")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Novel peptide myristoly-CM4 induces selective cytotoxicity in leukemia K562/MDR and Jurkat cells by necrosis and/or apoptosis pathway
Authors Zhang H, Han D, Lv T, Liu K, Yang Y, Xu X, Chen Y
Received 1 March 2019
Accepted for publication 9 May 2019
Published 2 July 2019 Volume 2019:13 Pages 2153—2167
DOI https://doi.org/10.2147/DDDT.S207224
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Huidan Zhang,* Dongju Han,* Tongtong Lv, Kehang Liu, Yunqing Yang, Xixi Xu, Yuqing Chen
Department of Biochemistry, Life Sciences College, Nanjing Normal University, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Purpose: There is an urgent need for the development of novel, effective, and less toxic drugs to treat leukemia. Antimicrobial peptides (AMPs) have received much more attention as alternative chemotherapeutic agents. This study aimed to examined the cytotoxicity of a novel AMP myristoly-CM4 against chronic myeloid leukemia cells (K562/MDR) and acute lymphocytic leukemia cells (Jurkat), and further investigated its selectivity to clarify the cytotoxic mechanism.
Materials and methods: In this study, the cytotoxicity and selectivity of myristoly-CM4 against K562/MDR and Jurkat cells were assessed in vitro, and the anticancer mechanism responsible for its cytotoxicity and selectivity was further investigated.
Results: Myristoly-CM4 was cytotoxic to these leukemia cell lines (IC50 2–4 μM) and was less cytotoxic to normal cells (HEK-293, L02 cells, peripheral blood mononuclear cells, and erythrocytes). Myristoyl-CM4 had stronger affinity to K562/MDR and Jurkat cells than to normal cells, while the contents of phosphatidylserine and sialic acids on the cell surfaces of K562/MDR and Jurkat cells were significantly higher than that of HEK293 cells. The myristoyl group effectively mediated the internalization of myristoyl-CM4 to leukemia cells. After internalization, myristoyl-CM4 could target mitochondria and affected mitochondrial function, including disruption of Δψm, increasing the accumulation of ROS, increasing the Bax/Bcl-2 ratio, activating caspase 9 and 3, and PARP to induce mitochondria-dependent apoptosis in both K562/MDR and Jurkat cells. Myristoyl-CM4 also induced K562/MDR cell necrosis by directive membrane disruption, and significantly decreased the level of P-glycoprotein in K562/MDR cells.
Conclusion: These results suggested that myristoyl-CM4 showed selective cytotoxicity to leukemia K562/MDR and Jurkat cells by apoptosis and/or necrosis pathway. Myristoyl-CM4, thus, appears to be a promising candidate for leukemia treatment, including multidrug-resistant leukemia.
Keywords: leukemia, multi-drug resistance, necrosis, apoptosis, myristoyl-CM4, selectivity
Introduction
Despite major advances over the past few years, cancer remains a serious threat to human survival. Leukemia is the most common form of cancer worldwide, with an estimated 437,033 new cases and 309,000 deaths predicted in 2018.1 Leukemia results from the clonal proliferation of bone marrow cells with impaired differentiation, regulation, and cell death. There have been breakthroughs in bone marrow transplantation for the treatment of childhood leukemias in recent decades, but chemotherapy remains the main treatment option for adults, with less therapeutic sucess, especially in elderly patients.2 Several chemotherapy drugs, such as tyrosine kinase inhibitors, are currently available for leukemia patients, representing major improvements in the treatment of chronic myeloid leukemia (CML) and acute lymphocytic leukemia (ALL). However, resistance to chemotherapy remains a common obstacle to treating leukemia patients.3,4 Furthermore, traditional chemotherapeutic drugs used in the clinic, such as doxorubicin and arsenic trioxide, show insufficient selectivity, resulting in deleterious side-effects.5,6 The development of novel safe and effective drugs, thus, remains an urgent priority for leukemia treatments.
Cationic antimicrobial peptides (AMPs) have shown antibacterial, antifungal, wound healing, antioxidant, and anticancer properties. About 200 natural and synthetic animal AMPs have demonstrated the ability to kill cancer cells, especially from solid tumors in vivo or in vitro.7 AMPs usually kill cancer cells by interacting with specific targets exposed in cancer cell plasma membranes, thus improving their selectivity for cancer cells compared with traditional chemotherapeutic drugs.8 However, although several AMPs have shown anticancer activity in leukemia cells, most were not effective enough.9–12
The AMP CM4 consists of 35 amino acids, and was initially isolated from the hemolymph of the Chinese silkworm Bombyx mori. CM4 was effective against bacteria and fungi, via affecting membrane interactions.13,14 It also showed anti-inflammatory effects mediated by neutralizing the endotoxin lipopolysaccharide.15 In addition to its antimicrobial activity, we previously reported that CM4 had selective anticancer activity in leukemia cells (K562, THP-1, and U937 cells), while exhibiting no cytotoxicity towards non-cancer cells, even at 120 μM.16 Targeting leukemia cells and disrupting their plasma membrane are major anticancer mechanisms. However, similar to most AMPs, the anticancer activity in leukemia cells was low (IC50 about 20–40 μM), thus hampering its use in leukemia therapy.
Covalent modification of proteins by fatty acids such as myristate or palmitate is a widely-recognized form of protein modification and plays an essential role in directing the cellular localization of proteins by facilitating protein–membrane interactions.17 Several groups have reported that the conjugation of fatty acids to AMPs improved their hydrophobicity, resulting in the enhancement, retention, or loss of antimicrobial activity.18–20 We previously showed that conjugation of a myristoyl group to the N-terminus of CM4 significantly enhanced its antitumor activity in breast cancer both in vitro and in vivo, resulting in the induction of mitochondria-dependent apoptosis in breast cancer cells.21 However, its anticancer activity against non-solid tumors such as leukemias remains unknown. In the current study, we assessed the cytotoxicity of myristoyl-CM4 against human chronic myeloid leukemia K562/MDR and T-cell acute leukemia Jurkat cells in vitro, and further investigated its selectivity to clarify the cytotoxic mechanism responsible for its action.
Materials and methods
Peptide synthesis, labeling, and reagents
CM4 (GRWKIFKKIEKVGQNIRDGIVKAGPAVAVVGQAATI-NH2), myristoyl-CM4, fluorescein isothiocyanate (FITC)-CM4, and FITC-lmyristoyl-CM4 were synthesized using solid-phase 9-fluorenylmethoxycarbonyl (Fmoc) methods by Biomatik Corporation (Ontario, Canada). The synthesized peptides used were all >95% homogeneous, as indicated by C18 reverse-phase high-performance liquid chromatography (HPLC) and electrospray ionization mass spectroscopy analysis.
RPMI-1640 and DMEM were purchased from Thermo Fisher Scientific (Waltham, MA, USA), FBS from Capricorn scientific (Hessen, Germany). Melittin was purchased from Synpeptide, Inc (Nanjing, China). MTT, rhodamine 123, and Triton-X-100 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Hoechst 33342/PI staining kit, ROS assay kit, and DCFH-DA were purchased from Beyotime Insititue of Biotechnology (Shanghai, China). JC-1 Detection kit, Annexin V/PI assay kit, and LDH assay kit were purchased from KeyGEN BioTECH, Inc. (Nanjing, China). FITC-SNA were purchased from Vector Laboratories (Burlingame, CA, USA). Bax(# 5023), Bcl-2(# 2870), cytochrome c(# 11940), caspase 9(# 9508), caspase 3(# 9662), PARP(# 9542), GAPDH(# 2118), β-actin(# 4967) and P-gp(# 13342) were purchased from Cell Signaling Technology (Boston, MA, USA). All the antibodies were used at a dilution multiple of 1:1,000. All other reagents were analytical grade reagents, and produced in China. All the reagents were used by the rules of standard bio-security and safety procedures of Nanjing Normal University.
Cell culture
HEK293, L02, K562/MDR, K562, and Jurkat cells were obtained from the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. L02, K562/MDR, K562, and Jurkat cells were maintained in RPMI 1640 and HEK293 cells were maintained in DMEM, at 37°C in a 5% CO2 humidified atmosphere, supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. K562/MDR cells was selected and identified by doxorubition.
MTT assay
K562/MDR, K562, Jurkat, L02, and HEK293 cells were incubated with different concentrations of myristoyl-CM4, and cytotoxic activity was detected by MTT assay. Briefly, 105 cells/well were seeded in 96-well plates in medium in the presence of different concentrations of CM4 (20, 30, 40 μM), myristoyl-CM4 (0.5, 1, 2, 4 μM), or without peptide (control). After 24 hours, 20 μL of 5 mg/ml MTT was added to each well, and the plates were incubated at 37°C for 4–8 hours. The supernatant was then removed, 200 μL dimethyl sulfoxide was added for 30 minutes to dissolve the formazan product that remained in the wells, and the absorbance was then measured using a BioTek SynergyTM 2 multi-mode microplate reader (BioTek Instruments, Inc., USA) at a test wavelength of 490 nm, with a reference wavelength of 570 nm. The final results were recorded as the average of at least three repeated experiments, with the viability of the control cells taken as 100% cell survival. The IC50 value for each cell line was the concentration of peptide causing a 50% reduction in absorbance compared with the control.
Trypan blue dye exclusion
Peripheral blood mononuclear cells (PBMCs) were isolated from mouse blood using the Ficoll gradient density method and then seeded in 12-well plates at a density of 5×105 cells/well. The cells were maintained in RPMI 1640 with 10% FBS and treated with different concentrations of myristoyl-CM4 (2, 4, 10 μM), with phosphate-buffered saline (PBS) and melittin (a hemolytic peptide from bee Apis mellifera) as controls. After incubation at 37°C for 24 hours, 0.4% Trypan Blue dye was added into the cell suspension for 5 minutes, followed by washing three times with PBS, and analysis under a fluorescence microscope. K562/MDR and Jurkat cells (5×105/well) were incubated with myristoyl-CM4 for 24 hours, stained with Trypan Blue dye, and observed by fluorescence microscopy.
Hemolytic activity assay
Erythrocytes were isolated from mouse blood cells by centrifugation at 1,000×g for 10 minutes, followed by washing three times with PBS. Hemolytic activity was evaluated as the method described previously.21 Briefly, erythrocytes (final concentration 4% v/v) were treated with myristoyl-CM4 or CM4 for 1 hour at 37°C followed by centrifugation at 1,000×g for 5 minutes. The absorbance of the supernatant was measured at 414 nm, with 0.1% TritonX-100 (v/v) and PBS w as 100% and 0% hemolysis controls, Melittin was also used as hemolytic control. The percentage of hemolysis was calculated as: (Apeptide−APBS)/(ATritonX-100−APBS)×100%, where A is the absorbance. Data were reported as the mean±standard error of the mean (SEM) of four-to-six independent experiments.
Peptide binding assay
Cellular uptake was measured in K562/MDR, Jurkat, HEK293, and erythrocytes by flow cytometry. Cells (1×106/ml) were collected and resuspended in PBS. The binding activities of the peptides were assessed by induction with FITC-myristoyl-CM4 or FITC-CM4 at 37°C for 30 minutes in the dark. The cells were washed with PBS and analyzed using a FACS Vantage SE flow cytometer (Becton-Dickinson, San Jose, CA, USA). The mean fluorescence of 10,000 cells was analyzed for each sample using BD flow cytometry software. Data were reported as the mean±SEM of three independent experiments.
Confocal laser scanning microscopy (CLSM)
K562/MDR and Jurkat cells were incubated with FITC-myristoyl-CM4 or FITC-CM4 for 30 minutes in the dark, harvested, rinsed three times with PBS, and analyzed immediately by CLSM with optical excitation using a 488 nm argon laser beam for the FITC signal. After treatment with FITC-labeled peptide, K562/MDR and Jurkat cells were then treated with 30 nM rhodamine 123 for 30 minutes in the dark, washed, and the distribution of fluorescence was analyzed immediately by Confocal laser scanning microscopy using 488 nm and 525 nm argon laser beams for the FITC and rhodamine 123 signals, respectively.
Annexin V/PI staining
Apoptosis was evaluated using two-color flow cytometric analysis with an Annexin V/PI assay kit. Briefly, 2.5×106 cells were seeded in 12-well plates in medium in the presence or absence of myristoyl-CM4. After incubation for 16 hours, the cells were collected, washed twice with PBS, and then re-suspended in 500 μL of binding buffer. FITC-conjugated Annexin V and PI were added to the cell suspension, and the mixture was incubated for 10 minutes on ice in the dark. The stained cells were analyzed by flow cytometry, and the percentage of apoptotic cells was calculated using Cell Quest software.
Measurement of the contents of phosphatidylserine and sialic acid
The contents of phosphatidylserine (PS) and sialic acid on the surface of K562/MDR, Jurkat, and HEK293 cells were determined using flow cytometry. Brifely, cells at concentrations above 1×106 cells/mL were stained with FITC–Annexin V for 15 minutes, or 10 μg/mL of FITC-SNA for 45 minutes at 4°C in the dark. The cells were washed with PBS and analyzed using a flow cytometer. The mean fluorescence of 10,000 cells was analyzed for each sample. Data were reported as the mean±SEM of three independent experiments.
Hoechst 33342/PI staining
Cells (1×105/mL) were seeded into 6-well plates and treated with myristoyl-CM4 (2 μM for K562/MDR, 3 μM for Jurkat) for 16 hours. Apoptotic nuclei were detected by staining with Hoechst 33342 (v/v at 1:200) and PI (v/v at 1:200) at 37°C for 20 minutes in the dark, followed by washing three times in PBS and examination by fluorescence microscopy.
Lactate dehydrogenase (LDH) release
The LDH assay was performed according to the manufacturer’s protocol. Briefly, K562/MDR and Jurkat cells (1×105/mL) were cultured in RPMI 1640 medium at 37°C for 4 hours and then incubated with different concentrations of myristoyl-CM4 or CM4 (3, 9, 30 μM) for different times. PBS and 2% Triton X-100 were used as controls. After centrifugation of the microtiter plates, 100 μL of cell-free culture supernatant was transferred into new microtiter plates and a tetrazolium salt reaction mixture was added to each plate, according to the manufacturer’s protocol. The absorbance of the colored formazan was determined using a microplate reader at 490 nm. LDH leakage (%) was calculated using the following equation: [(A−C)/(P−C)]×100, where A is the mean absorbance in the test wells, C is the PBS control, and P is the Triton X-100 control. Data were reported as mean±SEM of four independent experiments.
JC-1 assay
Change in mitochondrial membrane potential (Δψm) was detected using a mitochondria staining kit with JC-1 as a cationic fluorescent dye. Briefly, K562/MDR and Jurkat cells (2×106/mL) were cultured in the absence or presence of myristoyl-CM4 (3 μM for K562/MDR and 6 μM for Jurkat) for 12 hours. The cells were then collected, washed in ice-cold PBS, and incubated with 10 μg/mL for 30 minutes at room temperature. The cells were washed with JC-1 washing buffer and analyzed by flow cytometry. Flow cytometry was performed at 490 nm excitation and 530 nm emission wavelengths for JC-1 monomers and for 525 nm excitation and 590 nm emission wavelengths for JC-aggregates.
Detection of ROS accumulation
ROS accumulation was measured by detecting the fluorescence intensity of the oxidant-sensitive probe DCFH-DA. Briefly, K562/MDR and Jurkat cells (2×106/mL) were incubated with different concentrations of myristoyl-CM4 for 8 hours with Rosup as a positive control, followed by incubation with DCFH-DA (10 μM) for 30 minutes in the dark. The fluorescence intensity was then measured by flow cytometry at 488 nm to evaluate the production of ROS.
Western blotting analysis
K562/MDR and Jurkat cells were incubated with myristoyl-CM4 for 16 hours and the expression of P-glycoprotein (P-gp) was then detected. Briefly, the cells were harvested in cell lysis buffer containing protease inhibitors, followed by centrifugation at 15,000×g for 10 minutes. The supernatant was collected, separated by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto a polyvinylidene difluoride (PVDF) membrane. To detect the apoptosis pathway induced by the peptides, leukemia cells (5×106 cells) were cultured with different concentrations of myristoyl-CM4 for 16 hours, harvested in cell lysis buffer, and centrifuged at 15,000×g for 10 minutes. Supernatants were collected, separated by 12% SDS–PAGE, and transferred onto a PVDF membrane. The membranes were blocked with 5% bovine serum albumin and probed with polyclonal antibodies against P-gp, Bcl-2, Bax, caspase-9, caspase-3, PARP, β-actin, and GAPDH. Protein bands were visualized using the Odyssey infrared imaging system.
Statistical analysis
Values were expressed as mean±SEM from three-to-six independent experiments. Differences were analyzed by Two-tailed Student’s t-test and one-way ANOVA with Dunnett’s multiple comparison test. A value of P<0.05 was considered statistically significant. Statistical analysis was performed using SPSS (SPSS/PC 20.0; SPSS, Chicago, IL, USA).
Results
Myristoyl-CM4 inhibited the viability of leukemia cell lines
Drug-resistant K562/MDR cells were initially identified by doxorubicin assay and P-gp expression in the plasma membrane (Figures 1A and B). Compared with K562 cells, K562/MDR showed a certain resistence to doxorubicin. P-gp level was significantly higher in K562/MDR compared with Jurkat cells. Treatment with myristoyl-CM4 reduced the level of P-gp in a concentration-dependent manner. The anticancer activities of the peptides in the two leukemia cell lines (K562/MDR and Jurkat) were assessed by the standard MTT assay. More than 90% of K562/MDR and Jurkat cells were viable after treatment with 20 μM CM4, while myristoyl-CM4 significantly inhibited the viabilities of Jurkat and K562/MDR cells in concentration-dependent manners (Figure 1C). The reduction in cell viability was more effective in K562/MDR compared with Jurkat cells, with IC50 values of 2 μM and 3 μM, respectively. There was no difference in the toxicity of myristoyl-CM4 to K562 cells and K562/MDR cells, so K562/MDR cells were selected for further study (Figure 1D). The results of the Trypan Blue exclusion assay further supported the above results (Figure 1E), and very few viable cells were observed at a concentration of 8 μM.
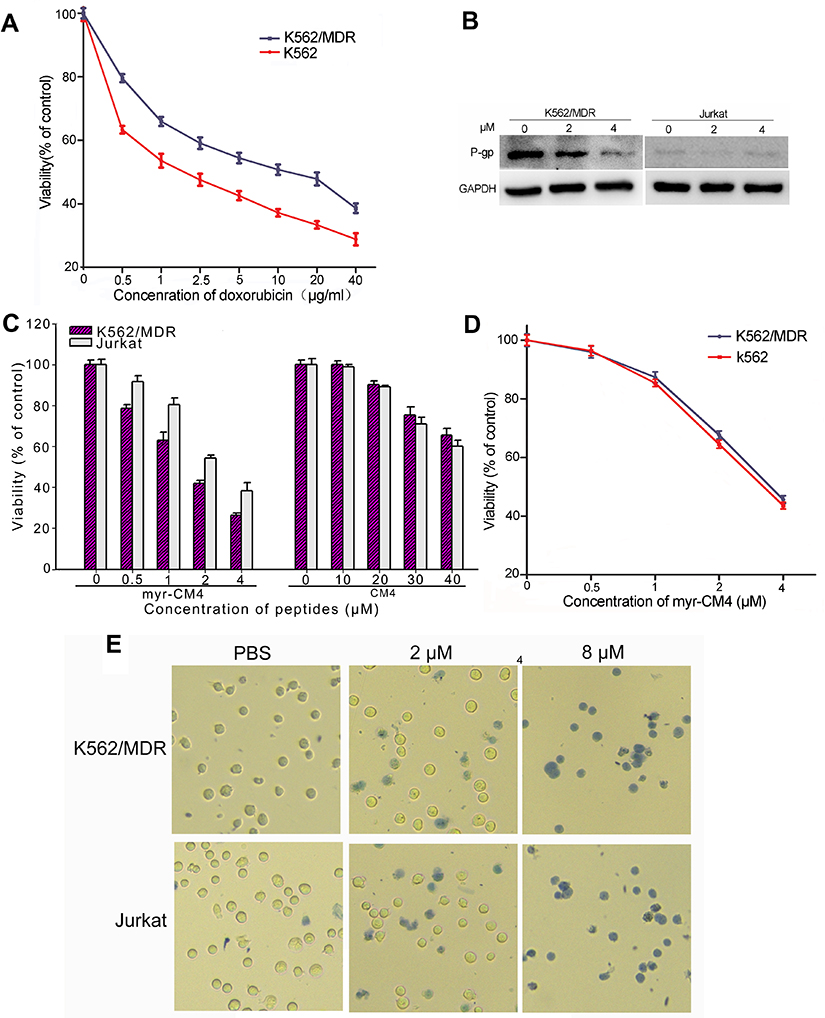 |
Figure 1 Cytotoxicity of myristoyl-CM4 in leukemia cells. K562/MDR cells were identified using doxorubicin by MTT assay (A) and P-gp expression by Western blotting (B). MTT results in K562/MDR and Jurkat cells after treatment with myristoyl-CM4 or CM4 for 24 hours (C). Cytotoxic comparison of myristoyl-CM4 in K562/MDR and K562 was conducted by MTT assay for 24 hours (D). The data represents the mean of four independent experiments (n=4±SEM). (E) Trypan Blue exclusion was observed by microscopy. |
Myristoyl-CM4 was less cytotoxic normal cells compared with leukemia cells
The cytotoxicities of CM4 and myristoyl-CM4 against normal mammalian cells (HEK293 and L02 cells) were detected by MTT assay (Figure 2A). More than 95% of cells remained viable after treatment with 10 μM myristoyl-CM4, and more than 60% after treatment with 40 μM, indicating that myristoyl-CM4 exhibited low cytotoxicity against HEK-293 and L02 cells. Hemolytic assay using mouse erythrocytes showed no hemolysis following treatment with 3 μM myristoyl-CM4 (Figure 2B), and only 2% hemolysis at a concentration of 10 μM; however, hemolysis increased to 20% at a concentration of 30 μM. The cytotoxicity of myristoyl-CM4 against mouse PBMCs was detected by Trypan Blue exclusion assay. The number of PBMCs was not reduced, and no dead PBMCs were observed after incubation for 24 hours with 2 μM myristoyl-CM4 (Figure 2C). In contrast, the AMP melittin isolated from honey bee (A. mellifera) venom resulted in 100% cytotoxicity of PBMCs at 2 μM. Treatment with 10 μM myristoyl-CM4 still resulted in more than 80% viable PBMCs, suggesting that myristoyl-CM4 showed selective cytotoxicity against leukemic K562/MDR and Jurkat cells.
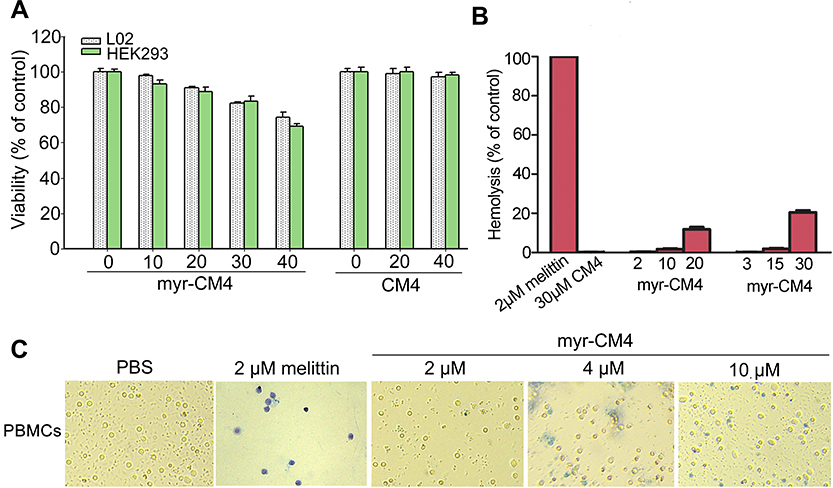 |
Figure 2 Cytotoxicity of myristoyl-CM4 in normal cells. (A) MTT results in L02 and HEK293 cells after treatment with myristoyl-CM4 for 24 hours. Results are the mean±SEM of four independent experiments. (B) Hemolytic activity was tested in mouse erythrocytes with melittin as a control. (C) PBMCs were isolated from mouse blood and incubated with myristoyl-CM4 for 24 hours, followed by the Trypan Blue exclusion assay. Abbreviation: PBMCs, peripheral blood mononuclear cells. |
Myristoyl-CM4 had higher affinity to leukemia cells compared with normal cells
The effect of the myristoyl group on the membrane affinity of myristoyl-CM4 was analyzed by FITC labeled peptide and fluorescence-activated flow cytometry. The fluorescence intensity was significantly higher than that of CM4 (Figure 3A). Compared with FITC-CM4, the FITC fluorescence intensities of myristoyl-CM4 were increased 14-fold and 19-fold in K562/MDR and Jurkat cells, respectively (Figure 3B). The fluorescence intensities of FITC-myristoyl-CM4 were very weak in normal HEK293 cells and mouse erythrocytes. The FITC fluorescence intensities in K562/MDR and Jurkat cells were 27.6-fold and 82.1-fold higher than in normal HEK293 cells, and 23.4-fold and 69.7-fold higher than in mouse erythrocytes. The binding activity was further analyzed by CLSM (Figure 3C). The FITC-myristoyl-CM4 signal on the cells was stronger than the FITC-CM4 signal after incubation for 30 minutes.
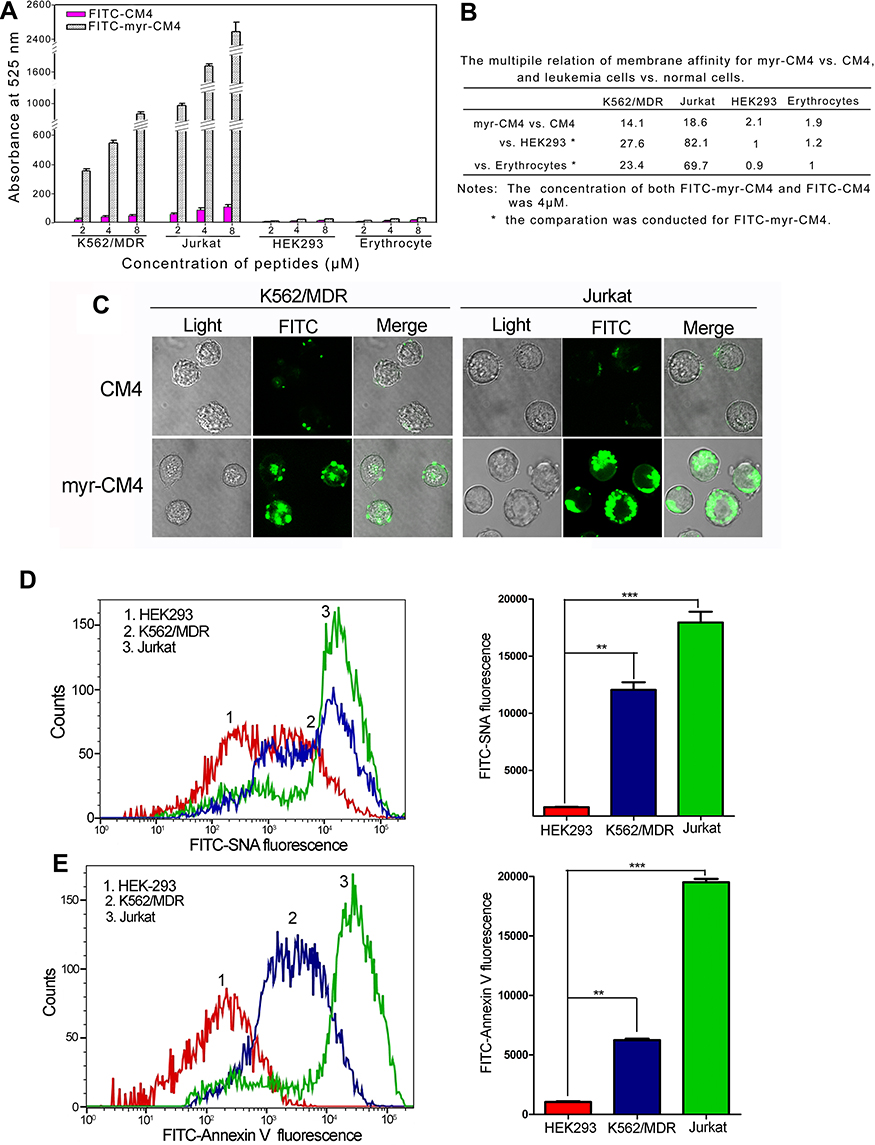 |
Figure 3 Comparison of binding activities of myristoyl-CM4 by fluorescence intensity. (A) K562/MDR, Jurkat, HEK293 cells and erythrocytes were incubated with FITC-labeled peptides for 30 minutes at 37°C and analyzed by flow cytometry. Results are mean±SEM of three different experiments. (B). Relative membrane affinities were deduced from (A). (C) Binding to K562/MDR and Jurkat cells was observed by CLSM after treatment with FITC-peptides for 30 minutes. Phosphatidylserine content (D) and sialic acid content (E) of K562/MDR, Jurkat, and HEK293 cells were detected by Annexin V-FITC staining and FITC-SNA staining, respectively, and the fluorescence was detected using flow cytometry. Results are the mean±SEM of three independent experiments. **P<0.01; ***P<0.001 between HEK293 cells and K562/MDR or Jurkat cells. Abbreviations: CLSM, confocal laser scanning microscopy; FITC, fluorescein isothiocyanate. |
The relative amounts of sialic acid on the surface of the three cell lines were determined by FITC-SNA, a specific lectin that binds to a-2,6 linked sialic acid. The a-2,6 linked sialic acid content in leukema cells (K562/MDR, and Jurkat cells) was significantly higher than that of HEK293 cells (Figure 3D). The relative amounts of PS on the surfaces of HEK-293, K562/MDR, and Jurkat cells were also determined by Annexin V-FITC, as a specific lectin that binds PS. The PS content of leukemia cells (K562/MDR and Jurkat) was significantly higher than that of HEK293 cells, as shown by flow cytometry (Figure 3E). These data suggested that myristoyl conjugation increased the membrane affinity of K562/MDR and Jurkat cells for myristoyl-CM4. Furthermore, the membrane affinities of myristoyl-CM4 to K562/MDR and Jurkat cells were markedly higher than its affinities to HEK293 cells and erythrocytes, while leukemic cells expressed more phosphatidylserine and sialylated glycoconjugate on the cell surface than normal cells.
Myristoyl-CM4 induced necrosis and/or apoptosis in leukemia cells
PI is a red fluorescent dye that selectively stains necrotic cells. Apoptosis/necrosis were analyzed by Annexin V-FITC/PI double staining. Treatment of K562/MDR and Jurkat cells with 2 μM and 3 μM myristoyl-CM4, respectively, resulted in apoptotic rates of 50.87% and 47.53% (Annexin V-FITC+), respectively (Figure 4A), while K562/MDR cells also showed 15.89% necrotic cells (Annexin V-FITC−/PI+) after myristoyl-CM4 treatment. Apoptosis and necrosis were further detected by Hoechst 33342/PI double staining. Cells undergoing apoptosis were characterized by chromatin condensation and changes in nuclear morphology, indicated by blue fluorescence of Hoechst 33342. Obvious changes in nuclear morphology and chromosome condensation were observed after myristoyl-CM4 treatment in both K562/MDR and Jurkat cells (Figure 4B). Several PI+ K562/MDR cells were also observed, but almost no PI+ Jurkat cells.
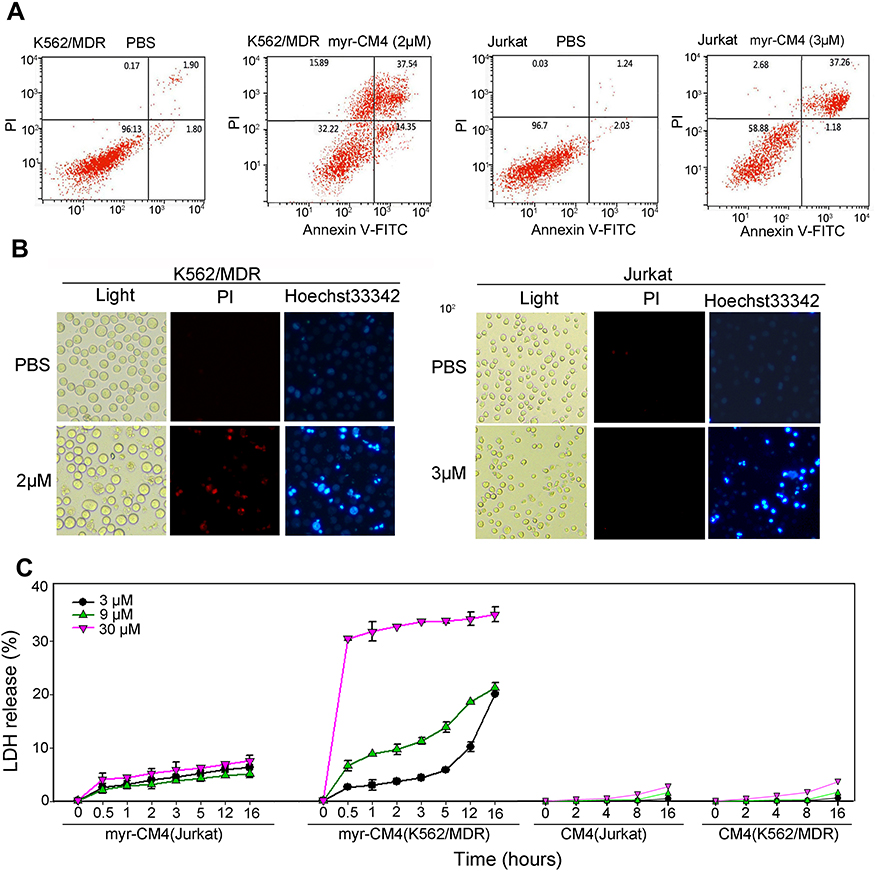 |
Figure 4 Necrosis and apoptosis of leukemia cells induced by myristoyl-CM4. (A) K562/MDR and Jurkat cells were treated with myristoyl-CM4 (2 μM for K562/MDR, 3 μM for Jurkat) for 16 hours, stained by Annexin V-FITC/PI, and analyzed by flow cytometry. (B) Cells were stained with Hoechst 33342/PI and then observed by fluorescence microscopy. (C) Membrane permeability changes were determined by monitoring LDH release from cells after incubation with different concentrations of myristoyl-CM4 (0, 3, 9, 30 μM) for different times at 37°C. CM4 was used as a control. Results are the mean±SEM of four different experiments. Abbreviations: FITC, fluorescein isothiocyanate; LDH, lactate dehydrogenase. |
LDH is an intracellular enzyme that is released when the cell membrane is disrupted. Treatment with myristoyl-CM4 for different times resulted in LDH release (Figure 4C). LDH release from Jurkat cells was less than 10% at all tested concentrations (3, 9, 30 μM), while LDH release from K562/MDR cells was about 20% after treatment with 3 μM myristoyl-CM4 for 16 hours, and 30% at 30 μM. Virtually no LDH was released from either K562/MDR or Jurkat cells after CM4 treatment. These results suggested that myristoyl-CM4 could induce both apoptosis and necrosis in K562/MDR cells, but only apoptosis in Jurkat cells.
Myristoyl-CM4 affected the mitochondria in leukemia cells
Cellular uptake of myristoyl-CM4 was examined using FITC-labeled peptides and rhodamine 123 as a specific probe for localizing mitochondria in living cells. Myristoyl-CM4 co-localized with mitochondria in K562/MDR and Jurkat cells (Figure 5A). After transport to the intracellular compartment, myristoyl-CM4 could be located on mitochondria. Disruption of mitochondrial integrity is one of the early events leading to apoptosis. We, therefore, examined changes in the mitochondrial membrane potential using the fluorescent dye, JC-1. JC-1 accumulated in intact mitochondria and formed aggregates with high Δψm, emitting red fluorescence. When the mitochondria collapse, the JC-1 fluorescence shifts from red to green. Green-fluorescence increased in K562/MDR and Jurkat cells after exposure to 2 μM and 3 μM myristoyl-CM4, respectively (Figure 5B), and the green/red ratio increased from 0.02 to 0.17 in K562/MDR cells and from 0.02 to 0.40 in Jurkat cells. These data showed an obvious shift in JC-1 fluorescence from red to green, indicating disruption of the Δψm induced by myristoyl-CM4. ROS were quantified in K562/MDR and Jurkat cells using DCFH-DA fluorescent dyes (Figure 5C). DCFH fluorescence increased after treatment with myristoyl-CM4, both in K562/MDR and Jurkat cells, compared with PBS cintrol cells, indicating obvious ROS generation in K562/MDR and Jurkat cells induced by myristoyl-CM4.
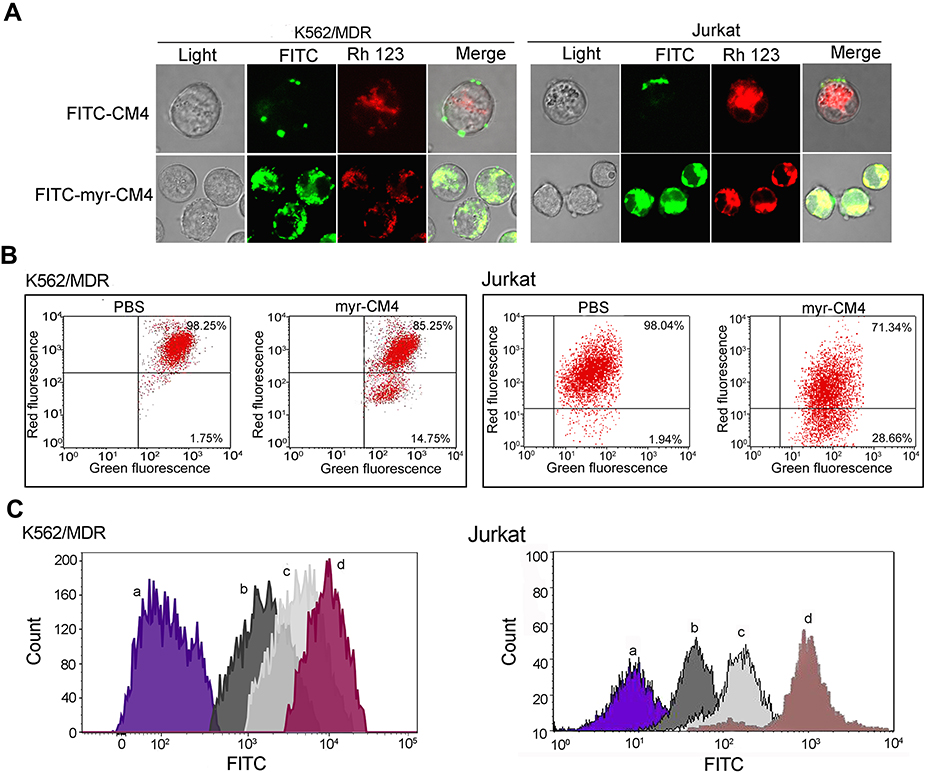 |
Figure 5 Effects of myristoyl-CM4 on the mitochondrial membrane in leukemia cells. (A) Confocal microscopic images of K562/MDR and Jurkat cells treated with FITC-myristoyl-CM4 and rhodamine 123. (B) Cells were treated with myristoyl-CM4 (2 μM K562/MDR and 3 μM Jurkat cells) for 6 hours, followed by staining with JC-1 and flow cytometry at 490 nm and 525 nm excitation. The emitted light was collected at 590 nm (red) and 530 nm (green). (C) K562/MDR were treated with 2 μM myristoyl-CM4 (b), 4 μM myristoyl-CM4 (c) for 12 hours, Jurkat cells were treated 3 μM myristoyl-CM4 (b), 6 μM myristoyl-CM4 (c) for 12 hours, and then stained with DCHF-DA (10 μM) for 30 minutes. PBS (a) and Rosup (dD) were used as controls. Cells were analyzed by flow cytometry at 488 nm excitation. Abbreviation: PBS, phosphate buffered saline. |
Myristoyl-CM4 induced mitochondria-dependent apoptosis
Bcl-2/Bax family proteins play major roles in modulating mitochondria-dependent apoptosis. Therefore, the expression of pro-apoptotic Bax, and anti-apoptotic Bcl-2 were detected in myristoyl-CM4-treated K562/MDR and Jurkat cells by Western blot analysis (Figure 6A). Myristoyl-CM4 increased the expression of pro-apoptotic Bax and decreased the expression of anti-apoptotic Bcl-2 in concentration-dependent manners in both K562/MDR and Jurkat cells (Figures 6B and C). The Bax/Bcl-2 ratio was significantly increased following myristoyl-CM4 treatment (P<0.001) (Figure 6D). These results implied that myristoyl-CM4 might induce apoptosis through a Bcl-2/Bax-mediated mitochondrial dysfunction pathway.
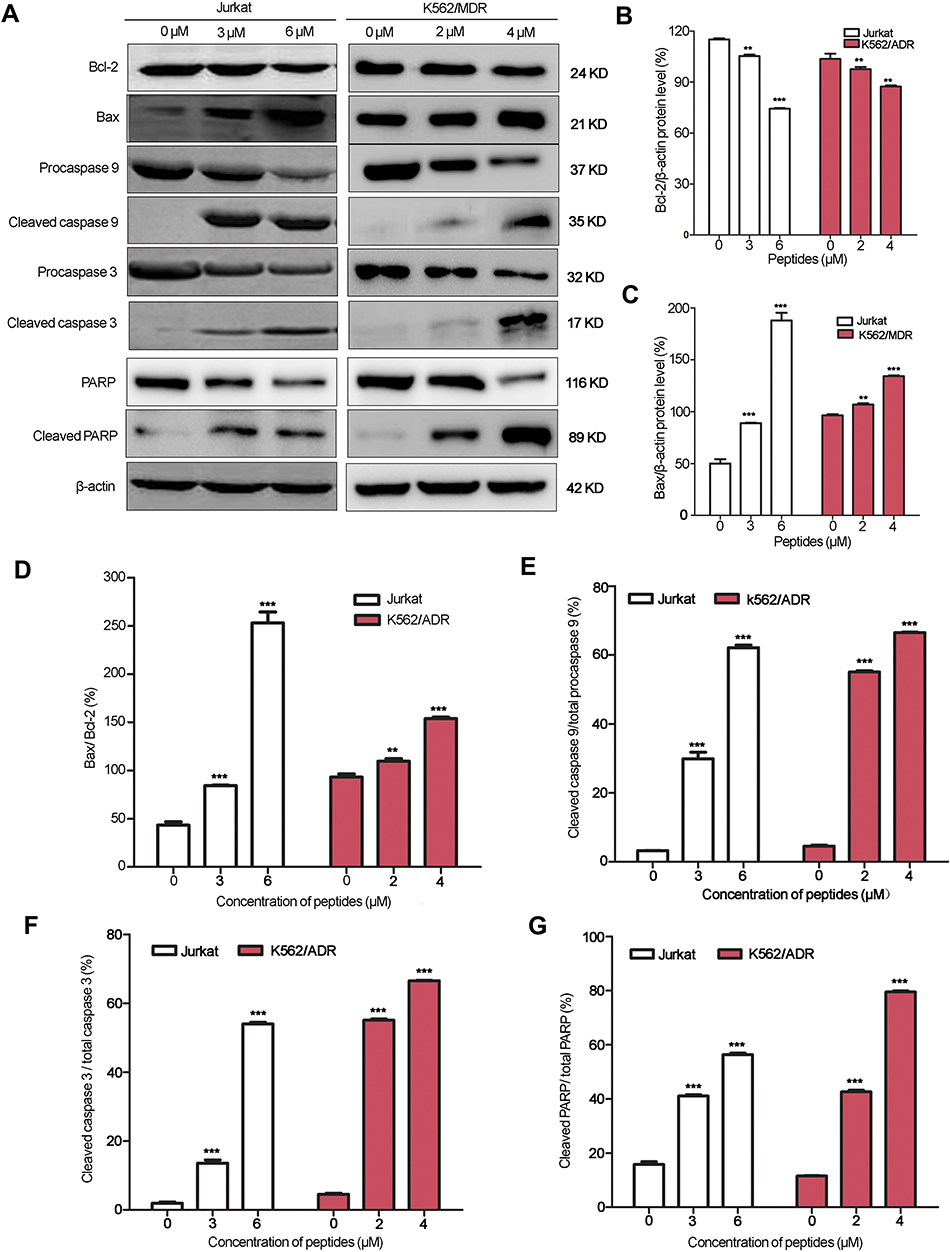 |
Figure 6 Apoptosis in leukemia cells induced by myristoyl-CM4. (A) Cells were treated with myristoyl-CM4 for 16 hours and the lysates were then harvested and apoptosis-related proteins were detected by Western blotting. The relative amounts of Bax, Bcl-2, active (cleaved) caspase 9, active (cleaved) caspase 3, and cleaved PARP relative β-actin control were determined by Western blotting and Image J densitometric analysis. The Bcl2 (B), Bax (C), Bax/Bcl-2 ratio (D), cleaved caspase 9/total caspase 9 ratio (E), cleaved caspase 3/total caspase 3 ratio (F), and cleaved PARP/total PARP ratio (G) were calculated. Results are the mean±SEM of three different experiments. **P<0.01; ***P<0.001 between PBS control and peptide treatment. Abbreviation: PBS, phosphate buffered saline. |
We further characterized the apoptotic pathway induced by myristoyl-CM4 by examining the activation of procaspase 9 (a caspase that initiates mitochondria-dependent apoptosis), and procaspase 3 (an important effector of mitochondria-mediated apoptosis pathway) by Western blot analysis. Myristoyl-CM4 significantly increased levels of active (cleaved) caspase 9, and active (cleaved) caspase 3. About 55% of procaspase 9 and 55% procaspase 3 were activated in K562/MDR cells after treatment with 2 μM myristoyl-CM4 (Figures 6E and F). The cleaved caspase 9/total caspase 9 and cleaved caspase 3/total caspase 9 ratios were both significantly increased in Jurkat cells after treatment with 3 μM or 6 μM myristoyl-CM4. More than 40% of PARP was finally cleaved by 3 μM myristoyl-CM4 in Jurkat cells or by 2 μM myristoyl-CM4 in K562/MDR cells (Figure 6G). These results indicated that myristoyl-CM4 could induce the mitochondria-dependent apoptotic signaling in both Jurkat and multi-drug resistant K562/MDR leukemia cells.
Discussion
In addition to their innate immune effect in pathogenic microbes, some AMPs can also target mammalian cells and may display selective activity towards tumor cells, making them attractive novel candidate anticancer agents.22 AMPs such as LfcinB, cecropin A, PaDef defensin, and Arminin 1α-C have shown anticancer activity against various leukemia cells, with IC50 values ranging from 10–60 μM.9–12 In the current study, CM4 showed very weak cytotoxicity against K562/MDR and Jurkat leukemia cells, but this cytotoxicity was increased by attaching a myristoyl to its N-terminus of CM4, with IC50 values of 2 μM and 3 μM in K562/MDR and Jurkat cells, respectively. As the first binding target, the plasma membrane plays an important role in the anticancer activity of AMPs, and interactions with the plasma membrane, leading to its disruption or permeation, represent a major anticancer mechanism for most AMPs. The myristoyl group is hydrophobic, and we speculated that the enhanced cytotoxicity of myristoyl-CM4 compared with CM4 may result from enhanced interaction between the peptide and the plasma membrane. Indeed, the binding of myristoyl-CM4 was increased 14-fold and 19-fold in K562/MDR and Jurkat cells, respectively, compared with CM4. As a widely-recognized form of protein fatty-acylation, myristoylation plays essential roles in directing the cellular localization of proteins by facilitating protein–membrane interactions. The myristoyl group may act as a lipid anchor in cell membranes for the N-myristoylated-protein, allowing peptides with high hydrophobicity to penetrate deeper into the hydrophobic core of the cell membrane.17,23 The current results were in accord with our previous study in breast cancer, which also found that myristoyl-CM4 showed enhanced binding to breast cancer cells.21 Myristoylation is, thus, an effective way of enhancing the hydrophobic interactions between AMPs and cancer cells. Moreover, the interaction between myristoyl-CM4 and K562/MDR was stronger than that with Jurkat cells, due to direct disruption of the K562/MDR cell membrane by myristoyl-CM4. Conventional chemotherapy for leukemia is hindered by the lack of selectivity and consequent targeting of healthy mammalian cells, leading to deleterious side-effects. However, the IC50 values indicated that myristoyl-CM4 had no cytotoxic effects in healthy human HEK293 and L02 cells, erythrocytes, or PBMCs. More than 95% of healthy HEK293, L02 cells, and mouse erythrocytes, and 80% of mouse PBMCs remained viable after treatment with myristoyl-CM4 at a concentration of five times to IC50 for K562/MDR cells, suggesting that myristoyl-CM4 showed selective cytotoxicity against K562/MDR and Jurkat cells. Further investigations showed that the binding activity of myristoyl-CM4 to HEK293 cells was only 1/27.6 and 1/82.1 of those to K562/MDR and Jurkat cells, and the binding activity to erythrocytes was only 1/23.4 and 1/69.7 of that in K562/MDR and Jurkat cells. This weak binding activity may be an important reason for its lack of cytotoxicity in healthy cells. We previously observed a similar phenomenon in breast cancer cells,21 however, myristoyl-CM4 showed stronger affinity to K562/MDR and Jurkat cells than to breast cancer cells. Sialic acid-containing molecules (such as O-mucin and gangliosides) are highly expressed in breast cancer cells and contribute to the binding of cationic AMPs to breast cancer cells.21,24 In the current study, we detected the sialic acid expression on the surface of leukemia cells, and found the sialic acid content in leukema cells (K562/MDR and Jurkat cells) was significantly higher than that of HEK293 cells. We also detected the phosphatidylserine content. Phosphatidylserine is a negatively charged phospholipid present in significant proportions in the inner monolayer of the plasma membrane of normal cells, Certain tumor cells show elevated levels of phosphatidylserine on the outer membrane leaflet, which may contribute to the selective cytotoxicity to cancer cells.25–27 Interestingly, we found that the PS contents of the outer membrane leaflets of both K562/MDR and Jurkat cells were higher than in normal PBMCs and HEK293 cells. Thus, the higher contents of sialic acid and PS may explain the increased binding activity of myristoyl-CM4 to K562/MDR and Jurkat leukemia cells compared with healthy normal cells. All our current data supported the observed higher cytotoxicity of myristoyl-CM4 against leukemia compared with healthy cells. The hydrophibicity, amphibicity, and length of native CM4 prevented it from internalizing in leukemia cells, whereas myristoyl-CM4 could enter into cells, including leukemia and breast cancer cells. This implied that myristoyl was crucial for cellular entry, highlighting the importance of hydrophobic interactions. Regarding the role of myristoylation in membrane binding, in order to achieve stable membrane binding, N-myristoylated proteins employ a second signal, such as polybasic regions, hydrophobic residues, another membrane-bound binding partner, or a second lipid modification.28,29 CM4 is a cationic molecule (net charge +5) with an amphipathic α-helix at the N-terminus and hydrophobic α-helix at the C-terminus.16 We speculated that the hydrophobic interaction of myristoyl-CM4 with the lipid bilayer membrane of leukemia cells may result from both the myristoyl group and the hydrophobic helix. Indeed, myristoyl-CM4 was shown to be more hydrophobic than CM4 by reverse phase-high-performance liquid chromatography (data not shown). Nelson et al30 revealed conjugated to the 14-carbon myristate group was efficiently loaded into lymphocyte-derived cells. Furthermore, myristoylated virus proteins played an important role in membrane fusion and host entry.31 Lipid modification was previously shown to enhance the passive transport of some drugs mediated by the lipid moiety, and increased antitumor efficacy could be achieved by increasing cell permeability and retention of anticancer agents using lipid-conjugation strategies.32 Myristoylation may, thus, be a useful tool for transporting AMPs into the interior of cancer cells.
Necrosis and apoptosis are the two main anticancer mechanisms of most α-helical AMPs.33 The present results showed that, after internalization into leukemia cells, myristoyl-CM4 could target mitochondria and affect their function, including disrupting the Δψm, increasing ROS accumulation, and the Bax/Bcl-2 ratio, then activating of caspases 9 and 3, and cleaving PARP to induce apoptosis. It has been suggested that leukemia is characterized by an impaired balance between the proliferation of blood cells and their capacity to undergo apoptosis, and the Bcl-2 family of anti-apoptotic (such as Bcl-2) and pro-apoptotic (such as Bax) proteins are critical regulators of apoptosis in leukemia.34 Both Bax and Bcl-2, as well as Bax/Bcl-2 ratio, have been regarded as prognostic markers in various cancers, including leukemia.35 We, therefore, examined the expression of Bax and Bcl-2 proteins. Bax levels were very low in Jurkat cells, and the Bax/Bcl-2 ratio was about 0.5. However, 6 μM myristoyl-CM4 increased the Bax level, and the Bax/Bcl-2 ratio increased to 2.5. Myristoyl-CM4 also increased the level of Bax and Bax/Bcl-2 ratio and decreased the level of Bcl-2 in K562/MDR cells. As a central cell death regulator, Bax is an indispensable gateway to mitochondrial dysfunction and a major pro-apoptotic member of the Bcl-2 family proteins.36 Increased levels of the Bax and an increased Bax/Bcl-2 ratio thus contributed to apoptosis of K562/MDR and Jurkat cells. In contrast, myristoyl-CM4 decreased the levels of Bcl-2 in K562/MDR and Jurkat cells, with Jurkat cells being more sensitive than K562/MDR cells. Among the Bcl-2 family, Bcl-2 is over-expressed in acute and chronic leukemias and seems to play an important role, not only in disease pathogenesis, but also in resistance to chemotherapy.37,38 Myristoyl-CM4 may, thus, be an effective candidate agent for activating anti-apoptotic Bax proteins.
Lower concentrations of AMPs have previously been reported to promote apoptosis of cancer cells, while higher concentrations primarily resulted in cancer cell death through membrane disruption.39 Wang and Zhou40 found that the AMPs temporin-1CEɑ could directly disrupt the plasma membrane and cause lysis of breast cancer cells at higher concentrations, but initiated cell death by an intracellular mechanism at lower concentrations. Interestingly, myristoyl-CM4 induced both mitochondria-mediated apoptosis and cell membrane disruption in K562/MDR cells at lower concentrations (3 μM), suggesting that it had a stronger interaction with the cell membrane in K562/MDR cells compared with Jurkat cells. Myristoyl-CM4 also exerted an obvious direct membrane-disrupting effect at higher concentrations (30 μM). Chemotherapy serves an essential role in leukemia treatment; however, it is frequently accompanied by the development of multidrug resistance, resulting in treatment failure. Overexpression of P-gp results in the export of chemotherapeutic agents from cancer cells, thus preventing the intracellular accumulation of active drug such as doxorubicin.41,42 We showed that K562/MDR cells were resistant to doxorubicin, and confirmed that they expressed high levels of P-gp in the plasma membrane, which were significantly decreased by myristoyl-CM4. Moreover, myristoyl-CM4 showed strong cytotoxicity against K562/MDR leukemia cells via at least two mechanisms (necrosis and apoptosis). These results implied that myristoyl-CM4 may be a potential candidate for overcoming P-gp-mediated multidrug resistance, leading to better outcomes for leukemia chemotherapy.
Conclusion
In summary, the current study showed that myristoyl-CM4 had effective cytotoxicity against leukemia cell lines (K562/MDR and Jurkat) and had less cytotoxicity to normal cells (HEK-293, L02 cells, mouse PMBCs, and erythrocytes). This anticancer activity may be attributable to the high affinity of myristoyl-CM4 to the leukemia cell membrane and its internalization within leukemia cells. The membrane affinities of myristoyl-CM4 to K562/MDR and Jurkat cells were markedly higher than its affinities to HEK293 and erythrocytes, and leukemic cells expressed more phosphatidylserine and sialylated glycoconjugate on the cell surface than normal cells, which could explain its selective cytotoxicity to cancer cells. Myristoyl-CM4 affected mitochondrial function to induce mitochondria-dependent apoptosis in both K562/MDR and Jurkat cells, but also induced necrosis in K562/MDR cells by direct membrane disruption. Myristoyl-CM4, thus, appears to be a promising candidate for an anti-leukemia agent, particularly in relation to multidrug-resistant leukemia.
Abbreviation list
MTT, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide; Rho123, Rhodamine 123; DAPI, 4,6-diamidino-2-phenylindole; DMEM, Dulbecco’s Modified Eagle’s Medium; ROS, Reactive Oxygen Species; DCFH-DA, 2ʹ,7ʹ-dichlorofluorescin-diacetate; PI, propidium iodide; LDH, Lactate dehydrogenase; ROS, Reactive oxygen species; PBMCs, Peripheral blood mononuclear cells; PBS, Phosphate buffer saline; PS, phosphatidylserine; PARP, poly-ADP ribose polymerase; GAPDH, lyceraldehyde 3-phosphate dehydrogenase.
Acknowledgments
This work was financially supported by a grant of the National Natural Science Foundation of China (Grant No. 81573337, 30900743), the Natural Science Foundation of Jiangsu Province of China (Grant No. BK20141446, BK2011368), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Szebeni GJ, Balog JA, Demjén A, et al. Imidazo[1,2-b]pyrazole-7-carboxamides induce apoptosis in human leukemia cells at nanomolar concentrations. Molecules. 2018;23(11):
3. Fava C, Morotti A, Dogliotti I, et al. Update on emerging treatments for chronic myeloid leukemia. Expert Opin Emerg Drugs. 2015;20(2):183–196. doi:10.1517/14728214.2015.1031217
4. Kayser S, Levis MJ. Advances in targeted therapy for acute myeloid leukaemia. Br J Haematol. 2018;180(4):484–500.5. doi:10.1111/bjh.15032
5. Talati C, Pinilla-Ibarz J. Resistance in chronic myeloid leukemia: definitions and novel therapeutic agents. Curr Opin Hematol. 2018;25(2):154–161.
6. Emadi A, Gore SD. Arsenic trioxide - An old drug rediscovered. Blood Rev. 2010;24(4–5):191–199.
7. Deslouches B, Di YP. Antimicrobial peptides with selective antitumor mechanisms: prospect for anticancer applications. Oncotarget. 2017;8(28):46635–46651.
8. Riedl S, Leber R, Rinner B, et al. Human lactoferricin derived di-peptides deploying loop structures induce apoptosis specifically in cancer cells through targeting membranous phosphatidylserine. Biochim Biophys Acta. 2015;1848:2918–2931.
9. Mader JS, Richardson A, Salsman J, et al. Bovine lactoferricin causes apoptosis in Jurkat T-leukemia cells by sequential permeabilization of the cell membrane and targeting of mitochondria. Exp Cell Res. 2007;313(12):2634–2650.
10. Cerón JM, Contreras-Moreno J, Puertollano E, et al. The antimicrobial peptide cecropin A induces caspase-independent cell death in human promyelocytic leukemia cells. Peptides. 2010;31(8):1494–1503. doi:10.1016/j.peptides.2010.05.008
11. Yan J, Liang X, Bai C, et al. NK-18, a promising antimicrobial peptide: anti-multidrug resistant leukemia cells and LPS neutralizing properties. Biochimie. 2018;147:143–152. doi:10.1016/j.biochi.2018.02.001
12. Flores-Alvarez LJ, Guzmán-Rodríguez JJ, López-Gómez R, et al. PaDef defensin from avocado (Persea americana var. drymifolia) is cytotoxic to K562 chronic myeloid leukemia cells through extrinsic apoptosis. Int J Biochem Cell Biol. 2018;99:10–18. doi:10.1016/j.biocel.2018.03.013
13. Zhang J, Wu X, Zhang SQ. Antifungal mechanism of antibacterial peptide, ABP-CM4, from Bombyx mori against Aspergillus niger. Biotechnol Lett. 2008;30(12):2157–2163. doi:10.1007/s10529-008-9819-x
14. Li JF, Zhang J, Xu XZ, et al. The antibacterial peptide ABP-CM4: the current state of its production and applications. Amino Acids. 2012;42(6):2393–2402. doi:10.1007/s00726-011-0982-3
15. Lin QP, Zhou LF, Li NN, et al. Lipopolysaccharide neutralization by the antibacterial peptide CM4. Eur J Pharmacol. 2008;596(1–3):160–165. doi:10.1016/j.ejphar.2008.08.017
16. Chen YQ, Min C, Sang M, et al. A cationic amphiphilic peptide ABP-CM4 exhibits selective cytotoxicity against leukemia cells. Peptides. 2010;31(8):1504–1510. doi:10.1016/j.peptides.2010.05.010
17. Martin DD, Beauchamp E, Berthiaume LG. Post-translational myristoylation: fat matters in cellular life and death. Biochimie. 2011;93(1):18–31. doi:10.1016/j.biochi.2010.10.018
18. Avrahami D, Shai Y. A new group of antifungal and antibacterial lipopeptides derived from non-membrane active peptides conjugated to palmitic acid. J Biol Chem. 2004;279(13):12277–12285. doi:10.1074/jbc.M312260200
19. Chu-Kung AF, Nguyen R, Bozzelli KN, Tirrell M. Chain length dependence of antimicrobial peptide-fatty acid conjugate activity. J Colloid Interface Sci. 2010;345(2):160–167. doi:10.1016/j.jcis.2009.11.057
20. Mathew B, Nagaraj R. Antimicrobial activity of human α-defensin 5 and its linear analogs: N-terminal fatty acylation results in enhanced antimicrobial activity of the linear analogs. Peptides. 2015;71:128–140. doi:10.1016/j.peptides.2015.07.009
21. Li C, Liu H, Yang Y, et al. N-myristoylation of antimicrobial peptide CM4 enhances its anticancer Aactivity by interacting with cell membrane and targeting mitochondria in breast cancer cells. Front Pharmacol. 2018;9:1297. doi:10.3389/fphar.2018.01297
22. Baxter AA, Lay FT, Poon IKH, et al. Tumor cell membrane-targeting cationic antimicrobial peptides: novel insights into mechanisms of action and therapeutic prospects. Cell Mol Life Sci. 2017;74:3809–3825. doi:10.1007/s00018-016-2440-6
23. Chen Y, Guarnieri MT, Vasil AI, et al. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob Agents Chemother. 2017;51:1398–1406. doi:10.1128/AAC.00925-06
24. Han YY, Han DJ, Zong XC, et al. Role of glycosylation in the anticancer activity of antibacterial peptides against breast cancer cells. Biochem Pharmacol. 2013;86(9):1254–1262. doi:10.1016/j.bcp.2013.08.008
25. Dong X, Shi J, Zhou J, et al. Chemotherapy induces enhanced procoagulant activity through phosphatidylserine exposure in acute lymphoblastic leukemia. Thromb Res. 2013;132(5):614–620. doi:10.1016/j.thromres.2013.09.010
26. Leite NB, Aufderhorst-Roberts A, Palma MS, et al. PE and PS lipids synergistically enhance membrane poration by apPeptide with anticancer properties. Biophys J. 2015;109(5):936–947. doi:10.1016/j.bpj.2015.07.033
27. Sharma B, Kanwar SS. Phosphatidylserine: a cancer cell targeting biomarker. Semin Cancer Biol. 2018;52:17–25. doi:10.1016/j.semcancer.2017.08.012
28. Resh MD. Covalent lipid modifications of proteins. Curr Biol. 2013;23(10):R431–R435. doi:10.1016/j.cub.2013.04.024
29. Resh MD. Fatty acylation of proteins: the long and the short of it. Prog Lipid Res. 2016;63:120–131. doi:10.1016/j.plipres.2016.05.002
30. Nelson AR, Borland L, Allbritton NL, et al. Myristoyl-based transport of peptides into living cells. Biochemistry. 2007;46(51):14771–14781. doi:10.1021/bi701295k
31. Moss B. Membrane fusion during poxvirus entry. Semin Cell Dev Biol. 2016;60:89–96. doi:10.1016/j.semcdb.2016.07.015
32. Zaro JL. Lipid-based drug carriers for prodrugs to enhance drug delivery. AAPS J. 2015;17(1):83–92. doi:10.1208/s12248-014-9670-z
33. Huang Y, Feng Q, Yan Q, Hao X, Chen Y. Alpha-helical cationic anticancer peptides: a promising candidate for novel anticancer drugs. Mini Rev Med Chem. 2015;15(1):73–81.
34. Scarfò L, Ghia P. Reprogramming cell death: BCL2 family inhibition in hematological malignancies. Immunol Lett. 2013;155(1–2):36–39. doi:10.1016/j.imlet.2013.09.015
35. Kulsoom B, Shamsi TS, Afsar NA, Memon Z, Ahmed N, Hasnain SN. Bax, Bcl-2, and Bax/Bcl-2 as prognostic markers in acute myeloid leukemia: are we ready for Bcl-2-directed therapy? Cancer Manag Res. 2018;10:403–416. doi:10.2147/CMAR.S154608
36. Liu Z, Ding Y, Ye N, Wild C, Chen H, Zhou J. Direct activation of Bax protein for cancer therapy. Med Res Rev. 2016;36(2):313–341. doi:10.1002/med.21379
37. Carter BZ, Mak PY, Mu H, et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci Transl Med. 2016;8(355):355ra117. doi:10.1126/scitranslmed.aaf0746
38. Tzifi F, Economopoulou C, Gourgiotis D, et al. The role of BCL2 family of apoptosis regulator proteins in acute and chronic leukemias. Adv Hematol. 2012;2012:524308. doi:10.1155/2012/524308
39. Paredes-Gamero EJ, Martin MN, Cappabianco FA, et al. Characterization of dual effects induced by antimicrobial peptides: regulated cell death or membrane disruption. Biochimica et Biophysica Acta. 2012;1820(7):1062–1072. doi:10.1016/j.bbagen.2012.02.015
40. Wang C, Zhou Y. Anticancer mechanisms of temporin-1CEa, an amphipathic α-helical antimicrobial peptide, in Bcap-37 human breast cancer cells. Life Sci. 2013;92:1004–1014.
41. Dewanjee S, Dua TK, Bhattacharjee N, et al. Natural products as alternative choices for P-glycoprotein (P-gp) inhibition. Molecules. 2017;22(6):
42. Min H, Niu M, Zhang W, et al. Emodin reverses leukemia multidrug resistance by competitive inhibition and downregulation of P-glycoprotein. PLoS One. 2017;12(11):e0187971.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.