")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Novel pharmacologic treatment in acute binge eating disorder – role of lisdexamfetamine
Authors Guerdjikova AI, Mori N, Casuto LS, McElroy S
Received 9 January 2016
Accepted for publication 22 February 2016
Published 18 April 2016 Volume 2016:12 Pages 833—841
DOI https://doi.org/10.2147/NDT.S80881
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Anna I Guerdjikova,1,2 Nicole Mori,1,2 Leah S Casuto,1,2 Susan L McElroy1,2
1Lindner Center of HOPE, Mason, OH, USA; 2Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati College of Medicine, Cincinnati, OH, USA
Abstract: Binge eating disorder (BED) is the most common eating disorder and an important public health problem. It is characterized by recurrent episodes of excessive food consumption accompanied by a sense of loss of control over the binge eating behavior without the inappropriate compensatory weight loss behaviors of bulimia nervosa. BED affects both sexes and all age groups and is associated with medical and psychiatric comorbidities. Until recently, self-help and psychotherapy were the primary treatment options for patients with BED. In early 2015, lisdexamfetamine dimesylate, a prodrug stimulant marketed for attention deficit hyperactive disorder, was the first pharmacologic agent to be approved by the US Food and Drug Administration for the treatment of moderate or severe BED in adults. This article summarizes BED clinical presentation, and discusses the pharmacokinetic profile, efficacy, and safety of lisdexamfetamine dimesylate in the treatment of BED in adults.
Keywords: binging, overeating, Vyvanse, stimulant, approved medication
Introduction to binge eating disorder (BED) – management challenges
BED is a newly recognized clinical entity in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5)1 and an important public health problem worldwide. BED is the most common eating disorder and recent data from the World Health Organization Mental Survey Study, which included community surveys of 24,124 adult respondents across 14 countries on four continents, found a lifetime prevalence rate of BED of 1.4%.2 In the USA, the lifetime prevalence of BED has been estimated to be 2.6%.2,3
The DSM-5 diagnostic criteria for BED, including indicators for severity, are listed in Table 1. BED is characterized by recurrent episodes of binge eating (BE), defined as eating in a discrete period of time (~2 hours) an amount of food larger than most people would eat under similar circumstances, and having a sense of loss of control over the eating, without the inappropriate compensatory behaviors of bulimia nervosa (BN), for example, purging, vomiting, or excessive use of diuretics or laxatives. The BE episodes occur on average at least once a week for 3 consecutive months, and are associated with feelings of guilt, depression, or distress. Patients with BED often eat in secrecy; they are embarrassed by the BE behavior and their perceived inability to control the urges to overeat. During a BE episode, the patient might eat more rapidly than normal, eat until feeling uncomfortably full, or eat large amounts of food when not feeling physically hungry.
 | Table 1 DSM-5 criteria for binge eating disorder (BED) |
BED co-occurs with a plethora of psychiatric disorders, most commonly with mood and anxiety disorders.4 Indeed, approximately four out of five adults with lifetime BED have at least one comorbid psychiatric disorder, and approximately one out of two adults has three or more comorbid psychiatric disorders.3
Obesity and its complications are among the medical comorbidities associated with BED. Growing evidence suggests that BED may independently increase the risk of development of certain components of metabolic syndrome, like diabetes, hypertension, and dyslipidemia, over and above the risk attributable to obesity alone.5 Preliminary data indicate that the cardiovascular system, reproductive system, and cortisol response might also be affected in BED patients.6
Psychological interventions have been recommended as first-line treatment for BED and are supported by meta-analytic reviews.7 Cognitive behavior psychotherapy, interpersonal therapy, and structured self-help, based mainly on cognitive behavioral techniques, are effective for reducing BE symptoms and associated psychopathology but not for weight loss.8 Various classes of medications, including antidepressants, antiepileptic drugs, antiobesity drugs, and medications approved for attention deficit hyperactive disorder (ADHD), have been tested in randomized, placebo-controlled trials in BED, and found helpful in improving BE behavior and eating related psychopathology.9 All medications used for BED until 2015 had limitations related to their efficacy or adverse event (AE) profiles. Additionally, they were prescribed “off label” and their use in general practice was limited.
On January 30, 2015, lisdexamfetamine dimesylate (LDX) received approval from the US Food and Drug Administration (FDA) for the treatment of moderate to severe BED in adults. LDX is the only medication currently approved for the treatment of BED10 and the second medication approved for the treatment of any eating disorder, after fluoxetine was approved for BN in 1997. The review below will summarize the pharmacology of LDX, the rationale for use of LDX in BED, and tolerability of LDX in BED. The clinical use of LDX in BED will also be discussed.
Pharmacology, mode of action, and pharmacokinetics of LDX
LDX (abbreviation from l-lysine-dextroamphetamine) is a novel prodrug of dextroamphetamine (d-amphetamine) covalently linked to the amino acid l-lysine. LDX itself is pharmacologically inactive and metabolized to d-amphetamine by a unique mechanism involving an enzymatic process predominantly associated with the red blood cells.
The pharmacology of d-amphetamine is complex and has been documented since the early 20th century when preparations containing d-amphetamine were used in World War II as a “go pill” to promote alertness and focus in Air Force pilots.11 In vitro, d-amphetamine is a moderately potent inhibitor of DAT, NET, and VMAT2, with much weaker affinity for the SERT. D-amphetamine is also a weak MAO inhibitor. The net effect of these multiple activities in vivo is increased catecholamine availability in the extracellular space.12
LDX (Vyvanse®) was developed by New River Pharmaceuticals (Radford, VA, USA) in the late 1990s with the intention of creating a longer-lasting formulation of d-amphetamine with lower abuse potential. New River Pharmaceuticals were bought by Shire Inc. (Dublin, Ireland) in 2007, a few months before LDX was marketed in adult ADHD.
Pharmacodynamics and pharmacokinetics
LDX has high aqueous solubility, low lipophilicity, and is rapidly absorbed intact after oral administration in animals and humans, attaining maximum plasma concentrations (Cmax) at 0.25–3 hours. It is inactive at receptors, transporters, and enzymes in vitro and its absorption occurs via an active transport process. Following absorption, LDX is hydrolyzed by peptidases associated with red blood cells to release the active drug, d-amphetamine, and a naturally occurring amino acid, l-lysine. Red blood cells have a high capacity for the metabolism of LDX to d-amphetamine, and substantial hydrolysis occurs even at low hematocrit levels.
LDX is not metabolized by CYP enzymes and does not cross the blood–brain barrier. Its metabolism is restricted to the formation of d-amphetamine and l-lysine. d-Amphetamine and its metabolites are eliminated largely in urine, with small amounts excreted in feces and bile.13
Indication
In the USA, LDX (Vyvanse®) received FDA approval for the treatment of ADHD in children in 2007, in adults with ADHD in 2008, and for maintenance treatment of adulthood ADHD in 2012. In early 2015, LDX was approved for the treatment of moderate and severe BED in adults. LDX has also been granted approval in Europe for the treatment of ADHD in children, adolescents, and adults. LDX is not marketed for BED in any other country beyond the USA.
Preliminary data suggest LDX might be effective as adjunctive therapy to antipsychotics in adults with clinically stable schizophrenia,14 to mood stabilizers in adults with bipolar depression,15 and to antidepressants in adults with major depressive disorder.16,17 LDX has also demonstrated potential to be an efficacious treatment for patients with multiple sclerosis with cognitive impairment.18
Rationale for LDX in BED
Preclinical, genetic, clinical, and neuroimaging data suggest that BE may involve dysfunction of the dopamine (DA) and norepinephrine (NE) systems. Those systems are important in regulating eating behavior and reward.19,20
BE rodents have lower D2-like DA receptor binding selectivity in the mesoaccumbens DA system.21 Methylphenidate reduced sucrose intake in an animal model of BE.21 A recent study demonstrated that LDX, via its metabolite, d-amphetamine, reduced chocolate binging in rats by 71%, partly by indirect activation of alpha1-adrenoceptors and perhaps D1 receptors.22
Eating disorders characterized by BE have been associated with the hypofunctional short allele of the 3′-UTR VNTR polymorphism of the DA transporter gene.23 Also, BE behavior was found to have a moderate association with the hypofunctional seven-repeat allele of the DA D4 receptor gene and increased maximal lifetime body mass index (BMI) in women with seasonal affective disorder, a condition characterized by overeating, carbohydrate craving, and weight gain.24
Preliminary data suggest that agents like LDX that facilitate DA and/or NE neurotransmission may reduce BE in humans. The selective NE reuptake inhibitor atomoxetine has been shown to reduce BE and body weight in one placebo-controlled study of BED in adults.25 Moreover, there are reports of stimulants reducing BE in patients with BN, a condition closely related to BED.
Neuroimaging studies demonstrated that food stimuli, when administered with methylphenidate to amplify DA signals, significantly increased DA in the caudate and putamen in obese binge eaters but not in obese non-binge eaters, and increases in DA in the caudate were significantly correlated with BE scores.26 Additionally, striatal DA release was significantly associated with the frequency of BE in a controlled positron emission tomography imaging study of 17 subjects with BN.27
In sum, the pharmacologically active d-amphetamine, released from LDX hydrolysis, inhibits reuptake of DA and NE from the synaptic cleft and simultaneously enhances the release of the DA, NE, and serotonin. By regulating these neurotransmitter systems which are involved in regulation of appetite, hunger, and eating behaviors, it was hypothesized that LDX might reduce pathological overeating and be an efficacious treatment for BED.
Efficacy studies of LDX in BED
Shire Inc. sponsored a BED clinical development program that included three randomized, placebo-controlled studies in acute adult BED: an 11-week Phase II proof-of-concept study and two identically designed 12-week Phase III trials. Basic study information, demographics, as well as primary and key secondary measures of the three studies are summarized in Table 2 and are described below.
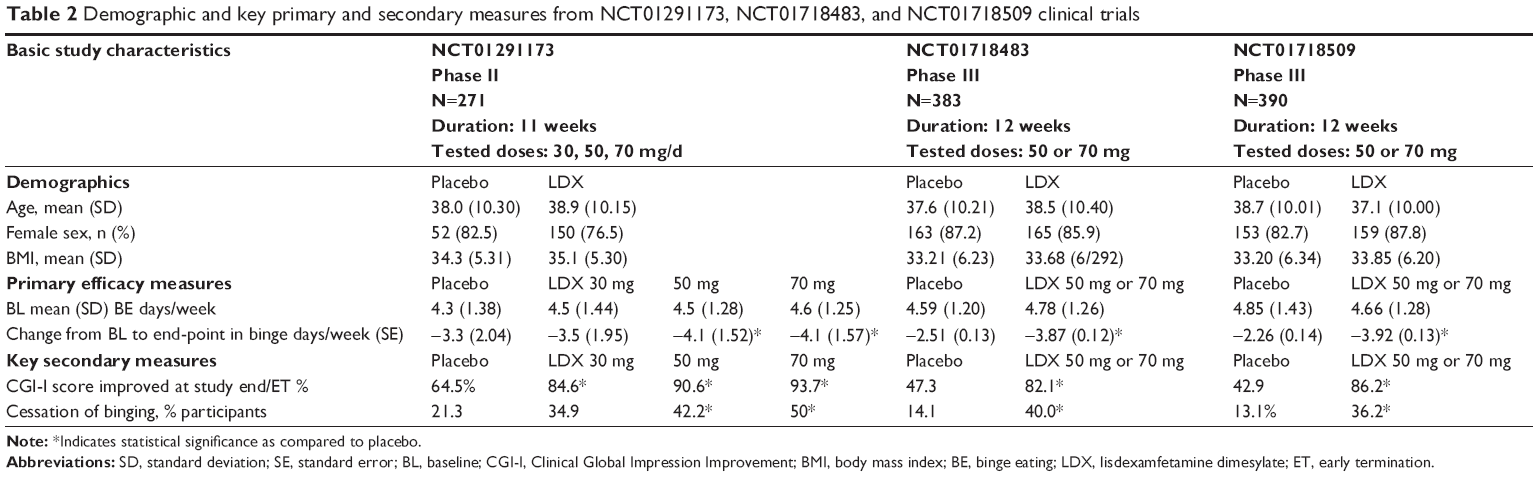 | Table 2 Demographic and key primary and secondary measures from NCT01291173, NCT01718483, and NCT01718509 clinical trials |
Phase II study (NCT01291173)
The Phase II study was an 11-week randomized, placebo-controlled, fixed-dose, parallel-group multicenter trial in 259 adults across 30 sites in the USA, aged 18–55 years, diagnosed with BED, and with a BMI between 25 and 40 kg/m2.28 Eligible subjects were randomized to receive LDX 30, 50 or 70 mg/day or placebo in 1:1:1:1 ratio. Intention-to-treat analyses included 255 subjects. To be randomized, the subjects were required to have moderate to severe BED, defined by experiencing at least three BE days per week for the 2 weeks before the baseline visit and verified with self-report take home diaries and clinical interview. Exclusion criteria included current BN or anorexia nervosa; a lifetime history of bipolar disorder, psychosis, or ADHD; significant clinical depression; use of a psychostimulant within 6 months of screening; a recent history of suspected substance abuse; or a lifetime history of psychostimulant abuse. Psychological or weight loss interventions initiated within 3 months of screening and history of diabetes or cardiovascular disease that might increase vulnerability to the sympathomimetic effects of stimulants were also exclusionary. Subjects with mild well-controlled hypertension on single antihypertensive agent were allowed in the study. The following medications were exclusionary: hypnotics, anxiolytics, antipsychotics, antidepressants, NE reuptake inhibitors, mood stabilizers, herbal preparations, and agents with weight-changing properties (eg, orlistat, topiramate, zonisamide, and antihistamines). After randomization, all treatment groups were initiated at the 30 mg/day dosage. Patients randomized to 50 or 70 mg/day were force-titrated weekly in increments of 20 mg/day to their assigned dosage. The 3-week forced-dose titration period was followed by an 8-week dose-maintenance period during which dose reductions were not permitted.
The primary efficacy measure was the number of BE days per week. A BE day was defined as a day when at least one BE episode occurred. BE episodes were recorded by subjects in a self-report diary and were confirmed during clinical interview with trained clinicians. A hierarchical testing procedure in descending order of LDX dosage was used for pairwise testing between LDX and placebo on the primary end-point measures because it was hypothesized that higher LDX dosage was more likely to be efficacious than lower dosages. Secondary efficacy measures included the number of BE episodes per week, 1-week BE episode response status, and 4-week cessation from BE (defined as no BE episodes in the last 4 weeks of double-blind treatment). Additional secondary measures included assessments of clinical global impression improvement, measured with the Clinical Global Impression – Improvement scale (CGI-I);29 BE psychopathology, measured with Three-Factor Eating Questionnaire (TFEQ);30 BE pathology as measured with the BE Scale (BES);31 obsessive–compulsive features of BED, measured with Yale Brown Obsessive Compulsive Scale modified for BE (YBOCS-BE);32 impulsivity, measured with version 11 of the Barratt Impulsiveness Scale (BIS-11);33 participant perceived quality of life, measured with 12-item Short Form Health Survey;34 depressive symptoms, measured with Montgomery-Asberg Depression Rating Scale,35 and anxiety symptoms, measured with Hamilton Anxiety Rating Scale.36 Safety assessments included treatment emergent AEs, suicidality assessment, vital signs, laboratory test results, electrocardiography, and body weight.
Seventy-eight percent of subjects completed the double-blind phase of the study. The primary efficacy end-point (least squares mean change from baseline to week 11 on log-transformed BE days per week) was significantly decreased in the 50 and 70 mg/day treatment groups, but not in the 30 mg/day treatment group compared with the placebo group. In the LDX 50 mg/day group, binge days/week (standard deviation [SD]) decreased from 4.5 (1.28) to 0.4 (0.86) (P=0.008); in the LDX 70 mg/day group binge days/week (SD) decreased from 4.6 (1.25) to 0.5 (1.25) (P<0.001). Similarly, in the 50 and 70 mg/day LDX groups, significant decreases in BE episodes per week and in measures of 1-week response status; as well as 4-week BE cessation were observed. Significant improvement in severity of illness (Clinical Global Impression Severity [CGI-S]) as compared to placebo was demonstrated at all LDX doses. The 30 mg dose was superior to placebo for the following measures: BES (P≤0.03), TFEQ restraint (P=0.04), disinhibition (P=0.03), and hunger subscales (P=0.02), and Y-BOCS-BE (all P=0.01). The aggregate change in the least squared mean score for the 12-item Short Form Health Survey Physical Health Component Summary was significant only for the 70 mg/day treatment group compared with the placebo group, and not for the other treatment groups. In this study, weight was assessed as a safety variable and the mean (SD) change in body weight was −3.1 kg (3.3), −4.9 kg (5.2), −4.9 kg (4.09), and −0.1 kg (3.09) for the 30, 50, and 70 mg/day LDX treatment groups and placebo, respectively.
An additional report provided descriptions of LDX effects on TFEQ, BES, Y-BOCS-BE, and BIS-11. Week 11 least squares mean treatment differences favored all LDX doses over placebo on the BES (P≤0.03), TFEQ disinhibition and hunger subscales (all P<0.05), Y-BOCS-BE total, obsessive, and compulsive scales (all P≤0.02) and on BIS-11 total score at 70 mg/day LDX (P=0.015), and the TFEQ cognitive restraint subscale at 30 and 70 mg/day LDX (both P<0.05). It was concluded that LDX decreased global BE severity and obsessive–compulsive and impulsive features of BED in addition to BE days.37
Phase III studies (NCT01718483 and NCT01718509)
The Phase III trials were two 12-week, randomized, placebo-controlled, parallel-group, dose-optimization, and multicenter studies (NCT01718483, referred to as study 1 hereafter; and NCT01718509, referred as study 2 hereafter).38 The two studies used identical designs and methods and were performed across 93 unique sites in the USA, Sweden, Germany, and Spain. A total of 773 subjects were enrolled across placebo (N=386) and LDX (N=387) treatment groups. In study 1, 187 subjects were randomized to placebo and 192 subjects were randomized to LDX; in study 2, 185 subjects were randomized to placebo and 181 subjects were randomized to LDX. The inclusion criteria were similar to those in the Phase II study with the following exceptions: at both screening and baseline, eligible participants had to have BMI ≥18 and ≤45 kg/m2 and BED severity was confirmed not only with a binge day frequency of 3 or more binge days/week for the 2 weeks between screening and baseline, but also with a CGI-S score of ≥4 (indicating at least a moderate severity of illness) at screening and baseline visits. Exclusion criteria different than the ones in the Phase II study included use of psychostimulants for fasting or dieting for BED ≤6 months before screening; and resting average sitting systolic blood pressure >139 mmHg or average diastolic blood pressure >89 mmHg at screening or baseline visits. Eligible subjects were randomized 1:1 to LDX 30 mg/day or placebo. The dose of study drug was titrated during the dose-optimization phase (weeks 1–4); 30 mg/day was increased to 50 mg/day after a 7-day period. After 7–14 days the dose was titrated up to 70 mg/day based on clinical need and tolerability. A single down-titration from 70 to 50 mg was allowed during the dose-optimization phase. After the 4th week of treatment, subjects continued with their established dose for the duration of the 8-week dose-maintenance period.
Similar to the Phase II study, the primary efficacy measure was the number of binge days per week obtained from the participants’ BE diaries and confirmed with clinician interview. Key secondary measures included CGI-I response at week 12/ET, 4-week binge cessation at week 12/ET, YBOCS-BE score, and change from baseline to week 12/early termination (ET) in body weight and fasting triglyceride levels. Hierarchical testing procedures were used, with statistical assessments made in the following order based on clinical importance: changes in BE days/week, CGI-I, 4-week BE cessation, percentage body weight changes, Y-BOCS-BE total score changes, and triglyceride changes. A later test was only significant if all earlier tests were significant. Safety assessments included treatment emergent AEs, suicidality assessment, amphetamine cessation symptom assessment, vital signs, laboratory test results, and electrocardiography.
Seventy-nine percent of subjects treated with LDX (study 1, N=158 of 192 and study 2, N=147 of 195 enrolled) and 79% of subjects treated with placebo (study 1, N=157 of 191 and study 2, N=147 of 195 enrolled) completed the double-blind phase.
The primary efficacy end-point (least squares mean change from baseline to week 12 on log-transformed BE days per week) was significantly decreased in the LDX treatment groups compared with the placebo in both studies (−2.51 [0.125] with placebo and −3.87 [0.124] with LDX in study 1, P<0.001, and −2.26 [0.137] with placebo, and −3.92 [0.135] with LDX in study 2, P<0.001).
All secondary measures showed significant improvement. Reduction in 4-week BE cessation at study end was observed in 38% of LDX treated subjects (40% in study 1 and 36.2% in study 2) compared to 13% in the placebo group (14.1% in study 1 and 13.1% in study 2). CGI-I (P<0.001 for both studies) and YBOCS-BE score change at study end (P<0.001 for both studies) also showed statistically significant treatment effect favoring LDX. Significant reductions in triglyceride levels were observed (P<0.001, effect size 1.03 for study 1 and P=0.002, effect size 1.11 for study 2). Percent change in weight from baseline in study 1 was −6.25% for the LDX group vs +0.11% for the placebo group (P<0.001, effect size 1.64). Percent change in weight from baseline in study 2 was −5.57% for the LDX group vs −0.15% for the placebo group (P<0.001, effect size 1.22).
Exploratory end-points included assessment of disability with the Sheehan Disability Scale39 and health-related quality of life (with the EuroQoL 5-dimension 5-level questionnaire).40 Post hoc analyses were performed to determine the relationship between LDX therapy and disability, BE days per week and disability, and BE episodes per week and disability. Results indicated that LDX therapy had a positive effect on Sheehan Disability Scale scores. Reduction in BE days per week and BE episodes per week was associated with improvement in disability over 12 weeks.41 LDX also had a positive effect on health-related quality of life which was indirect and mediated in part by LDX effects on BE frequency, disability, and daily functioning.42
Limitations of Phase II and Phase III studies
In all three studies, participants were mainly women, white, overweight, or obese, and by design did not have any current psychiatric comorbidities or cardiovascular conditions. Generalizing the results to a more heterogeneous population of individuals with BED warrants caution. Potential sex differences in LDX efficacy in BED patients were not specifically explored, but the percentage of male subjects across the studies was low. Additionally, the studies were relatively short in duration and this limits extrapolations to the long-term efficacy, tolerability, and safety of LDX in individuals with BED. Ongoing studies are addressing these issues.
Safety and tolerability
LDX is contraindicated in patients with known hypersensitivity to amphetamine products or other ingredients of LDX. Anaphylactic reactions, Stevens-Johnson Syndrome, angioedema, and urticaria have been observed in post-marketing reports. LDX should not be administered along with MAO inhibitors or within 14 days of the last MAO inhibitor dose as hypertensive crisis can occur. In the USA, LDX is categorized as a Schedule II medication by the Drug Enforcement Administration. Schedule II encompasses medications from various classes with high abuse potential. The LDX prescribing insert contains a boxed warning informing about risk of abuse and dependence. The box urges for the risk of abuse to be assessed prior to prescribing, and to further monitor for signs of abuse and dependence while on therapy. Per LDX prescribing insert, the most frequent adverse reactions leading to LDX discontinuation at a rate at least twice that of placebo in adults with ADHD were insomnia (2%), tachycardia (1%), irritability (1%), hypertension (1%), headache (1%), anxiety (1%), and dyspnea (1%). Adverse reactions reported by 3% or more of adult patients with ADHD taking LDX and at least twice the incidence compared to patients taking placebo included decreased appetite, insomnia, dry mouth, diarrhea, nausea, anxiety, anorexia, feeling jittery, agitation, increased blood pressure, hyperhidrosis, restlessness, and decreased weight. LDX is pregnancy category C and should be prescribed only if potential benefits justify the potential risk to the fetus.
In the Phase II BED study, discontinuing rate due to AEs was 3.1%, and due to serious AEs was 1.5%. AEs reported by >10% of subjects in the LDX treatment group and at a rate greater than placebo included dry mouth, decreased appetite, headache, and insomnia. One subject died because of toxicology findings consistent with a methamphetamine overdose and this event was not considered related to the study drug. Mean (SD) changes in pulse and blood pressure measurement from baseline to study end were observed in the LDX treated group (increase of 3.8 [11.75] bpm for pulse rate and 0.1 [9.85] mmHg for systolic blood pressure; diastolic blood pressure decreased with treatment, −0.7 [7.32]). No clinically meaningful trends were observed for clinical laboratory results or electrocardiography interval data.
In the Phase III BED studies, discontinuation rate due to AEs was 6.3% for study 1 and 3.9% for study 2 for LDX group as compared with 2.7% in study 1 and 2.2% in study 2 in the placebo group, respectively. AEs reported by >10% of subjects in the LDX treatment group and at a rate greater than placebo included dry mouth, headache, and insomnia. No deaths occurred in either study. Other serious AEs were rare and with similar incidence in LDX and placebo groups. Minimal increases in pulse and blood pressure from baseline to study end were observed in the LDX treated group (4.41–6.31 bpm for pulse rate, 0.2–1.45 mmHg for systolic blood pressure, and 1.06–1.83 mmHg for diastolic blood pressure). No clinically meaningful trends were observed for clinical laboratory results or electrocardiography interval data.
The safety profile of LDX in adults with moderate to severe BED was consistent with data from ADHD studies. No suicidality or misuse was reported across the three trials.
Administration and optimal dose
LDX is available as 10, 20, 30, 40, 50, 60, and 70 mg capsules. It is indicated for the treatment of ADHD and moderate to severe BED, but not for weight loss. The prescribing information warns that the safety and effectiveness of LDX for the treatment of obesity have not been established. LDX is to be taken by mouth in the morning with or without food. Afternoon doses are to be avoided because of risk for insomnia.
The recommended starting dose in BED treatment is 30 mg/day to be titrated in increments of 20 mg at approximately weekly intervals to achieve the recommended target dose of 50 to 70 mg/day. The maximum dose is 70 mg/day. An adequate trial is 11 to 12 weeks or 50–70 mg for 8 weeks. The medication should be discontinued if there is no improvement.
Patient focused perspectives
BED is an unrecognized and undertreated condition. Indeed, recent data from an international survey indicated that less than 10% of respondents with BED received treatment for their eating disorder within the last year.2 Patients rarely spontaneously disclose BE symptoms because of embarrassment or shame. BE behavior is often overlooked and treatment commonly focuses on obesity and its complications as the presenting problem.
In routine clinical practice, the administration of a brief self-report measure like the SCOFF or the Eating Attitudes Test might assist the diagnostic process if BED is suspected.43 Shire Inc. has developed a validated self-report instrument (Binge Eating Disorder Screener-7 [BEDS-7]), that consists of seven “yes” or “no” questions and is available free of charge (https://www.bingeeatingdisorder.com/hcp/content/media/BingeEatingDisorder_Screener.pdf) on BED informational portal. The BED informational portal supported by Shire Inc. (https://www.bingeeatingdisorder.com/) also provides current information on clinical characteristics and functional consequences of BED along with expert videos discussing the illness and helpful links for further self-education. Additional resources to assist in patients’ screening, diagnostics, psychoeducation, and treatment can be found on Binge Eating Disorder Association (bedaonline.com), Alliance for Eating Disorders Awareness (www.allianceforeatingdisorders.com), and National Eating Disorder Association (www.nationaleatingdisorders.org) websites.
Patients can be offered self-help tools or psychotherapy as first-line of treatment, especially if BED symptomatology appears to be mild. Numerous applications (apps) for mobile devices have been developed in the recent years as self-help tools or to enhance treatment in eating disorders in general,44 and in BED in particular.45 Most of the currently available eating disorders-focused apps provide means of regular self-assessment and real-time monitoring of eating habits. As the apps’ functionalities grow, there is the possibility that in the future some could deliver entire personalized BED-focused interventions. In moderate and severe BED cases, pharmacotherapy can be considered monotherapy or as adjunct to psychoeducation and psychological interventions. Importantly, patient preference needs to be considered when making treatment decisions.
LDX is the first medication to receive regulatory approval for the treatment of BED in the world. It is specifically approved for moderate and severe BED in adults at 50–70 mg/day. LDX dosed at 50 or 70 mg significantly reduced BE symptoms as measured by weekly binge day frequency as well as improved obsessive compulsive features associated with the BE behaviors, and had a positive effect on disability. LDX is not approved as weight loss medication and thorough assessment of the binging behavior with clinical interview and/or review of food logs and self-report measures on eating pathology is paramount in making the correct diagnosis and further guiding treatment. In adult BED patients, LDX was generally well tolerated. No studies comparing LDX with other psychological and pharmacological treatments in BED have as yet been conducted. Therefore, no comments can be made about the relative efficacy and the tolerability of LDX as compared to self-help treatment, cognitive behavior psychotherapy, interpersonal therapy, antidepressants, antiepileptic, or any obesity drugs. Data are lacking regarding the efficacy and tolerability of LDX in adults with mild BED, youth, or elderly, and in BED patients with certain comorbid conditions such as mood, anxiety, or substance use disorders; clinically significant or unstable hypertension; cardiovascular disease, or diabetes. It might not be appropriate in adults with BED who also have bipolar disorder as it might exacerbate manic symptoms. However, in an 8-week placebo-controlled study of adjunctive LDX in bipolar depression, LDX was associated with significant improvement in BE, measured with BES, and no adverse psychiatric effects were observed.15 LDX should not be prescribed in BED if drug or alcohol abuse is suspected because of its abuse potential, and in uncontrolled hypertension or cardiovascular disease.
Long-term studies are essential to extend the results of the Phase II and Phase III studies. It would be of interest to examine LDX efficacy in mild BED and in adolescents as well as in older adults. Validation of BED and approval of the first medication for its treatment mark the beginning of a new era in management of eating disorders in general, and in pharmacotherapy of BED in particular.
Disclosure
Susan L McElroy is a consultant to or member of the scientific advisory boards of Alkermes, Bracket, Corcept, F. Hoffmann-La Roche Ltd., MedAvante, Myriad, Naurex, Novo Nordisk, Shire, Sunovion, and Teva. She is a principal or co-investigator on studies sponsored by the Agency for Healthcare Research & Quality (AHRQ), Alkermes, AstraZeneca, Cephalon, Eli Lilly and Company, Forest, Marriott Foundation, National Institute of Mental Health, Naurex, Orexigen Therapeutics, Inc., Pfizer, Shire, Takeda Pharmaceutical Company Ltd., and Transcept Pharmaceuticals. She is also an inventor on United States Patent No. 6,323,236 B2, Use of Sulfamate Derivatives for Treating Impulse Control Disorders, and along with the patent’s assignee, University of Cincinnati, Cincinnati, Ohio, has received payments from Johnson & Johnson, which has exclusive rights under the patent. The other authors have no conflicts of interest to disclose.
References
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. American Psychiatric Publishing; 2013. | ||
Kessler RC, Berglund PA, Chiu WT, et al. The prevalence and correlates of binge eating disorder in the World Health Organization World Mental Health Surveys. Biol Psychiatry. 2013;73(9):904–914. | ||
Hudson JI, Hiripi E, Pope HG Jr, Kessler RC. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiatry. 2007;61(3):348–358. | ||
Javaras KN, Pope HG, Lalonde JK, et al. Co-occurrence of binge eating disorder with psychiatric and medical disorders. J Clin Psychiatry. 2008;69(2):266–273. | ||
Hudson JI, Lalonde JK, Coit CE, et al. Longitudinal study of the diagnosis of components of the metabolic syndrome in individuals with binge-eating disorder. Am J Clin Nutr. 2010;91(6):1568–1573. | ||
Mitchell JE. Medical comorbidity and medical complications associated with binge-eating disorder. Int J Eat Disord. Epub 2015 Aug 27. | ||
Vocks S, Tuschen-Caffier B, Pietrowsky R, et al. Meta-analysis of the effectiveness of psychological and pharmacological treatments for binge eating disorder. Int J Eat Disord. 2010;43(3):205–217. | ||
McElroy SL, Guerdjikova AI, Mori N, Munoz MR, Keck PE. Overview of the treatment of binge eating disorder. CNS Spectr. 2015;20(6):546–556. | ||
McElroy SL, Guerdjikova AI, Mori N, Keck PE Jr. Psychopharmacologic treatment of eating disorders: emerging findings. Curr Psychiatry Rep. 2015;17(5):35. | ||
US Food and Drug Administration. FDA expands uses of Vyvanse to treat binge-eating disorder [press release]. US Food and Drug Administration; 2015 [January 30]. Available from: http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm432543.htm. Accessed February 24, 2016. | ||
Rasmussen N. Speed. The many lives of amphetamine. New York University Press; 2008. | ||
Hutson PH, Pennick M, Secker R. Preclinical pharmacokinetics, pharmacology and toxicology of lisdexamfetamine: a novel d-amphetamine pro-drug. Neuropharmacology. 2014;87:41–50. | ||
Steer C, Froelich J, Soutullo CA, Johnson M, Shaw M. Lisdexamfetamine dimesylate: a new therapeutic option for attention-deficit hyperactivity disorder. CNS Drugs. 2012;26(8):691–705. | ||
Lasser RA, Dirks B, Nasrallah H, et al. Adjunctive lisdexamfetamine dimesylate therapy in adult outpatients with predominant negative symptoms of schizophrenia: open-label and randomized-withdrawal phases. Neuropsychopharmacology. 2013;38(11):2140–2149. | ||
McElroy SL, Martens BE, Mori N, et al. Adjunctive lisdexamfetamine in bipolar depression: a preliminary randomized, placebo-controlled trial. Int Clin Psychopharmacol. 2015;30(1):6–13. | ||
Trivedi MH, Cutler AJ, Richards C, et al. A randomized controlled trial of the efficacy and safety of lisdexamfetamine dimesylate as augmentation therapy in adults with residual symptoms of major depressive disorder after treatment with escitalopram. J Clin Psychiatry. 2013;74(8):802–809. | ||
Madhoo M, Keefe RS, Roth RM, et al. Lisdexamfetamine dimesylate augmentation in adults with persistent executive dysfunction after partial or full remission of major depressive disorder. Neuropsychopharmacology. 2014;39(6):1388–1398. | ||
Morrow SA, Smerbeck A, Patrick K, et al. Lisdexamfetamine dimesylate improves processing speed and memory in cognitively impaired MS patients: a phase II study. J Neurol. 2013;260(2):489–497. | ||
Wellman PJ. Modulation of eating by central catecholamine systems. Curr Drug Targets. 2005;6(2):191–199. | ||
Palmiter RD. Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci. 2007;30(8):375–381. | ||
Bello NT, Hajnal A. Acute methylphenidate treatments reduce sucrose intake in restricted-fed bingeing rats. Brain Res Bull. 2006;70(4–6):422–429. | ||
Vickers SP, Hackett D, Murray F, Hutson PH, Heal DJ. Effects of lisdexamfetamine in a rat model of binge-eating. J Psychopharmacol. 2015;29(12):1290–1307. | ||
Shinohara M, Mizushima H, Hirano M, et al. Eating disorders with binge-eating behaviour are associated with the s allele of the 3′-UTR VNTR polymorphism of the dopamine transporter gene. J Psychiatry Neurosci. 2004;29(2):134–137. | ||
Levitan RD, Masellis M, Basile VS, et al. The dopamine-4 receptor gene associated with binge eating and weight gain in women with seasonal affective disorder: an evolutionary perspective. Biol Psychiatry. 2004;56(9):665–669. | ||
McElroy SL, Guerdjikova A, Kotwal R, et al. Atomoxetine in the treatment of binge-eating disorder: a randomized placebo-controlled trial. J Clin Psychiatry. 2007;68(3):390–398. | ||
Wang GJ, Geliebter A, Volkow ND, et al. Enhanced striatal dopamine release during food stimulation in binge eating disorder. Obesity (Silver Spring). 2011;19(8):1601–1608. | ||
Broft A, Shingleton R, Kaufman J, et al. Striatal dopamine in bulimia nervosa: a PET imaging study. Int J Eat Disord. 2012;45(5):648–656. | ||
McElroy SL, Hudson JI, Mitchell JE, et al. Efficacy and safety of lisdexamfetamine for treatment of adults with moderate to severe binge-eating disorder: a randomized clinical trial. JAMA Psychiatry. 2015;72(3):235–246. | ||
Guy W. ECDEU Assessment Manual for Psychopharmacology. Rockville, MD: US National Institute of Health, Psychopharmacology Research Branch; 1976. | ||
Stunkard AJ, Messick S. The three-factor eating questionnaire to measure dietary restraint, disinhibition and hunger. J Psychosom Res. 1985;29(1):71–83. | ||
Gormally J, Black S, Daston S, Rardin D. The assessment of binge eating severity among obese persons. Addict Behav. 1982;7(1):47–55. | ||
Deal LS, Wirth RJ, Gasior M, Herman BK, McElroy SL. Validation of the yale-brown obsessive compulsive scale modified for binge eating. Int J Eat Disord. 2015;48(7):994–1004. | ||
Patton JH, Stanford MS, Barratt ES. Factor structure of the Barratt impulsiveness scale. J Clin Psychol. 1995;51(6):768–774. | ||
Ware J Jr, Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity. Med Care. 1996;34(3):220–233. | ||
Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–389. | ||
Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. | ||
McElroy SL, Mitchell JE, Wilfley D, et al. Lisdexamfetamine dimesylate effects on binge eating behaviour and obsessive-compulsive and impulsive features in adults with binge eating disorder. Eur Eat Disord Rev. Epub 2015 Dec 1. | ||
McElroy SL, Hudson J, Ferreira-Cornwell MC, et al. Lisdexamfetamine dimesylate for adults with moderate to severe binge eating disorder: results of two pivotal phase 3 randomized controlled trials. Neuropsychopharmacology. Epub 2015 Sep 9. | ||
Sheehan DV, Harnett-Sheehan K, Raj BA. The measurement of disability. Int Clin Psychopharmacol. 1996;11 Suppl 3:89–95. | ||
Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727–1736. | ||
Pawaskar M, Voko Z, Agh T, et al. Longitudinal modeling the effect of lisdexamfetamine dimesylate and changes in binge eating frequency on disability in patients with binge eating disorder. Value Health. 2015;18(7):A407. | ||
Pawaskar M, Agh T, Radewonuk J, et al. Longitudinal modeling of the relationship between lisdexamfetamine dimesylate and health-related quality of life in adults with moderate to severe binge eating disorder. Value Health. 2015;18(7):A407. | ||
Whiteford HA, Degenhardt L, Rehm J, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet. 2013;382(9904):1575–1586. | ||
Fairburn CG, Rothwell ER. Apps and eating disorders: a systematic clinical appraisal. Int J Eat Disord. 2015;48(7):1038–1046. | ||
Juarascio AS, Goldstein SP, Manasse SM, Forman EM, Butryn ML. Perceptions of the feasibility and acceptability of a smartphone application for the treatment of binge eating disorders: qualitative feedback from a user population and clinicians. Int J Med Inform. 2015; 84(10):808–816. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.