were first rinsed with PBS, then added to the JC-1 dye working solution and shaken evenly. Next, the cells were incubated at room temperature for 15 min, washed with JC-1 staining buffer twice, and analyzed by flow cytometry.
Western-Blotting Analysis
U266 and RPMI8226 cells were cultured with various concentrations of AE-848 (2.5, 5, and 10 µM) for 12 h. Drug-treated cells were lysed in RIPA buffer supplemented with a phosphatase inhibitor cocktail (Roche, Mannheim, Germany) and 1 mM PMSF on ice for 20 min. Cytoplasmic and nuclear proteins were isolated using the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Protein concentrations were measured using a BCA Protein Assay kit (Pierce, Thermo Scientific, Waltham, MA, USA) following the manufacturer’s instructions. Protein samples were separated by polyacrylamide-SDS gels and electroblotted onto nitrocellulose membranes. After blocking, the membrane was incubated overnight with the following primary antibodies: Caspase-8, Caspase-3, cleaved-PARP, P65, NF-κB2 P100/P52, NF-κB1 P105/P50, Rel-B, c-Rel, PI3K, AKT1, mTOR, GAPDH and β-actin (Caspase-8, mouse polyclonal, 1:1000, CST; Caspase-3, mouse polyclonal, 1:1000, CST; Cleaved-PARP, rabbit polyclonal, 1:1000, CST; Cleaved-PARP, rabbit polyclonal, 1:1000, CST; P65, rabbit polyclonal, 1:1000, CST; NF-κB2 P100/P52, rabbit polyclonal, 1:1000, Abcam; NF-κB1 P105/P50, rabbit polyclonal, 1:1000, Abcam; Rel-B, rabbit polyclonal, 1:1000, Abcam; c-Rel, rabbit polyclonal, 1:1000, Abcam; PI3K, rabbit polyclonal, 1:1000, CST; AKT1, rabbit polyclonal, 1:1000, CST; mTOR, rabbit polyclonal, 1:1000, CST; GAPDH, mouse polyclonal, 1:1000, CST; β-actin, rabbit polyclonal, 1:1000, Abcam; Histone-3, rabbit polyclonal, 1:1000, Abcam), followed by a secondary antibody for 1 h at room temperature.
Cell Cycle Analysis
Cell cycle distribution was analyzed using flow cytometric assay, as previously described.14 In brief, U266 and RPMI8226 cells treated with AE-848 (2.5, 5, and 10 µM) were harvested, washed twice with PBS, and fixed overnight with 75% ethanol. U266 and RPMI8226 cells were incubated with 500 μL PI/RNase staining buffer at 37 °C for 30 min, followed by FACS analyses.
Mouse Xenograft Model
The animal study was approved by the Second Hospital of Shandong University of Medicine Institutional Animal Care & Use Committee (approval no. KYLL-2018(LW)019). The study obeyed the principles of the ethical guidelines outlined by the International Council for Laboratory Animal Science (ICLAS).15 Female NOD/SCID mice (6–8 weeks, weighing 18–20 g) were purchased from the Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China) and raised under a specific-pathogen-free (SPF) environment. Mice were subcutaneously injected with 1×107 RPMI8226 cells suspended in 100 μL normal saline (NS) in the right foreleg. Based on our pilot evaluations, we selected a concentration of 12.5 mg/kg via intraperitoneal injection for subsequent in vivo experiments.
Approximately 3 weeks after RPMI8226 cell injection, when the tumor reached a size of approximately 200 mm3, 12 immunized mice were randomly divided into two groups: the control (n=6) and treatment groups (n=6). The mice in the control group were intraperitoneally injected with NS containing DMSO and Cremophor EL for 14 consecutive days, while the mice in the treatment group were additionally administered AE-848 (12.5 mg/kg). When the mice reached the endpoint of the observations, which was defined as when the tumor size exceeded 2.0 cm in any direction or when a mouse was unable to creep for food and/or water, the mice were humanely euthanized by cervical dislocation. Changes in mice weight and tumor volume were monitored in the control and treatment groups every 3 days. The administration of compounds was carried out as a blind experiment, and all information about the expected outputs and the nature of compounds used were kept from the animal technicians. Tumors were measured using calipers, and volumes were calculated using the formula V = long diameter × (short diameter) 2 × π/6.16
Statistical Analysis
The data were analyzed using SPSS 19.0, and one-way analysis of variance (ANOVA) and Bonferroni’s test were used to compare differences among different treatment groups. Survival rate was analyzed by Kaplan–Meier analysis. A P-value of < 0.05 was considered statistically significant.
Results
Chemistry
AE-848 was prepared by following the expected reaction route in Figure 1. The starting material, 2-chloro-1-methyl-1H-benzimidazole (1) as the starting material, was reacted with hydrazine hydrate to obtain intermediate 2, which was then reacted with 5-bromo-2-hydroxy-1,3 benzenedicarboxaldehyde (4). The reaction produced the target product (5). The NMR spectra of AE-848 are shown in Supplementary Figure 1.
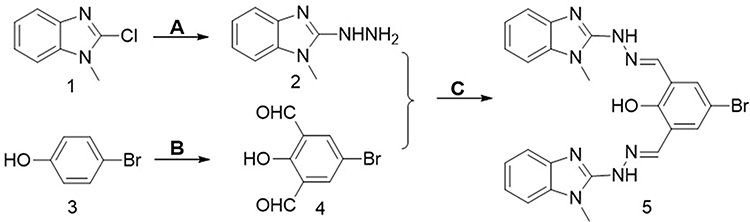 |
Figure 1 Synthetic route of AE-848. (A) Hydrazine, Ethanol. (B) Trifluoroacetic Acid, Hexamethylenetetramine. (C) Ethanol. |
Inhibitory Effect of AE-848 on U266 and RPMI8226 Cell Viability
AE-848 is a synthesized small-molecule compound (Figure 2A). To investigate whether AE-848 affects the viability of U266 and RPMI8226 cells, we conducted an MTT assay. As presented in Figure 2B and D, AE-848 suppressed the viability of U266 and RPMI8226 cells in a concentration- and time-dependent manner. More specifically, after treatment with 2.5, 5, 10, and 20 μM AE-848 for 12 h, the inhibition rates of U266 and RPMI8226 increased gradually. The IC50 values of U266 and RPMI8336 cells were 4.3 ± 2.5 μM and 5.1 ± 3.5 μM, respectively (Figure 2B and D). As shown in Figure 2C and E, after exposure to 5 μM AE-848 for 12, 24, and 48 h, the remaining viable U266 cells were 52.9 ± 4.2%, 32.0 ± 1.8%, and 18.4 ± 1.2%, respectively. Meanwhile, the viable RPMI8336 cells were 53.1 ± 0.7%, 35.0 ± 1.3% and 6.1 ± 4.2%, respectively.
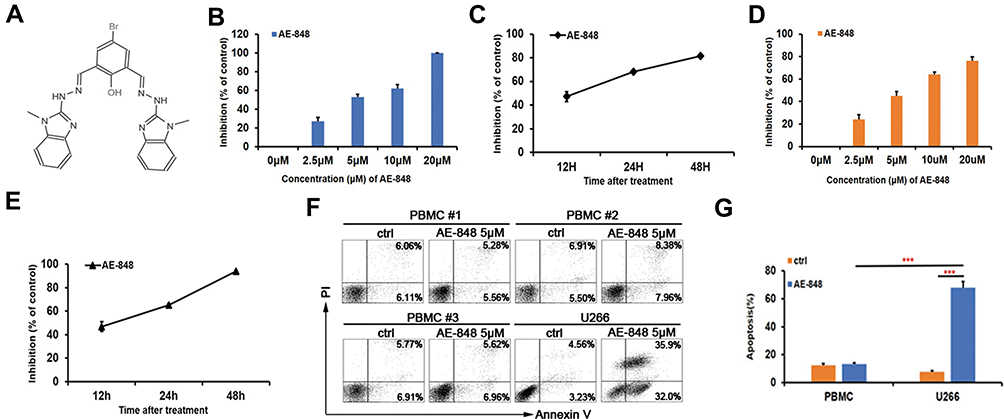 |
Figure 2 The chemical structure of AE-848. AE-848 inhibits MM cell viability in a dose- and time-dependent manner, and AE-848 induces minimal cytotoxicity to peripheral blood mononuclear cells (PBMCs). (A) The chemical structure of AE-848. (B) U266 cells were treated with AE-848 (2.5, 5, 10, and 20 µM) for 12 h, and cell viability was then assessed using MTT. (C) U266 cells were treated with 5 µM AE-848 for 12, 24, and 48 h, and the viability was determined using MTT. (D) RPMI8226 cells were treated with different concentrations of AE-848 (2.5, 5, 10, and 20 µM) for 12 h followed by cell viability detection. (E) RPMI8226 cells were treated with AE-848 (5 µM) for 12, 24, and 48 h, and cell viability was then analyzed. (F) Apoptosis of PBMCs and U266 cells treated with 5 µM AE-848 was analyzed using flow cytometry. (G) Apoptosis of PBMCs and U266 cells. ***P < 0.001 (untreated control vs AE-848-treated cells). Results were expressed as the mean ± SEM from three independent experiments. |
AE-848 Has Lower Cytotoxicity Against Peripheral Blood Mononuclear Cells
To evaluate the toxicity of AE-848 to normal cells, we collected PBMCs from normal human peripheral blood, and compared apoptosis between PBMCs and U266 cells incubated with AE-848 for 12 h. Annexin V/PI staining results showed that when incubated with 5 μM AE-848 for 12 h, AE-848 exhibited negligible toxicity to normal PBMCs, while a great killing effect was observed on U266 cells (13.3 ± 1.1%, PBMCs vs 68.0 ± 4.3%, U266; P < 0.001) (Figure 2F and G).
Induction of Cell Apoptosis by AE-848
U266 and RPMI8226 cells were stained with Annexin-V FITC and PI to test apoptosis by flow cytometry. The total number of AV+PI− and AV+ PI+ cells were counted as apoptotic cells. As shown in Figure 3A, the induction of apoptosis by AE-848 increased with its concentration, and the proportion of Annexin V-positive cells increased. More specifically, compared to the control group (11.4 ± 4.5%), the apoptosis of U266 cells increased to 36.3 ± 5.4% at 2.5 μM, 62.6 ± 4.6% at 5 μM, and 85.0 ± 2.5% at 10 μM. Meanwhile, the apoptotic RPMI8226 cells (baseline 6.6 ± 1.7%) increased to 13.6 ± 4.6% at 2.5 μM, 35.2 ± 5.0% at 5 μM, and 66.4 ± 2.3% at 10 μM (Figure 3B). A similar pattern was observed in Figure 3C and D, in which markedly increased apoptosis occurred after incubation with 5 μM AE-848 for 24 or 48 h. Apoptosis was induced in both U266 and RPMI8226 cells in a dose- and time-dependent manner, consistent with our findings in the MTT assay.
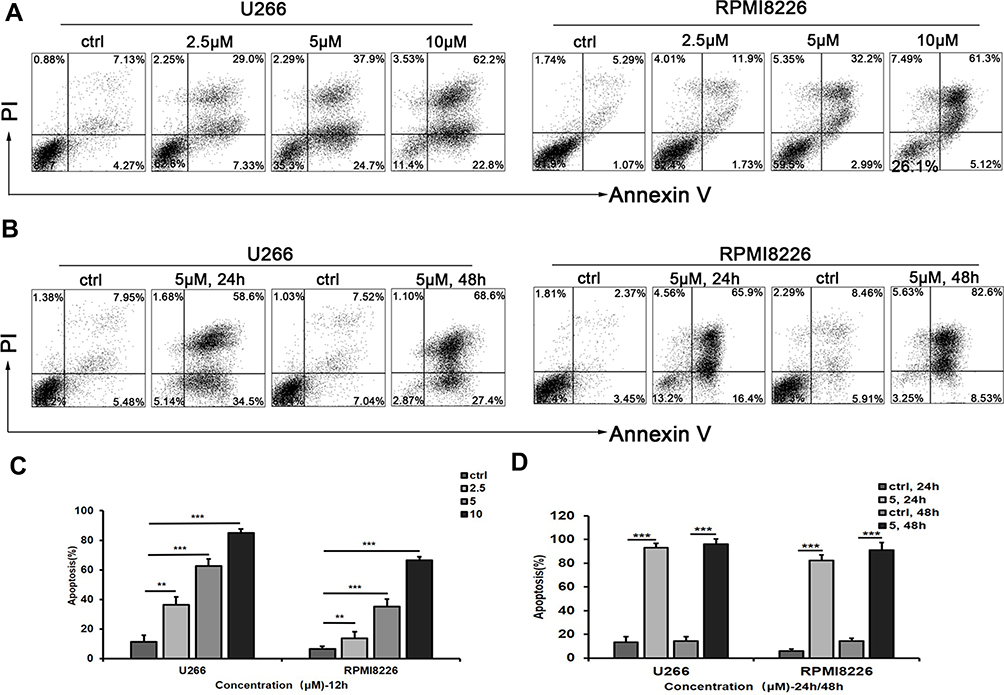 |
Figure 3 AE-848 significantly induces apoptosis of U266 and RPMI8226 cells. (A) U266 and RPMI8226 cells were treated with AE-848 (2.5, 5, and 10 µM) or vehicle control for 12 h, and cells were then stained with Annexin V/PI. (B) Apoptotic U266 and RPMI8226 cells were measured using flow cytometry after being treated with AE-848 (5 µM) for 24 and 48 h, respectively. (C) After exposure to AE-848 (2.5, 5, and 10 µM) for 12 h, the apoptosis rates of U266 and RPMI8226 cells robustly increased in a dose-dependent manner. (D) After treatment with 5 µM AE-848 for 24 and 48 h, the apoptosis rates of U266 and RPMI8226 cells significantly increased in a time-dependent manner (n=3). **P < 0.01, and ***P < 0.001, respectively (untreated control vs AE-848-treated group). |
To further evaluate the toxic effect of AE-848 on primary MM cells obtained from the bone marrow of newly diagnosed or relapsed/refractory patients, we treated the cells with 5 μM AE-848 for 12 h. Consistent with the results obtained from MM cell lines, AE-848 significantly promoted the apoptosis of primary MM cells in comparison to the control group (74.2 ± 21.3% vs 20.7 ± 10.6%, P < 0.001) (Figure 4).
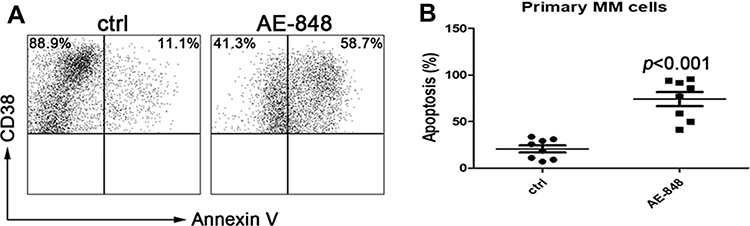 |
Figure 4 AE-848 induced apoptosis of primary MM cells. (A) Purified patient MM cells (CD38+) were treated with AE-848 or vehicle control, and the cell apoptosis rate was then assessed. (B) Percentage of Annexin-V expression in the control and AE-848 groups. Data are presented as the mean ± SD (n=8). |
AE-848 Induced Loss of MMP in MM Cells
JC-1 dye-related techniques were used to assess MMP in MM cells. The relative proportion of red and green fluorescence is commonly used to measure the degree of mitochondrial depolarization. A decrease in the red/green ratio is indicative of apoptosis. When JC-1 staining was applied to investigate the possible involvement of the mitochondrial apoptosis pathway, we found that after exposure to 5 µM AE-848 for 12 h, a dramatic drop in MMP was observed in both U266 and RPMI8226 (Figure 5A and B) (U266, treated vs untreated, 87.0 ± 3.1% vs 8.0 ± 3.1%, P < 0.001; RPMI8226, treated vs untreated, 82.5 ± 2.7% vs.15.7 ± 3.5%, P < 0.001). In addition, a stronger green fluorescence was observed in AE-848-treated groups, in line with our flow cytometry data (Figure 5C). These results suggest that the mitochondrial-related intrinsic apoptosis pathway is involved in AE-848-induced apoptosis.
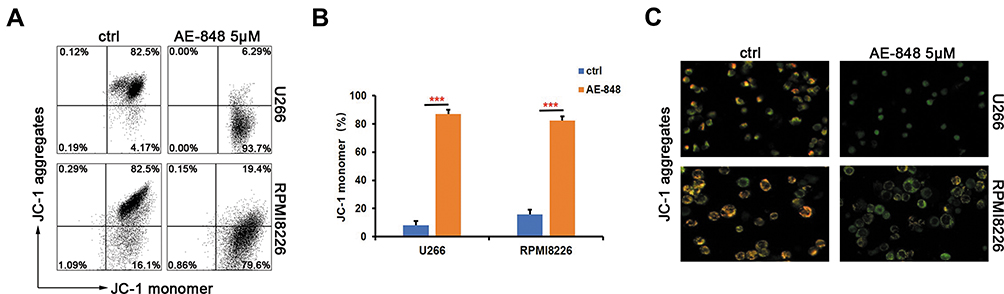 |
Figure 5 AE-848 markedly induces loss of MMP. (A) After exposing U266 and RPMI8226 cells to 5 µM AE-848 for 12 h, the loss of MMP was detected by using JC-1 flow cytometry. (B) Fluorescence intensity of the AE-848 treatment groups (***P < 0.001 compared to the control). (C) Fluorescence images of U266 and RPMI8226 cells stained with JC-1. JC-1 red and JC-1 green represent the JC-1 polymer and JC-1 monomer, respectively. |
AE-848 Induced Cleavage of Caspase 8, Caspase 3, and Cleaved PARP in MM Cells
Western blotting analysis was performed to study the underlying mechanisms of the related proteins associated with mitochondria-mediated intrinsic apoptosis in U266 cells. This included pro-caspase-3 and pro-caspase-8, two key proteases in the apoptosis pathway. Processing of both pro-caspase-3 and pro-caspase-8 increased in AE-848-treated U266 and RPMI8226 cells (Figure 6A and B). In addition, the cleavage of the nuclear protein PARP, a caspase-3-dependent protein, was also observed in AE-848-treated U266 and RPMI8226 cells.
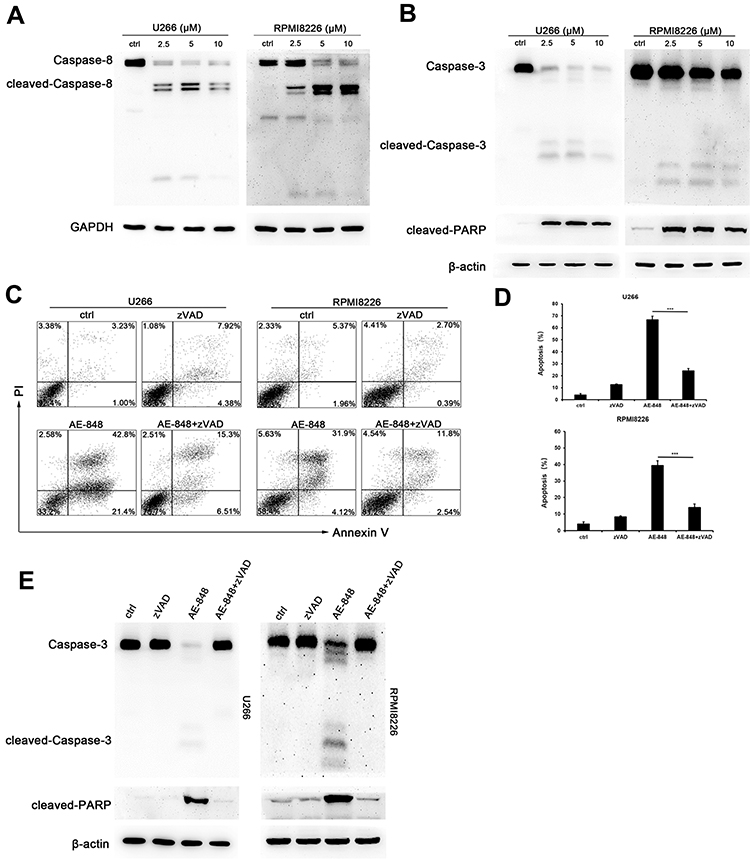 |
Figure 6 AE-848 induced cleavage of caspase-8, caspase-3, and PARP. (A) The protein expression of cleaved caspase-8 in U266 and RPMI8226 cells was determined using Western blotting. (B) The protein expression of cleaved-caspase-3 and cleaved-PARP in U266 and RPMI8226 cells was assessed using Western blotting. (C) U266 and RPMI8226 cells were treated with 20 μM zVAD for 1 h prior to adding 5 µM AE-848 for 12 h. Percentages of AV- and PI-positive cells are indicated in the plots. (D) Apoptosis of U266 and RPMI8226 cells treated with AE-848 in the presence or absence of zVAD (***P < 0.001, compared to the control). (E) The protein expression of caspase-3 and cleavage of PARP in U266 and RPMI8226 cells treated with AE-848 in the presence or absence of zVAD at 12 h. |
AE-848 Induces Apoptosis in MM Cells in a Caspase-Dependent Manner
In this study, we observed caspase activation in MM cells treated with AE-848. To further reveal the importance of caspase activation for AE-848-induced apoptosis, we applied Z-VAD-FMK (zVAD), a pan-caspase inhibitor, to cell culture at 20 μM for 1 h, followed by the addition of AE-848 (5 μM). Interestingly, flow cytometry results showed that zVAD significantly attenuated AE-848-induced apoptosis (24.0 ± 2.1% vs 66.7 ± 3.0% in U266 cells; 14.0 ± 2.0% vs 39.3 ± 3.1%, RPMI8226 cells; P < 0.001; Figure 6C and D). Consistently, Western blotting revealed that zVAD markedly reduced the cleavage of caspase-3 and PARP (Figure 6E). Taken together, the extrinsic cell apoptosis pathway is involved in AE-848-induced apoptosis in a caspase-dependent manner.
AE-848 Induced U266 and RPMI8226 Cell Apoptosis by Inhibiting the NF-κB and PI3K/Akt/mTOR Signaling Pathways
We then sought to determine whether the NF-κB and PI3K/Akt/mTOR signaling pathways are involved in AE-848-induced U266 and RPMI8226 cell apoptosis. P65, NF-κB2 P100/P52, NF-κB1 P105/P50, Rel-B, c-Rel, PI3K, Akt1, and mTOR protein expression were assessed. As presented in Figure 7A–D, the cytoplasmic and nuclear protein expression of P65, NF-κB2 P100/P52, NF-κB1 P105/P50, Rel-B, and c-Rel were significantly decreased in U266 and RPMI8226 cells, indicating that the NF-κB pathway was inhibited in the AE-848-treated MM cells. Moreover, AE-848 markedly reduced PI3K, mTOR and AKT1 protein expression (Figure 7E and F), indicating that down-regulation of PI3K/Akt/mTOR signaling pathways is involved in MM cell apoptosis induced by AE-848.
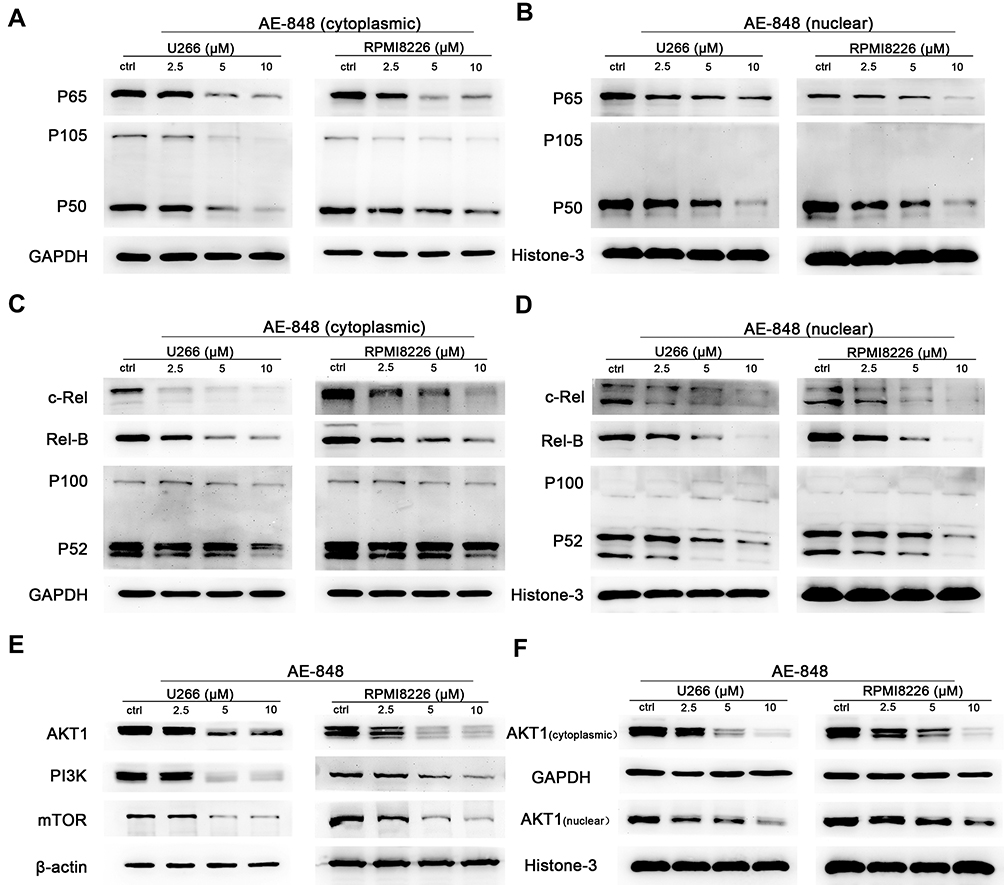 |
Figure 7 AE-848 inhibits the NF-κB and PI3K/Akt/mTOR signaling pathways. (A–D), Effects of AE-848 (2.5, 5, and 10 µM) on the cytoplasmic and nuclear protein expression of NF-κB pathway-associated proteins (P65, NF-κB2 P100/P52, NF-κB1 P105/P50, Rel-B, and c-Rel) in U266 and RPMI8226 cells. (E) Effects of AE-848 (2.5, 5, and 10 µM) on the protein expression of PI3K/AKT/mTOR pathway-associated proteins in U266 and RPMI8226 cells. (F) Cytoplasmic and nuclear AKT1 levels were measured at the indicated concentrations of AE-848. |
AE-848 Induced Cell Cycle Arrest in MM Cells
We next conducted flow cytometry assays to test whether AE-848 could cause cell cycle arrest. As shown in Figure 8A–C, after AE-848 treatment for 12 h, the percentage of cells in G2/M phase increased from 23.5% to 29.3% for U266 (Figure 8B) and from 46.6% to 62.0% for RPMI8226 (Figure 8C).
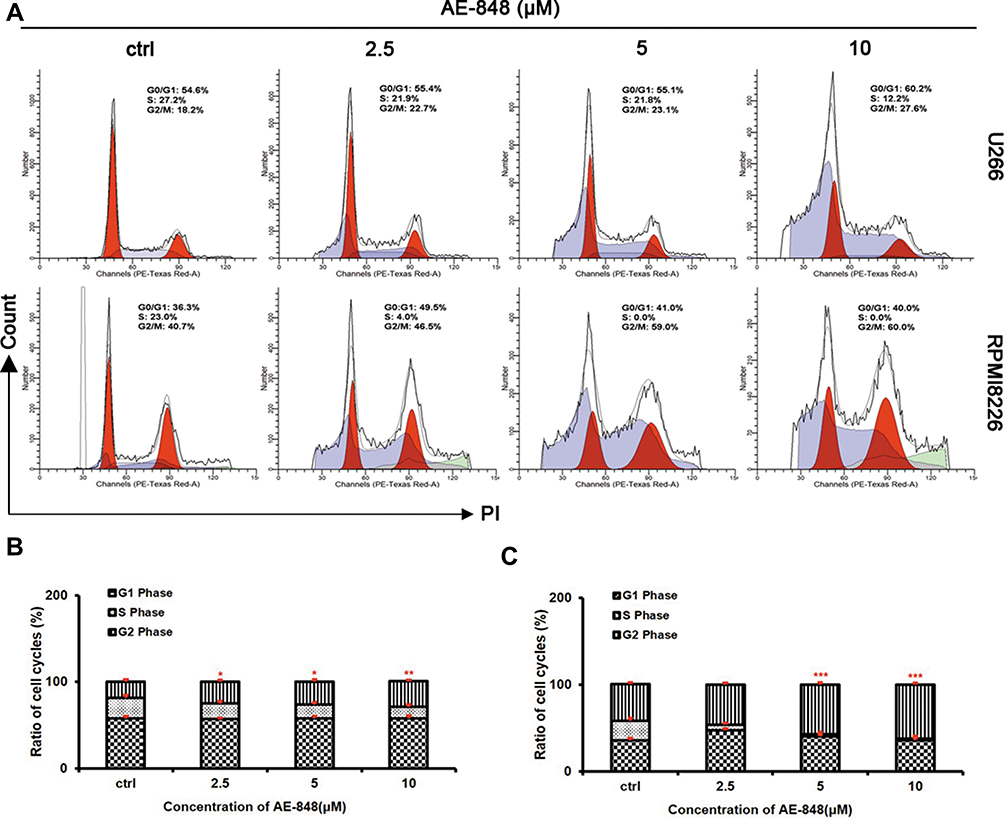 |
Figure 8 AE-848 elicits cell cycle arrest in MM cells. (A) Cell cycle analysis was performed in U266 and RPMI8226 cells treated with and without AE-848 (2.5, 5, and 10 μM) for 12 h. (B and C) Cell cycle distribution in U266 and RPMI8226 cells. The histogram shows the percentage of cells in the G1, S, and G2/M phases. *P < 0.05, **P < 0.01, and ***P < 0.001, respectively (compared with the control). |
AE-848 Suppressed Tumor Growth and Prolonged Overall Survival of MM-Bearing Mice
To examine the therapeutic effects of AE-848 on MM in vivo, MM cell-bearing mice were treated with AE-848 or NS containing DMSO and Cremophor EL by intraperitoneal injection every day for 14 days. As shown in Figure 9A–C, AE-848 administration inhibited tumor growth, in terms of tumor weight and tumor volume. Kaplan–Meier curves (Figure 9D) showed that AE-848 treatment significantly prolonged the survival time of MM cell-bearing mice (23.5 vs 17.0 days, P < 0.001). Taken together, AE-848 selectively inhibited tumor growth and prolonged survival of MM cell-bearing mice in vivo.
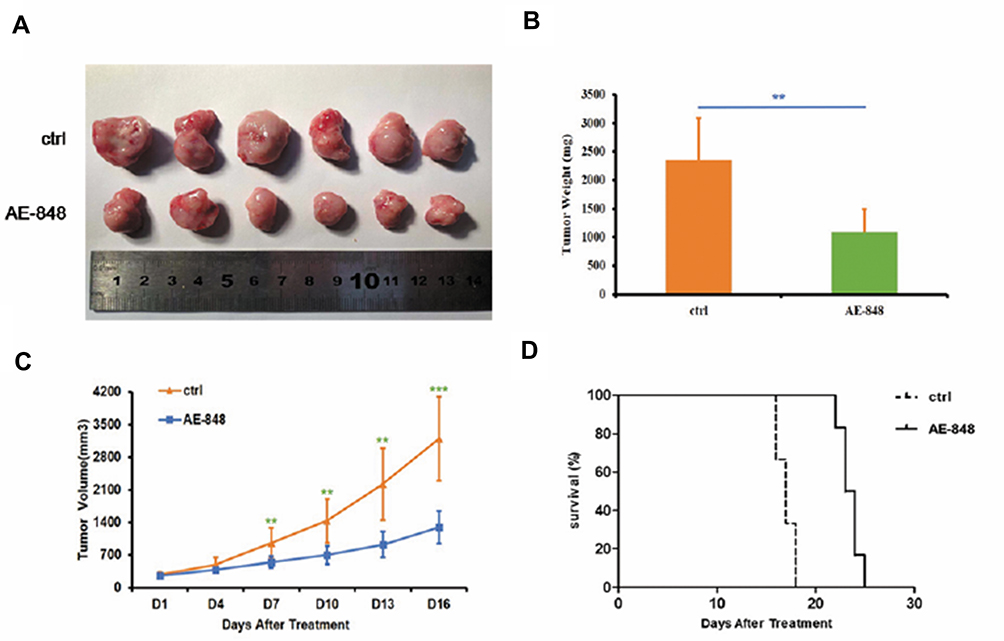 |
Figure 9 AE-848 significantly inhibits tumor growth and prolongs overall survival of MM cell-bearing mice. (A) Xenograft images. (B) AE-848 treatment significantly reduced the average tumor weight of the MM cell-bearing mice. **P < 0.01 (compared with the control group). (C) The tumor volumes were measured every day for 16 days, and data represent the mean ± SD (**P < 0.01, ***P < 0.001). (D) AE-848 treatment significantly prolonged survival time of MM cell-bearing mice. |
Discussion
Multiple myeloma (MM) is a malignant plasma cell disease, often accompanied by multiple osteolytic lesions, hypercalcemia, anemia, and kidney damage.17 Although proteasome inhibitors and immunoregulatory drugs significantly improve the treatment efficiency and prognosis of MM patients, patients eventually relapse; MM therefore remains an incurable malignancy. Additionally, side effects created by the current regimens affect the quality of life of patients.1,18 Thus, the development of new therapeutic drugs with high efficacy and minimal side effects is urgently required. By screening a small molecular library (Specs_SC), we identified twenty compounds and found that only AE-848 significantly induced apoptosis in MM cells but not in normal cells.
Apoptosis is a kind of programmed death, which is crucial for cells to maintain the balance of the body.19 As cancer cells have abnormal proliferation and survival characteristics, inducing apoptosis is one of the main mechanisms for many anti-tumor drugs.20 A novel aryl-guanidino compound AE-848 was synthesized in our study, and our results demonstrate its potent anti-MM efficacy. We used an MTT assay to evaluate the viability of MM cells. AE-848 inhibited U266 and RPMI8226 cell viability in a dose- and time-dependent manner. Moreover, the same concentration of AE-848 and treatment times had negligible toxicity in normal PBMCs, providing a therapeutic window for AE-848 in MM treatment. Moreover, exposure to AE-848 remarkably induced apoptosis of U266, RPMI8226, and primary MM cells. Consistently, in vivo experiments using MM cell-bearing mice showed that tumor weight and tumor volume were significantly reduced following AE-848 treatment.
Mitochondria-mediated apoptosis is considered an important apoptotic pathway. The loss of MMP leads to mitochondrial depolarization, which in turn promotes the release of apoptotic factors and ultimately triggers cell apoptosis.21 In our study, treatment with AE-848 sharply decreased the MMP in U266 and RPMI8266 cells, which was consistent with our immunofluorescence results. Mitochondria-mediated apoptosis involves many factors, such as caspase-3, caspase-8, and PARP.22 Caspase-8 (initiator caspase) and caspase-3 (executor caspase) are core components of apoptosis resulting from exogenous or endogenous apoptotic signals.23 Meanwhile, PARP is the main shear substrate of caspase-3, which is considered an important indicator of caspase-3 activation. Western blotting results showed that treatment with AE-848 induced the cleavage of caspase-3, caspase-8, and PARP. When zVAD was added, the apoptosis rate of U266 cells was significantly reduced, coupled with diminished cleavage of caspase-3.
Uncontrolled cell proliferation is one of the most important hallmarks of tumor cells. Moreover, an aberrant cell cycle accounts for dysregulated cell growth, which ultimately leads to tumor formation.24 In this study, we demonstrated that AE-848 inhibited the growth of U266 and RPMI8226 cells by inducing cell cycle arrest at the G2/M phase.
As a transcription factor, NF-κB is composed of dimeric complexes of p50 (NF-κB1) or p52 (NF-κB2), usually associated with members of the Rel family (P65, c-Rel, Rel-B). NF-κB plays an important role in regulating the inflammatory response and cellular proliferation, and is generally inactive in normal cells.25 IκB, an inhibitor of NF-κB, prevents NF-κB from transferring into the nucleus. Phosphorylated IκB releases NF-κB, which then enters the nucleus and triggers the activation of downstream genes and participates in a series of biological processes, including MM.26 In the present study, the cytoplasmic and nuclear protein expression of P65, NF-κB2 P100/P52, NF-κB1 P105/P50, Rel-B, and c-Rel in U266 and RPMI8226 cells were significantly inhibited by AE-848, indicating that AE-848 inhibits the NF-κB signaling pathway in MM cells.
PI3K/Akt/mammalian target of rapamycin (mTOR) is an important intracellular signaling pathway directly related to cell dormancy, proliferation, and longevity.22 Increasing numbers of studies have suggested that inhibition of PI3K/Akt/mTOR is crucial for the anti-proliferation effect of MM cells.27 Akt is the downstream target of PI3K. Upon PI3K activation, Akt is phosphorylated and activated to localize in the plasma membrane. Activated Akt regulates cell function by phosphorylating downstream factors, including various enzymes, kinases, and transcription factors.28,29 mTOR is an important downstream target of PI3K/Akt, and participates in the regulation of tumor cell proliferation.30 It has been shown that inhibition of PI3K/Akt/mTOR signaling pathway prolongs the life cycle and improves the quality of life of MM patients.31 In our study, a decreased protein expression of PI3K, Akt, and mTOR was observed in MM cells, which was consistent with the results of a previous study.32 Here, in our study, a decreased protein expression of PI3K, Akt and mTOR were observed in MM cells treated by AE-848, suggesting that PI3K/Akt/mTOR pathway is involved in AE-848 induced MM apoptosis.
Conclusion
AE-848 induces apoptosis of MM cells in vitro and in vivo, and exerts this effect on primary MM cells through its inhibitory effects on the NF-κB and PI3K/Akt/mTOR signaling pathways. Given its minimal toxicity to normal blood cells, AE-848 may thus be a promising candidate drug to develop for the treatment of MM.
Acknowledgments
The present study was supported by the Science and Technology Innovation Project of Shandong Province (grant nos. 2019JZZY011115, 2017GSF18136 and 2018GSF118034), the National Natural Science Foundation of China (grant nos. 81600176, 81602694 and 81372545), and the National Natural Science Foundation of Shandong Province (grant no. ZR2016HB71). We would like to thank Editage for English language editing.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Zeng C, Li X, Li A, et al. Knockdown of NUPR1 inhibits the growth of U266 and RPMI8226 multiple myeloma cell lines via activating PTEN and caspase activation-dependent apoptosis. Oncol Rep. 2018;40(3):1487–1494.
2. Feng X, Zhang L, Nie S, et al. The effect of ras homolog C/Rho-associated coiled-protein kinase (Rho/ROCK) signaling pathways on proliferation and apoptosis of human myeloma cells. Med Sci Mon. 2019;25:7605–7616. doi:10.12659/MSM.915998
3. Furukawa Y, Kikuchi J. Molecular pathogenesis of multiple myeloma. Int J Clin Oncol. 2015;20(3):413–422. doi:10.1007/s10147-015-0837-0
4. Xiang Y, Remily-Wood ER, Oliveira V, et al. Monitoring a nuclear factor-kappaB signature of drug resistance in multiple myeloma. Mol Cell Proteomics. 2011;10(11).
5. Liu S, Zheng LL, Zhu YM, et al. Knockdown of REGgamma inhibits the proliferation and migration and promotes the apoptosis of multiple myeloma cells by downregulating NF-kappaB signal pathway. Hematology. 2018;23(5):277–283. doi:10.1080/10245332.2017.1385194
6. Keppler-Noreuil KM, Parker VE, Darling TN, Martinez-Agosto JA. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am J Med Genet C Semin Med Genet. 2016;172(4):402–421. doi:10.1002/ajmg.c.31531
7. Mao X, Liang SB, Hurren R, et al. Cyproheptadine displays preclinical activity in myeloma and leukemia. Blood. 2008;112(3):760–769. doi:10.1182/blood-2008-02-142687
8. Min YH, Eom JI, Cheong JW, et al. Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: its significance as a prognostic variable. Leukemia. 2003;17(5):995–997. doi:10.1038/sj.leu.2402874
9. Kazandjian D. Multiple myeloma epidemiology and survival: a unique malignancy. Semin Oncol. 2016;43(6):676–681. doi:10.1053/j.seminoncol.2016.11.004
10. Landgren O, Iskander K. Modern multiple myeloma therapy: deep, sustained treatment response and good clinical outcomes. J Intern Med. 2017;281(4):365–382. doi:10.1111/joim.12590
11. Schain F, Dominicus A, Borgsten F, Mozart M, Bjorkholm M. Real-word data on autologous stem cell transplantation in older patients with multiple myeloma. Ann Hematol. 2019.
12. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi:10.1016/0022-1759(83)90303-4
13. Xie B, Xu Z, Hu L, et al. Pterostilbene inhibits human multiple myeloma cells via ERK1/2 and JNK pathway in vitro and in vivo. Int J Mol Sci. 2016;17(11):1927. doi:10.3390/ijms17111927
14. Cai B, Wang S, Huang J, Lee CK, Gao C, Liu B. Cladribine and bendamustine exhibit inhibitory activity in dexamethasone-sensitive and -resistant multiple myeloma cells. Am J Transl Res. 2013;5(1):36–46.
15. Osborne N, Avey MT, Anestidou L, Ritskes‐Hoitinga M, Griffin G. Improving animal research reporting standards. EMBO Rep. 2018;19(5):5. doi:10.15252/embr.201846069
16. Guo Y, Feng X, Jiang Y, et al. PD1 blockade enhances cytotoxicity of in vitro expanded natural killer cells towards myeloma cells. Oncotarget. 2016;7(30):48360–48374. doi:10.18632/oncotarget.10235
17. Bird JM, Owen RG, D’Sa S, et al. Guidelines for the diagnosis and management of multiple myeloma 2011. Br J Haematol. 2011;154(1):32–75.
18. Mateos MV, Ludwig H, Bazarbachi A, et al. Insights on multiple myeloma treatment strategies. HemaSphere. 2019;3(1):e163. doi:10.1097/HS9.0000000000000163
19. Zhang FH, Yan Y-L, Wang Y, Liu Z. Lactucin induces potent anti-cancer effects in HL-60 human leukemia cancer cells by inducing apoptosis and sub-G1 cell cycle arrest. Bangladesh J Pharmacol. 2016;11(2):478–484. doi:10.3329/bjp.v11i2.26729
20. Fulda S. Targeting apoptosis for anticancer therapy. Semin Cancer Biol. 2015;31:84–88. doi:10.1016/j.semcancer.2014.05.002
21. Wang C, Li S, Ren H, et al. Anti-proliferation and pro-apoptotic effects of diosmetin via modulating cell cycle arrest and mitochondria-mediated intrinsic apoptotic pathway in MDA-MB-231 cells. Med Sci Mon. 2019;25:4639–4647. doi:10.12659/MSM.914058
22. Kong Y, Li B, Chang S, et al. DCZ0814 induces apoptosis and G0/G1 phase cell cycle arrest in myeloma by dual inhibition of mTORC1/2. Cancer Manag Res. 2019;11:4797–4808. doi:10.2147/CMAR.S194202
23. Kim SJ, Kim ES, Kim S, et al. Antitumoral effect of arsenic compound, sodium metaarsenite (KML001), on multiple myeloma cells. Int J Oncol. 2017;51(6):1739–1746. doi:10.3892/ijo.2017.4161
24. Jiang J, Huang J, Wang XR, Quan YH. MicroRNA-202 induces cell cycle arrest and apoptosis in lung cancer cells through targeting cyclin D1. Eur Rev Med Pharmacol Sci. 2016;20(11):2278–2284.
25. D’Ignazio L, Batie M, Rocha S. Hypoxia and inflammation in cancer, focus on HIF and NF-kappaB. Biomedicines. 2017;5(2).
26. Chen ZJ. Ubiquitination in signaling to and activation of IKK. Immunol Rev. 2012;246(1):95–106. doi:10.1111/j.1600-065X.2012.01108.x
27. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:64. doi:10.3389/fonc.2014.00064
28. Du W, Pang C, Xue Y, Zhang Q, Wei X. Dihydroartemisinin inhibits the Raf/ERK/MEK and PI3K/AKT pathways in glioma cells. Oncol Lett. 2015;10(5):3266–3270. doi:10.3892/ol.2015.3699
29. Wu XY, Tian F, Su MH, et al. BF211, a derivative of bufalin, enhances the cytocidal effects in multiple myeloma cells by inhibiting the IL-6/JAK2/STAT3 pathway. Int Immunopharmacol. 2018;64:24–32. doi:10.1016/j.intimp.2018.08.016
30. Zhang L, Wang H, Zhu J, Xu J, Ding K. Mollugin induces tumor cell apoptosis and autophagy via the PI3K/AKT/mTOR/p70S6K and ERK signaling pathways. Biochem Biophys Res Commun. 2014;450(1):247–254. doi:10.1016/j.bbrc.2014.05.101
31. Liu TJ, Koul D, LaFortune T, et al. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther. 2009;8(8):2204–2210. doi:10.1158/1535-7163.MCT-09-0160
32. Ramakrishnan V, Kumar S. PI3K/AKT/mTOR pathway in multiple myeloma: from basic biology to clinical promise. Leuk Lymphoma. 2018;59(11):2524–2534. doi:10.1080/10428194.2017.1421760
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.