")
Back to Archived Journals » Cell Health and Cytoskeleton » Volume 7
Novel perspectives on the origins of the hepatic myofibroblasts
Authors Xu J, Liu X, Brenner D, Kisseleva T
Received 18 December 2014
Accepted for publication 13 January 2015
Published 20 March 2015 Volume 2015:7 Pages 111—119
DOI https://doi.org/10.2147/CHC.S57679
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Denis Wirtz
Jun Xu,1,2 Xiao Liu,1,2 David A Brenner,1 Tatiana Kisseleva2
1Department of Medicine, 2Department of Surgery, University of California, San Diego, CA, USA
Abstract: Liver fibrosis results from chronic liver injury that causes hepatocellular damage. Damaged hepatocytes apoptose, and release factors that facilitate recruitment of leukocytes to the site of injury, which in turn mediate recruitment and activation of liver- resident (Kupffer cells) and bone marrow (BM)-derived macrophages. Activated macrophages secrete TGF-ß1, the major profibrogenic cytokine, which activates hepatic myofibroblasts, which are not present in the liver under physiological conditions. Several sources of myofibroblasts have been identified, but it is believed that liver-resident hepatic stellate cells (HSCs) and portal fibroblasts (PFs) are the major source of hepatic myofibroblasts in fibrotic liver. Fibrocytes, designated as BM-derived collagen Type I producing cells, were also implicated in liver fibrosis; hence, their contribution to liver fibrosis remains controversial. Upon removal of the etiological agent, myofibroblasts either undergo apoptosis or inactivate into a quiescent-like state, followed by resorbtion of the fibrous scar. However, prolonged/repeated liver injury triggers irreversible cross-linking of collagen fibers that prevents fibrous scar from collagenase-mediated degradation. This review will discuss several types of fibrogenic cells contributing to the myofibroblast population, and the signaling pathways regulating their activation and collagen deposition.
Keywords: Liver fibrosis, TGF-ß1 signaling, hepatic stellate cells, portal fibroblasts, fibrocytes, collagen Type I deposition
Introduction
Liver fibrosis results from chronic liver injury, and is characterized by tissue remodeling, formation of fibrous scar, and activation of myofibroblasts. Activated myofibroblasts produce extracellular matrix (ECM), mainly collagen Type I. In addition, myofibroblasts express stromal markers, alpha smooth muscle actin (α-SMA), nonmuscle myosin, and fibronectin and exhibit spindle-like morphology.1–3 They contain rough endoplasmic reticulum (rER) and Golgi apparatus that are required for ECM production. Data obtained from patient biopsies and experimental models implicate myofibroblasts in the pathogenesis of liver fibrosis, and demonstrate that increased numbers of myofibroblasts correlate with severity of liver fibrosis.3 Furthermore, clinical and experimental studies have suggested that liver fibrosis can regress upon cessation of liver injury. Reversibility of liver fibrosis is associated with the disappearance of hepatic myofibroblasts.4,5 Therefore, myofibroblasts become an attractive target for antifibrotic therapy. Although all myofibroblasts share similar characteristics, such as expression of α-SMA, myofibroblasts may originate from different sources. Heterogeneity of the myofibroblast population may provide challenges for drug design. Five sources of myofibroblasts have been suggested to contribute to liver fibrosis: 1) Hepatic stellate cells (HSCs), which were reported to be a major source of myofibroblasts during hepatoxic liver injury,6,7 2) Portal fibroblasts (PFs), which arise in fibrotic liver in response to cholestatic liver injury, 3) Fibrocytes, a population of bone marrow (BM)-derived collagen Type I producing cells that are capable of differentiating into myofibroblasts, 4) Epithelial-to-mesenchymal transition, a mechanism described during embryonic development when epithelial cells undergo transdifferentiation into myofibroblasts, and 5) Endothelial cells that may undergo similar changes via endothelial-to-mesenchymal transition (EndMT). Hence, most recent cell fate mapping-based studies did not support a concept of EMT contribution to myofibroblasts. Although the composition of myofibroblasts varies depending on the etiology of liver fibrosis, their cellular origin can be identified by the expression of unique lineage markers. Identification of the origin and composition of myofibroblasts is critical for their targeting by antifibrotic therapies. In this review, we will discuss the characteristics of myofibroblasts of different origins, and the methodologies to isolate and target each population.
Hepatic stellate cells
HSCs, liver-resident mesenchymal cells, are considered to be a major source of myofibroblasts in fibrotic liver in response to toxic liver injury.7–9 In the healthy liver, HSCs exhibit a quiescent phenotype: they reside in the space of Disse (located between hepatocytes and sinusoidal endothelial cells), contain lipid droplets, and serve as a major storage of vitamin A in the mammalian body.10 Under physiological conditions, quiescent HSCs do not express collagen, but can be identified by the expression of neuronal markers (glial fibrillar acidic protein, GFAP, synemin, and neural growth factor p75) and type III intermediate filament, desmin. In response to liver injury, they downregulate vitamin A droplets, migrate to the injury foci, and elevate the expression of collagens and α-SMA for ECM deposition. The activated HSCs obtain a myofibroblast phenotype,5 and upregulate α-SMA, Col1a1, TIMP1, Crlf1, Spp1, IL1r1, IL-17ra, and Itga5.4,5 In agreement, partial deletion of HSCs in mice (achieved by overexpression of the herpes simplex virus thymidine kinase gene under the control of GFAP promoter) results in inhibition of CCl4-induced liver fibrosis.11
Furthermore, cell fate mapping experiments that utilize the Cre-loxp system in mice have identified a novel phenotype of HSCs. Thus, in addition to quiescent and activated phenotypes (aHSCs), HSCs can acquire an inactivated phenotype (iHSCs) during regression of liver fibrosis. Upon the removal of underlying etiological agents, liver fibrosis can regress. During fibrosis resolution, activated HSCs can either undergo apoptosis or become inactivated. Inactivation of HSCs is characterized by downregulation of fibrogenic gene mRNA, such as col1a1 and α-SMA, vimentin, SSP1, and TIMP1, and partial re-expression of lipogenesis genes, such as PPARg, Insig1, Grap, IL-10RA, and BAMBI.4,5 It is unknown why some aHSCs undergo apoptosis, while others become inactivated. However, survival and inactivation of HSCs is associated with transient upregulation of heat shock proteins 1a/b (Hsps1a/b) that may protect these cells from apoptosis.5
Activation of HSCs is orchestrated by a complex mechanism that includes damage to hepatocytes, reactive oxygen species (ROS) formation, recruitment of inflammatory cells, and upregulation of proinflammatory and profibrogenic cytokines. Much less is known about the mechanism of iHSC inactivation. Some of the pathways that promote HSCs activation/inactivation are discussed below.
TGF-β1 is a potent profibrogenic cytokine
Activation of hepatic myofibroblasts occurs in response to increased levels of proinflammatory and profibrogenic cytokines. The fibrogenic cytokine TGF-β1 is considered the most potent activator of HSCs/myofibroblasts in the damaged liver (Figure 1). Kupffer cells (KCs) and recruited macrophages are a major source of TGF-β1 in fibrotic liver.8 In addition, other fibrogenic cytokines and growth factors, such as PDGF, CTGF, and FGF, contribute to HSC/myofibroblast activation. Furthermore, proinflammatory cytokines, IL-6, TNF-α, IL-1β, IL-17A, MIP-1, and MIP-2, also facilitate the activation of HSCs/myofibroblasts. They also mediate the recruitment of inflammatory cells to the site of injury, and mount a local and systemic immune response, including activation of BM-derived macrophages and KCs (liver-resident macrophages). IL-6, IL-17A, and leptin can directly activate hepatic myofibroblasts to produce collagen.
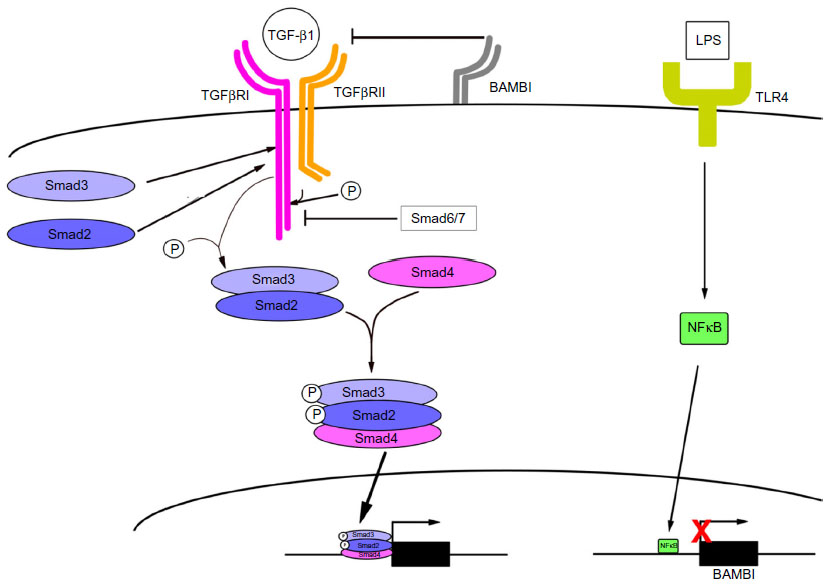 | Figure 1 TGF-β1 signaling plays a critical role in activation of HSCs (hepatic stellate cells). |
The binding of TGF-β1 to its corresponding receptors, TGF-β1 receptors type I and type II (TGFβRI and II), results in receptor heterodimerization, and subsequently serine phosphorylation of Smad2 and Smad3 (that dock at the intracellular domain of TGFβRI) at the serine residue (SSXS motif). Phosphorylation of cytoplasmic Smad2/3 (pSmad3) at the docking site of TGFβRI is the major event that triggers TGF-β1 signaling in aHSCs (Figure 1). Phosphorylated Smad2/3 form a complex with Smad4, a transporter molecule that facilitates nuclear translocation of Smad2/3/4 complex, where it binds the cis-regulatory elements of the target genes and triggers their transcription. Transcription of Col1a1, Smad7, and PAI-1 is directly regulated by the Smad2/3/4 complex, and therefore these genes are considered as TGF-β1-target genes.12,13 Smad7 forms a stable complex with TGFβRI to block further Smad3 phosphorylation and prevent TGF-β1 signaling.14 Remarkably, the transcriptional activity of Smad2/3/4 complex is negatively regulated by Smad7 in quiescent HSCs, but such an inhibitory effect has not been observed in HSC-derived myofibroblasts. Loss of inhibitory function of Smad7 may also contribute to “sensitization” of aHSCs to TGF-β1 signaling, causing their activation.15–17 The middle-linker region of Smad3 is phosphorylated by mitogen-activated protein kinase (MAPK), which activates an alternative pathway of TGF-β1-induced Smad3 phosphorylation and subsequent activation of collagen Type I expression.18 The Smad3 and MAPK pathway produce an additive effect on collagen Type I expression. Inhibition of either Smad3 or MAPK can only partially reduce collagen Type I expression in TGF-β1 aHSCs, but inhibition of both pathways significantly reduces ECM expression in aHSCs.19 TGF-β1 signaling is also regulated by other mechanisms during HSCs activation. The TGF-β1 pseudoreceptor BMP and activin membrane bound inhibitor (BAMBI) exhibits similarity to the TGF-β1 receptor type II, but lacks the intracellular serine threonine-kinase domain. BAMBI interferes with TGF-β1 signaling by blocking the ability of TGFβR I to form heterodimers with the TGFβR II.20 Therefore, repression of BAMBI expression during HSC activation augments TGF-β1 signaling. During regression of liver fibrosis, BAMBI is one of the factors that is re-expressed in inactivated human (h) HSCs to block TGF-β1 signaling. Inactivated HSCs slowly regain BAMBI expression.4 Regulation of BAMBI expression in HSCs is under the control of toll-like receptor (TLR)4 receptor signaling and NFκB signaling.21,22 aHSCs express a high level of TLR4 and its coreceptors MD2 and CD14.23 In response to LPS, TLR4 signaling promotes the translocation of NFκB into the nucleus. The p50 subunit recognizes the cis-regulatory element of NFκB, which locates within the 5′ promoter region of BAMBI locus, and facilitates the recruitment of histone deacetylase 1 (HDAC1) responsible for epigenetic changes within the BAMBI promoter region that results in suppression of BAMBI transcription in the activated HSCs.22
Activation of HSCs is regulated by pro/anti-inflammatory macrophages
Progression of liver fibrosis is associated with the overproduction of inflammatory cytokines. Macrophages, including KCs and BM-recruited macrophages, are the source of proinflammatory and profibrogenic cytokines in the injured liver. KCs represent a large group of liver-resident macrophages located in close proximity to hepatic sinusoids.24 KCs are strategically positioned to trap, phagocytose, and clear microbes from the circulation, and act as a first line of immune defense. KCs are F4/80highCD11low and CD68 (macrosialin) positive irradiation-resistant cells that modulate immediate immunoinflammatory responses in the damaged liver. BM-derived macrophages express similar markers, and upon recruitment to the injured liver can be identified by F4/80lowCD11high markers and increased expression of costimulatory molecule CD80 (B7-1) (reviewed by Sica et al24). An attempt has been made to determine whether KC and BM-recruited macrophages perform specialized functions, but owing to difficulties with distinguishing KC and BM-derived macrophages and their high plasticity, this question remains unresolved.
Macrophages are traditionally divided into two groups on the basis of the ability to produce specific subsets of cytokines. Chronic liver injury causes damage to hepatocytes, increased gut permeability, leakage of intestinal LPS into circulation, and disruption of the endothelial barrier.6 In response to TLR ligands and IFN-γ or IL-4/IL-13, macrophages undergo differentiation (polarization) and acquire M1 (classical) or M2 (alternative) phenotypes.24 The M1 phenotype is characterized by the expression of high levels of inflammatory cytokines, high production of ROS, promotion of Th1 response, and activity against microbes and tumors. In addition, M1 macrophages express CD14, CD11c, CD68, CD115, Marco, NFκB, STAT1, IRF3, IRF4, IRF5, and IRF8, Socs1 and Socs2 markers, TNF-α, IL-1β, IL-6, IL-12, IL-15, IL-18, IL-23 cytokines, CCL5 and 9, CXCL1 and 2 chemokines, secrete MMP1,3 and 9,25 activation of NFκB and AP-1.26 In contrast, M2 macrophages are involved in the protection against parasites, promote tissue remodeling and tumor progression, and have immunoregulatory functions. They exhibit high phagocytic activity; express scavenging, mannose, and galactose receptors; produce ornithine and polyamines through the arginase pathway; and have an IL-12lowIL-10highIL1decoyRhiIL-1RAhi phenotype (reviewed by Sica et al,24 Yi and Jeong,27 and Heymann et al28). M2 macrophages upregulate Arg1, Mcr2, Mgl1, CD115, Klf4, Dectin-1, Chi3l2 markers, IL-4, IL-10, IL-13 cytokines, and express MMP2, 9, 12, and 1325,29,30 with activation of Irf4, Socs3, Stat3, Stat5, Stat6, and PPARs.26 Polarization of M2 macrophages requires M-CSF, IL-4, and IL-13, and is inhibited in the presence of TNF-α and INF-γ.
The classical activated M1 macrophages are the major hepatic ROS source by expression of induced nitric oxide synthase (iNOS). The iNOS metabolizes L-arginine to nitric oxide (NO), which could directly activate HSCs, and promotes collagen expression.31 In turn, M2 macrophages exhibit stronger phagocytic activity, express Argenase-1, which competes with iNOS for L-arginine, but metabolizes L-arginine into urea and L-ornithine.32 M1 macrophages also secrete inflammatory cytokines, IL-1β, IL-6, and TNF-α that directly regulate HSCs activation. Macrophage-derived IL-1β promotes the expression of intercellular adhesion molecule 1 (ICAM-1) and TIMP-1 in aHSCs. Macrophage IL-1β triggers the secretion of MIP-2 by BM-derived cells, which further promotes recruitment and activation of inflammatory cells.33,34 Macrophage-derived IL-6 plays a critical role in the pathogenesis of liver fibrosis. IL-6 signal pathway is mainly mediated by gp130 and IL-6R, which are ubiquitously expressed in HSCs, hepatocytes, KCs, and sinusoid endothelial cells. Upon activation, Janus kinases (JAKs) are recruited to the IL-6/receptor complex and activated (phosphorylated). Activated JAKs phosphorylate STAT3. p-STAT3 dissociates from the receptor complex and forms a homodimer, which is then translocated to the nucleus, binds to DNA, and facilitates transcription of its target genes. IL-6 is a potent cytokine for activation of TGF-β1 production in KCs. In addition, IL-6 is one of the few inflammatory cytokines that can directly activate HSCs to synthesize collagen Type I. Interestingly, upon activation, HSCs start producing IL-6, which further promotes their activation via a paracrine mechanism.35 IL-17A, a cytokine produced by T helper 17 cells (Th17), stimulates IL-6 production by aHSCs. Both IL-6 and TGF-β1 regulate differentiation of Th17 from naive T-cells. Elevated levels of IL-17A are linked to liver fibrosis. IL-17A is also implicated in direct induction of collagen Type I production in aHSCs. Finally, macrophage-derived TNF-α mediates HSCs activation mainly via p38 MAPK and NFκB pathways;36,37 TNF-α signaling in aHSCs results in inhibition of BAMBI expression, and subsequent augmentation of TGF-β1 signaling.22 TNF-α also increases production of ROS in aHSCs.38
T cell cytokines regulate liver fibrosis
In addition to macrophages, T-cells, eosinophils, and plasma cells are recruited to the injured liver (reviewed by Wynn39). T-cells regulate recruitment and activation of macrophages by releasing unique cytokines that sustain liver fibrosis during chronic liver injury. Th2 and Th17 cells promote liver fibrosis by secreting cytokines IL-4/IL-5/IL-13 and IL-17A/IL-17F separately, while the interferon-γ-producing Th1 and IL-10-producing Treg cells inhibit fibrosis progression.35,40–46 T-cell-derived cytokines can directly regulate activation of HSCs. IL-4 and IL-13 exhibit about 30% similarities in amino acid (AA) sequences,47 and their cellular effects are mediated by the type II IL-4 receptor, following phosphorylation of STAT6 and expression of pSTAT6 downstream genes.48 HSCs express type II IL-4RA and IL-13RA1.49 Neutralization of IL-4 by anti-IL-4 antibodies (Abs) reduces collagen deposition in mouse models of schistosomiasis-induced liver fibrosis.50 Administration of IL-4 to cultured fibroblasts stimulates collagen synthesis.51 IL-13 is a more potent fibrogenic cytokine than IL-4. Neutralization of IL-13 with anti-IL-13 Abs attenuates schistosomiasis-induced liver fibrosis (specifically reduces collagen deposition around granulomas).52,53 Furthermore, administration of IL-13 to HSCs induces STAT6 phosphorylation, collagen expression and proliferation.49 IL-17A and IL-17F are secreted by Th17 cells. In patients with liver fibrosis, and mouse models of liver fibrosis, hepatic IL-17A, IL-17F, and IL-17RA are elevated. Disruption of IL-17 signaling protects IL-17RA−/− mice from liver fibrosis.35,54
The role of B cells in liver fibrosis of different etiologies has been suggested. B cell activation, differentiation, and proliferation occurs in the lymphoid follicles of secondary lymphoid organs, such as germinal centers of spleen. Intrahepatic B cells (IHB cells) are phenotypically similar to splenic B2 cells but express lower levels of CD23 and CD21 and higher levels of CD5.55 IHB cells proliferate as well as splenic B cells in response to anti-IgM and LPS stimulation in vitro. The original study by Novobrantseva et al implicated B cells in the pathogenesis of hepatic fibrogenesis,55 which demonstrated that while the acute inflammatory responses to repeated injections of CCl4 were similar in wildtype and B-cell-deficient mice, collagen deposition was decreased in animals with B cell deficiency. Recently, there has been increased interest in the role of B cells in the pathogenesis of primary biliary cirrhosis (PBC). Depletion of B cells was associated with reduced fibrosis in mouse models of cholestatic liver injury.56
Portal fibroblasts
Biliary fibrosis is characterized by cholestasis, and is accompanied by dysregulated cholangiocyte proliferation, often referred to as ductular reaction.57 Bile duct ligation (BDL), which mimics primary biliary cirrhosis in patients, is a widely used mouse model to study biliary fibrosis. Activated portal fibroblasts (aPFs) are implicated in the pathogenesis of biliary fibrosis. aPFs are shown to serve as a major source of myofibroblasts at the onset of injury.58,59 Under physiological conditions, PFs surround the portal vein to maintain the integrity of the portal tract.58 aPFs are the first “responders” to liver injury caused by biliary obstruction60 and differentiate into α-SMA-expressing myofibroblasts that synthesize ECM.58,61,62 HSCs (and to a lesser extent fibrocytes) also contribute to myofibroblast populations with the disease progression.63,64 The contribution of aPFs to liver fibrosis in response to different etiologies is not well understood, mainly because of the difficulties of isolation and in vivo tracking of the PFs. The most widely used method of aPF isolation from rat liver is based on in vitro culturing of the isolated bile duct segments and outgrowth of PFs from cultured bile duct.65–67 By utilizing collagen-α1(I) promoter-driven GFP (Col-GFP) transgenic mouse,61 our group established a flow cytometry-based method to isolate activated PFs from BDL-injured liver.64 In the Col-GFP mouse, all myofibroblasts can be identified by the expression of GFP, and the origin of each subset of myofibroblasts can be identified by phenotyping of GFP+ myofibroblasts for expression of specific markers. As HSC-specific markers have been discussed previously (vitamin A, desmin, GFAP, p75, etc), PF-derived myofibroblasts can be distinguished from other myofibroblasts by the expression of Thy1, Elastin (TE-7), Fibulin 2, and NTPD258,59,68,69 and lack of vitamin A. Gene expression profiling of isolated PFs identified new markers (calcitonin α, mesothelin, uroplakin 1β, basonuclin 1, aspirin, proteoglycan 4, glipican, and CD200) that distinguish BDL-derived activated PFs from activated HSCs.64
Origin of portal fibroblasts
Recent studies suggest that during embryonic development, PFs, HSCs, and vascular smooth muscle cells (VSMC) originate from the same precursor in the septum transversum.70,71 Lineage tracing of the mesothelial precursor cells using MesP1Cre × Rosa26-flox-Stop-flox-reporter mice provides evidence that the septum transversum originates from the mesoderm. At day 9.5 (E9.5) of embryogenesis, when the hepatic bile duct and sinusoids start to form, the septum transversum is invaded by the foregut endoderm. At E12.5, the perihepatic membrane, mesothelial cells, and submesothelial cells give rise to the mesenchymal cell precursors that invade the liver parenchyma.72,73 Several cell markers of these mesenchymal precursors have been identified, including mesothelin, desmin, p75NTR, Wt1, and ALCAM. These mesenchymal precursors interact with endodermal hepatoblasts to induce differentiation of the hepatic parenchyma. It remains unclear at which time point and specific stage of embryonic development the PF and HSC precursors diverge from each other and retain expression of cell-specific markers throughout adulthood. One explanation is that positioning during hepatic lobule formation may affect functional specialization of PFs and HSCs, leading to their divergence. In concordance, fetal PFs secrete BMPs, Jagged1, and Hedgehog ligands to induce signaling critical for differentiation of hepatoblasts into cholangiocytes during bile canaliculi and lumen formation,58,74 while fetal HSCs are implicated in supporting proliferation of hepatoblasts by producing HGF, pleiotrophin, and FGF10.75,76
TGF signaling activates PFs
In the cases of cholestatic liver injury, PFs respond rapidly to increasing levels of TGF-β177 by upregulation of fibrogenic genes, Col-α1(I), α-SMA, TIMP1, TGF-β2,60 PAI-1, elastin,58 fibronectin,68,78 and of CD73 ectoenzyme.78–80 At the same time, activated PFs give rise to myofibroblasts.62,81,82 BDL-activated PFs also secrete TGF-β1 and TGF-β2, which induce TGF-β1 signaling and transduction in nearby HSCs.60 FGF-2 facilitates the activation of PFs as well.83 Binding of FGF-2 to its tyrosine kinase receptors FGFRs and subsequent activation of Ras-MEK-Erk1/2 signaling cause proliferation and migration of aPFs.84 Proliferation and activation of aPFs are shown to be blocked by curcumin, a nonsteroidal yellow pigment found in rhizomes of the perennial herb Curcuma longa, which blocks ERK1/2 phosphorylation in aPFs, and thus has the potential to reduce cholestasis-induced fibrogenesis.85
Bone marrow-derived collagen-producing cells (fibrocyte)
Fibrocytes are bone marrow-derived collagen-producing cells in the circulating blood with dual characteristics of both hematopoietic and stromal cells.86–88 They are derived from a subset of monocytes labeled with CD11b, CD115, and Gr1 in a T-cell-dependent manner.89 Outgrowth of fibrocytes from monocytes is controlled by T-cell-released cytokines. IL-4 and IL-13 from Th2 cells promote outgrowth of fibrocytes from CD14+ precursors, while interferon-γ and IL-12 from Th1 inhibited fibrocytes outgrowth.90 The circulating fibrocytes are multipotent and can differentiate into several cell types.91 In the injured organs, fibrocytes commit to the myofibroblast fate and contribute to the ECM deposition and tissue remodeling. At the same time, tissue fibrocytes are involved in adaptive immunity and serve as antigen-presenting cells. They express antigen-presenting membrane proteins MHC I, II, CD80, and CD86, and are able to recognize antigen and promote T-cell proliferation.92 Hepatic fibrocytes were implicated in collagen Type I deposition in fibrotic liver. However, they contribute only to 5%–6% of total collagen expressing myofibroblasts, and, therefore, the roles of hepatic fibrocytes in the pathogenesis of liver fibrosis remain controversial. A significant population of fibrocytes was shown to be recruited into the spleen in response to liver and kidney injury, LPS, bacterial infection, or TGF-β1. While splenic fibrocytes are capable of antigen presentation and differentiation into myofibroblasts (similar to hepatic fibrocytes), they exhibit unique functions. Gene expression profiling of splenic fibrocytes revealed that they also contribute to the innate immunity by mounting an elaborate immune response: splenic fibrocytes are not capable of phagocytosis, but can kill bacteria by the formation of DNA-based extracellular traps containing the antimicrobial peptide cathelicidin (CRAMP).93
Conclusion
Despite extensive studies, there is no effective antifibrosis therapy for human patients. Recent improvements in mouse models of hepatic fibrogenesis are providing new insights into the pathogenesis of the liver fibrosis and the origin of myofibroblasts, which may provide new targets for therapy.
Acknowledgments
This work was supported by National Institutes of Health (DK088837, U01AA022614, AI0777802, and P50 AA011999).
Disclosure
The authors report no conflicts of interest in this work.
References
Eyden B. The myofibroblast: phenotypic characterization as a prerequisite to understanding its functions in translational medicine. J Cell Mol Med. 2008;12:22–37. | |
Watsky MA, Weber KT, Sun Y, Postlethwaite A. New insights into the mechanism of fibroblast to myofibroblast transformation and associated pathologies. Int Rev Cell Mol Biol. 2010;282:165–192. | |
Brenner DA, Kisseleva T, Scholten D, et al. Origin of myofibroblasts in liver fibrosis. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S17. | |
Liu X, Xu J, Brenner DA, Kisseleva T. Reversibility of liver fibrosis and inactivation of fibrogenic myofibroblasts. Curr Pathobiol Rep. 2013;1:209–214. | |
Kisseleva T, Cong M, Paik Y, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:9448–9453. | |
Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol. 2006;21(Suppl 3):S84–S87. | |
Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. | |
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. | |
Cassiman D, Libbrecht L, Desmet V, Denef C, Roskams T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J Hepatol. 2002;36:200–209. | |
Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis. 2001;21:311–335. | |
Stewart RK, Dangi A, Huang C, et al. A novel mouse model of depletion of stellate cells clarifies their role in ischemia/reperfusion- and endotoxin-induced acute liver injury. J Hepatol. 2014;60:298–305. | |
Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol. 1991;30:77–87. | |
Schnabl B, Kweon YO, Frederick JP, Wang XF, Rippe RA, Brenner DA. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology. 2001;34:89–100. | |
Nakao A, Afrakhte M, Moren A, et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389:631–635. | |
Tahashi Y, Matsuzaki K, Date M, et al. Differential regulation of TGF-beta signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology. 2002;35:49–61. | |
Stopa M, Anhuf D, Terstegen L, Gatsios P, Gressner AM, Dooley S. Participation of Smad2, Smad3, and Smad4 in transforming growth factor beta (TGF-beta)-induced activation of Smad7. The TGF-beta response element of the promoter requires functional Smad binding element and E-box sequences for transcriptional regulation. J Biol Chem. 2000;275:29308–29317. | |
Stopa M, Benes V, Ansorge W, Gressner AM, Dooley S. Genomic locus and promoter region of rat Smad7, an important antagonist of TGFbeta signaling. Mamm Genome. 2000;11:169–176. | |
Furukawa F, Matsuzaki K, Mori S, et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003;38:879–889. | |
Tsukada S, Westwick JK, Ikejima K, Sato N, Rippe RA. SMAD and p38 MAPK signaling pathways independently regulate alpha1(I) collagen gene expression in unstimulated and transforming growth factor-beta-stimulated hepatic stellate cells. J Biol Chem. 2005;280:10055–10064. | |
Sekiya T, Oda T, Matsuura K, Akiyama T. Transcriptional regulation of the TGF-beta pseudoreceptor BAMBI by TGF-beta signaling. Biochem Biophys Res Commun. 2004;320:680–684. | |
Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. | |
Liu C, Chen X, Yang L, Kisseleva T, Brenner DA, Seki E. Transcriptional repression of the transforming growth factor beta (TGF-beta) Pseudoreceptor BMP and activin membrane-bound inhibitor (BAMBI) by Nuclear Factor kappaB (NF-kappaB) p50 enhances TGF-beta signaling in hepatic stellate cells. J Biol Chem. 2014;289:7082–7091. | |
Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37: 1043–1055. | |
Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. 2014;59:2034–2042. | |
Butcher MJ, Galkina EV. Phenotypic and functional heterogeneity of macrophages and dendritic cell subsets in the healthy and atherosclerosis-prone aorta. Front Physiol. 2012;3:44. | |
Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. | |
Yi HS, Jeong WI. Interaction of hepatic stellate cells with diverse types of immune cells: foe or friend? J Gastroenterol Hepatol. 2013;28(Suppl 1):99–104. | |
Heymann F, Trautwein C, Tacke F. Monocytes and macrophages as cellular targets in liver fibrosis. Inflamm Allergy Drug Targets. 2009;8:307–318. | |
Schwartz Y, Svistelnik AV. Functional phenotypes of macrophages and the M1–M2 polarization concept. Part I. Proinflammatory phenotype. Biochemistry (Mosc). 2012;77:246–260. | |
Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. | |
Li JT, Liao ZX, Ping J, Xu D, Wang H. Molecular mechanism of hepatic stellate cell activation and antifibrotic therapeutic strategies. J Gastroenterol. 2008;43:419–428. | |
Lech M, Anders HJ. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim Biophys Acta. 2013;1832:989–997. | |
Hellerbrand C, Jobin C, Licato LL, Sartor RB, Brenner DA. Cytokines induce NF-kappaB in activated but not in quiescent rat hepatic stellate cells. Am J Physiol. 21998;75:G269–G278. | |
Gieling RG, Wallace K, Han YP. Interleukin-1 participates in the progression from liver injury to fibrosis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1324–G1331. | |
Meng F, Wang K, Aoyama T, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143:765–776, e761–e763. | |
Thirunavukkarasu C, Watkins SC, Gandhi CR. Mechanisms of endotoxin-induced NO, IL-6, and TNF-alpha production in activated rat hepatic stellate cells: role of p38 MAPK. Hepatology. 2006;44:389–398. | |
Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–594. | |
Han D, Ybanez MD, Ahmadi S, Yeh K, Kaplowitz N. Redox regulation of tumor necrosis factor signaling. Antiox Redox Signal. 2009;11:2245–2263. | |
Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4:583–594. | |
Wynn TA, Cheever AW, Jankovic D, et al. An IL-12-based vaccination method for preventing fibrosis induced by schistosome infection. Nature. 1995;376:594–596. | |
Hoffmann KF, Cheever AW, Wynn TA. IL-10 and the dangers of immune polarization: excessive type 1 and type 2 cytokine responses induce distinct forms of lethal immunopathology in murine schistosomiasis. J Immunol. 2000;164:6406–6416. | |
Gurujeyalakshmi G, Giri SN. Molecular mechanisms of antifibrotic effect of interferon gamma in bleomycin-mouse model of lung fibrosis: downregulation of TGF-beta and procollagen I and III gene expression. Exp Lung Res. 1995;21:791–808. | |
Oldroyd SD, Thomas GL, Gabbiani G, El Nahas AM. Interferon-gamma inhibits experimental renal fibrosis. Kidney Int. 1999;56:2116–2127. | |
Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev. 2014;14:181–194. | |
Knolle PA, Thimme R. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology. 2014;146:1193–1207. | |
Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med. 2013;19:859–868. | |
Minty A, Chalon P, Derocq JM, et al. Interleukin-13 is a new human lymphokine regulating inflammatory and immune responses. Nature. 1993;362:248–250. | |
Wynn TA. IL-13 effector functions. Annu Rev Immunol. 2003;21:425–456. | |
McHedlidze T, Waldner M, Zopf S, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. 2013;39:357–371. | |
Cheever AW, Williams ME, Wynn TA, et al. Anti-IL-4 treatment of Schistosoma mansoni-infected mice inhibits development of T cells and non-B, non-T cells expressing Th2 cytokines while decreasing egg-induced hepatic fibrosis. J Immunol. 1994;153:753–759. | |
Tiggelman AM, Boers W, Linthorst C, Sala M, Chamuleau RA. Collagen synthesis by human liver (myo)fibroblasts in culture: evidence for a regulatory role of IL-1 beta, IL-4, TGF beta and IFN gamma. J Hepatol. 1995;23:307–317. | |
Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;104:777–785. | |
Chiaramonte MG, Cheever AW, Malley JD, Donaldson DD, Wynn TA. Studies of murine schistosomiasis reveal interleukin-13 blockade as a treatment for established and progressive liver fibrosis. Hepatology. 2001;34:273–282. | |
Tan Z, Qian X, Jiang R, et al. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol. 2013;191:1835–1844. | |
Novobrantseva TI, Majeau GR, Amatucci A, et al. Attenuated liver fibrosis in the absence of B cells. J Clin Invest. 2005;115:3072–3082. | |
Marra F, Aleffi S, Galastri S, Provenzano A. Mononuclear cells in liver fibrosis. Semin Immunopathol. 2009;31:345–358. | |
Jhandier MN, Kruglov EA, Lavoie EG, Sevigny J, Dranoff JA. Portal fibroblasts regulate the proliferation of bile duct epithelia via expression of NTPDase2. J Biol Chem. 2005;280:22986–22992. | |
Dranoff JA, Wells RG. Portal fibroblasts: underappreciated mediators of biliary fibrosis. Hepatology. 2010;51:1438–1444. | |
Clouzeau-Girard H, Guyot C, Combe C, et al. Effects of bile acids on biliary epithelial cell proliferation and portal fibroblast activation using rat liver slices. Lab Invest. 2006;86:275–285. | |
Wells RG, Kruglov E, Dranoff JA. Autocrine release of TGF-beta by portal fibroblasts regulates cell growth. FEBS Lett. 2004;559:107–110. | |
Yata Y, Scanga A, Gillan A, et al. DNase I-hypersensitive sites enhance alpha1(I) collagen gene expression in hepatic stellate cells. Hepatology. 2003;37:267–276. | |
Desmouliere A, Darby I, Costa AM, et al. Extracellular matrix deposition, lysyl oxidase expression, and myofibroblastic differentiation during the initial stages of cholestatic fibrosis in the rat. Lab Invest. 1997;76:765–778. | |
Goddard CJ, Smith A, Hoyland JA, et al. Localisation and semiquantitative assessment of hepatic procollagen mRNA in primary biliary cirrhosis. Gut. 1998;43:433–440. | |
Iwaisako K, Jiang C, Zhang M, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci U S A. 2014;111:E3297–E3305. | |
Wen JW, Olsen AL, Perepelyuk M, Wells RG. Isolation of rat portal fibroblasts by in situ liver perfusion. J Vis Exp. 2012;29(64). | |
Uchio K, Tuchweber B, Manabe N, Gabbiani G, Rosenbaum J, Desmouliere A. Cellular retinol-binding protein-1 expression and modulation during in vivo and in vitro myofibroblastic differentiation of rat hepatic stellate cells and portal fibroblasts. Lab Invest. 2002;82:619–628. | |
Kruglov EA, Jain D, Dranoff JA. Isolation of primary rat liver fibroblasts. J Investig Med. 2002;50:179–184. | |
Knittel T, Kobold D, Saile B, et al. Rat liver myofibroblasts and hepatic stellate cells: different cell populations of the fibroblast lineage with fibrogenic potential. Gastroenterology. 1999;117:1205–1221. | |
Dranoff JA, Kruglov EA, Robson SC, Braun N, Zimmermann H, Sevigny J. The ecto-nucleoside triphosphate diphosphohydrolase NTPDase2/CD39L1 is expressed in a novel functional compartment within the liver. Hepatology. 2002;36:1135–1144. | |
Asahina K, Tsai SY, Li P, et al. Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology. 2009;49:998–1011. | |
Suzuki K, Tanaka M, Watanabe N, Saito S, Nonaka H, Miyajima A. p75 Neurotrophin receptor is a marker for precursors of stellate cells and portal fibroblasts in mouse fetal liver. Gastroenterology. 2008;135: 270–281, e273. | |
Asahina K, Zhou B, Pu WT, Tsukamoto H. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology. 2011;53: 983–995. | |
Rinkevich Y, Mori T, Sahoo D, Xu PX, Bermingham JR Jr, Weissman IL. Identification and prospective isolation of a mesothelial precursor lineage giving rise to smooth muscle cells and fibroblasts for mammalian internal organs, and their vasculature. Nat Cell Biol. 2012;14: 1251–1260. | |
Fabris L, Strazzabosco M. Epithelial-mesenchymal interactions in biliary diseases. Semin Liver Dis. 2011;31:11–32. | |
Asahina K, Sato H, Yamasaki C, et al. Pleiotrophin/heparin-binding growth-associated molecule as a mitogen of rat hepatocytes and its role in regeneration and development of liver. Am J Pathol. 2002;160: 2191–2205. | |
Berg T, Rountree CB, Lee L, et al. Fibroblast growth factor 10 is critical for liver growth during embryogenesis and controls hepatoblast survival via beta-catenin activation. Hepatology. 2007;46:1187–1197. | |
Liu C, Gaca MD, Swenson ES, Vellucci VF, Reiss M, Wells RG. Smads 2 and 3 are differentially activated by transforming growth factor-beta (TGF-beta) in quiescent and activated hepatic stellate cells. Constitutive nuclear localization of Smads in activated cells is TGF-beta-independent. J Biol Chem. 2003;278:11721–11728. | |
Ji J, Yu F, Ji Q, et al. Comparative proteomic analysis of rat hepatic stellate cell activation: a comprehensive view and suppressed immune response. Hepatology. 2012;56:332–349. | |
Fausther M, Sheung N, Saiman Y, Bansal MB, Dranoff JA. Activated hepatic stellate cells upregulate transcription of ecto-5′-nucleotidase/CD73 via specific SP1 and SMAD promoter elements. Am J Physiol Gastrointest Liver Physiol. 2012;303:G904–G914. | |
Li Z, Dranoff JA, Chan EP, Uemura M, Sevigny J, Wells RG. Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology. 2007;46:1246–1256. | |
Desmouliere A. Hepatic stellate cells: the only cells involved in liver fibrogenesis? A dogma challenged. Gastroenterology. 2007;132: 2059–2062. | |
Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13:7–12. | |
Horiguchi N, Lafdil F, Miller AM, et al. Dissociation between liver inflammation and hepatocellular damage induced by carbon tetrachloride in myeloid cell-specific signal transducer and activator of transcription 3 gene knockout mice. Hepatology. 2010;51:1724–1734. | |
Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445–489. | |
Baghdasaryan A, Claudel T, Kosters A, et al. Curcumin improves sclerosing cholangitis in Mdr2-/- mice by inhibition of cholangiocyte inflammatory response and portal myofibroblast proliferation. Gut. 2010;59:521–530. | |
Bucala R. Circulating fibrocytes: cellular basis for NSF. J Am Coll Radiol. 2008;5:36–39. | |
Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81. | |
Grieb G, Steffens G, Pallua N, Bernhagen J, Bucala R. Circulating fibrocytes-biology and mechanisms in wound healing and scar formation. Int Rev Cell Mol Biol. 2011;291:1–19. | |
Niedermeier M, Reich B, Rodriguez Gomez M, et al. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci U S A. 2009;106:17892–17897. | |
Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D. Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J Leukoc Biol. 2008;83:1323–1333. | |
Suga H, Rennert RC, Rodrigues M, et al. Tracking the elusive fibrocyte: identification and characterization of collagen-producing hematopoietic lineage cells during murine wound healing. Stem Cells. 2014;32:1347–1360. | |
Chesney J, Bacher M, Bender A, Bucala R. The peripheral blood fibrocyte is a potent antigen-presenting cell capable of priming naive T cells in situ. Proc Natl Acad Sci U S A. 1997;94:6307–6312. | |
Kisseleva T, von Kockritz-Blickwede M, Reichart D, et al. Fibrocyte-like cells recruited to the spleen support innate and adaptive immune responses to acute injury or infection. J Mol Med. 2011;89: 997–1013. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.