")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Novel purine thioglycoside analogs: synthesis, nanoformulation and biological evaluation in in vitro human liver and breast cancer models
Authors Abu-Zaied MA, Loutfy SA , Hassan AE, Elgemeie GH
Received 11 January 2019
Accepted for publication 5 June 2019
Published 26 July 2019 Volume 2019:13 Pages 2437—2457
DOI https://doi.org/10.2147/DDDT.S201249
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sukesh Voruganti
This paper has been retracted.
Mamdouh A Abu-Zaied,1 Samah A Loutfy,2,3 Ashraf E Hassan,2,4 Galal H Elgemeie4
1Green Chemistry Department, National Research Centre, Dokki, Giza, Egypt; 2Virology & Immunology Unit, Cancer Biology Department, National Cancer Institute, Cairo University, Cairo, Egypt; 3Nanotechnology Research Center, British University, Cairo, Egypt; 4Chemistry Department, Faculty of Science, Helwan University, Helwan, Cairo, Egypt
Background: A series of novel pyrazolopyrimidine and pyrazololpyridine thioglycosides were synthesized and confirmed via their spectral analyses.
Purpose: To evaluate the effect of these anti-metabolic compounds against proliferation of Huh-7 and Mcf-7 as in vitro models of human liver and breast cancers, respectively. Vero cells were used as an example of normal green monkey kidney cells.
Methods: The most promising compound was subjected to a nanoformulation by its encapsulation into chitosan nanoparticles to increase its anti-cancerous activity. Nanoformulation was confirmed by TEM and FT-IR to ensure encapsulation and screened for their cytotoxicity against Huh-7 and Mcf-7 cells using MTT colorimetric assay and morphological examination. Genotoxic effect was performed by cellular DNA fragmentation assay. Simulated CompuSyn software (linear interaction effect) was conducted to predict the possible synergistic effect of nanocomposite as anticancerous activity. Apoptotic effect was further analyzed by detection of apoptotic proteins using ELISA assay.
Results: The nano preparation was successfully prepared by encapsulation of compound 14 into chitosan nanoparticles, controlled to a size at 105 nm and zeta charges at 40.2 mV. Treatment of Huh-7 and Mcf-7 showed that compound 14 was the most cytotoxic compound on both cancer cell lines where IC50 was 24.59 (9.836 μg/mL) and 12.203 (4.8812 μg/mL) on Huh-7 and Mcf-7 respectively. But IC50 of the nano preparation was 37.19 and 30.68 μg/mL on Huh-7 and Mcf-7, respectively, indicating its aggressiveness on human breast cancer cells as confirmed by DNA fragmentation assay and theoretically by CompuSyn tool.
Conclusion: A novel series of pyrazolopyrimidine thioglycosides and pyrazolopyridine thioglycosides were synthesized. Nanoformulation of compound 14 into chitosan nanoparticles demonstrated anticancer activity and can be used as a drug delivery system, but further studies are still required.
Keywords: purine thioglycoside analogs, chitosan nanoparticles, human breast cancer cells, human liver cancer cells, anticancer agents, antimetabolites
Introduction
Interest in the synthesis of antipyrine derivatives has been intensified following the discovery of their various biological applications. They have been reported to have diverse pharmacological activities, such as antimicrobial,1 antiviral,2 anticancer, antioxidant, antipyretic, analgesic and anti-inflammatory activities.3 Cancer disease has been turned out to be one of the world’s primary general medical issues. Early attack, invulnerable escape, early metastasis and other natural exhibitions are the inconvenience of tumor treatment. At the point when the tumor has been found in the dynamic stage, the tumor has the ability to be exchanged to a phase in which it became so difficult to be eradicated by medical procedure and simple to relapse after medical procedure. The old treatment methods are surgery, radiation4 and chemical treatment,5 which is associated with high-risk destruction to neighboring tissues, infection, pain and relapse of cancer. A less aggressive choice is radiation therapy, showed side effects such as skin changes,6 faucal incontinence,7 diarrhea,8 nausea and vomiting9 which obviously affects public health. Further down the line in terms of non-invasiveness is chemotherapy, which properly is the first choice in the administration of cancer. However, chemotherapy is also connected with side effects such as pain sores in the mouth10 and throat,11 nausea and vomiting as well as blood disorders.12 In order to relieve some of the side effects due to chemotherapy, there was a growing interest in the use of nanotechnology as an alternative approach for cancer treatment. The purpose of overcoming the adverse effects of drugs is by encapsulating the chemotherapeutic drug into the nano-delivery system.13 Advancement has been made in the treatment of cancer by conjugating chemotherapeutic agents into nanocarriers. These nanocarriers are often made from polymeric materials such as poly(lactide-co-glycolide) (PLGA),14 chitosan15–17 and poly-hydroxyethyl methacrylate/ stearic acid.18 Chitosan (Cs) is the essential polysaccharide elucidated by deacetylation of chitin, which must be the most copious common biopolymer on the earth with the exception of cellulose.19 Chitosan has been broadly utilized in the pharmaceutical production due to its low lethality, biodegradability,20 and biocompatibility.21 In recent years, Chitosan was significantly used for the delivery of active pharmaceutical compounds. It tends to be utilized to shape nanocarriers for drug delivery, in particular feebly dissolvable therapies or biotechnology-based medications. The two frameworks can protect the medication from being broken down inside the body. Polymeric conveyance frameworks can change the pharmacokinetics of medication, prompting a propelled helpful list by diminishing the symptoms and developing viability. Small size and large surface area to volume ratio of the chitosan nanoparticles gave it its unique characteristics. Chitosan nanoparticles showed to be promising carriers for several drugs specially hydrophobic drugs in cancer drug delivery application.22 Chitosan and sodium alginate were utilized to uploading 5-FU (anti-metabolic medication) by ion gelation method and had maintained discharge of 5-FU in a controlled way.23 Karolyn Infanta and David have performed their in vitro study by successful encapsulating quercetin and 5-FU into chitosan nanoparticles and hence its sustainable release.24 Such strategies created an impressive consideration because of its associated efficacy, improving drug properties, helpful and controllable.25 Ionic gelation procedure depends on the ionic communications between the decidedly charged essential amino gatherings of chitosan and the contrarily charged gatherings of polyanion, for example, sodium tripolyphosphate (TPP), which is the most broadly utilized particle cross-connecting operator because of its non-dangerous and multivalent properties.26,27 This physical cross-connecting strategy not just keeps away from the utilization of substance cross-connecting and emulsifying agents which are regularly dangerous to the organ, in addition stays away from the likelihood of drugs impairment. In the light of the abovementioned results along with our previous reports for synthesizing novel antimetabolic agents28–32 and heterocyclic thioglycosides.33–48 The purpose of this work is to design, synthesize and investigate the anti-tumor activity of antipyrine derivative and fused antipyrine bearing carbohydrate moieties by forming S-glycosidic bonds. To our knowledge, this is the first method in which the preparation of such a novel class of 4-aminoantipyrines will be reported. Also, in this study, we choose to use LMW chitosan with high degree of deacetylation, and concentration on their producible construction of (Chitosan–TPP) and (Chitosan-14- TPP) nanoparticles, which desired to stimulate the development of (Chitosan–TPP) nanoparticles in the applications of drug delivery for our synthetic compounds.
Materials and methods
Low molecular weight chitosan (Cs, Deacetylation degree of ≥75%, viscosity 20ـــ 300 cps and molecular weight of 50ــ 190 kDa), sodium tripolyphosphate (TPP) were purchased from Sigma Aldrich (St. Louis, MO, USA), acetic acid glacial.
Preparation of (Cs–14) nanoparticles
Chitosan nanoparticles were prepared according to a modified method of Calvo et al22 based on the ionic gelation of chitosan with TPP anions. Briefly, a specific amount of Cs (1 mg/mL) was dissolved in 1% acetic acid solution and TPP solution (10 mg/mL). Nanoparticles were obtained by mixing Cs solution with TPP for a volume ratio of [3:1], respectively. Compound 14 powder which was accurately weighed dissolved in DMSO to prepare a concentration of (1 mg/mL) solution. A 300 µL aliquot solution from the prepared compound 14 solution was then slowly dropped into the Cs solution using a micro-syringe under mechanical stirring for (600 rpm) for (30 mins). Finally, sodium tripolyphosphate solution was added to the above mixture under mechanical stirring (600 rpm) for (1 hr). The reaction was kept for 1.5 hrs at room temperature. Chitosan nanoparticles (Cs NPs) without compound 14 were prepared by the same method.
Nanoparticles characterization
Size and zeta potential measurement
Particle size and zeta potential, Z-average diameter of the prepared nanoparticles and their zeta potential were determined by the Malvern particle size analyzer (Model-Nano ZS, Malvern Instruments limited, UK). Triplicate samples were analyzed and the mean value was reported.
Measurement of particle size and morphology by TEM
Nanoparticles morphology was examined by TEM apparatus (JOEL-JEM–1010, Japan) at an accelerating voltage of 100 kV. Nanoparticles suspension was properly diluted. A drop was withdrawn with a micropipette then placed on a carbon-coated copper grid. The excess of the suspension was removed by blotting the grid with a filter paper. Then, the deposit was left to dry before analysis.
Fourier transform infrared spectroscopy (FTIR)
In order to confirm the potential chemical interaction happening between (Cs–TPP) NPs and (Cs–compound 14–TPP) NPs, FTIR spectra of compound 14, (Cs–TPP) nanoparticles and compound 14 loaded Cs nanoparticles were taken using (Perkin Elmer, USA) Fourier transform infrared spectrophotometer.
Cell culture
Established human liver cancer cell line (Huh-7) and human hormonal breast cancer cell line (Mcf-7) were acquired from American Tissue Culture Collection, USA. The cells were kept up in DMEM media (Lonza) supplemented with heat inactivated 10% fetal bovine serum (Gibco), and anti-microbials (2% penicillin-streptomycin [Gibco] and 0.5% fungizone [seralab]). The cells were kept up in monolayer culture at 37 °C under a humidified air of 5% CO2. The cells were passaged by trypsinization (0.025% trypsin and 0.0025% EDTA; Biowest), and kept up in tissue culture laboratory of Virology & Immunology Unit, Cancer Biology Dept. at the National Cancer Institute, Cairo University, Egypt. Cell numbers and viability were observed by standard Trypan blue color (Gibco).
Cytotoxicity assay (MTT)
The anti-cancerous activity of the synthesized compounds was determined against a human liver cancer cell line (Huh-7) and breast cancer (Mcf-7) using Doxorubicin as a reference drug and (Serva electrophoresis, Germany) MTT assay based on the ability of active mitochondrial dehydrogenase enzyme of living cells to cleave the tetrazolium rings of the yellow MTT and form a dark violet insoluble formazan crystals.58 Data generated were used to plot a dose–response curve from which the concentration of test compounds required to kill 50% of cell population (IC50) was determined. Cells were seeded in 96-well plate at a cell concentration (6×103) cells per well in 100 μL of growth medium. Fresh medium containing different concentrations of the test sample was added after 24 hrs of seeding. Serial two-fold dilutions of the tested chemical compound were added to confluent cell monolayers dispensed in 96-well, flat-bottomed microtiter plates using a multichannel pipette. The microtiter plates were incubated at 37°C in a humidified incubator with 5% CO2 for 48 h. Four wells (quadruplicate) were used for each concentration of the test sample. Control cells were incubated without test sample with DMSO (0.1% v/v). After incubation of the cells for 24 hrs at 37°C, various concentrations of sample (100, 50, 25, 12.5 and 6.25 μM/mL) were added, the incubation was continued for 48 hrs. At the end of experiment, MTT solution in PBS (5 mg/mL) was then added to all wells, and left to incubate for 2 hrs. The formation of formazan crystals were visually confirmed using phase contract microscopy. DMSO (100 μL/well) was added to dissolve the formazan crystals with shaking for 10 mins, after which the absorbance was read at 570 nm on ELISA microplate reader (Tecan Sunrise ELISA Reade, USA). Cell proliferation was calculated comparing the OD values of the DMSO control wells and those of the samples both represented as % viability to the control.
DNA fragmentation
Fragmentation of cellular DNA was investigated following treatment of Mcf-7 and Huh-7 cell lines with compounds 3, 4, 8a, 6b, 8b, 9b, 13, 15, 14 and 16 at high concentrations (100 µM) and (Cs–14) NPs at concentration 100 µg. A fixed amount (350 ng) of cellular DNA (GeneJET Genomic DNA purification kit, Thermo Scientific) extracted from treated and untreated cells was subjected to 1.5% agarose gel electrophoresis in TEA buffer, stained with 5 µL ethidium bromide. The bands were examined under UV transillumination and photographed. Smearing, or presence of many low molecular weight DNA fragments, is a characteristic feature of apoptotic cells.59
Drug combination analysis using median effect principle
The nature of interaction of both compounds (Cs NPs), compound 14 in vitro was determined by treatment of (Mcf-7 and Huh-7) cells with specific concentration. The cells were seeded at a density of 6,000 cells/well and allowed to adhere overnight. All compounds were administered at a concentration (100 µg) and incubated for 48 hrs in a humidified chamber at 37°C. After incubation, MTT assay was performed as described previously. All possible combinations of the two compounds within this concentration were analyzed for any additive, synergistic or antagonistic effects. The data were analyzed for combination index using CompuSyn software.
Detection of apoptotic caspase-3 and anti-apoptotic Bcl-2 proteins levels
Caspase-3 activity was measured after treatment of cells (Mcf-7 and Huh-7) with compound 14 at concentration of 100 µg. Caspase-3 activity was measured using colorimetric Bender Med System (Caspase 3 assay kit; BMS2012INST) while Bcl-2 activity was measured using colorimetric abcam (Human Bcl-2 ELISA Kit (ab119506). Six well plates were seeded with (3×104) cells and incubated overnight under optimum culture conditions before treatments with the estimated (IC50) of the selected compounds in relation to doxorubicin as standard chemotherapeutic agents. The cells were harvested and total proteins were isolated. Protein levels of the apoptotic (Caspase-3) and anti-apoptotic marker (Bcl-2) were then measured using ELISA according to the manufacturers’ instructions (eBiosience, USA).Standard curves were drawn for each kit. The reaction products were measured at 450 nm using ELISA reader (Tecan sunrise microplate reader, Awareness Technology Inc, Minnesota, USA).
Results and discussion
Chemistry
The synthesis was started by the reaction of 4-aminoantipyrine 1 and sodium cyanocarbonimidodithioate salt 2 in acetic acid (Scheme 1). The reaction mixture was refluxed for 5 mins producing compound 3 in a quantitative yield. The structure of the synthesized compound 3 was elucidated on the basis of its spectroscopic data. The latter reacts with α-acetobromoxylose 5a and α-acetobromoarabinose 5b in acetone-KOH at room temperature to give the corresponding S-xyloside 6a or S-arabinoside 6b (Scheme 1). The structures of reaction products 6a,b were confirmed by their spectroscopic data and basic analysis (13C NMR, 1H NMR and IR). The 1H NMR spectrum revealed the anomeric proton of 6a as a doublet at δ 5.42 ppm. The coupling constant (J1’-’2=8.2 Hz) indicated H-1’ to be trans-diaxial to H-2’, and the other five xylose protons resonated at 3.56–5.13 ppm and showed three acetyl groups as three singlets at δ 1.98–2.06 ppm. The 13C NMR spectrum of 6a contained a signal at δ 78.54 corresponding to the -C-1’ atom and four signals appearing at δ 60.36, 68.61, 75.47 and 76.73 which were set to -C-5’, -4’, C-3’ and C-2’, respectively. When glycosides 6a, b were reacted with NH3-MeOH at 0–5C° for 10 mins provided the free hydroxyl derivatives 8a, b (Scheme 1), the structures of which were confirmed based on their spectroscopic data. Thus, 1H NMR spectra of 8a shows the anomeric proton as doublet at δ 5.34 (J 1’-2’=10.22 Hz), indicating only the 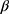 D-configuration and three hydroxy groups of xylose resonance at δ 4.99–5.12 (exchangeable by D2O). Cyclization of compound 3 in the boiling of EtOH/HCl resulted in the formation of the 7-imino-5-mercapto-pyrazolo [4, 3-d] pyrimidin-3(2H)-one 4 (by mass spectra). The 1H NMR spectrum revealed an imine group (D2O exchangeable) and two different methyl protons, demonstrating that the methyl group at C-5 did not participate in the cyclization. A similar intramolecular cyclization reaction of this type was reported by us.40 In another experiment, compound 4 reacted with α-acetobromoxylose 5a and α-acetobromoarabinose 5b in acetone at room temperature to give the corresponding S-xyloside 7a or S-arabinoside 7b, respectively. It was suggested that cis (α) sugar be reacted by a simple SN2 reaction to give a
D-configuration and three hydroxy groups of xylose resonance at δ 4.99–5.12 (exchangeable by D2O). Cyclization of compound 3 in the boiling of EtOH/HCl resulted in the formation of the 7-imino-5-mercapto-pyrazolo [4, 3-d] pyrimidin-3(2H)-one 4 (by mass spectra). The 1H NMR spectrum revealed an imine group (D2O exchangeable) and two different methyl protons, demonstrating that the methyl group at C-5 did not participate in the cyclization. A similar intramolecular cyclization reaction of this type was reported by us.40 In another experiment, compound 4 reacted with α-acetobromoxylose 5a and α-acetobromoarabinose 5b in acetone at room temperature to give the corresponding S-xyloside 7a or S-arabinoside 7b, respectively. It was suggested that cis (α) sugar be reacted by a simple SN2 reaction to give a 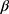 glycoside product. The structures of the 7a,b reaction products were confirmed by elemental analyses and spectral data (13C NMR, 1H NMR, IR). Thus, the 1H NMR spectrum for 7a showed the anomeric proton as a doublet at δ 5.56 ppm with a spin–spin coupling constant of 10.6 Hz which corresponds to the diaxial orientation of H-1’ and H-2’ protons indicating the
glycoside product. The structures of the 7a,b reaction products were confirmed by elemental analyses and spectral data (13C NMR, 1H NMR, IR). Thus, the 1H NMR spectrum for 7a showed the anomeric proton as a doublet at δ 5.56 ppm with a spin–spin coupling constant of 10.6 Hz which corresponds to the diaxial orientation of H-1’ and H-2’ protons indicating the 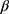 configuration. The formation of 1,2-trans glycosides is strongly favored by the neighboring group involved. Typically, the use of a co-replacement substituent at C-2 is a support to justify stereoselective 1,2-trans glycosylation.41,42 The 13C NMR spectrum of 7a contained a signal at δ=79.58 corresponding to the -C-1’ atom of the
configuration. The formation of 1,2-trans glycosides is strongly favored by the neighboring group involved. Typically, the use of a co-replacement substituent at C-2 is a support to justify stereoselective 1,2-trans glycosylation.41,42 The 13C NMR spectrum of 7a contained a signal at δ=79.58 corresponding to the -C-1’ atom of the 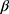 -configuration. Four signals appearing at δ=62.11, 69.27, 71.86 and 73.75 were set to C-5’, C-4’, C-3’ and C-2’ respectively. After the removal of protecting groups of compounds 6a, b and 7a,b with a NH3/MeOH solution at room temperature, the final free glycosides 8a,b and 9a,b were obtained and their structures were confirmed on the basis of their spectroscopic data. Thus, the 1H NMR spectrum of 9a shows the anomeric proton as a doublet at δ 4.66 (J1’-2’=10.2 Hz) indicating that only the
-configuration. Four signals appearing at δ=62.11, 69.27, 71.86 and 73.75 were set to C-5’, C-4’, C-3’ and C-2’ respectively. After the removal of protecting groups of compounds 6a, b and 7a,b with a NH3/MeOH solution at room temperature, the final free glycosides 8a,b and 9a,b were obtained and their structures were confirmed on the basis of their spectroscopic data. Thus, the 1H NMR spectrum of 9a shows the anomeric proton as a doublet at δ 4.66 (J1’-2’=10.2 Hz) indicating that only the 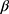 D-configuration exists and the other five protons of xylose appear at δ 3.78–4.41, while the three hydroxyl xylose signals are observed at δ 4.91–5.23 (exchangeable by D2O). The 13C NMR spectrum of 9a showed a signal at δ 81.93 corresponding to the -C-1’ atom of
D-configuration exists and the other five protons of xylose appear at δ 3.78–4.41, while the three hydroxyl xylose signals are observed at δ 4.91–5.23 (exchangeable by D2O). The 13C NMR spectrum of 9a showed a signal at δ 81.93 corresponding to the -C-1’ atom of 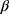 -D-xylopyranose, and four other signals at δ 62.41, 69.28, 71.62 and 74.92 are assigned to C-5’, C-4’, C-3’ and C-2’ respectively. In order to investigate the scope of this reaction, and in order to determine whether the reaction of sodium salts of cyanocarbonimidodithioate or cyanoketene dithioacetals with 4-amino-pyrazol-3(2H)-5-one could be extended to provide a general approach to fused pyrazole thioglycosides, we studied the reaction of 4-aminoantipyrene 1 with functioned ketene dithioacetals (Scheme 2). Thus, compound 1 reacts with sodium 2,2-dicyanoethene-1,1-bis(thiolate) 10 in refluxing acetic acid for 5 mins to yield the acyclic structures 11. The latter undergoes intramolecular cyclization on refluxing in EtOH/HCl to afford the corresponding 5- mercapto-pyazolo[4,3-b]pyridines 12. The structure of 12 was confirmed on the basis of its spectroscopic data (13C NMR, 1H NMR, and IR). The 13C NMR spectrum of 12 was characterized by two methyl carbons at 12.13 (CH3) and 40.42 (N-CH3) ppm. Compounds 11 and 12 bear a mercapto group which may be useful for the synthesis of 4-aminoantipyrine thioglycoside and their corresponding pyazolo[4,3-b]pyrimidine-5-thioglycosides. Thus, it has been found that the reaction of the structures 11 and 12 with α-acetobromoxylose 5a and α-acetobromoarabinose 5b in acetone/ KOH at room temperature gives the corresponding thioglycosides 13 and 14. The structures of 13 and 14 were confirmed based on spectral data (13C NMR, 1H NMR, IR). Thus, the 1H NMR spectrum for compound 14 showed the ring imine proton at δ 5.66 ppm and the anomeric xylose proton as a doublet at δ 5.53 ppm with a spin–spin coupling constant of 10.4 Hz which corresponds to the diaxial orientation of H-1’ and H-2’ protons indicating the
-D-xylopyranose, and four other signals at δ 62.41, 69.28, 71.62 and 74.92 are assigned to C-5’, C-4’, C-3’ and C-2’ respectively. In order to investigate the scope of this reaction, and in order to determine whether the reaction of sodium salts of cyanocarbonimidodithioate or cyanoketene dithioacetals with 4-amino-pyrazol-3(2H)-5-one could be extended to provide a general approach to fused pyrazole thioglycosides, we studied the reaction of 4-aminoantipyrene 1 with functioned ketene dithioacetals (Scheme 2). Thus, compound 1 reacts with sodium 2,2-dicyanoethene-1,1-bis(thiolate) 10 in refluxing acetic acid for 5 mins to yield the acyclic structures 11. The latter undergoes intramolecular cyclization on refluxing in EtOH/HCl to afford the corresponding 5- mercapto-pyazolo[4,3-b]pyridines 12. The structure of 12 was confirmed on the basis of its spectroscopic data (13C NMR, 1H NMR, and IR). The 13C NMR spectrum of 12 was characterized by two methyl carbons at 12.13 (CH3) and 40.42 (N-CH3) ppm. Compounds 11 and 12 bear a mercapto group which may be useful for the synthesis of 4-aminoantipyrine thioglycoside and their corresponding pyazolo[4,3-b]pyrimidine-5-thioglycosides. Thus, it has been found that the reaction of the structures 11 and 12 with α-acetobromoxylose 5a and α-acetobromoarabinose 5b in acetone/ KOH at room temperature gives the corresponding thioglycosides 13 and 14. The structures of 13 and 14 were confirmed based on spectral data (13C NMR, 1H NMR, IR). Thus, the 1H NMR spectrum for compound 14 showed the ring imine proton at δ 5.66 ppm and the anomeric xylose proton as a doublet at δ 5.53 ppm with a spin–spin coupling constant of 10.4 Hz which corresponds to the diaxial orientation of H-1’ and H-2’ protons indicating the 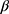 configuration. The formation of 1,2-trans glycosides is strongly favored by the neighboring group involved. Typically, the use of a co-replacement substituent at C-2 is a support to justify stereoselective 1,2-trans glycosylation [43,44]. The 13C NMR spectrum of 14 contained a signal at δ=78.81 corresponding to the -C-1’ atom of the
configuration. The formation of 1,2-trans glycosides is strongly favored by the neighboring group involved. Typically, the use of a co-replacement substituent at C-2 is a support to justify stereoselective 1,2-trans glycosylation [43,44]. The 13C NMR spectrum of 14 contained a signal at δ=78.81 corresponding to the -C-1’ atom of the 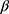 configuration. Four signals appearing at δ=62.63, 68.74, 72.64 and 74.22 were assigned to C-5’, C-4’, C-3’ and C-2’ respectively. The IR spectrum of compound 14 showed the three ester CO groups at
configuration. Four signals appearing at δ=62.63, 68.74, 72.64 and 74.22 were assigned to C-5’, C-4’, C-3’ and C-2’ respectively. The IR spectrum of compound 14 showed the three ester CO groups at  1751 cm−1 and the ring CO at
1751 cm−1 and the ring CO at  1630 cm−1. The deprotection of the protected thioglycosides 13 and 14 with a solution of NH3/MeOH at room temperature lead to the formation of the final free thioglycosides 15, 16. The structures of compounds 15, 16 were proved according to their spectral data and chemical analysis. Thus, the 1H NMR spectrum for the free pyrazolopyrimidine thioglycoside 16 revealed the anomeric xylose proton as a doublet at δ 5.24 (J1ʹ-2’=10.23 Hz), indicating the existence of the β-D-configuration [43,44]. The other five xylose protons appear as a multiplet at δ 3.63–4.55, while the three hydroxyl groups of xylose moiety resonated at δ 4.91–5.01 (exchangeable by D2O). Antitumor activity of the synthesized compounds against human tumor cell lines has been tested; liver (Huh-7) and breast (Mcf-7).
1630 cm−1. The deprotection of the protected thioglycosides 13 and 14 with a solution of NH3/MeOH at room temperature lead to the formation of the final free thioglycosides 15, 16. The structures of compounds 15, 16 were proved according to their spectral data and chemical analysis. Thus, the 1H NMR spectrum for the free pyrazolopyrimidine thioglycoside 16 revealed the anomeric xylose proton as a doublet at δ 5.24 (J1ʹ-2’=10.23 Hz), indicating the existence of the β-D-configuration [43,44]. The other five xylose protons appear as a multiplet at δ 3.63–4.55, while the three hydroxyl groups of xylose moiety resonated at δ 4.91–5.01 (exchangeable by D2O). Antitumor activity of the synthesized compounds against human tumor cell lines has been tested; liver (Huh-7) and breast (Mcf-7).
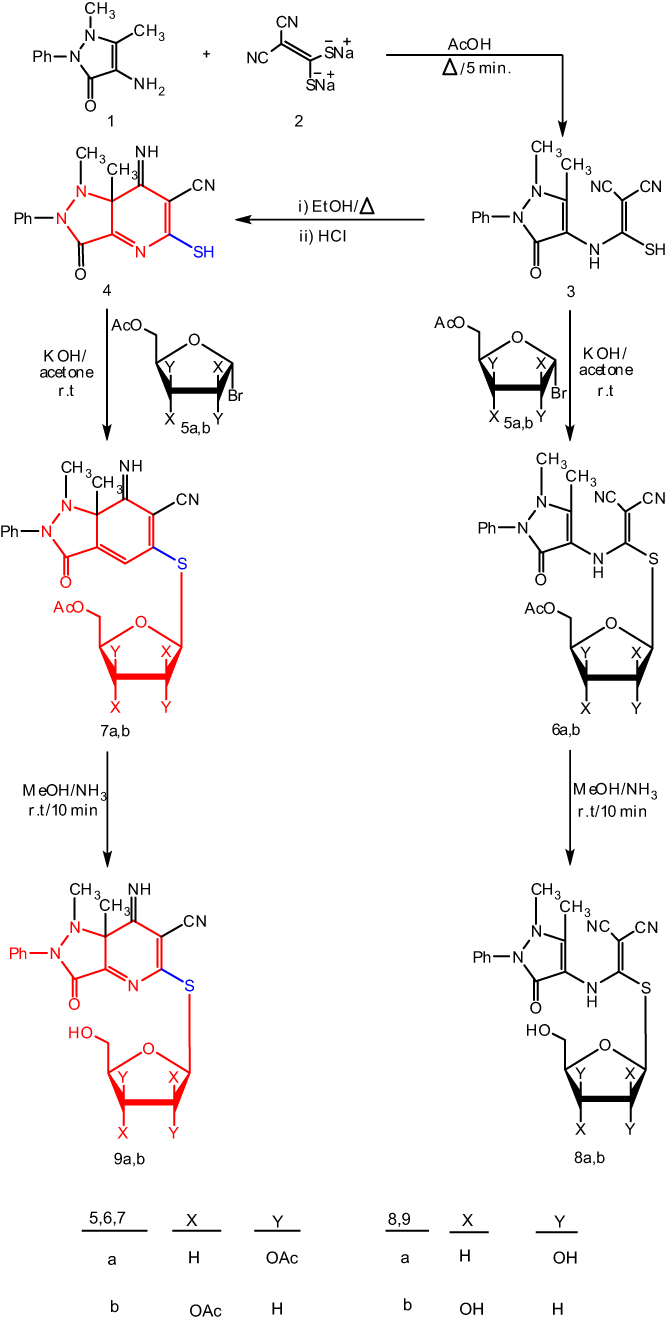 |
Scheme 1 A synthetic pathway for pyrazolopyridine thioglycosides 7a,b and 9a,b. |
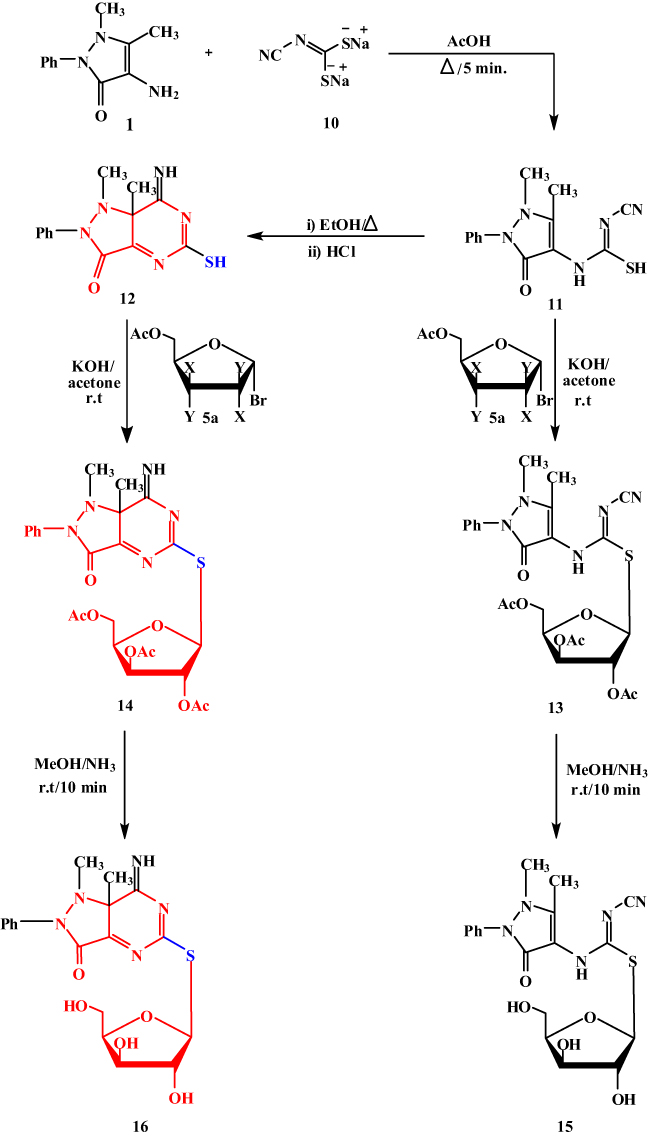 |
Scheme 2 A synthetic pathway for pyrazolopyrimidine thioglycosides 14 and 16. |
Fourier transform infrared spectroscopy (FTIR) characterization
The FTIR spectra of the synthetic compound 14 and formulated nanoparticles are presented in Figure 1. In the spectra (a), stretching vibrations of (–C=O), and (=N–H) of compound 14 appears at 1,751 and 3,438 cm−1, respectively. For chitosan nanoparticles, the peak of amide I (NH2 bending) appeared at 1,643 cm−1, d peaks appeared at 1,226 (C–O–C stretch) and 1,543 cm−1 (amide II), implying the complex formation via electrostatic interaction between NH3+ groups of chitosan and phosphoric groups of TPP within the nanoparticles.49,50 The characteristic peak at 1,086 cm−1 is assigned to (P=O) groups of TPP, while the one at 897 cm−1 is related to the (P-O-P) asymmetric stretching.51 These bands were all present in both the formulations (Cs NPs) and (Cs–14) NPs, spectra (b and c, respectively). We may conclude that these groups are not typically involved in covalent chemical bonding with the other components during the formulation process. The (Cs–14) NPs) FTIR spectrum is slightly similar to the (Cs NPs) spectrum except for a slight shifting of the amine peak at 1,633 cm−1 which is attributed to the loading of compound 14 on (Cs NPs). Furthermore, the peak attributed to compound 14 is absent in the (Cs–14) NPs spectrum, which assures loading compound 14 in the latter.
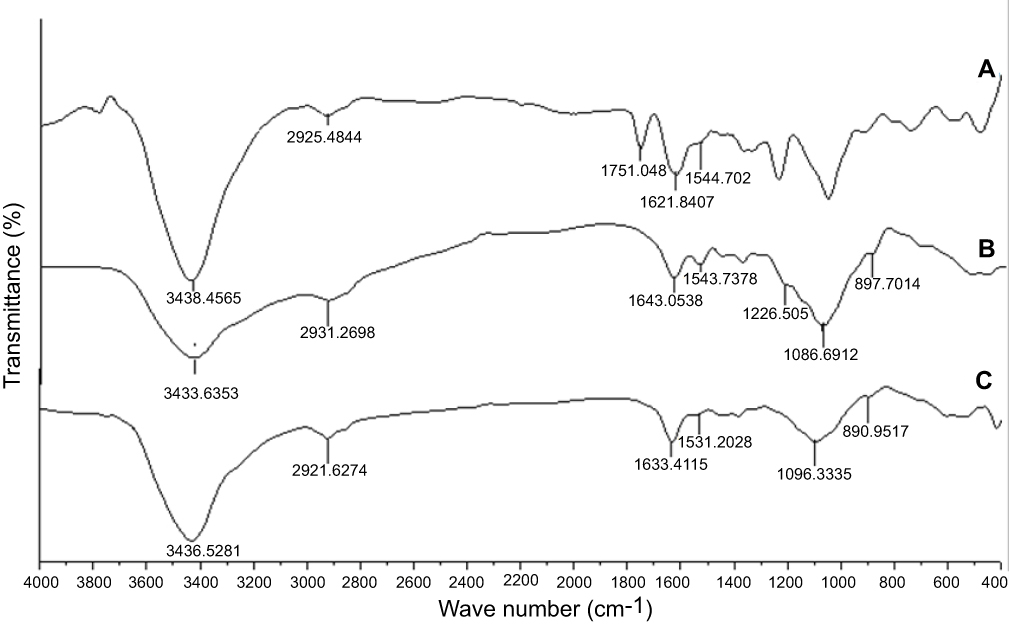 |
Figure 1 FTIR spectra of compound (14) (A), chitosan NPs (B), and (Cs –14) NPs (C). |
Determination of particle size by zeta-sizer and stability studies by zeta potential measurements
The particle size distribution, PDI and zeta potential of (Cs NPs) were measured by Zeta-Sizer instrument, which shows a size 44 nm, 0.457 and 38.5 mV, respectively while those for (Cs–14) NPs were 105 nm, 0.418 and 40.2 mV. The particle size distribution via Zeta- Sizer for the compound 14 loaded (Cs NPs) nanoparticle is represented in Figure 2. These zeta potential values confirm the good stability of both (Cs NPs) and compound 14 loaded (Cs NPs) nanoparticle and the positive zeta potential reveals the positive surface charge in both of the systems. The zeta potential of (Cs NPs) nanoparticles was increased with compound 14 loading; this can be elucidated by the effective interaction chemistry between compound 14 and the (Cs NPs) nanoparticles. Once loaded within the (Cs NPs) nanoparticles, compound 14 enhanced an additional positive charge to the loaded (Cs NPs) nanoparticles as it can have a net positive charge (outcomes from the protonation of the (=N–H)). Function group in the pyrimidine ring within compound 14 as well, this added up the surface charge. The interesting result here is that, the zeta potential itself is a good evidence for the conceivable interaction chemistry of compound 14 with (Cs NPs) nanoparticles.
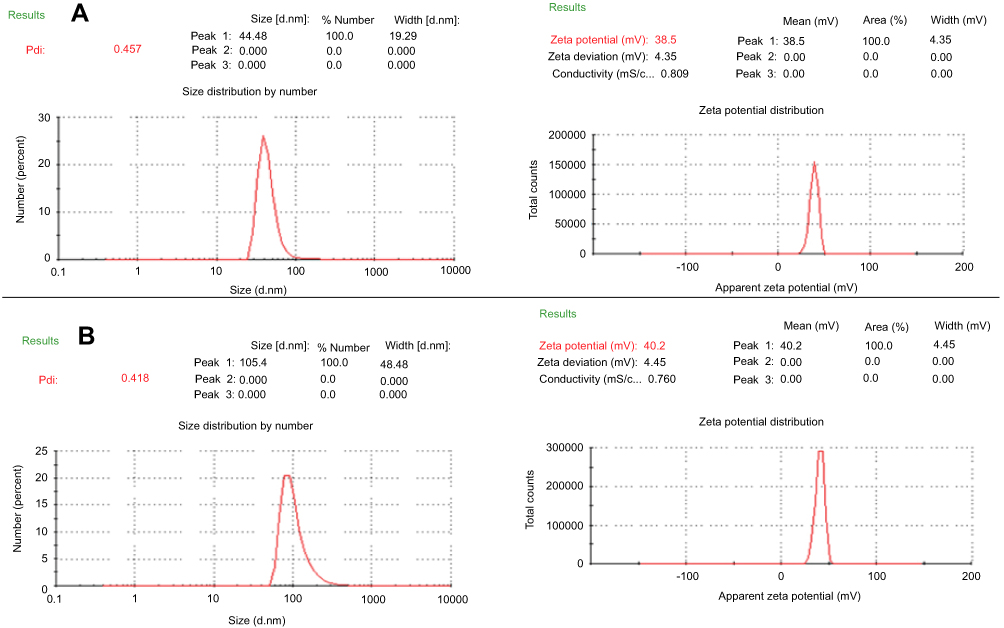 |
Figure 2 The particle size distribution ion and zeta potential of (A) chitosan NPs and (B) (Cs–14) NPs. |
Morphology of (Cs NPs) and (Cs–14) nanoparticles
The morphology of the particles was detected by transmission electron microscopy (TEM). The TEM images of the (Cs NPs) and (Cs–14) NPs exhibited regular distribution and spherical shape that show and have a particle size distribution in the range (20–35) and (40–70) nm, respectively, as shown in Figure 3. This divergence in the size of (Cs NPs) and (Cs–14) NPs between Zeta-sizer and TEM may be according to that (Cs NPs) and (Cs–14) NPs expand in aqueous media and Zeta-sizer provides a hydrodynamic diameter of nanoparticles, whilst TEM gives a definite diameter of NPs in dry state. Whilst the aggregation of the (Cs NPs) and (Cs–14) NPs is possible because that the hydrogen bonding contacts amongst (Cs NPs) gradually become dominant in the drying process.52
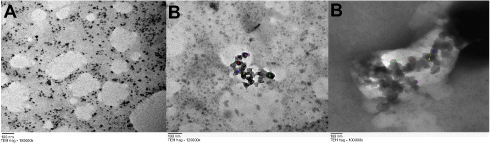 |
Figure 3 TEM image of (Cs NPs) (A), (Cs–14) NPs (B). |
Biological activity
Evaluation of the cytotoxic effect of the 16 novel synthesized compounds comparing to doxorubicin (as a reference antitumor drug) was performed in vitro for their anticancer activities against human cancer cells according to the standard procedures as described in the experimental part. The tumor cell lines have been used included; human hepatocellular carcinoma (Huh-7), breast cancer cell line (Mcf-7) and green monkey kidney cell lines (VERO) as examples of the normal cells. Our result showed that compound 14 was the most cytotoxic compound that could inhibit proliferation of human liver cancer cells Huh-7 where IC50 was found to be 24.59 μM, followed by compounds 16 and 15 where their IC50 values were 26.64 and 30.67 μM, respectively. Whereas the following compounds; 6a, 6b, 8b, 7a and 13 displayed moderate and mild cytotoxic effect on Huh-7 giving IC50 values of 89.43, 64.52, 83, 92.42 and 42.9 μM, respectively. But upon testing those compounds on human breast cancer cells (Mcf-7), our results revealed that compounds 14, 16 and 13 were the most cytotoxic compounds against Mcf-7 with IC50 values of 12.203, 14.84 and 26.66 μM, respectively. Whereas compound 15 showed moderate cytotoxic effect with IC50 of 55.026 μM. But compound 8b showed a mild cytotoxic effect with IC50 of 71.38 μM. Regarding the remaining compounds, they revealed no marked cytotoxic effect IC50>100 μM (Table 1). While the cytotoxicity effect of (Cs–14) NPs showed us moderate effect on both cell lines (Huh-7 and Mcf-7) with IC50 on the two cell lines 37.19 µg, 30.68 µg for (Cs–14) NPs, respectively, as shown in Table 2
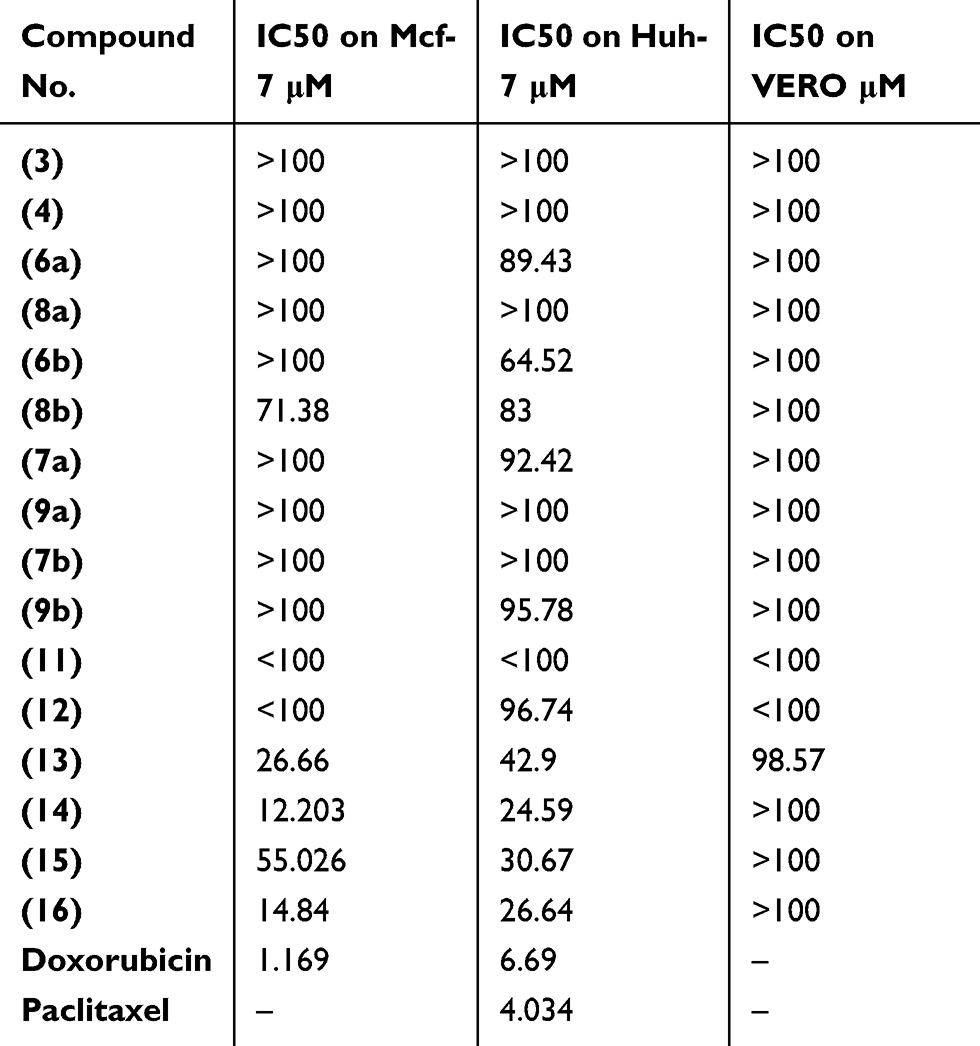 |
Table 1 Cytotoxicity of the synthesized candidates on breast (Mcf-7) and liver (Huh-7) cancer cell lines |
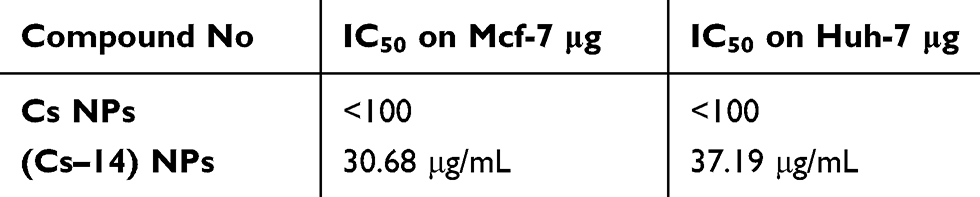 |
Table 2 Cytotoxicity of compound (14) as a nanoformulation loaded on chitosan NPs (Cs–14) NPs on breast (Mcf-7) and liver (Huh-7) cancer cell lines |
Notably, the structure–activity relationshipof the 16 newly synthesized compounds revealed that the cyclized thioglycosides compounds 14 and 16 showed higher anti-proliferative activity against both Huh-7 and Mcf-7 cell lines than the acyclic thioglycosides 13 and 15. In addition, pyrazolopyrimidine thioglycosides 14 and 16 had higher antiproliferative activity than pyrazolopyridine thioglycosides 7a, 9a, 7b and 9b against the same cell lines. This indicates that xylose’s derivatives had higher antitumor activities than the corresponding arabinose’s derivatives as shown in Figure 4. Cells treated with compounds 3, 4, 8a, 9b, 6b and 8b showed no significant morphology alteration compared to untreated cells (Figure 5). Whereas the cells treated with compounds 13, 15, 14, 16 and (Cs–14)NPs showed moderate changes in the cell morphology from adherent cells to shrieked/rounded cellular features and smearing at most of the bands as illustrated in Figure 5. Such changes were observed after treatment of either Huh-7 or Mcf-7 with these compounds. These changes might be considered to be hallmark of initial stage of apoptosis.
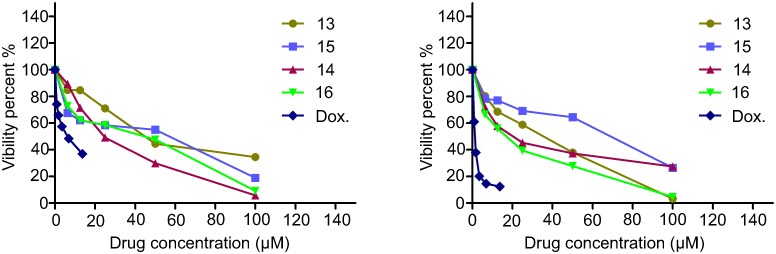 |
Figure 4 Representative graph showing survival of Huh-7 and MCF-7 cell grown compared to Doxorubicin in the presence of increasing concentrations of compounds 13, 14, 15 and 16. |
 |
Figure 5 Morphological examination of cell lines treated with the selected compounds (3, 4, 8a, 6b, 8b, 9b,13, 14, 15, 16 (Cs–14) NPs) treatment on (A) Huh-7 and (B) Mcf-7 cell lines. Light microscopy (phase contrast, magnification power of 400X) 24 hrs exposure 100 μM (high dose from each compound). |
Drug combination analysis using median effect principle
Nature of the combination of two medications was determined by median effect rule based on Chou principle.53 This was performed by plotting the dose-effect curves for each drug in choice and wither constant or nonconstant ratios for each compound as previously demonstrated by Chou and Talalay.54 The combination index indicates whether the two compounds interact in an additive (CI=1), synergistic (CI<1) or antagonistic (CI>1) manner.55 Tables 3 and 4 and Figures 6 and 7 elaborate the CI grades appointed for different combinations of (Cs NPs) and compound 14 on both cell lines (Huh-7 and Mcf-7). The arranging for the CI values has been achieved as recommended by Chou.56 The median effect analysis and calculation of combination index of free (Cs NPs) and compound 14 using the CompuSyn software showed that (Cs NPs) and compound 14 produced different results with different cell lines.
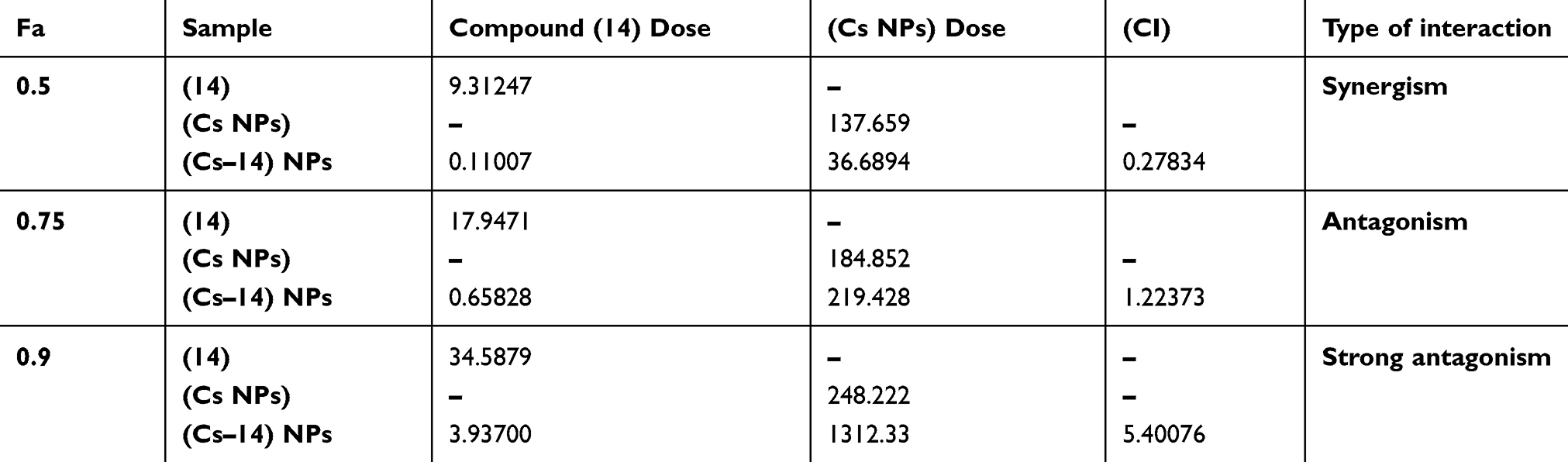 |
Table 3 Combination index of chitosan and compound (14) nanocomposite in Huh-7 cell line and its different cytotoxic effects as calculated by CompuSyn software |
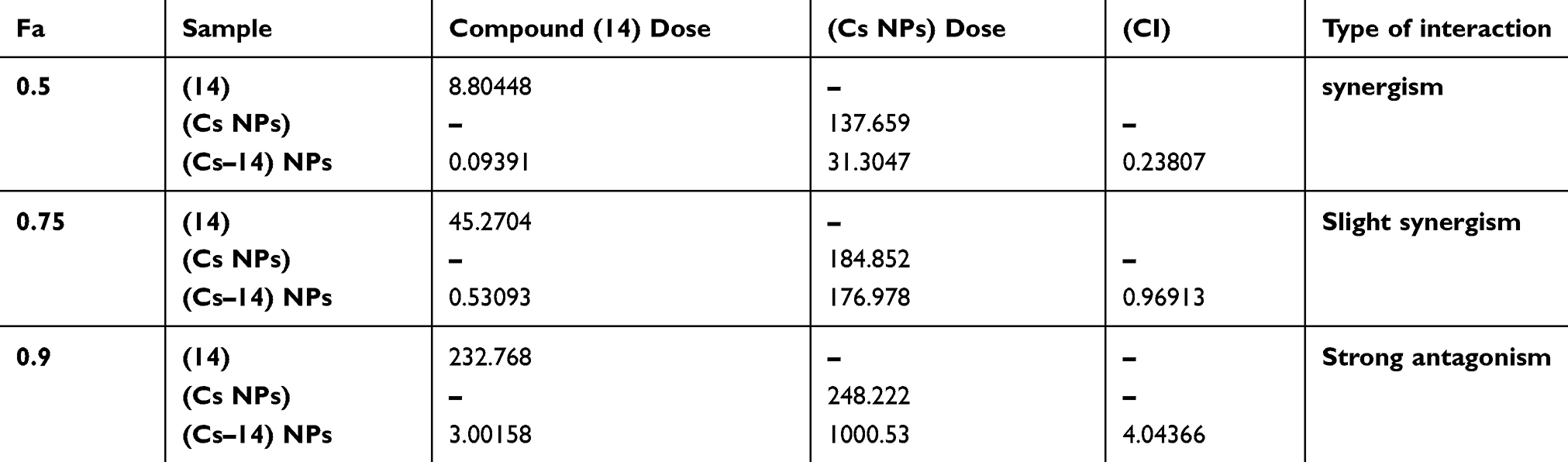 |
Table 4 Combination index of chitosan and compound (14) Nanocomposite in Mcf-7 cell line and its different cytotoxic effects as calculated by CompuSyn software |
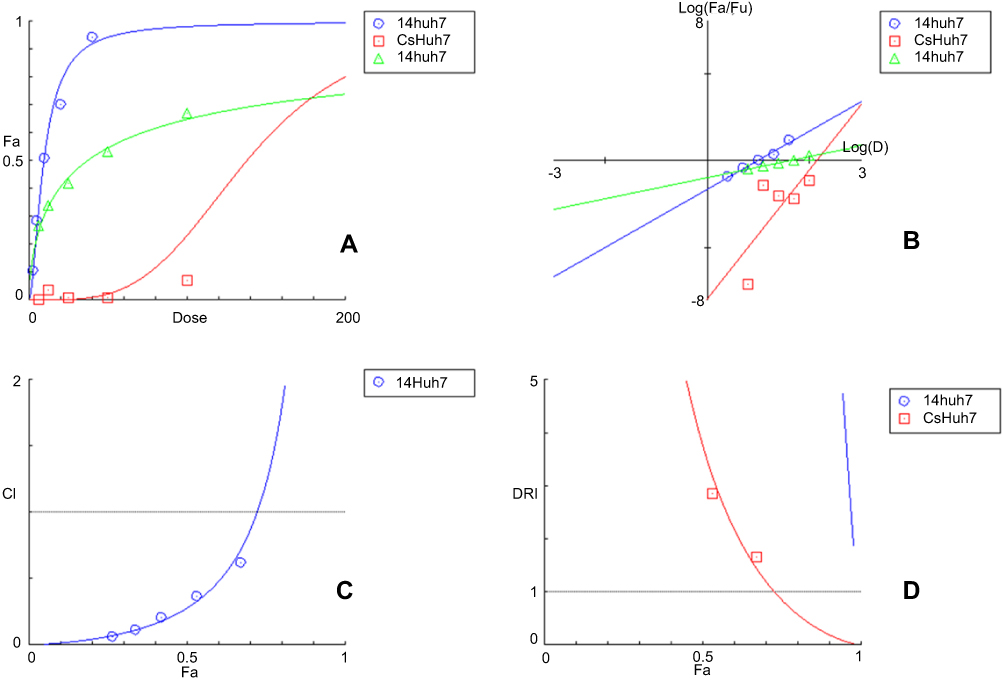 |
Figure 6 The simulated (A) Dose Effect Curve (B) Median-effect Plot (C) Combination Index Plot (D) DRI Plot (Cs–14) NPs on Huh-7 cell line calculated by CompuSyn Software. |
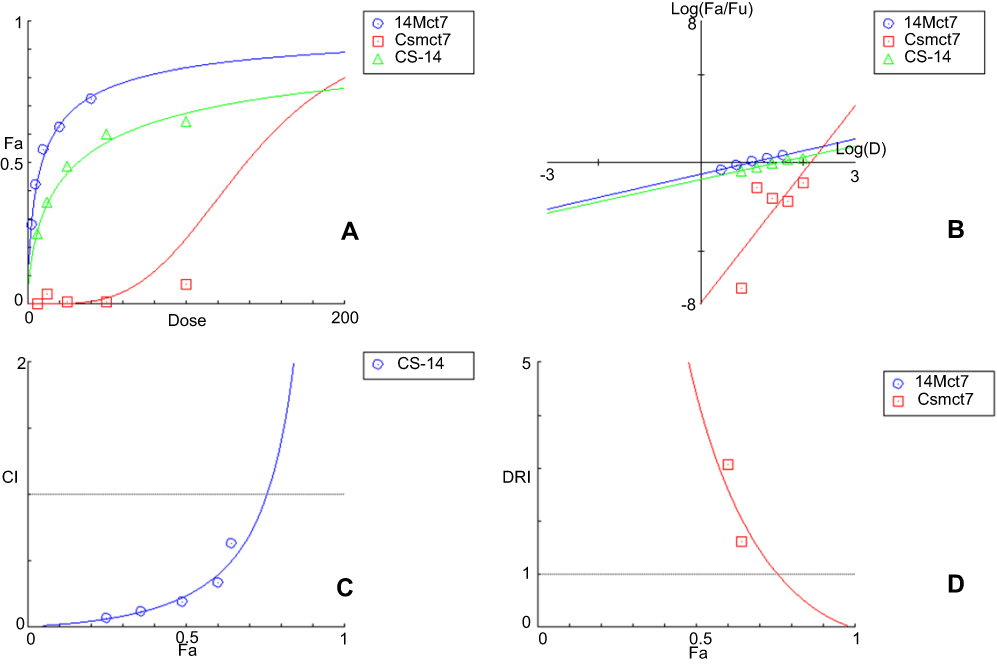 |
Figure 7 The simulated (A) Dose Effect Curve (B) Median-effect Plot (C) Combination Index Plot (D) DRI Plot (Cs–14) NPs on Mcf-7 cell line calculated by CompuSyn Software. |
Cellular DNA fragmentation assay
Further investigation on a molecular level was applied by DNA fragmentation assay to investigate genotoxic effect which in turn demonstrates the late apoptotic effect.57 After treatment of both types of the cancer cell lines with the compounds 3, 4, 8a, 6b, 8b, 9b, 13, 15, 14 and 16 at concentration of 100 μM and (Cs–14)NPs at concentration 100 µg for 24 hrs. Our results showed that there were no much differences in the concentration of DNA between treated and untreated cells for compounds 3, 4, 8a, 6b, 8b and 9b, as shown in Tables 5 and 6, and Figures 8–10. In contrary, the other compounds 13, 15, 14, 16 and (Cs–14) NPs showed a notable alteration in the DNA concentration between the treated and untreated cells (Mcf-7 and Huh-7). Data are illustrated in (Table 7). There was no significant change in bands shape between cells treated with our selected compounds 3, 4, 8a, 6b, 8b and 9b and the untreated ones (control cell).
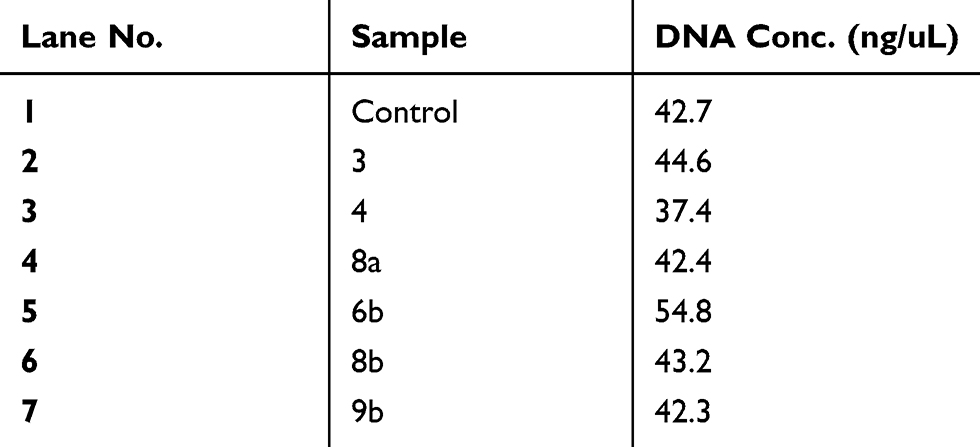 |
Table 5 Genomic DNA content of DNA fragmentation assay after treatment of Mcf-7 cells with compounds (CC, 3, 4, 8a, 6b, 8b, 9b) at high concentration (100 μM) of each compound after 24 hrs of cell exposure |
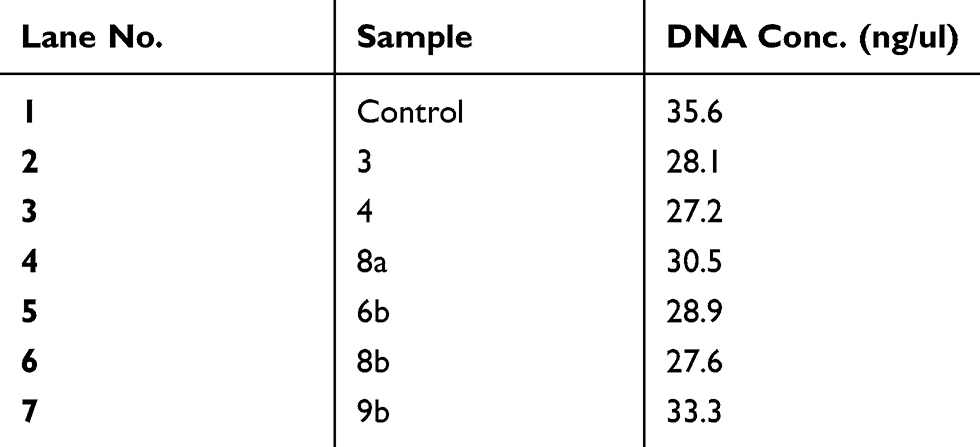 |
Table 6 Genomic DNA content of DNA fragmentation assay after treatment of Huh-7 cells with compounds (CC, 3, 4, 8a, 6b, 8b, 9b) at high concentration (100 μM) of each compound for 24 hrs of cell exposure |
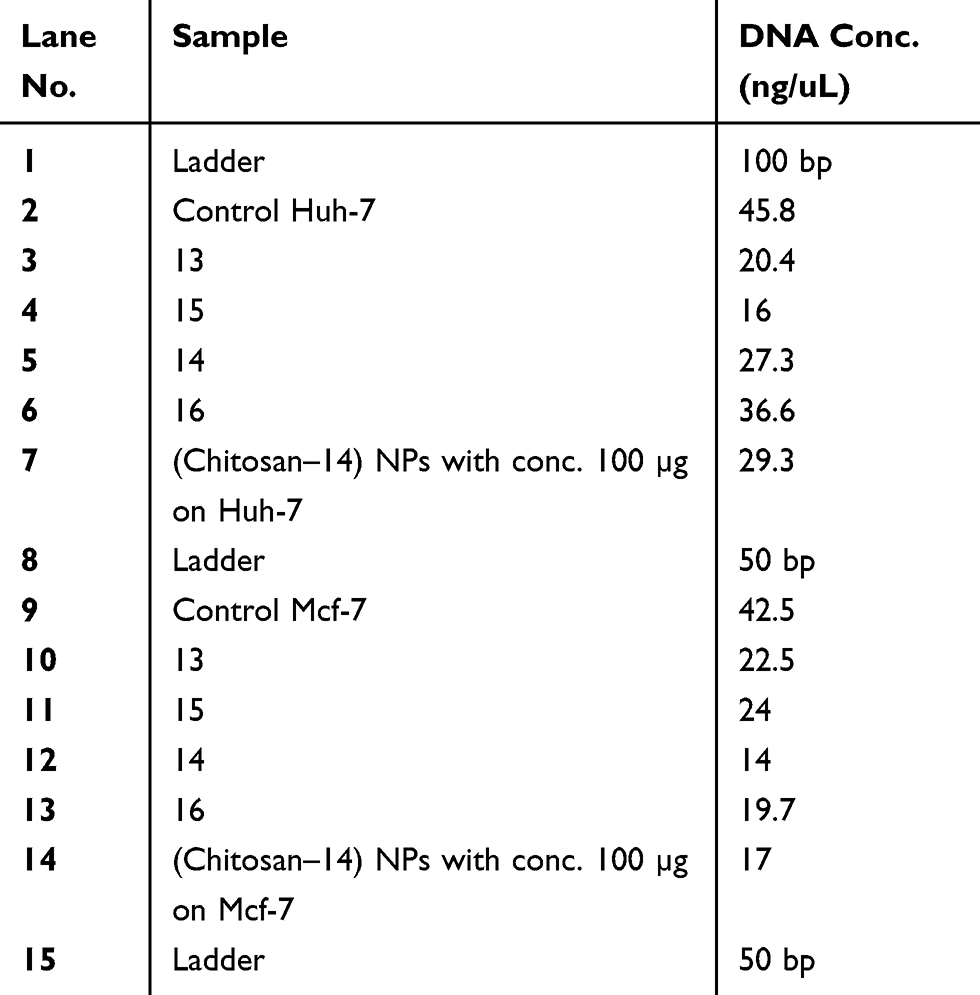 |
Table 7 Genomic DNA content of DNA fragmentation assay after treatment of Huh-7 and Mcf-7 cells with compounds (CC, 13, 15, 14, 16 (Cs–14) NPs) at high concentration (100 μM) of each compound for 24 hrs of cell exposure |
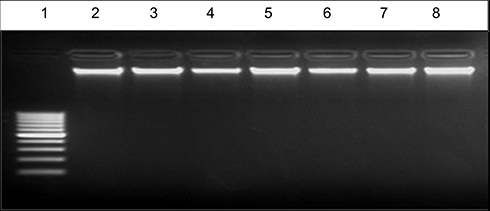 |
Figure 8 EB – stained gel electrophoresis of genomic DNA extraction at high concentration 300 ng from untreated and treated Mcf-7 cell line with lane 1:100 bp ladder, Lane 2: control, lane 3: compound 3, lane 4: compound 4, lane 5: compound 8a, lane 6: compound 6b, lane 7: compound 8b, lane 8: compound 9b with high concentration (100 μM)of each compound. |
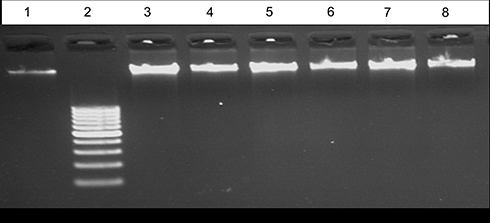 |
Figure 9 EB – stained gel electrophoresis of genomic DNA extraction at high concentration 300 ng from untreated and treated Huh-7 cell line with Lane 1: control, lane 2:100 bp ladder, lane 3: compound 3, lane 4: compound 4, lane 5: compound 8a, lane 6: compound 6b, lane 7: compound 8b, lane 8: compound 9b with high concentration 100 μM of each compound. |
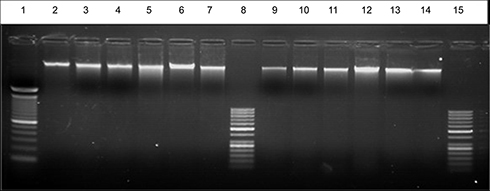 |
Figure 10 EB – stained gel electrophoresis of genomic DNA extraction at high concentration 300 ng from untreated and treated Huh-7 and Mcf-7 cell line with lane 1:100 bp ladder, lane 2: control Huh-7, lane 3: compound 13, lane 4: compound 14, lane 5: compound 15, lane 6: compound 16, lane 7: (Cs– 14) NPs on Huh-7. lane 8: 50 bp ladder, lane 9: control Mcf-7, lane10: compound 13, lane11: compound 14, lane12: compound 15, lane 13: compound 16lLane14: (Cs –14)NPs) on Mcf-7, lane15: 50 bp ladder with high concentration 100 μM of each compound. |
Detection of Caspase-3, BcL-2 protein levels
This investigation was performed to evaluate the apoptotic activity of the newly synthesized compounds, the most compound revealed cytotoxic activity on Huh-7 and Mcf-7 cell lines, compound 14 after treatment of cells with 100 μM for 24 hrs. Caspase-3 and Bcl-2 proteins levels were measured and the results showed a marked increase in the Caspase-3 activity on both cell lines compared to the untreated one. In contrast, the level of the anti-apoptotic protein level of Bcl-2 showed marked decrease in the protein levels compared to the untreated cells as shown in Figure 11. These results confirm the apoptotic activity of compound 14 and its promising application as an antitumor drug after further chemical and biological investigations in vivo model.
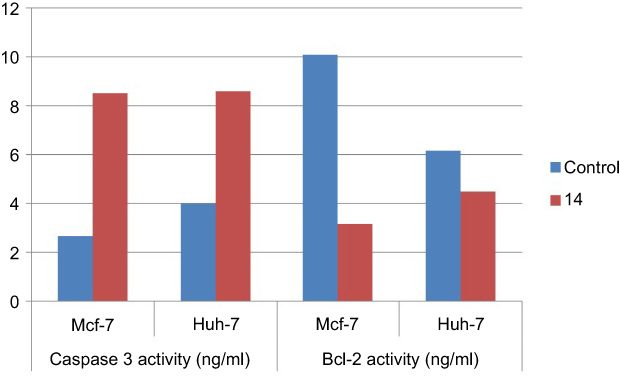 |
Figure 11 Caspase-3 and Bcl-2 activity for compound (14) on both (Mcf-7 and Huh-7) cell lines. |
Conclusion
A novel series of pyrazolopyrimidine thioglycosides and pyrazolopyridine thioglycosides were synthesized and their anticancer activities were evaluated. Even most of the newly synthesized products revealed moderate anticancer activity against human liver cancer (Huh-7) and breast cancer (Mcf-7) cell lines. There were some promising compounds which had a high cytotoxic effect on both cancer cell lines (Huh-7 and Mcf-7), compound 14 was the most cytotoxic compound followed by compound 16, 13 and 15. (Cs––14) nanoparticles were successfully prepared by the ionic gelation method, as confirmed by instrumental analytical techniques (FTIR and TEM). The particle size was spherical in shape with a smooth surface, size 105 nm and zeta potential 40.2 mV. Results of anticancer activity via MTT and (Caspase-3 and Bcl-2) assays proved the toxicity of our compounds and (Cs–14) NPs toward breast and liver cancer cell lines. Therefore, our results indicated that the use of Cs–14 nanoparticle system in breast and liver cancer can be used as drug delivery in which the side effects of conventional chemotherapy could be reduced. This was confirmed by DNA fragmentation and determination of protein activity for both Caspase-3 and Bcl-2. This will open further biological research to extensively evaluate the mechanism of antitumor activity of these compounds on biomedical application. Moreover, in vivo studies required to use this system for future applications.
Experimental
Chemistry
All melting points were measured with a Gallenkamp melting point apparatus. The 1H NMR and 13C NMR spectra were measured on a Jeol-500 MHz spectrometer in DMSO-d6 or CDCl3 using Si(CH3)4 as an internal standard at the National Research Centre, Cairo, Egypt. The IR spectra were recorded on a Pye Unicam Spectra-1000 (KBr disk). Elemental analyses were obtained from the Micro analytical Data Center at Cairo University. Progress of the reactions was monitored by TLC using aluminum sheets coated with silica gel F254 (Merck). Viewing under a short-wavelength UV lamp effected detection, biological studies were performed at the National Cancer Institute, Cairo University and at the National Research Center, Dokki, Giza, Egypt.
General procedure for synthesizing compounds 3 and 11
To a solution of 4-aminoantipyrine 1 (2.03 g, 10 mmol) in glacial acetic acid (10 mL), sodium 2,2 dicyanoethene-1,1-bis(thiolate) 2 (1.86 g, 10 mmol) or sodium cyanocarbonimidodithioate 10 (1.62 g, 10 mmol) were added, and warmed for 5 mins. A precipitate was formed, filtered off and recrystallized from the appropriate solvent to give compounds 3, 11.
Yield: (2.49 g, 80%) as yellow solid, m.p. 190–191°C; IR (KBr, cm−1 ):  3,415 (NH), 3,063 (CH, aromatic) 2,928 (CH3), 2,217, 2,189 (2CN), 1,653 (CO), 1,615 (C=C); 1H NMR (500 MHz, DMSO-d6): δ 2.24 (s, 3H, CH3), 3.09 (s, 3H, CH3), 7.48–7.31 (m, 5H, C6H5), 9.55 (s, 1H, NH), 13.45 (s, 1H, SH). 13C NMR: δ 12.50 (CH3), 34.00 (CH3), 61.00 (C=(CN)2), 110.00 (C-4), 112.45 (CN),115.15 (CN),133–123.55 (7C, 6Ar-C, C-5), 161.00 (CO), 163.50 (C=C). Anal. Calcd. For. C15H13N5OS (311.36): C% 57.86; H% 4.21; N% 22.49; S% 10.30. Found: C% 57.78; H% 4.15; N% 22.36; S% 10.22.
3,415 (NH), 3,063 (CH, aromatic) 2,928 (CH3), 2,217, 2,189 (2CN), 1,653 (CO), 1,615 (C=C); 1H NMR (500 MHz, DMSO-d6): δ 2.24 (s, 3H, CH3), 3.09 (s, 3H, CH3), 7.48–7.31 (m, 5H, C6H5), 9.55 (s, 1H, NH), 13.45 (s, 1H, SH). 13C NMR: δ 12.50 (CH3), 34.00 (CH3), 61.00 (C=(CN)2), 110.00 (C-4), 112.45 (CN),115.15 (CN),133–123.55 (7C, 6Ar-C, C-5), 161.00 (CO), 163.50 (C=C). Anal. Calcd. For. C15H13N5OS (311.36): C% 57.86; H% 4.21; N% 22.49; S% 10.30. Found: C% 57.78; H% 4.15; N% 22.36; S% 10.22.
Yield: (2.30 g, 80%) as yellow solid, m.p. 178–179°C; IR (KBr,cm−1):  3,477 (NH), 3,072 (CH), 2,205 (CN), 1,640 (CO), 1,532 (C=N); 1H NMR (500 MHz, DMSO-d6): δ 2.13 (s, 3H, CH3), 3.08 (s, 3H, CH3), 7.47–7.31 (m, 5H, C6H5), 9.41 (s,1H, NH), 10.90 (s,1H, SH); 13C NMR :δ 12.26 (CH3), 35.23 (CH3), 117.45 (C-4), 118.21 (CN), 135.33–124.45 (7C, 6Ar-C, C-5), 164.26 (CO), 166.42 (C=N). Anal. Calcd. For. C13H13N5OS (287.34): C% 54.34; H% 4.56; N% 24.37; S% 11.16. Found: C% 54.22; H% 4.44; N% 24.22; S% 11.11.
3,477 (NH), 3,072 (CH), 2,205 (CN), 1,640 (CO), 1,532 (C=N); 1H NMR (500 MHz, DMSO-d6): δ 2.13 (s, 3H, CH3), 3.08 (s, 3H, CH3), 7.47–7.31 (m, 5H, C6H5), 9.41 (s,1H, NH), 10.90 (s,1H, SH); 13C NMR :δ 12.26 (CH3), 35.23 (CH3), 117.45 (C-4), 118.21 (CN), 135.33–124.45 (7C, 6Ar-C, C-5), 164.26 (CO), 166.42 (C=N). Anal. Calcd. For. C13H13N5OS (287.34): C% 54.34; H% 4.56; N% 24.37; S% 11.16. Found: C% 54.22; H% 4.44; N% 24.22; S% 11.11.
General procedure for synthesizing compounds 4 and 12
A solution of 3 or 11 (3.11 or 2.87 g, 10 mmol) in ethanol (20 mL) was treated with concentrated hydrochloric acid (1 mL). The reaction mixture was heated under reflux for 30 mins, and then evaporated under reduced pressure. The resulting solid product was collected by filtration and recrystallized from methanol to give 4 and 12.
Yield: (2.43 g, 78%) as yellow solid, m.p. 240–242°C (MeOH); IR (KBr, cm−1):  3,299 (NH), 3,925 (CH3), 2,211 (CN), 16,262 (CO), 1,615 (C=N);1H NMR (500 MHz, DMSO-d6): δ 2.16 (s, 3H, CH3), 2.71 (s, 3H, CH3), 6.34 (s, br, 1H, =NH), 7.45–7.13 (m, 5H, C6H5), 13.24 (s, 1H, SH); 13C NMR: δ 20.24 (CH3), 41.64 (N-CH3), 50.37 (C-7a), 101.21 (C-6), 117.46 (CN), 138.42–123.45 (6C, Ar-C), 154.42 (C-5), 160.62 (CO), 166.25 (C-3a), 167.73 (C-7). Anal. Calcd. For. C15H13N5OS (311.36): C% 57.86; H% 4.21; N% 22.49; S% 10.30. Found: C% 57.77; H% 4.10; N% 22.36; S% 10.24.
3,299 (NH), 3,925 (CH3), 2,211 (CN), 16,262 (CO), 1,615 (C=N);1H NMR (500 MHz, DMSO-d6): δ 2.16 (s, 3H, CH3), 2.71 (s, 3H, CH3), 6.34 (s, br, 1H, =NH), 7.45–7.13 (m, 5H, C6H5), 13.24 (s, 1H, SH); 13C NMR: δ 20.24 (CH3), 41.64 (N-CH3), 50.37 (C-7a), 101.21 (C-6), 117.46 (CN), 138.42–123.45 (6C, Ar-C), 154.42 (C-5), 160.62 (CO), 166.25 (C-3a), 167.73 (C-7). Anal. Calcd. For. C15H13N5OS (311.36): C% 57.86; H% 4.21; N% 22.49; S% 10.30. Found: C% 57.77; H% 4.10; N% 22.36; S% 10.24.
Yield: (2.24 g, 78%) as yellow solid, mp 225–227°C (MeOH); IR (KBr, cm−1 ):  3,329 (NH), 3,025 (CH aromatic), 2,973 (CH3), 1,642 (CO); 1H NMR (500 MHz, DMSO-d6): δ 2.04 (s, 3H, CH3), 3.01 (s, 3H, CH3), 6. 21 (s, br, 1H, =NH), 7.37–7.28 (m, 5H, C6H5), 12.46 (s, 1H, SH); 13C NMR: δ 12.13 (CH3), 40.42 (N-CH3), 61.47 (C-7a), 138.21–124.22 (6C, Ar-C), 161.23 (CO), 163.64 (C-3a), 165.24 (C-7), 176.28 (C-5). Anal. MS, m/z (%): 287 [M+] (45). Calcd. For. C13H13N5OS (287.34): C% 54.34; H% 4.56; N% 24.37; S% 11.16. Found: C% 54.22; H% 4.45; N% 24.24; S% 11.10.
3,329 (NH), 3,025 (CH aromatic), 2,973 (CH3), 1,642 (CO); 1H NMR (500 MHz, DMSO-d6): δ 2.04 (s, 3H, CH3), 3.01 (s, 3H, CH3), 6. 21 (s, br, 1H, =NH), 7.37–7.28 (m, 5H, C6H5), 12.46 (s, 1H, SH); 13C NMR: δ 12.13 (CH3), 40.42 (N-CH3), 61.47 (C-7a), 138.21–124.22 (6C, Ar-C), 161.23 (CO), 163.64 (C-3a), 165.24 (C-7), 176.28 (C-5). Anal. MS, m/z (%): 287 [M+] (45). Calcd. For. C13H13N5OS (287.34): C% 54.34; H% 4.56; N% 24.37; S% 11.16. Found: C% 54.22; H% 4.45; N% 24.24; S% 11.10.
General procedure for synthesizing compounds 6a,b
To a solution of 3 (3.11 g, 10 mmol) in aqueous KOH [0.56 g (0.01 mol) in 6 mL of distilled water], a solution of 2,3,5 tri-O-acetyl-α-D-xylo- or arabino-furanosyl bromide 5a,b (4.52 g, 0.011 mol) in acetone (30 mL) was added. The reaction mixture was stirred at room temperature until the reaction was judged complete by TLC (1–3 hrs), using chloroform: ether 4:1, v/v (Rf 0.66–0.70 region), then evaporated under reduced pressure at 60°C and the residue was washed with distilled water to remove KBr. The product was dried prior to crystallization from methanol to give compounds 6a,b.
Yield: (3.99 g, 70%) as yellow solid, m.p. 216–217°C (EtOH); IR (KBr, cm−1 ):  3,395 (NH), 3,028 (CH aromatic), 2,954 (CH), 2,221, 2,207 (2CN), 1,746 (4CO), 1,639 (CO); 1H NMR (500 MHz, DMSO-d6):δ 1.98 −2.06 (3s, 9H, 3xOAc), 2.28 (s, 3H, CH3), 3.24 (s, 3H, CH3), 3.56–3.58 (m, 2H, 2H-5ʹ ), 4.62–4.64 (m, 1H, H-4ʹ ), 4.82 (t, J=4.9 Hz, 1H, H-3ʹ), 5.13 (t, J=9.3 Hz, 1H, H-2ʹ ), 5.42 (d, J1ʹ −2’ =8.2 Hz, 1H, H-1ʹ ), 7.26–7.37 (m, 5H, C6H5), 11.27 (s, br, D2O exch., 1H, NH); 13C NMR: δ 10.21 (CH3), 21.42 (3CH3CO), 34.88 (N-CH3), 43.45 (=C-(CN)2), 60.36 (C-5ʹ ), 68.61 (C-4ʹ ), 75.47 (C-3ʹ ), 76.73 (C-2ʹ ), 78.54 (C-1ʹ ), 102.67 (C-4), 118.114 (2CN), 124.26–138.29 (7C, Ar-C, C-5), 158.89 (CO), 170.26 (4CO), 183.47 (=C-S), Anal. Calcd. For Chemical Formula: C26H27N5O8S (569.59): C% 54.83; H% 4.78; N% 12.30; S% 5.63. Found: C% 54.74; H% 4.80; N% 12.25; S% 5.52.
3,395 (NH), 3,028 (CH aromatic), 2,954 (CH), 2,221, 2,207 (2CN), 1,746 (4CO), 1,639 (CO); 1H NMR (500 MHz, DMSO-d6):δ 1.98 −2.06 (3s, 9H, 3xOAc), 2.28 (s, 3H, CH3), 3.24 (s, 3H, CH3), 3.56–3.58 (m, 2H, 2H-5ʹ ), 4.62–4.64 (m, 1H, H-4ʹ ), 4.82 (t, J=4.9 Hz, 1H, H-3ʹ), 5.13 (t, J=9.3 Hz, 1H, H-2ʹ ), 5.42 (d, J1ʹ −2’ =8.2 Hz, 1H, H-1ʹ ), 7.26–7.37 (m, 5H, C6H5), 11.27 (s, br, D2O exch., 1H, NH); 13C NMR: δ 10.21 (CH3), 21.42 (3CH3CO), 34.88 (N-CH3), 43.45 (=C-(CN)2), 60.36 (C-5ʹ ), 68.61 (C-4ʹ ), 75.47 (C-3ʹ ), 76.73 (C-2ʹ ), 78.54 (C-1ʹ ), 102.67 (C-4), 118.114 (2CN), 124.26–138.29 (7C, Ar-C, C-5), 158.89 (CO), 170.26 (4CO), 183.47 (=C-S), Anal. Calcd. For Chemical Formula: C26H27N5O8S (569.59): C% 54.83; H% 4.78; N% 12.30; S% 5.63. Found: C% 54.74; H% 4.80; N% 12.25; S% 5.52.
Yield: (4.27 g, 75%) as yellow solid, m.p. 206–207°C (EtOH); IR (KBr, cm−1 ):  3,386 (NH), 3,029 (CH aromatic), 2,972 (CH3), 2,218, 2,214 (2CN), 1,753 (4CO), 1,642 (CO); 1H NMR (500 MHz, DMSO-d6): δ 1.99–2.17 (3s, 9H, 3xOAc), 2.411 (s, 3H, CH3), 3.35 (s, 3H, CH3), 4.12–4.15 (m, 2H, 2H-5ʹ ), 4.64 −4.65 (m, 1H, H-4ʹ ), 4.68 (t, J=9.6 Hz, 1H, H-3ʹ ), 5.26 (t, J=8.4 Hz, 1H, H-2ʹ ), 5.42 (d, J1ʹ −2’=6.5 Hz, 1H, H-1ʹ ), 7.34–7.67 (m, 5H, C6H5), 11.56 (s, D2O exch., 1H, NH). Anal. Calcd. For Chemical Formula: C26H27N5O8S (569.59): C% 54.83; H% 4.78; N% 12.30; S% 5.63. Found: C% 54.75; H% 4.74; N% 12.25; S% 5.55.
3,386 (NH), 3,029 (CH aromatic), 2,972 (CH3), 2,218, 2,214 (2CN), 1,753 (4CO), 1,642 (CO); 1H NMR (500 MHz, DMSO-d6): δ 1.99–2.17 (3s, 9H, 3xOAc), 2.411 (s, 3H, CH3), 3.35 (s, 3H, CH3), 4.12–4.15 (m, 2H, 2H-5ʹ ), 4.64 −4.65 (m, 1H, H-4ʹ ), 4.68 (t, J=9.6 Hz, 1H, H-3ʹ ), 5.26 (t, J=8.4 Hz, 1H, H-2ʹ ), 5.42 (d, J1ʹ −2’=6.5 Hz, 1H, H-1ʹ ), 7.34–7.67 (m, 5H, C6H5), 11.56 (s, D2O exch., 1H, NH). Anal. Calcd. For Chemical Formula: C26H27N5O8S (569.59): C% 54.83; H% 4.78; N% 12.30; S% 5.63. Found: C% 54.75; H% 4.74; N% 12.25; S% 5.55.
General procedure for synthesizing compound 13
To a solution of 11 (2.87 g, 10 mmol) in aqueous KOH [0.56 g (0.01 mol) in 6 mL of distilled water], a solution of 2,3,5 tri-O-acetyl-α-D-xylo-furanosyl bromide 5a,b (4.52 g, 0.011 mol) in acetone (30 mL) was added. The reaction mixture was stirred at room temperature until the reaction was judged complete by TLC (1–3 hrs), using chloroform: ether 4:1, v/v (Rf 0.66–0.70 region), then evaporated under reduced pressure at 60°C and the residue was washed with distilled water to remove KBr. The product was dried prior to crystallization from methanol to give compound 13.
Yield: (3.93 g, 72%) as yellow solid, m.p. 206–207°C (EtOH); 1H NMR (500 MHz, DMSO-d6): δ 1.98–2.11 (3s, 9H, 3xOAc), 2.27 (s, 3H, CH3), 3.41 (s, 3H, CH3), 4.35–4.36 (m, 2H, 2H-5ʹ ), 4.52–4.53 (m, 1H, H-4ʹ ), 4.83 (t, J=9.8 Hz, 1H, H-3ʹ ), 5.14 (t, J=10.3 Hz, 1H, H-2ʹ ), 5.33 (d, J1ʹ −2’=10.6 Hz, 1H, H-1ʹ ), 7.35–7.48 (m, 5H, C6H5), 10.52 (s, D2O exch., 1H, NH); 13C NMR: δ 7.96 (CH3), 22.45 (3CH3CO), 35. 64 (CH3), 61.65 (C-5ʹ ), 69.42 (C-4ʹ ), 72.22 (C-3ʹ ), 75.16 (C-2ʹ ), 76.25(C-1ʹ), 116.11 (C-4), 117.57 (CN), 123.67–136.23 (7C, Ar-C, C-5), 161.63 (CO), 166.29 (-C=N), 172.24 (4CO). Anal. Calcd. For C24H27N5O8S (545.56): C% 52.84; H% 4.99; N% 12.84; S% 5.88. Found: C% 52.75; H% 4.82; N% 12.66; S% 5.80.
General procedure for synthesizing compounds 7a,b
To a solution of 4 (3.11 g 10 mmol) in aqueous KOH [0.56 g (0.01 mol) in 6 mL of distilled water], a solution of 2,3,5-tri-O-acetyl-a-D-xylo- or arabino-furanosyl bromide 5a,b (0.011 mol) in acetone (30 mL) was added. The reaction mixture was stirred at room temperature until the reaction was judged complete by TLC (1–3 hrs), using chloroform: ether 4:1, v/v (Rf 0.66–0.70 region), then evaporated under reduced pressure at 40°C and the residue was washed with distilled water to remove KBr. The product was dried prior to crystallization from methanol to give compounds 7a,b.
Yield: (4.50 g, 79%) as yellow solid, m.p. 208–209°C (EtOH); IR (KBr, cm−1 ):  3,394 (NH), 3,035 (CH, aromatic), 2,952 (CH), 2,210 (CN), 1,743 (4CO), 1,638 (CO), 1,592 (C=N); 1H NMR (500 MHz, DMSO-d6): δ 1.97–2.12 (3s, 9H, 3xOAc), 2.36 (s, 3H, CH3), 2.62 (s, 3H, CH3), 4.13–4.19 (m, 3H, 2H-5ʹ), 4.36–4.37 (m, 1H, H-4’ ), 4.62 (t, J =9.3 Hz, 1H, H-3ʹ ), 4.98 (t, J =9.1 Hz, 1H, H-2ʹ), 5.56 (d, J1ʹ-2’ =10.6 Hz, 1H, H-1ʹ), 6.64 (s, D2O exch.,1H, NH), 7.34–7.76 (m, 5H, C6H5); 13C NMR: δ 19.16 (CH3), 21.36 (3CH3CO), 38.95 (CH3), 51.24 (C-7a), 62.11 (C-5’ ), 69.27 (C-4ʹ), 71.86 (C-3ʹ), 73.75 (C-2ʹ ), 79.58 (C-1ʹ ), 82.31 (C-6), 116.38 (CN), 125.21–139.51 (6C, Ar-C), 160.54 (CO), 165.61 (C-3a), 165.14 (C-7), 167.22 (C-5), 171.42 (4CO). Anal. Calcd. For C26H27N5O8S (569.59): C% 54.83; H% 4.78; N% 12.30; S% 5.63. Found: C% 54.75; H% 4.70; N% 12.26; S% 5.54.
3,394 (NH), 3,035 (CH, aromatic), 2,952 (CH), 2,210 (CN), 1,743 (4CO), 1,638 (CO), 1,592 (C=N); 1H NMR (500 MHz, DMSO-d6): δ 1.97–2.12 (3s, 9H, 3xOAc), 2.36 (s, 3H, CH3), 2.62 (s, 3H, CH3), 4.13–4.19 (m, 3H, 2H-5ʹ), 4.36–4.37 (m, 1H, H-4’ ), 4.62 (t, J =9.3 Hz, 1H, H-3ʹ ), 4.98 (t, J =9.1 Hz, 1H, H-2ʹ), 5.56 (d, J1ʹ-2’ =10.6 Hz, 1H, H-1ʹ), 6.64 (s, D2O exch.,1H, NH), 7.34–7.76 (m, 5H, C6H5); 13C NMR: δ 19.16 (CH3), 21.36 (3CH3CO), 38.95 (CH3), 51.24 (C-7a), 62.11 (C-5’ ), 69.27 (C-4ʹ), 71.86 (C-3ʹ), 73.75 (C-2ʹ ), 79.58 (C-1ʹ ), 82.31 (C-6), 116.38 (CN), 125.21–139.51 (6C, Ar-C), 160.54 (CO), 165.61 (C-3a), 165.14 (C-7), 167.22 (C-5), 171.42 (4CO). Anal. Calcd. For C26H27N5O8S (569.59): C% 54.83; H% 4.78; N% 12.30; S% 5.63. Found: C% 54.75; H% 4.70; N% 12.26; S% 5.54.
Yield: (4.49 g, 70%) as yellow solid, m.p. 215–216°C (EtOH); IR (KBr, cm−1 ):  3,422 (NH), 3,029 (CH, aromatic), 2,933 (CH3), 2,216 (CN), 1,754 (4CO), 1,638 (CO), 1,602 (C=N); 1H NMR (500 MHz, DMSO-d6): δ 2.11–2.16 (3s, 9H, 3xOAc), 2.25 (s, 3H, CH3), 2.64 (s, 3H, CH3), 3.92–3.94 (m, 2H, 2H-5ʹ ), 4.28–4.31 (m, 1H, H-4ʹ ), 4.46 (t, J =9.3 Hz, 1H, H-3ʹ ), 4.84 (t, J =8.6 Hz, 1H, H-2ʹ ), 5.64 (d, J1ʹ −2’ =6.8 Hz, 1H, H-1’ ), 6.19 (s, D2O exch., 1H, NH), 7.38–7.61 (m, 5H, C6H5); 13C NMR: d 18.24 (CH3), 22.31 (3CH3CO), 41.56 (CH3), 50.47 (C-7a), 62.13 (C-5ʹ ), 66.25 (C-4ʹ), 72.34 (C-3ʹ ), 74.62 (C-2ʹ), 78.92 (C-1ʹ ), 80.43 (C-6), 116.23 (CN), 128.26–140.22 (6C, Ar-C), 159.34 (CO), 162.41(C-7), 164.11(C-3a), 166.64 (C-5), 172.18 (4CO). Anal. Calcd. For C26H27N5O8S (569.59): C% 54.83; H% 4.78; N% 12.30; S% 5.63. Found: C% 54.76; H% 4.72; N% 12.26; S% 5.58.
3,422 (NH), 3,029 (CH, aromatic), 2,933 (CH3), 2,216 (CN), 1,754 (4CO), 1,638 (CO), 1,602 (C=N); 1H NMR (500 MHz, DMSO-d6): δ 2.11–2.16 (3s, 9H, 3xOAc), 2.25 (s, 3H, CH3), 2.64 (s, 3H, CH3), 3.92–3.94 (m, 2H, 2H-5ʹ ), 4.28–4.31 (m, 1H, H-4ʹ ), 4.46 (t, J =9.3 Hz, 1H, H-3ʹ ), 4.84 (t, J =8.6 Hz, 1H, H-2ʹ ), 5.64 (d, J1ʹ −2’ =6.8 Hz, 1H, H-1’ ), 6.19 (s, D2O exch., 1H, NH), 7.38–7.61 (m, 5H, C6H5); 13C NMR: d 18.24 (CH3), 22.31 (3CH3CO), 41.56 (CH3), 50.47 (C-7a), 62.13 (C-5ʹ ), 66.25 (C-4ʹ), 72.34 (C-3ʹ ), 74.62 (C-2ʹ), 78.92 (C-1ʹ ), 80.43 (C-6), 116.23 (CN), 128.26–140.22 (6C, Ar-C), 159.34 (CO), 162.41(C-7), 164.11(C-3a), 166.64 (C-5), 172.18 (4CO). Anal. Calcd. For C26H27N5O8S (569.59): C% 54.83; H% 4.78; N% 12.30; S% 5.63. Found: C% 54.76; H% 4.72; N% 12.26; S% 5.58.
General procedure for synthesizing compound 14
To a solution of 12 (2.87 g, 10 mmol) in aqueous KOH [0.56 g (0.01 mol) in 6 mL of distilled water], a solution of 2,3,5-tri-O-acetyl-α-D-xylo-furanosyl bromide 5a (0.011 mol) in acetone (30 mL) was added. The reaction mixture was stirred at room temperature until the reaction was judged complete by TLC (1–3 hrs), using chloroform: ether 4:1, v/v (Rf 0.66–0.70 region), then evaporated under reduced pressure at 40°C and the residue was washed with distilled water to remove KBr. The product was dried prior to crystallization from methanol to give compound 14.
Yield: (4.32 g, 70%) as yellow solid, m.p. 222–223°C (EtOH); IR (KBr, cm−1 ):  3,436 (NH), 3,028 (CH, aromatic), 2,925 (CH), 1,751 (CO), 1,630 (CO); 1H NMR (500 MHz, DMSO-d6): δ 1.98–2.09 (3s, 9H, 3xOAc), 2.36 (s, 3H, CH3), 2.95 (s, 3H, CH3), 4.02–4.13 (m, 2H, 2H-5ʹ ), 4.37–4.45 (m, 1H, H-4ʹ ), 4.62 (t, J=9.2 Hz, 1H, H-3ʹ ), 5.04 (t, J =8.6 Hz, 1H, H-2ʹ ), 5.53 (d, J1ʹ −2’ =10.4 Hz, 1H, H-1ʹ ), 5.66 (s, br, D2O exch.,1H, =NH), 7.21–7.58 (m, 5H, C6H5); 13C NMR: δ 18.41 (CH3), 22.63 (3CH3CO), 39.68 (CH3), 55.31 (7a), 62.63 (C-5ʹ ), 68.74 (C-4ʹ ), 72.64 (C-3ʹ ), 74.22 (C-2ʹ ), 78.81 (C-1ʹ ), 124.46–143.21 (6C, Ar-C), 161.47 (CO), 166.83 (3a), 169.33 (C-7), 173.19 (4CO), 178.52 (C-5). Anal. Calcd. For C24H27N5O8S (545.56): C% 52.84; H% 4.99; N% 12.84; S% 5.88. Found: C% 52.69; H% 4.86; N% 12.77; S% 5.80.
3,436 (NH), 3,028 (CH, aromatic), 2,925 (CH), 1,751 (CO), 1,630 (CO); 1H NMR (500 MHz, DMSO-d6): δ 1.98–2.09 (3s, 9H, 3xOAc), 2.36 (s, 3H, CH3), 2.95 (s, 3H, CH3), 4.02–4.13 (m, 2H, 2H-5ʹ ), 4.37–4.45 (m, 1H, H-4ʹ ), 4.62 (t, J=9.2 Hz, 1H, H-3ʹ ), 5.04 (t, J =8.6 Hz, 1H, H-2ʹ ), 5.53 (d, J1ʹ −2’ =10.4 Hz, 1H, H-1ʹ ), 5.66 (s, br, D2O exch.,1H, =NH), 7.21–7.58 (m, 5H, C6H5); 13C NMR: δ 18.41 (CH3), 22.63 (3CH3CO), 39.68 (CH3), 55.31 (7a), 62.63 (C-5ʹ ), 68.74 (C-4ʹ ), 72.64 (C-3ʹ ), 74.22 (C-2ʹ ), 78.81 (C-1ʹ ), 124.46–143.21 (6C, Ar-C), 161.47 (CO), 166.83 (3a), 169.33 (C-7), 173.19 (4CO), 178.52 (C-5). Anal. Calcd. For C24H27N5O8S (545.56): C% 52.84; H% 4.99; N% 12.84; S% 5.88. Found: C% 52.69; H% 4.86; N% 12.77; S% 5.80.
General procedure for deacetylation of compounds 8a,b and 15
Dry gaseous ammonia was passed through a solution of protected nucleoside 6a,b or 13 (6.41 g or 6.17 g, 10 mmol) in dry methanol (20 mL) for 10 mins with cooling and stirring, then the reaction mixture was stirred at room temperature until the reaction was judged complete by TLC (9–10 hrs) using (CHCl3/MeOH 9:1) (Rf, 0.56–0.58). The resulting mixture was concentrated under reduced pressure to afford a solid residue which washed several times by boiling chloroform. The residue was dried, purified by column chromatography using chloroform/methanol (9:1) and crystallized from the appropriate solvent to give compound 8a,b and 15.
Yield:(3.22 g, 68%) as yellow solid,m.p.211–212°C (EtOH); IR (KBr, cm−1):  3,532 (OH), 3,413 (NH), 3,069 (CH aromatic), 2,216 (CN), 2,210 (CN), 1,642 (CO); 1H NMR (500 MHz, CDCl3) δ 2.26 (s, 3H, CH3), 3.14 (s, 3H, CH3), 3.53–5.54 (m,, 2H, H-5ʹ), 3.86–3.87 (m, 2H, H-4ʹ), 4.01 (t, J=8.4 Hz, 1H, H-3ʹ), 4.33 (t, J =9.1 Hz, 1H, H-2ʹ ), 4.99 (s, D2O exch., 1H, 5ʹ -OH), 5.11–5.12 (m, D2O exch., 2H, 2ʹ-OH and 3ʹ-OH), 5.34 (d, J1ʹ −2’ =10.22 Hz, 1H, H-1ʹ ), 7.21–7.57 (m, 5H, C6H5), 10.26 (s,D2O exch., 1H, NH); 13C NMR: δ 8.21 (CH3), 37.41 (CH3), 43.65 (=C-CN), 62.46 (C-5ʹ ), 72.82 (C-4ʹ ), 74.23 (C-3ʹ ), 75.73 (C-2ʹ ), 81.46 (C-1ʹ), 106.25 (C-4), 116.47 (2CN), 128.13–136.78 (7C, Ar-C, C-5), 164.25 (CO), 182.18 (=C-S). Anal. Calcd. For C20H21N5O5S (443.48): C% 54.17; H% 4.77; N% 15.79; S% 7.23. Found: C% 54.11; H% 4.72; N% 14.68; S% 7.18.
3,532 (OH), 3,413 (NH), 3,069 (CH aromatic), 2,216 (CN), 2,210 (CN), 1,642 (CO); 1H NMR (500 MHz, CDCl3) δ 2.26 (s, 3H, CH3), 3.14 (s, 3H, CH3), 3.53–5.54 (m,, 2H, H-5ʹ), 3.86–3.87 (m, 2H, H-4ʹ), 4.01 (t, J=8.4 Hz, 1H, H-3ʹ), 4.33 (t, J =9.1 Hz, 1H, H-2ʹ ), 4.99 (s, D2O exch., 1H, 5ʹ -OH), 5.11–5.12 (m, D2O exch., 2H, 2ʹ-OH and 3ʹ-OH), 5.34 (d, J1ʹ −2’ =10.22 Hz, 1H, H-1ʹ ), 7.21–7.57 (m, 5H, C6H5), 10.26 (s,D2O exch., 1H, NH); 13C NMR: δ 8.21 (CH3), 37.41 (CH3), 43.65 (=C-CN), 62.46 (C-5ʹ ), 72.82 (C-4ʹ ), 74.23 (C-3ʹ ), 75.73 (C-2ʹ ), 81.46 (C-1ʹ), 106.25 (C-4), 116.47 (2CN), 128.13–136.78 (7C, Ar-C, C-5), 164.25 (CO), 182.18 (=C-S). Anal. Calcd. For C20H21N5O5S (443.48): C% 54.17; H% 4.77; N% 15.79; S% 7.23. Found: C% 54.11; H% 4.72; N% 14.68; S% 7.18.
Yield: (3.02 g, 68%) as yellow solid, m.p. 224–225°C (EtOH); IR (KBr, cm−1)  3,514 (OH), 3,421 (NH), 3,065 (CH aromatic), 2,213 (CN), 2,207 (CN), 1,646 (CO); 1H NMR (500 MHz, CDCl3) δ 2.34 (s, 3H, CH3), 3.21 (s, 3H, CH3), 3.62–3.63 (m, 2H, 2H-5’), 3.84 (m, 1H, 4ʹ), 4.02 (t,, J=9.1 Hz, 1H, H-3ʹ), 4.35 (t, J=6.1 Hz, 1H, H-2ʹ), 4.66 (d, J1ʹ −2’ =9.53 Hz, 1H, H-1ʹ ), 5.14 (s, D2O exch., 1H, 5ʹ-OH), 5.22–5.23 (m, D2O exch., 2H, 2ʹ-OH and 3ʹ-OH,), 7.35–7.86 (m, 5H, C6H5) 11.33 (s, D2O exch., 1H, NH); 13C NMR: δ 9.35 (CH3), 36.55 (CH3), 44.67 (=C-CN), 62.36 (C-5ʹ ), 73.25 (C-4ʹ ), 72.62 (C-3ʹ), 74.51 (C-2ʹ ), 82.79 (C-1ʹ), 107.36 (C-4), 118.36 (2CN), 124.53–136.25 (7C, Ar-C, C-5), 163.37 (CO), 182.52 (=C-S). Anal. Calcd. For C20H21N5O5S (443.48): C% 54.17; H% 4.77; N% 15.79; S% 7.23. Found: C% 54.10; H%, 4.80; N% 15.70; S% 7.12.
3,514 (OH), 3,421 (NH), 3,065 (CH aromatic), 2,213 (CN), 2,207 (CN), 1,646 (CO); 1H NMR (500 MHz, CDCl3) δ 2.34 (s, 3H, CH3), 3.21 (s, 3H, CH3), 3.62–3.63 (m, 2H, 2H-5’), 3.84 (m, 1H, 4ʹ), 4.02 (t,, J=9.1 Hz, 1H, H-3ʹ), 4.35 (t, J=6.1 Hz, 1H, H-2ʹ), 4.66 (d, J1ʹ −2’ =9.53 Hz, 1H, H-1ʹ ), 5.14 (s, D2O exch., 1H, 5ʹ-OH), 5.22–5.23 (m, D2O exch., 2H, 2ʹ-OH and 3ʹ-OH,), 7.35–7.86 (m, 5H, C6H5) 11.33 (s, D2O exch., 1H, NH); 13C NMR: δ 9.35 (CH3), 36.55 (CH3), 44.67 (=C-CN), 62.36 (C-5ʹ ), 73.25 (C-4ʹ ), 72.62 (C-3ʹ), 74.51 (C-2ʹ ), 82.79 (C-1ʹ), 107.36 (C-4), 118.36 (2CN), 124.53–136.25 (7C, Ar-C, C-5), 163.37 (CO), 182.52 (=C-S). Anal. Calcd. For C20H21N5O5S (443.48): C% 54.17; H% 4.77; N% 15.79; S% 7.23. Found: C% 54.10; H%, 4.80; N% 15.70; S% 7.12.
Yield: (2.89 g, 69%) as yellow solid, m.p. 182–183°C (EtOH); IR (KBr, cm−1)  3,543 (OH), 3,415 (NH), 3,062 (CH aromatic), 2,217 (CN), 1,645 (CO), 1,591 (C=N); 1H NMR (500 MHz, CDCl3) δ 2.28 (s, 3H,CH3), 3.04 (s, 3H, CH3), 3.54–3.95 (m, 5H, 2H-6ʹ, H-5ʹ, H-4ʹ, H-3ʹ,H-2ʹ), 4.99 (d, 1H, J1ʹ-2’=5.8 Hz, H-1ʹ), 5.02 (s, D2O exch., 1H, 5ʹ-OH), 5.15–5.245 (m, D2O exch., 2H, 2ʹ-OH, 3ʹ-OH), 6.72 (s, D2Oexch., 1H, NH), 7.06–7.36 (m, 5H, C6H5); 13C NMR: δ 8.28 (CH3), 35.84 (CH3), 62.36 (C-5ʹ), 69.27 (C-4ʹ), 71.52 (C-3ʹ), 74.51 (C-2ʹ), 83.43 (C-1ʹ), 118.22 (CN), 120.31 (C-4), 124.37–135.72 (6C, Ar-C, C-5), 164.42 (CO), 165.61 (-C=N). Anal. Calcd. For C18H21N5O5S (419.45.): C% 51.54; H% 5.05; N% 16.70; S% 7.64. Found: C% 51.46; H% 5.10; N% 16.62; S% 7.52.
3,543 (OH), 3,415 (NH), 3,062 (CH aromatic), 2,217 (CN), 1,645 (CO), 1,591 (C=N); 1H NMR (500 MHz, CDCl3) δ 2.28 (s, 3H,CH3), 3.04 (s, 3H, CH3), 3.54–3.95 (m, 5H, 2H-6ʹ, H-5ʹ, H-4ʹ, H-3ʹ,H-2ʹ), 4.99 (d, 1H, J1ʹ-2’=5.8 Hz, H-1ʹ), 5.02 (s, D2O exch., 1H, 5ʹ-OH), 5.15–5.245 (m, D2O exch., 2H, 2ʹ-OH, 3ʹ-OH), 6.72 (s, D2Oexch., 1H, NH), 7.06–7.36 (m, 5H, C6H5); 13C NMR: δ 8.28 (CH3), 35.84 (CH3), 62.36 (C-5ʹ), 69.27 (C-4ʹ), 71.52 (C-3ʹ), 74.51 (C-2ʹ), 83.43 (C-1ʹ), 118.22 (CN), 120.31 (C-4), 124.37–135.72 (6C, Ar-C, C-5), 164.42 (CO), 165.61 (-C=N). Anal. Calcd. For C18H21N5O5S (419.45.): C% 51.54; H% 5.05; N% 16.70; S% 7.64. Found: C% 51.46; H% 5.10; N% 16.62; S% 7.52.
General procedure for synthesizing compounds 9a,b and 16
Dry gaseous ammonia was passed through a solution of protected nucleoside 7a,b or 14 (5.70 or 5.46 g, 10 mmol) in dry methanol (20 mL) for 10 mins with cooling and stirring, then the reaction mixture was stirred at room temperature until the reaction was judged complete by TLC (9–10 hrs) using (CHCl3/MeOH 9:1) (Rf, 0.56–0.58). The resulting mixture was concentrated under reduced pressure to afford a solid residue which washed several times with boiling chloroform. The residue was dried, purified by column chromatography using chloroform/methanol (9:1) and crystallized from appropriate solvent to give compounds 9a,b and 16.
Yield: (3.30 g, 70%) as yellow solid, m.p. 192–193°C (EtOH); IR (KBr, cm−1)  3,514 (OH), 3,439 (NH), 3,078 (CH aromatic), 1,649 (CO), 1,599 (C=N); 1H NMR (500 MHz, CDCl3) δ 2.16 (s, 3H, CH3), 3.11 (s, 3H, CH3), 3.78–3.79 (m, 2H, 2H-5ʹ), 3.92–3.94 (m, 1H, H-4ʹ), 4.24 (t, J=8.7 Hz, 1H, H-3ʹ), 4.41 (t, J=6.5 Hz, 1H, H-2ʹ ), 4.66 (d, 1H, J1ʹ −2’ =10.2 Hz, H-1ʹ ), 4.91 (s, D2O exch., 1H, 5ʹ -OH), 5.23–5.23 (m, D2O exch., 2H, 2ʹ-OH and 3ʹ-OH), 6.28 (s, D2O exch., 1H, NH), 7.53–7.87 (m,5H,C6H5); 13C NMR: δ 20. 47 (CH3), 41.24 (CH3), 51.49 (7a), 62.41 (C-5ʹ ), 69.28 (C-4ʹ), 71.62 (C-3ʹ), 74.92 (C-2ʹ), 81.93 (C-1ʹ), 86.25 (C-6), 117.44 (CN), 123.68–140.37 (6C, Ar-C), 160.61 (CO), 168.15(3a), 169.61 (C-7), 170.13 (C-5). Anal. Calcd. For C20H21N5O5S (443.48): C% 54.17; H% 4.77; N% 15.79; S% 7.23. Found: C% 54.15; H% 4.64; N% 15.68; S% 7.16.
3,514 (OH), 3,439 (NH), 3,078 (CH aromatic), 1,649 (CO), 1,599 (C=N); 1H NMR (500 MHz, CDCl3) δ 2.16 (s, 3H, CH3), 3.11 (s, 3H, CH3), 3.78–3.79 (m, 2H, 2H-5ʹ), 3.92–3.94 (m, 1H, H-4ʹ), 4.24 (t, J=8.7 Hz, 1H, H-3ʹ), 4.41 (t, J=6.5 Hz, 1H, H-2ʹ ), 4.66 (d, 1H, J1ʹ −2’ =10.2 Hz, H-1ʹ ), 4.91 (s, D2O exch., 1H, 5ʹ -OH), 5.23–5.23 (m, D2O exch., 2H, 2ʹ-OH and 3ʹ-OH), 6.28 (s, D2O exch., 1H, NH), 7.53–7.87 (m,5H,C6H5); 13C NMR: δ 20. 47 (CH3), 41.24 (CH3), 51.49 (7a), 62.41 (C-5ʹ ), 69.28 (C-4ʹ), 71.62 (C-3ʹ), 74.92 (C-2ʹ), 81.93 (C-1ʹ), 86.25 (C-6), 117.44 (CN), 123.68–140.37 (6C, Ar-C), 160.61 (CO), 168.15(3a), 169.61 (C-7), 170.13 (C-5). Anal. Calcd. For C20H21N5O5S (443.48): C% 54.17; H% 4.77; N% 15.79; S% 7.23. Found: C% 54.15; H% 4.64; N% 15.68; S% 7.16.
Yield: (3.20 g, 72%) as yellow solid, m.p. 200–202°C (EtOH); IR (KBr, cm−1 ) 3,545 (OH), 3,436 (NH), 3,062 (CH aromatic), 1649 (CO), 1,600 (C=N); 1H NMR (500 MHz, CDCl3) δ 2.41 (s, 3H, CH3),3.14 (s, 3H, CH3), 3.42–3.45 (m, 2H, 2H-5ʹ), 3.55–3.58 (m, 1H, H-4’), 3.76 (t, J=8.8 Hz, 1H, H-3ʹ), 3.89 (t, J=6.9 Hz, 1H, H-2ʹ), 4.62 (d,1H, J1ʹ-2’ =6.62 Hz, H-1’), 4.93 (s, D2O exch., 1H, 5ʹ-OH), 5.21–5.24 (m, D2O exch., 2H, 2ʹ-OH and 3ʹ-OH), 5.53 (s, D2O exch., 1H,NH), 7.47–7.86 (m, 5H, C6H5). Anal. Calcd. For C20H21N5O5S (443.48): C% 54.17; H% 4.77; N% 15.79; S% 7.23. Found: C% 54.12; H% 4.72; N% 15.71; S% 7.15.
3,545 (OH), 3,436 (NH), 3,062 (CH aromatic), 1649 (CO), 1,600 (C=N); 1H NMR (500 MHz, CDCl3) δ 2.41 (s, 3H, CH3),3.14 (s, 3H, CH3), 3.42–3.45 (m, 2H, 2H-5ʹ), 3.55–3.58 (m, 1H, H-4’), 3.76 (t, J=8.8 Hz, 1H, H-3ʹ), 3.89 (t, J=6.9 Hz, 1H, H-2ʹ), 4.62 (d,1H, J1ʹ-2’ =6.62 Hz, H-1’), 4.93 (s, D2O exch., 1H, 5ʹ-OH), 5.21–5.24 (m, D2O exch., 2H, 2ʹ-OH and 3ʹ-OH), 5.53 (s, D2O exch., 1H,NH), 7.47–7.86 (m, 5H, C6H5). Anal. Calcd. For C20H21N5O5S (443.48): C% 54.17; H% 4.77; N% 15.79; S% 7.23. Found: C% 54.12; H% 4.72; N% 15.71; S% 7.15.
Yield: (3.14 g, 70%) as yellow solid m.p. 188–189°C (EtOH); IR (KBr, cm−1)  3,547 (OH), 3,429 (NH), 3,088 (CH aromatic), 1,641 (CO), 1,593 (C=N); 1H NMR (500 MHz, CDCl3) δ 1.63 (s, 3H, CH3), 2.46 (s,3H, CH3), 3.63–3.65 (m, 2H, H-5ʹ), 4.24-4.27 (m,1H, H-4’), 4.46 (t, J=6.4 Hz, 1H, H-3ʹ ), 4.55 (t, J=9.2 Hz, 1H, H-2ʹ), 4.91 (s, D2O exch., 1H, 5ʹ-OH), 5.01–5.12 (m, D2O exch., 2H, 2ʹ-OH and 3ʹ-OH), 5.24–5.25 (1H, J1ʹ-2’ =10.23 Hz, H-1’), 5.52 (s, D2O exch., 1H,NH), 7.12–7.51 (m, 5H, C6H5); 13C NMR: δ 17.48 (CH3), 38.26 (CH3), 53.64 (7a), 62.33 (C-5ʹ), 67.93 (C-4ʹ), 68.98 (C-3ʹ), 73.96 (C-2ʹ), 81.25 (C-1ʹ), 124.34–136.45 (6C, Ar-C), 161.28 (CO), 164.26 (3a), 166.64 (C-7), 179.15 (C-5). Anal. Calcd. For C18H21N5O5S (419.45): C% 51.54; H% 5.05; N% 16.70; S% 7.64. Found: C% 51.46; H% 5.15; N%16.64; S% 7.60%.
3,547 (OH), 3,429 (NH), 3,088 (CH aromatic), 1,641 (CO), 1,593 (C=N); 1H NMR (500 MHz, CDCl3) δ 1.63 (s, 3H, CH3), 2.46 (s,3H, CH3), 3.63–3.65 (m, 2H, H-5ʹ), 4.24-4.27 (m,1H, H-4’), 4.46 (t, J=6.4 Hz, 1H, H-3ʹ ), 4.55 (t, J=9.2 Hz, 1H, H-2ʹ), 4.91 (s, D2O exch., 1H, 5ʹ-OH), 5.01–5.12 (m, D2O exch., 2H, 2ʹ-OH and 3ʹ-OH), 5.24–5.25 (1H, J1ʹ-2’ =10.23 Hz, H-1’), 5.52 (s, D2O exch., 1H,NH), 7.12–7.51 (m, 5H, C6H5); 13C NMR: δ 17.48 (CH3), 38.26 (CH3), 53.64 (7a), 62.33 (C-5ʹ), 67.93 (C-4ʹ), 68.98 (C-3ʹ), 73.96 (C-2ʹ), 81.25 (C-1ʹ), 124.34–136.45 (6C, Ar-C), 161.28 (CO), 164.26 (3a), 166.64 (C-7), 179.15 (C-5). Anal. Calcd. For C18H21N5O5S (419.45): C% 51.54; H% 5.05; N% 16.70; S% 7.64. Found: C% 51.46; H% 5.15; N%16.64; S% 7.60%.
Acknowledgments
We would like to thank the Egyptian Academy of Scientific Research & Technology (ASRT) Program of “Scientists for Next Generation” for awarding such a grant.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bayrak H, Demirbas A, Karaoglu SA, Demirbas N. Synthesis of some new 1, 2, 4-triazoles, their Mannich and Schiff bases and evaluation of their antimicrobial activities. Eur J Med Chem. 2009;44(3):1057–1066. doi:10.1016/j.ejmech.2008.06.019
2. Moiseev IK, Kon’kov SA, Ovchinnikov KA, et al. Synthesis and antiviral activity of new adamantane derivatives. Pharmaceutical Chemistry Journal. 2012;45(10):588–592.
3. Bornstein BA, Recht A, Connolly JL, et al. Results of treating ductal carcinoma in situ of the breast with conservative surgery and radiation therapy. Cancer. 1991;67(1):7–13.
4. Sağir T, Huysal M, Durmus Z, Kurt BZ, Senel M, Isık S. Preparation and in vitro evaluation of 5-flourouracil loaded magnetite–zeolite nanocomposite(5-FU-MZNC) for cancer drug delivery applications. Biomed Pharmacother. 2016;77:182–190. doi:10.1016/j.biopha.2015.12.025
5. Bourgier C, Levy A, Vozenin MC, Deutsch E. Pharmacological strategies to spare normal tissues from radiation damage: useless or overlooked therapeutics? Cancer Metastasis Rev. 2012;31(3–4):699–712. doi:10.1007/s10555-012-9381-9
6. Sia J, Joon DL, Viotto A, et al. Toxicity and long-term outcomes of dose-escalated intensity modulated radiation therapy to 74Gy for localised prostate cancer in a single Australian centre. Cancers. 2011;3(3):3419–3431. doi:10.3390/cancers3033419
7. van Gijn W, Marijnen CA, Nagtegaal ID, Kranenbarg EMK, Putter H, Wiggers T; Dutch Colorectal Cancer Group. Preoperative radiotherapy combined with total mesorectal excision for resectable rectal cancer: 12-year follow-up of the multicentre, randomised controlled TME trial. Lancet Oncol. 2011;12(6):575–582. doi:10.1016/S1470-2045(11)70097-3
8. Bruheim K, Tveit KM, Skovlund E, et al. Sexual function in females after radiotherapy for rectal cancer. Acta Oncol (Madr). 2010;49(6):826–832. doi:10.3109/0284186X.2010.486411
9. Farrell C, Brearley SG, Pilling M, Molassiotis A. The impact of chemotherapy-related nausea on patients’ nutritional status, psychological distress and quality of life. Supportive Care Cancer. 2013;21(1):59–66. doi:10.1007/s00520-012-1493-9
10. Henry DH, Langer CJ, McKenzie RS, et al. Hematologic outcomes and blood utilization in cancer patients with chemotherapy-induced anemia (CIA) pre-and post-national coverage determination (NCD): results from a multicenter chart review. Supportive Care Cancer. 2012;20(9):2089–2096. doi:10.1007/s00520-011-1318-2
11. Qi F, Li A, Inagaki Y, et al. Chinese herbal medicines as adjuvant treatment during chemoor radio-therapy for cancer. Biosci Trends. 2010;4:6.
12. Yallapu MM, Gupta BK, Jaggi M, Chauhan SC. Fabrication of curcumin encapsulated PLGA nanoparticles for improved therapeutic effects in metastatic cancer cells. J Colloid Interface Sci. 2010;351(1):19–29. doi:10.1016/j.jcis.2010.05.022
13. Kankala RK, Zhang YS, Wang SB, Lee CH, Chen AZ. Supercritical fluid technology: an emphasis on drug delivery and related biomedical applications. Adv Healthc Mater. 2017;6(16):1700433.
14. Park JH, Saravanakumar G, Kim K, Kwon IC. Targeted delivery of low molecular drugs using chitosan and its derivatives. Adv Drug Deliv Rev. 2010;62(1):28–41. doi:10.1016/j.addr.2009.10.003
15. Kumar SSD, Mahesh A, Mahadevan S, Mandal AB. Synthesis and characterization of curcumin loaded polymer/lipid based nanoparticles and evaluation of their antitumor effects on MCF-7 cells. Biochim et Biophys Acta (BBA)-Gen Subj. 2014;1840(6):1913–1922. doi:10.1016/j.bbagen.2014.01.016
16. Kankala RK, Lin XF, Song HF, et al. Supercritical fluid-assisted decoration of nanoparticles on porous microcontainers for codelivery of therapeutics and inhalation therapy of diabetes. ACS Biomater Sci Eng. 2018;4(12):4225–4235.
17. Xu PY, Kankala RK, Pan YJ, Yuan H, Wang SB, Chen AZ. Overcoming multidrug resistance through inhalable siRNA nanoparticles-decorated porous microparticles based on supercritical fluid technology. Int J Nanomedicine. 2018;13:4685. doi:10.2147/IJN.S177627
18. Zhu L, Ma J, Jia N, Zhao Y, Shen H. Chitosan-coated magnetic nanoparticles as carriers of 5-fluorouracil: preparation, characterization and cytotoxicity studies. Colloids Surf B. 2009;68(1):1–6. doi:10.1016/j.colsurfb.2008.07.020
19. Arias JL, López-Viota M, Gallardo V, Adolfina Ruiz M. Chitosan nanoparticles as a new delivery system for the chemotherapy agent tegafur. Drug Dev Ind Pharm. 2010;36(6):744–750. doi:10.3109/03639040903517914
20. Liu CG, Kankala RK, Liao HY, Chen AZ, Wang SB. Engineered pH‐responsive Hydrazone‐carboxylate complexes‐encapsulated 2D matrices for Cathepsin‐mediated apoptosis in cancer. J Biomed Mater Res Part A. 2019. doi:10.1002/jbm.a.36610
21. Rajan M, Raj V. Potential drug delivery applications of chitosan based nanomaterials. Int Rev Chem Eng. 2013;5(2):145–155.
22. Calvo P, Remuñán-López C, Vila-Jato JL, Alonso MJ. Novel hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers. J Appl Polym Sci. 1997;63(1):125–132. doi:10.1002/(SICI)1097-4628(19970103)63:1<125::AID-APP13>3.0.CO;2-4
23. Nagarwal RC, Kumar R, Pandit JK. Chitosan coated sodium alginate–chitosan nanoparticles loaded with 5-FU for ocular delivery: in vitro characterization and in vivo study in rabbit eye. Eur J Pharm Sci. 2012;47(4):678–685. doi:10.1016/j.ejps.2012.08.008
24. David KI, Jaidev LR, Sethuraman S, Krishnan UM. Dual drug loaded chitosan nanoparticles—sugar-coated arsenal against pancreatic cancer. Colloids surf B. 2015;135:689–698. doi:10.1016/j.colsurfb.2015.08.038
25. Agnihotri SA, Mallikarjuna NN, Aminabhavi TM. Recent advances on chitosan-based micro-and nanoparticles in drug delivery. J controlled release. 2004;100(1):5–28. doi:10.1016/j.jconrel.2004.08.010
26. Shu XZ, Zhu KJ. The influence of multivalent phosphate structure on the properties of ionically cross-linked chitosan films for controlled drug release. Eur J Pharm Biopharm. 2002;54(2):235–243.
27. Berger J, Reist M, Mayer JM, Felt O, Peppas NA, Gurny R. Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications. Eur J Pharm Biopharm. 2004;57(1):19–34.
28. Elgemeie G, Altalbawy F, Alfaidi M, Azab R, Hassan A. Synthesis, characterization, and antimicrobial evaluation of novel 5-benzoyl-N-substituted amino-and 5-benzoyl-N-sulfonylamino-4-alkylsulfanyl-2-pyridones. Drug Des Devel Ther. 2017;11:3389. doi:10.2147/DDDT.S149615
29. Azzam RA, Elgemeie GH, Ramadan R, Jones PG. Crystal structure of 2-cyano-3, 3-bis (ethylsulfanyl)-No-tolylacrylamide. Acta Crystallogr Sect E. 2017;73(5):752–754. doi:10.1107/S2056989017005783
30. Azzam RA, Elgemeie GH, Elsayed RE, Jones PG. Crystal structure of N′-[2-(benzo [d] thiazol-2-yl) acetyl]-4-methylbenzenesulfonohydrazide. Acta Crystallogr Sect E. 2017;73(7):1041–1043. doi:10.1107/S2056989017008738
31. Azzam RA, Elgemeie GH, Elsayed RE, Jones PG. Crystal structure of N-[6-amino-5-(benzo [d] thiazol-2-yl)-3-cyano-4-methylsulfanyl-2-oxo-1, 2-dihydropyridin-1-yl]-4-methylbenzenesulfonamide dimethylformamide monosolvate. Acta Crystallogr Sect E. 2017;73(12):1820–1822. doi:10.1107/S2056989017015778
32. Abdallah AE, Elgemeie GH. Design, synthesis, docking, and antimicrobial evaluation of some novel pyrazolo [1, 5-a] pyrimidines and their corresponding cycloalkane ring-fused derivatives as purine analogs. Drug Des Devel Ther. 2018;12:1785. doi:10.2147/DDDT.S159310
33. Elgemeie G, Fathy N, Zaghary W, Farag A. S-glycosides in medicinal chemistry: novel synthesis of cyanoethylene thioglycosides and their pyrazole derivatives. Nucleosides. Nucleotides Nucleic Acids. 2017;36(3):198–212. doi:10.1080/15257770.2016.1257807
34. Elgemeie GH, Jones PG. Crystal structure of 1-amino-2-oxo-2, 5, 6, 7, 8, 9-hexahydro-1H-cyclohepta [b] pyridine-3-carbonitrile. Acta Crystallogr E Crystallogr Commun. 2016;72(9):1239–1241.
35. Elgemeie GH, Salah AM, Abbas NS, Hussein HA, Mohamed RA. Nucleic acid components and their analogs: design and synthesis of novel cytosine thioglycoside analogs. Nucleosides. Nucleotides Nucleic Acids. 2017;36(2):139–150. doi:10.1080/15257770.2016.1231318
36. Elgemeie GH, Farag AB. Design, synthesis, and in vitro anti-hepatocellular carcinoma of novel thymine thioglycoside analogs as new antimetabolic agents. Nucleosides Nucleotides Nucleic Acids. 2017;36(5):328–342.
37. Elgemeie GH, Fathy NM, Farag AB, Alkursani SA. Antimetabolites: design, synthesis, and cytotoxic evaluation of novel dihydropyridine thioglycosides and pyridine thioglycosides. Nucleosides Nucleotides Nucleic Acids. 2017;36(5):355–377.
38. Elgemeie GH, Abu-Zaied MA. Heterocyclic thioglycosides in carbohydrate research: synthesis of thiophene thioglycosides. Nucleosides. Nucleotides Nucleic Acids. 2017;36(8):511–519. doi:10.1080/15257770.2017.1332371
39. Masoud DM, Hammad SF, Elgemeie GH, Jones PG. Crystal structure of 4, 6-dimethyl-2-{[3, 4, 5-trihydroxy-6-(hydroxymethyl) tetrahydro-2H-pyran-2-yl] sulfanyl} nicotinonitrile. Acta Crystallogr Sect E. 2017;73(11):1751–1754. doi:10.1107/S2056989017015213
40. Abu-Zaied MA, Elgemeie G. Thiazoles in glycosylation reactions: novel synthesis of thiazole thioglycosides. Heteroat Chem. 2017;28(6):e21404. doi:10.1002/hc.21404
41. Abu-Zaied MA, Elgemeie GH. Synthesis of the first novel pyrazole thioglycosides as deaza ribavirin analogues. Nucleosides Nucleotides Nucleic Acids. 2017;36(12):713–725. doi:10.1080/15257770.2017.1378817
42. Abu-Zaied MA, Elgemeie GH. A facile synthesis of novel pyrazolopyrimidine thioglycosides as purine thioglycoside analogues. Nucleosides. Nucleotides Nucleic Acids. 2018;37(1):67–77. doi:10.1080/15257770.2017.1419254
43. Elgemeie GH, Abu-Zaied MA, Nawwar GA. First novel synthesis of triazole thioglycosides as ribavirin analogues. Nucleosides Nucleotides Nucleic Acids. 2018;37(2):112–123. doi:10.1080/15257770.2017.1423079
44. Elgemeie GH, Fathy NM, Farag AB, Alkhursani SA. Design, synthesis, molecular docking and anti-hepatocellular carcinoma evaluation of novel acyclic pyridine thioglycosides. Nucleosides Nucleotides Nucleic Acids. 2018;37(3):186–198. doi:10.1080/15257770.2018.1450508
45. Hammad SF, Masoud DM, Elgemeie GH, Jones PG. Crystal structure of racemic 2-[(β-arabinopyranosyl) sulfanyl]-4, 6-diphenylpyridine-3-carbonitrile. Acta Crystallogr Sect E. 2018;74(6):853–856. doi:10.1107/S2056989018007284
46. Elgemeie GH, Elzanate AM, Elghandour AH, Ahmed SA. Novel intramolecular cyclization of pyrazolone ketene S, N-acetals for the construction of methylsulfanylpyrazolo-[4, 3-b] pyridines. Synth Commun. 2002;32(22):3509–3517. doi:10.1081/SCC-120014787
47. Davis BG. Recent developments in oligosaccharide synthesis. J Chem Soc Perkin Tran. 2000;1(14):2137–2160. doi:10.1039/a809774g
48. Ferrier RJ, Hay RW, Vethaviyasar N. A potentially versatile synthesis of glycosides. Carbohydr Res. 1973;27(1):55–61. doi:10.1016/S0008-6215(00)82424-6
49. Yoksan R, Jirawutthiwongchai J, Arpo K. Encapsulation of ascorbyl palmitate in chitosan nanoparticles by oil-in-water emulsion and ionic gelation processes. Colloids Surf B. 2010;76(1):292–297. doi:10.1016/j.colsurfb.2009.11.007
50. Jingou J, Shilei H, Weiqi L, Danjun W, Tengfei W, Yi X. Preparation, characterization of hydrophilic and hydrophobic drug in combine loaded chitosan/cyclodextrin nanoparticles and in vitro release study. Colloids Surf B. 2011;83(1):103–107. doi:10.1016/j.colsurfb.2010.11.005
51. Alkhader E, Billa N, Roberts CJ. Mucoadhesive chitosan-pectinate nanoparticles for the delivery of curcumin to the colon. AAPS PharmSciTech. 2017;18(4):1009–1018. doi:10.1208/s12249-016-0623-y
52. Fan W, Yan W, Xu Z, Ni H. Formation mechanism of monodisperse, low molecular weight chitosan nanoparticles by ionic gelation technique. Colloids and Surfaces B. Biointerfaces. 2012;90:21–27. doi:10.1016/j.colsurfb.2011.09.042
53. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. doi:10.1158/0008-5472.CAN-09-1947
54. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621–681. doi:10.1124/pr.58.3.10
55. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55.
56. Chou TC. Assessment of synergistic and antagonistic effects of chemotherapeutic agents in vitro. Contríb Gynecol Obstet. 1994;19:91–107.
57. Siddique YH, Ara G, Beg T, Gupta J, Afzal M. Assessment of cell viability, lipid peroxidation and quantification of DNA fragmentation after the treatment of anticancerous drug mitomycin C and curcumin in cultured human blood lymphocytes. Exp Toxicol Pathol. 2010;62(5):503–508. doi:10.1016/j.etp.2009.06.010
58. Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119(2):203–210.
59. Loutfy SA, El-Din HMA, Elberry MH, Allam NG, Hasanin MTM, Abdellah AM. Synthesis, characterization and cytotoxic evaluation of chitosan nanoparticles: in vitro liver cancer model. Adv Nat Sci. 2016;7(3):035008.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.