")
Back to Journals » International Journal of General Medicine » Volume 14
Novel Mutations in NPC1 are Associated with Pelizaeus-Merzbacher-Like Disease: A Case Report
Received 25 November 2020
Accepted for publication 22 February 2021
Published 9 March 2021 Volume 2021:14 Pages 797—803
DOI https://doi.org/10.2147/IJGM.S293675
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Hongling Fu,1,2 Qiu Wang,2,3 Hanmin Liu1,2
1Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, Sichuan, People’s Republic of China; 2Key Laboratory of Birth Defect and Related Diseases of Women and Children (Sichuan University), Ministry of Education, West China Second University Hospital, Sichuan University, Chengdu, Sichuan, People’s Republic of China; 3Department of Rehabilitation Medicine, West China Second University Hospital, Sichuan University, Chengdu, Sichuan, People’s Republic of China
Correspondence: Hanmin Liu
Department of Pediatrics, West China Second University Hospital, Sichuan University, No. 20, 3rd Section, Ren Min Nan Lu, Chengdu, 610041, People’s Republic of China
Email [email protected]
Abstract: Pelizaeus-Merzbacher-like disease (PMLD) is an autosomal recessive hypomyelinating leukodystrophy with clinical symptoms and imaging manifestations similar to those of Pelizaeus-Merzbacher disease (PMD), an X-linked recessive hypomyelinating leukodystrophy. Typical manifestations of PMLD are nystagmus, dysmyotonia, ataxia, progressive motor dysfunction, and diffuse leukodystrophy on magnetic resonance imaging (MRI). This report identified novel mutations in NCP1 causing PMLD. A 7-month-old male patient was referred to our hospital because he could not lift his head until that time. He had symptoms including congenital nystagmus, hypotonia, and developmental delay. According to the MRI scan, there were signs of leukodystrophy. According to the clinical manifestations and the results of whole-exome sequencing (compound heterozygote mutations in NPC1 (p. G911S, c2731G>A and p. D128H, c382G>C)), the diagnosis of PMLD was considered, and his parents were determined to be carriers of mutant genes. He began rehabilitation training at the age of 1 year old. After 5 years of training, he was still experiencing global developmental delay, equivalent to the developmental level of a nine-month-old child. PMLD is a disease that seriously affects the quality of life of children and can result from mutations in different genes. In this report, we expand the gene spectrum of PMLD and suggest early genetic counselling for suspected patients and their patients.
Keywords: PMLD, hypomyelinating leukodystrophy, NPC, compound heterozygote mutations
Introduction
Pelizaeus-Merzbacher disease (PMD; MIM#312080) is an X-linked recessive hypomyelinating leukodystrophy-1 (HLD1) that presents in infancy with hypotonia, nystagmus, cognitive impairment, ataxia, severe spasticity, and respiratory distress.1 The disorder is caused by mutations or rearrangements in the PLP1 gene, located on chromosome Xq22 and encoding myelin proteolipid protein – a major and essential component of the CNS.1,2 The prevalence of PMD is estimated to be 1: 300,000–1: 500,000 among American males. Pelizaeus-Merzbacher-like disease (PMLD; MIM#608804), with clinical manifestations associated with hypomyelination, is an autosomal recessive disorder. To date, PMLD has been termed a disorder with a clinically similar phenotype associated with hypomyelination but not linked to PLP1.3 Therefore, PMLD can show genetic heterogeneity depending on different mutant genes.
In our case, we found mutations in NPC1 (p. G911S, c2731G>A and p. D128H, c382G>C) in a patient with similar clinical characteristics of PMLD (Figures 1 and 2). NPC1, encoding a transmembrane protein of the acidic compartment, has been associated with Niemann-Pick type C1 (NPC1) disease. Niemann-Pick disease type C (NPC) is an autosomal recessive lysosomal lipid storage disease caused by mutations of the NPC1 gene.3,4 It mainly affects the internal organs and nervous system. Persistent neonatal cholestatic jaundice, hepatosplenomegaly, dystaxia, dysphagia, dysarthria and dysgnosia are characteristic manifestations of the disorder.5 Patients vary in age from newborn to adult, though the disease manifests more often in childhood. Moreover, due to the age of onset, the range of lesions involved is not consistent, and the clinical manifestations have obvious heterogeneity.6 Generally, the earlier the age of onset, the more serious the state of the disease. Although the first neurological symptoms are different and there is obvious clinical heterogeneity, prolonged neonatal cholestasis, hepatosplenomegaly or isolated cataplexy, splenomegaly, and vertical supranuclear gaze palsy are typical symptoms of the disease. The dysfunction of NPC genes, such as NPC1 and NPC2, leads to impaired cholesterol trafficking and thus causes intracellular accumulation of cholesterol and glycolipids in late endosomes and lysosomes.7 Dysfunctional cellular autophagy, defective lysosomal calcium homeostasis and oxidative stress play important roles in the pathophysiological processes of the disease.
 |
Figure 1 c2731G>A in exon 18 (chr18: 21119839) caused point mutation which resulted in serine replaced by glycine (p.G911S). |
 |
Figure 2 c382G>C in exon 4 (chcr18: 21148868) caused point mutation which resulted in histidine replaced by aspartic acid (p.D128H). |
In our case, we noticed a male child who had a typical PMLD phenotype; on an MRI scan, there were signs of leukodystrophy. We performed whole-exome sequencing (WES) in this patient and found mutations in NPC1 rather than variants in genes attributed to PMD or PMLD, such as PLP1 or GJC2. Moreover, no mutations related to leukodystrophy were identified. Therefore, our findings expand the genetic spectrum of PMLD.
Ethical Approval
The patients’ parents (as his legal guardian) provided signed informed consent for genetic analysis and publication. Our study was approved by the Ethics Committee of West China Second Hospital of Sichuan University, Chengdu, China.
Case Presentation
The patient was G2P1 (the mother had a history of induced abortion), born by vaginal delivery. His father was 27 years old and his mother was 26 years old when he was born. During pregnancy, the mother had threatened abortion before 12 weeks, and she took Chinese medicine as antiparturition therapy. The delivery went smoothly. The patient’s gestational age was 39+2 W, and his birth weight was 3350 g. Moreover, the patients denied a history of neonatal asphyxia, jaundice, convulsion, and related symptoms during the patient’s neonatal period, but when mouth feeding, the patient often choked on milk and spit milk and experienced overflow. The patient was first referred to neurology when he was 7 months old because he could not lift his head until that time. Our doctor performed a physical examination, which revealed that he could not hold his head to maintain a neutral position or sit by himself. He also had horizontal nystagmus and hypotonia. MRI of the patient’s brain at 7 months of age showed abnormal symmetry signals of the bilateral pallidus, diffuse symmetry abnormal white matter signals of the bilateral cerebral hemispheres and cerebellum, and delayed development of the brain myelin sheath, suggesting the possibility of hereditary metabolic encephalopathy (Figures 3 and 4). Urine metabolism testing showed that there were increases in Leu/Ala and C4/C3 and decreases in Thr, C0/C3 and C3/C2. No specific fatty acid metabolism abnormalities were suspected. The peripheral nerve function of the patient’s lower limbs was normal. DNA microarray examination showed no chromosomal abnormalities at 1440 genetic sites on 46 chromosomes. Whole-exome sequencing (WES) showed no mutations in genes related to PMD and PMLD (such as PLP1 and GJC2) or variants in genes related to leukodystrophy, such as HSPD1. The MRI scans were repeated when he was 9 months and 18 months old. The images at the age of 9 months displayed diffusely and symmetrically abnormal signals in the white matter in the bilateral cerebral hemispheres, cerebellar abnormality, and bilateral pallidus, suggesting myelin formation disorder (Figures 5 and 6).
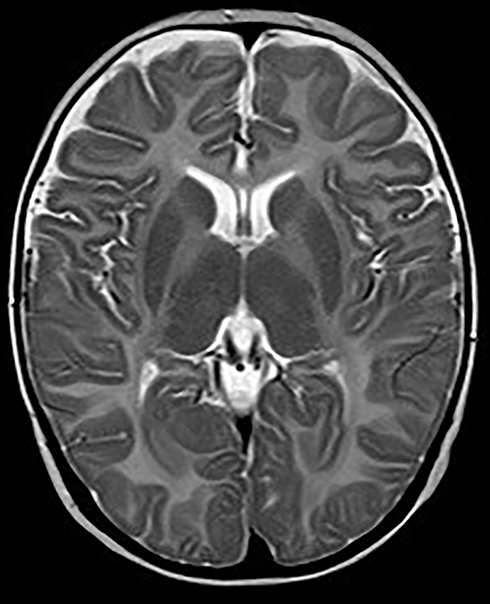 |
Figure 3 Axial MRI images at age of 7 months showing abnormal symmetry signals of bilateral pallidus. |
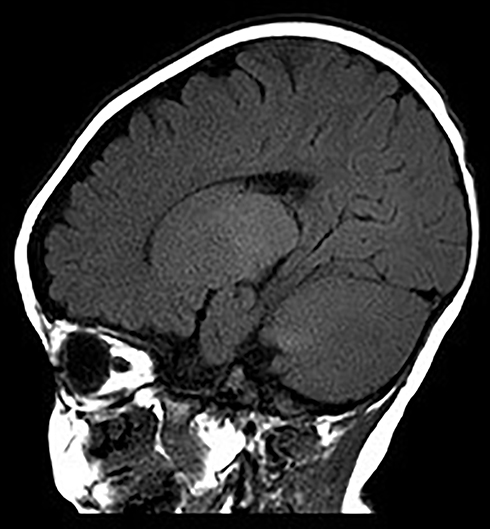 |
Figure 4 Sagittal MRI images at age of 7 months showing diffuse symmetry abnormal white matter signals of bilateral cerebral hemispheres and cerebellum, and delayed development of brain myelin sheath. |
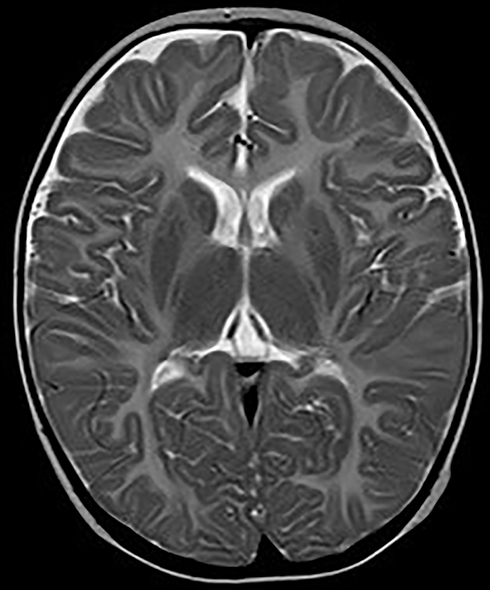 |
Figure 5 Axial MRI images at age of 9 months showing displayed diffusely and symmetrically abnormal signal in bilateral pallidus. |
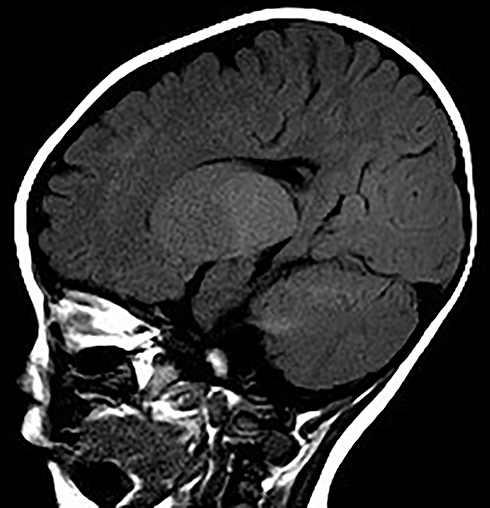 |
Figure 6 Sagittal MRI images at age of 9 months showing displayed diffusely and symmetrically abnormal signal in the white matter in bilateral cerebral hemispheres, cerebellar. |
The patient began rehabilitation training at the age of 1 year old. He is 6 years old now and has been treated for 5 years. Rehabilitation assessment suggests that his motor and mental scales are lagging significantly behind. To date, he can only say 1–2 meaningful words, such as consciously call mom and dad, but the pronunciation is not very clear. He can understand simple instruction; for example, he can roll over with difficulty when he hears the instruction “roll over”. Sitting alone remains impossible, and eating is completely dependent on help. The symptom of nystagmus has improved. Other symptoms include hypermyotonia of both lower extremities, talipes varus and inflexion.
As mentioned above, the patient had early-onset nystagmus, dysmyotonia, motor and mental retardation and absence of myelination. All of the above features tended to suggest the diagnosis of PMD; however, whole-exome sequencing did not provide the genetic basis for PMD; thus, then we established Pelizaeus-Merzbacher-like disease (PMLD) as the patient’s primary diagnosis.
Genetic Results
Whole-exome sequencing in our patient revealed compound heterozygous mutations in NCP1 (p. G911S, c2731G>A and p.D128H, c382G>C). c2731G>A in exon 18 (chr18: 21119839) caused a point mutation that resulted in replacement of glycine with serine at position 911 (p.G911S). Point mutation of c382G>C in exon 4 (chcr18: 21148868) caused replacement of aspartic acid with histidine at position 128. The variants have not been reported according to the HGMDpro database. Moreover, we found no mutations in hypomyelinating leukodystrophy-associated genes, such as PLP1, GJC2, TUBB4A, GLB1, HEXA, HEXB, ERCC2, POLR3A, or RARS.8 According to the family’s genetic screening, the patient’s father had a mutation in NPC1 – c2731G>A in exon 18 (chr18: 21119839), and the patient’s mother also had a mutation in NPC1 – c382G>C in exon 4 (chr18: 21148868) [Figures 7 and 8]. Thus, the complex heterozygous mutations in our patient were inherited from his father and mother, complying with Mendel’s law of inheritance.
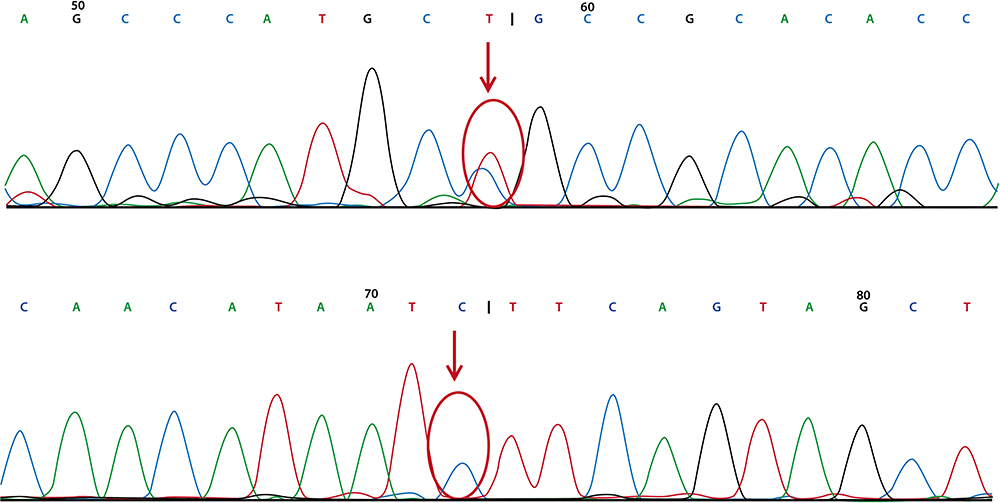 |
Figure 7 The patient’s father had genetic mutation in chr18: 21119839 (upper red arrow) but no mutation in chr18: 211488868 (under red arrow). |
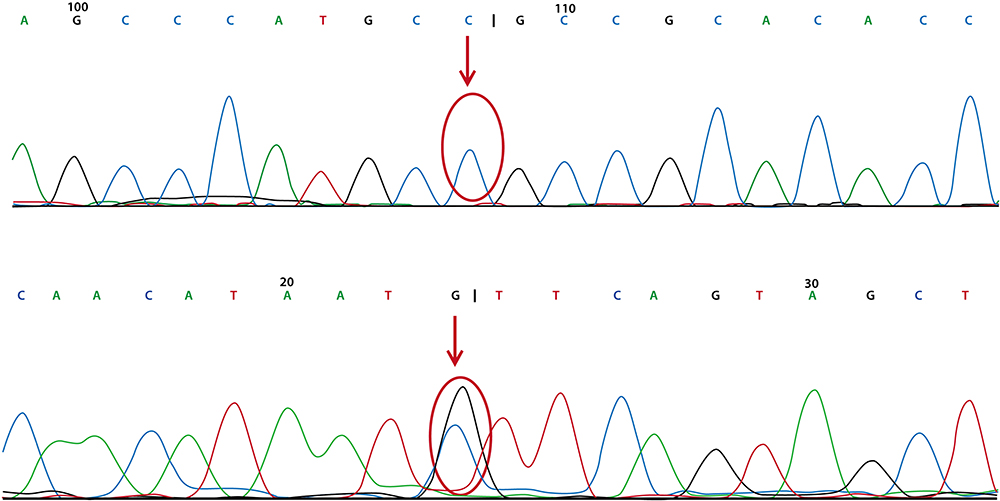 |
Figure 8 The patient’s mother had genetic mutation in chr18: 211488868 (under red arrow) but no mutation in chr18: 21119839 (upper red arrow). |
Discussion
In our report, we found compound heterozygous mutations in NCP1 (p. G911S, c2731G>A and p. D128H, c382G>C), which are thought to be associated with Pelizaeus-Merzbacher-like disease (PMLD).
As previously reported, variants in NCP1 are thought to be related to Niemann-Pick C disease. The dominant biochemical feature of Niemann-Pick type C is abnormal accumulation of low-density lipoprotein (LDL), which produces unesterified cholesterol in endosomes and lysosomes.9 The NPC1 (MIM 607623; chr 18q11-q12) gene encodes a membrane glycoprotein of 1278 amino acids, including an N-terminal domain (NTD), a middle luminal domain (MLD), a C-terminal domain (CTD), and 13 transmembrane helices (TMs).10 Moreover, studies on abnormal NPC1 protein have demonstrated that this protein is indeed a necessary part of intracellular cholesterol trafficking.9 To date, almost 490 mutations in the NPC1 gene have been reported, causing Niemann-Pick disease type C. The clinical symptoms consist of abnormal visceral function (liver, spleen, or lung) and neurologic manifestations.7 However, the phenotype of our patient who showed mutations in the NPC1 gene was consistent with Pelizaeus-Melizaeus disease (PMD) rather than Niemann-Pick disease type C. The symptoms of our patient included nystagmus, dysmyotonia, ataxia, progressive motor dysfunction, mental retardation, and diffuse leukodystrophy on magnetic resonance imaging (MRI). All of the clinical signs and imaging manifestations strongly suggest that the diagnosis of our patient with mutations in the NPC1 gene is more likely to be PMLD, although these mutations used to be related to Niemann-Pick disease type C.
The NPC1 protein is an essential component of intracellular cholesterol trafficking and is localized in endosomes and lysosomes.9
The NPC1 protein is a membrane glycoprotein that consists of 13 transmembrane domains and three large loops localized to endosomes and lysosomes. These domains and loops play roles in regulating lipid metabolism when adequate levels of cholesterol are transmitted to the cell.9,10 Mutations in the NPC1 gene could result in abnormal lipid metabolism, causing large amounts of nonesterified cholesterol and glucoside sphingolipids to deposit in lysosomes. All these pathologic changes lead to a decrease in the self-renewal ability of neural stem cells and neurodegeneration. The myelin sheath is the membrane that surrounds the axon of a nerve cell and has a high lipid content – almost 70–75% of its dry weight.11 Therefore, abnormal lipid metabolism caused by mutations in the NPC1 gene could influence myelin formation, which was observed in our patient.
Llaci et al described a 12-year-old Caucasian/Hispanic male with pathogenic mutations in SNAP29 (p. Leu119AlafsX15, c.354DupG and p.0?, c.2T>C), which have been associated with CEDNIK syndrome.12 Nafisinia et al reported two siblings with clinical symptoms of “PMD”, while WES analysis confirmed a homozygous variant in the arginyl-tRNA synthetase (RARS) gene: NM_002887.3: c. [5A4G]; p.(Asp2Gly), which is possibly the cause of these symptoms.13
Numerous examples indicate that even the same exact mutation in the same zygosity can cause different diseases.14 Our patient with compound mutations in NPC1 could add a new example to the different diseases caused by the same gene.
Concluding Remarks
We found compound heterozygous mutations in the NPC1 gene in a patient with clinical symptoms of PMLD. Our findings expand the spectrum of NPC1-associated disorders and the gene spectrum of PMLD.
Abbreviations
PMLD, Pelizaeus-Merzbacher-like disease; PMD, Pelizaeus-Merzbacher disease; MRI, magnetic resonance imaging; WES, whole exome sequencing; NPC, Niemann-Pick disease type C; HLD1, hypomyelinating leukodystrophy-1; CNS, central nervous system; PLP1, proteolipid protein 1; GJC2, Gap junction protein gamma-2; Leu, leucine; Ala, alanine; Thr, threonine; NTD, N-terminal domain; MLD, middle luminal domain; CTD, C-terminal domain; CEDNIK, Cerebral Dysgenesis, Neuropathy, Ichthyosis and Keratoderma; RARS, arginyl-tRNA synthetase.
Acknowledgments
We thank the patient and his family for their informed consent for publication of this case.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research is supported by the National Key R&D Program of China (No. 2017YFC0211705) and Science & Technology Department Program of Sichuan Province (No.2019YFS0144).
Disclosure
The authors have no conflicts of interest to disclose.
References
1. Lossos A, Elazar N, Lerer I, et al. Myelin-associated glycoprotein gene mutation causes Pelizaeus-Merzbacher disease-like disorder. Brain. 2015;138:2521–2536. doi:10.1093/brain/awv204
2. Inoue K. Pelizaeus-Merzbacher disease: molecular and cellular pathologies and associated phenotypes. Adv Exp Med Biol. 2019;1190:201–216.
3. Pouwels PJW, Vanderver A, Bernard G, et al. Hypomyelinating leukodystrophies: translational research progress and prospects. Ann Neurol. 2014;76:5–19. doi:10.1002/ana.24194
4. Lloyd-Evans E, Morgan A, Xingxuan H, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nature Med. 2008;14:1247–1255. doi:10.1038/nm.1876
5. Davies JP, Chen FW, Ioannou YA. Transmembrane molecular pump activity of Niemann-Pick C1 protein. Science. 2000;290:2295–2298. doi:10.1126/science.290.5500.2295
6. Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16. doi:10.1186/1750-1172-5-16
7. Zervas M, Somers KL, Thrall MA, et al. Critical role for glycosphingolipids in Niemann-Pick disease type C. Current Biol. 2001;11:1283–1287. doi:10.1016/S0960-9822(01)00396-7
8. Ji H, Li D, Wu Y, et al. Hypomyelinating disorder in China: the clinical and genetic heterogeneity in 199 patients. PLoS One. 2018;13(2):e0188869. doi:10.1371/journal.pone.0188869
9. Scott C, Ioannou YA. The NPC1 protein: structure implies function. Biochim Biophys Acta. 2004;1685:8–13. doi:10.1016/j.bbalip.2004.08.006
10. Andrea D, Stefania Z, Cinzia G, et al. Molecular genetics of Niemann–Pick type C disease in Italy: an update on 105 patients and description of 18 NPC1 novel variants. J Clin Med. 2020;9:679. doi:10.3390/jcm9030679
11. Nave K-A, Werner HB. Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol. 2014;30:503–533. doi:10.1146/annurev-cellbio-100913-013101
12. Llaci L, Ramsey K, Belnap N, et al. Compound heterozygous mutations in Snap29 is associated with Pelizaeus-Merzbacher-like disorder (PMLD). Hum Genet. 2019. doi:10.1007/s00439-019-02077-7
13. Nafisinia M, Sobreira N, Riley L, et al. Mutations in RARS cause a hypomyelination disorder akin to Pelizaeus-Merzbacher disease. Eur J Human Gene. 2017;26:1–8. doi:10.1038/s41431-017-0024-z
14. Zhu X, Need AC, Petrovski S, Goldstein DB. One gene, many neuropsychiatric disorders: lessons from Mendelian disease. Nat Neurosci. 2014;17(6):773–781. doi:10.1038/nn.3713.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.