")
Back to Journals » International Journal of Nanomedicine » Volume 14
Novel lipophilic SN38 prodrug forming stable liposomes for colorectal carcinoma therapy
Authors Xing J, Zhang X, Wang Z, Zhang H, Chen P, Zhou G, Sun C, Gu N, Ji M
Received 12 February 2019
Accepted for publication 27 June 2019
Published 12 July 2019 Volume 2019:14 Pages 5201—5213
DOI https://doi.org/10.2147/IJN.S204965
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Jing Xing,*,1,2 Xiquan Zhang,*,3 Zhe Wang,4 Huanqing Zhang,3 Peng Chen,1,2 Gaoxin Zhou,5 Chunlong Sun,6 Ning Gu,1,2 Min Ji1,2
1School of Biological Science and Medical Engineering, Southeast University, Nanjing 210096, People’s Republic of China; 2School of Biological Science and Medical Engineering and Collaborative Innovation Center of Suzhou Nano Science and Technology, Southeast University, Suzhou 215123, People’s Republic of China; 3Nanjing Institute of Pharmaceutical Research and Development, Chia-Tai Tianqing Pharmaceutical Group Co. Ltd, Nanjing 210023, People’s Republic of China; 4Emergency Department, The Second Affiliated Hospital of Nanjing Medical University, Nanjing 210029, People’s Republic of China; 5School of Biomedical Engineering, Shenzhen University, Shenzhen 518071, People’s Republic of China; 6College of Biological and Environmental Engineering, Binzhou University, Binzhou 256603, People’s Republic of China
*These authors contributed equally to this work
Background: SN38 (7-ethyl-10-hydroxy camptothecin), as a potent metabolite of irinotecan, is highly efficacious in cancer treatment. However, the clinical utility of SN38 has been greatly limited due to its undesirable properties, such as poor solubility and low stability.
Materials and methods: In order to overcome these weaknesses, moeixitecan, a lipophilic SN38 prodrug containing a SN-38, a trolox, a succinic acid linker, and a hexadecanol chain, was loaded into liposomal nanoparticles by ethanol injection method.
Results: Experiments showed that the moeixitecan-loaded liposomal nanoparticles (MLP) with a diameter of 105.10±1.49 nm have a satisfactory drug loading rate (90.54±0.41%), high solubility and stability, and showed sustained release of SN38. Notably, MLP exhibited better antitumor activity against human colon adenocarcinoma cells than irinotecan, a FDA-approved drug for the treatment of advanced colorectal cancer. Furthermore, xenograft model results showed that MLP outperformed irinotecan in terms of pharmacokinetics, in vivo therapeutic efficacy and safety. Finally, we used molecular dynamic simulations to explore the association between the structure of MLP and the physical and functional properties of MLP, moeixitecan molecules in MLP folded themselves inside the hydrocarbon chain of the lipid bilayer, which led an increased acyl chain order of the lipid bilayer, and therefore enhanced the lactone ring stability protecting it from hydrolysis.
Conclusion: Our MLP constructing strategy by liposome engineering technology may serve a promising universal approach for the effective and safe delivery of lipophilic prodrug.
Keywords: SN38, lipophilic prodrug, liposomes, molecular dynamic simulations, cancer therapy
Introduction
SN38 (7-ethyl-10-hydroxy camptothecin) is a potent antineoplastic agent. As a member of the camptothecin family, SN38 inhibits DNA topoisomerase I, prevents DNA religation and replication via irreversibly binding to topo-I and DNA, and ultimately causes apoptosis in tumor cells.1,2 The high potency of SN38 makes it a great candidate against many malignancies, such as colorectal carcinoma, lung tumors, gastric, cervical and ovarian cancers.3 However, the clinical application of SN38 is limited due to its poor solubility in most pharmaceutically acceptable solvents and side effects caused by uncontrolled conversion from active lactone ring to inactive carboxylate form at pH>6.4–6 Then, a water-soluble prodrug of SN38, irinotecan (CPT-11) was developed and has been widely used as clinical a first- or second-line drug to treat the advanced colorectal cancer.7–9 However, only 2–8% of CPT-11 administrated transforms into the active metabolite.10,11 Moreover, the active lactone ring of CPT-11 is readily converted to the inactive carboxylate form after infusion in humans.10,12 Thus, unpredictable therapeutic side effects and dose-limiting toxicity usually happen when treating patients with CPT-11.
To overcome these problems, numerous drug delivery systems, such as prodrugs, polymeric micelles, and liposome-based formulations, have been extensively developed.13–17 The macromolecular prodrugs and small-molecule prodrugs with amphiphilic characteristic can self-assemble into nanoscale particle or be wrapped with nanoscale vehicles.17–19 Although the prodrugs-assembled nanoparticles were effective in high drug loading, enhancing drug delivery to tumor tissue and suppressing tumor growth, their inherent shortcomings must be considered, especially inadequate pharmacokinetics. Employing liposomes as the drug vector of SN38 and its derivatives is an effective strategy to prolong the systemic circulation of their in the bloodstream and optimal concentration in tumor tissues.20 Liposomes are lipid bilayer vesicles.21,22 And as one of the best drug delivery systems, they were investigated for delivery of both lipophilic and hydrophilic drugs. Especially the nanoliposomes have some advantages, such as increased solubility and stability, enhanced cellular uptake, improved bioavailability, and specific targeting of organs or tissues for the controlled release of drugs.23,24 Due to the robust biocompatibility, several liposomal formulations are available for chemotherapeutic application in clinic.25 Encapsulation of drugs within liposomes often confers the drugs with high stability to avoid chemical degradation. However, it is a challenge to employ liposomes to deliver SN38 because of its unfavorable physicochemical properties and low drug entrapment efficiency.4,5 Increasing the lipophilicity through lipophilic conjugate of SN-38 has multiple benefits, including enhancement of drug loading in lipid-based formulations, possible stabilization of the active lactone form, and improvement of the permeability of the drug through cell membranes.26,27 Indeed, many research have focused on the biological activities of the liposomal formulations of lipophilic prodrug of SN38.28 A few studies concern on the effect of lipophilic prodrug on the membrane of liposome formulation.29–32 However, there have been no reports on the influence of encapsulation of the lipophilic prodrug of SN38 on the structure and the stability of the lipid bilayer.
Motivated by this rationale, we have recently developed a liposome-based delivery system for encapsulation of a lipophilic SN38 prodrug (designated as moeixitecan) to improve its stability and biological activities.33,34 In our previous study, we already synthesized moeixitecan, which contained a SN-38 component, a trolox component, a succinic acid linker, and a hexadecanol chain, with excellent properties in vivo and in the clinic. The physiochemical properties of moeixitecan-loaded nanoliposomes were characterized and the antitumor efficacy were evaluated in colorectal tumor models in vitro and in vivo, respectively. Meanwhile, molecular dynamics (MD) simulations were performed to expound the underlying molecular mechanisms of the influence of moeixitecan molecules on the structure and the stability of the phospholipid bilayer. Finally, the in vivo biodistribution of moeixitecan-loaded nanoliposomes and a safety evaluation compared to CPT11 were examined in mouse.
Materials and methods
Materials
2-(hexadecyloxycarbonyl)-2, 5, 7, 8-tetramethylchroman-6-yl 7-ethyl-camptothecin-10-yl succinate (moeixitecan) obtained internally by Chia-tai Tianqing Pharmaceutical (Nanjing, People's Republic of China). 7-Ethyl-10-hydroxycamptothecin (SN38) was provided by Sigma-Aldrich (Shanghai). Dipalmitoyl phosphatidylcholine (DPPC), hydrogenated soybean phospholipids (HSPC), and distearoyl phosphoethanolamine-PEG2000 (DSPE-PEG2000) were purchased from Southeast Pharmaceuticals Co. (Soochow, People's Republic of China). Chloroform and methanol were purchased from Shanghai Chemical Reagent Company.
Preparation of moeixitecan (SN38 prodrug)-loaded liposomal nanoparticles (MLP)
Liposomes loaded with moeixitecan were prepared on the basis of ethanol injection as described previously. Briefly, DPPC, HSPC, and DSPE-PEG2000 were first dissolved in ethanol at a molar ratio of 75:20:5. The moeixitecan was added to the above ethanolic solution with the ratio of lipids to moeixitecan fixed at 20:1 (w/w) and incubated at 60°C. The mixture (1 mL) was then rapidly injected into 5% dextrose solution (9 mL) at 60°C. Finally, the resulting vesicles suspension was extruded through polycarbonate filters (Avanti Polar Lipids, Alabaster, AL, USA) of 200 and 100 nm (five times each) for MLP.
Characterization of MLP
The particle size and surface potential of MLP were measured at 25±0.1°C by Nano-ZS 90 Nanosizer (Malvern, UK). The morphology and structure were observed by transmission electron microscope (JEOL, Japan) with negative stain method. Before analysis, the samples were prepared by placing a drip onto 300-mesh copper-grid, and a drip of 3 wt% phosphotungstic acid was dropped on the samples, then kept the sample at room temperature overnight for drying.
The encapsulation efficiencies (EE) of moeixitecan in liposomes were determined. Firstly, the nanoparticles were extracting 1 mL of MLP with 10 mL acetonitrile for 1 hr while stirring. Then, the samples were then filtered and analyzed by HPLC. A C18 ODS reverse-phase column (5 mm, 250 mm×4.6 mm, Shimadzu, Japan) was used for HPLC analysis at room temperature. The mobile phase consisted of methanol and 1% formic acid aqueous solution (80:20, v/v) and the flow rate was maintained at 1 mL/min. The injection volume was 20 μL and the determined wavelength was 220 nm. The EE of moeixitecan were calculated according to the following formula EE (%)=(Moeixitecan total–Moeixitecan free)/Moeixitecan total×100%.
In vitro drug release from liposomes
The drug release behavior from free moeixitecan and MLP was evaluated using the dialysis method against PBS (pH =7.4) containing 0.2% Tween 80. Free moeixitecan solutions and MLP solutions were infused into dialysis bags (1 mL solution per bag at an SN38 equivalent concentration of 0.5 mg/mL) and submerged into 50 mL of PBS containing 0.2% Tween 80 (w/v) and maintained at 37°C under 100 rpm shaking for 96 hrs. At pre-determined time intervals, 1 mL of the release media was collected and equal volumes of fresh media were supplemented. After freeze-drying and redissolution with 1.5 mL methanol, the concentration of moeixitecan and SN38 was analyzed by HPLC. Finally, the cumulative amount of drug released into the media at each time point was calculated as the percentage of total released drug to the initial amount of the drug.
Physical stability of MLP
The physical stability of MLP was estimated by measuring the size change of liposomes in the rat plasma as well as PBS buffer (pH 7.4) with or without 20% FBS at 37°C. Lastly, the MLP prepared with the highest physical stability was further evaluated for any size change during storage at 4°C for 3 months.
MD simulations of the effect of moeixitecan on the structure of lipid membranes
We performed atomistic MD simulations for six model systems (Table 1) containing lipid bilayers with 0–25 mol% of moeixitecan. Each system was added with 4000 water molecules and Na+ or Cl− ions were added in order to achieve physiological concentration.
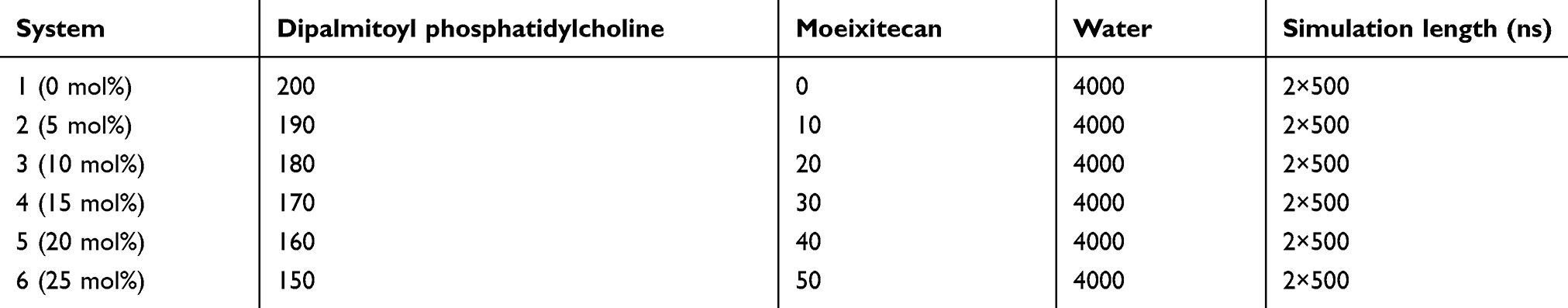 |
Table 1 Summary of the simulated systems |
Simulations were carried out using the GROMACS 4.6 software package. The all-atom OPLS force field was employed with a recent extension for lipids to parameterize all lipid molecules. The TIP3P model, compatible with the OPLS-AA force field, was used for water. The time step was set to 2 fs, and the simulations were carried out at 1 bar and 300 K. The Nosé-Hoo-ver method was used to couple the temperature with separate heat baths for the membrane and the rest of the system with time constants of 0.4 ps. The linear constraint solver algorithm was used to preserve covalent bond lengths. Prior to all MD simulations, the steepest-descent algorithm was used to minimize the energy of the initial configurations.
Cell lines and cell culture
Human colon adenocarcinoma cells (HT-29) were used in this study provided by Cell Bank of Shanghai Institute of Cell Biology, Chinese Academy of Sciences. HT29 cells were maintained in McCoy’s 5A (modified) medium, supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C.
In vitro cytotoxicity
The cytotoxicity of MLP was assessed against HT-29 cells by the CCK-8 assays. Briefly, HT29 cells were seeded in 96-well plates at a density of 5000 cells per well and incubated for 24 hrs. Then, MLP at various concentrations was added to the culture media. Moeixitecan and CPT-11 were used as controls. After incubation for 24and 48 hrs, the cell viability was determined using CCK8 assays. At the end of the incubation period, 10 μL of CCK-8 reagent (Beyotime, Shanghai, People's Republic of China) was added to the cells and further incubated for 2 hrs. Afterward, the 96-well plate was analyzed at a 450 nm wavelength in an Infinite 200 PRO plate reader (Tecan, Switzerland) to determine cell viability.
Apoptosis analysis using flow cytometry
The apoptosis of HT29 cells was measured by Annexin-V-FITC/PI double staining method. HT29 cells were seeded in 6-well plates at a density of 2×105 cells per well and incubated overnight. Then, moeixitecan, CPT-11, and MLP (with the equal SN38 dose of 2 μg/mL) were added to the culture media. Cells that were untreated were used as controls. After 24 or 48 hrs of treatment, the HT29 cells were collected and detected on FACS CantoII flow cytometry (BD, MA) by using an Annexin-V-FITC Apoptosis Detection Kit (Beyotime).
Animals and tumor model
The animal experiments were carried out according to the protocol approved by the Ministry of Health in People’s Republic of China (document no. 55, 2001) and the guidelines for the care and use of Laboratory Animals of Southeast University. Female BALB/C nude mice (20±2 g, 6–8 weeks old) were purchased from Nanjing model animal research institute (Nanjing, People’s Republic of China) and received care under standard housing conditions. HT-29 cells (6×106/200 μL PBS) were administered by subcutaneous injection into the flank region of the mice. Two weeks after post-inoculation, the xenograft tumor model was built. Tumor volume was calculated as (tumor length)×(tumor width)2/2.
In vivo pharmacokinetic studies
In vivo pharmacokinetic study was performed according to the previous report. Briefly, the Sprague-Dawley rats (250±20 g) were injected with a single dose of 2.5 mg/kg (SN38 equivalent) of CPT11, free moeixitecan, MLP via tail vein (n=3), respectively. Blood samples were collected into heparinized tubes after 0.5, 1, 3, 6, 12, and 24 hrs. Subsequently, whole blood samples were centrifuged at 3000 rpm for 10 mins at 4°C to separate the plasma. The plasma was stored at 80°C prior to analysis by LC/MS/MS. An aliquot 5 μL of the processed samples were injected to LC/MS/MS system and the sample was separated on an Agilent Eclipse Plus C18 (4.6×100 mm, 3.5 μm, Agilent Technologies, USA), which was kept at room temperature (25± 2°C). In the current study, SN38 were separated on a gradient elution, which was performed by varying the proportion of mixture A (5 mM ammonium acetate with 0.2% formic acid) and mixture B (acetonitrile) as follows: 0 min (75% A–25% B), at 1.0 min (50% A–50% B), at 1.5 mins (75% A–25% B), at 1.6 mins (25% A–75% B), and at 2.2 mins (25% A–75% B) was used and delivered at a rate of 0.60 mL min−1 into the mass spectrometer’s electrospray ionization chamber. SN38 were detected by triple-quadrupole LC/tandem mass spectrometric detection (A Shimadzu-LC-30AD from Shimadzu, Tokyo, Japan equipped with MS/MS; AB Sciex triple quad 6500, from Sciex, Framingham, MA) with electrospray ionization interface running in positive ionization mode. After extracting, samples were analyzed using a LC/MS/MS analysis.
In vivo distribution by near-infrared (NIR) imaging
A NIR region probe (iDSPE)-embeded liposomes were used to evaluate the in vivo distribution of MLP in HT29 tumor-bearing mice.35 The NIR probe, iDSPE was co-encapsulated with moeixitecan in iDSPE-embeded MLP using the same method. The tumor-bearing mice were intravenously injected 0.2 mL of ir623 and iDSPE-embeded MLP, respectively. The equivalent ir623 dose was kept at 0.5 mg/kg. The NIR images of mice were obtained at 0.5, 2, 4, 8, 24, and 48 hrs postinjection using the ex/in vivo imaging system (Caliper Life Science, USA) with a 630 nm excitation wavelength and 710 nm filter. After imaging, the mice were immediately sacrificed and organs were harvested for the ex vivo imaging under the same conditions as described above.
In vivo antitumor efficiency
The antitumor activity was conducted using HT29 tumor-bearing mice. When the tumor volume reached about 100 mm3, the mice were randomly divided into four groups (n=5 per group) and treated with PBS, CPT11, free moeixitecan, MLP (with the equal SN38 dose of 2.5 mg/kg). Drugs were administered intravenously via the tail vein at designated times. The tumor sizes were measured with a caliper, and the tumor volumes were calculated according to the formula (length×width2)×1/2. The body weight of mice in each group was also monitored. At the end of the “antitumor activity study”, the mice were sacrificed for humane reasons. Meanwhile, the tumors and major organs were excised and fixed with 4% formalin. Then, the organs sections were mounted on glass slides and stained with H&E and observed using a digital microscope.
Safety evaluation
The blood collected from the HT29 tumor-bearing mice were used to perform hematological tests using a blood counter (Sinnowa, HBVet-5 model) with adapted dilutions. Hematological analysis involves assessment of white blood cell count, red blood cell (RBC) count, hematocrit, % neutrophils, % lymphocyte, % monocyte, % eosinophil, and % basophile.
Results and discussion
Preparation and characterization of MLP
The structure of moeixitecan is 2-(hexadecyloxycarbonyl)-2,5,7,8-tetramethylchroman-6-yl 7-ethyl-camptothecin-10-yl succinate, containing an SN-38 component, a trolox component, a succinic acid linker, and a hexadecanol chain, as shown in Figure 1A. MLP were prepared by ethanol injection method, the lipids were composed of DPPC, HSPC, and DSPE-PEG2000, as depicted in Figure 1A. Then, the physiochemical characters of MLP were systematically evaluated and the results are presented in Table 2, including mean diameter, polydispersity index (PDI) value, zeta potentials, and EE. The average hydrodynamic diameter of MLP was 105.10±1.49 nm, which was a little lower than the blank liposomes (117±2.49 nm), as shown in Figure 1C. Compared with the blank liposomes, MLP had a much lower surface charge, indicating a good stability of the formulations against vesicle aggregation and fusion. As expected, when moeixitecan was added, the negative zeta potential increased significantly, which would maintain the stability of the liposome solution by electrostatic repulsion. In addition, the EE of MLP was 90.54±0.41%, which was higher than that (34.62–60.11%) of CPT11-loaded nanoliposomes reported in the literature.
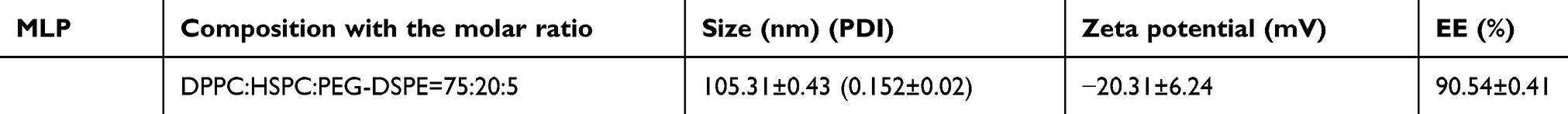 |
Table 2 Composition and physical properties of moeixitecan-loaded liposomal nanoparticles (MLP) |
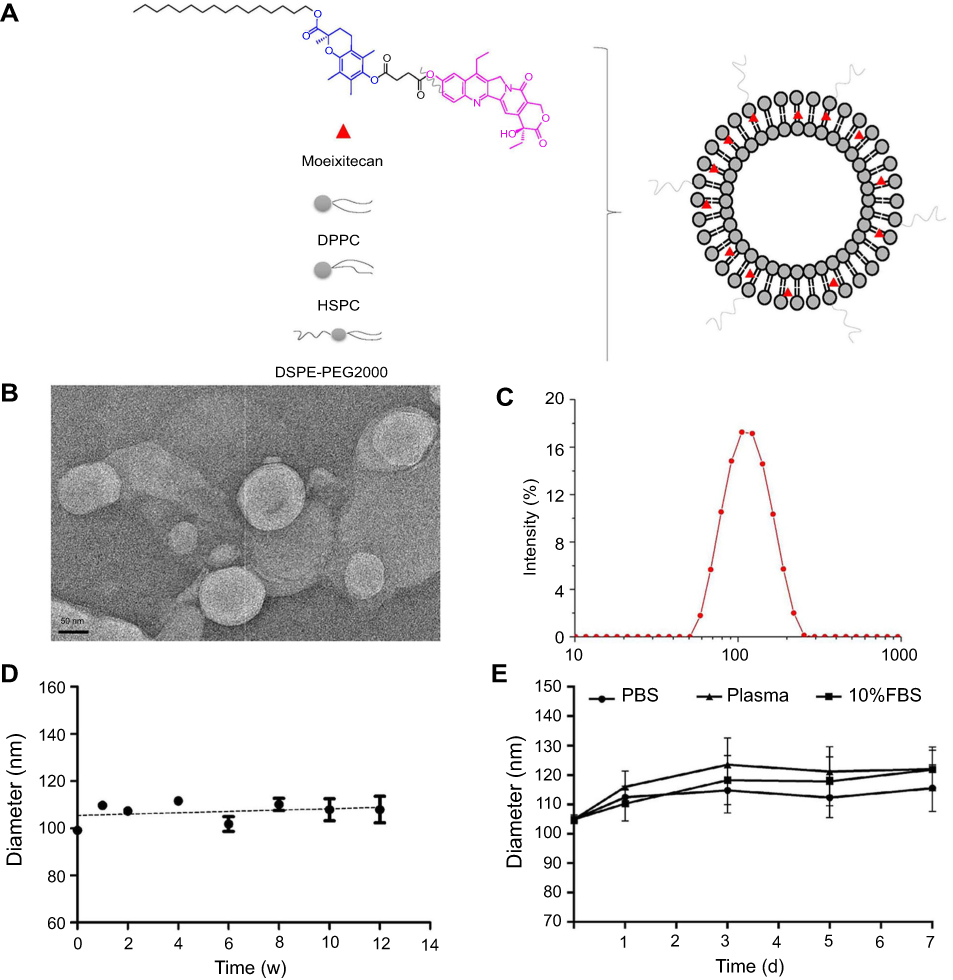 |
Figure 1 The characterization and physical stability of moeixitecan-loaded liposomal nanoparticle (MLP). (A) Schematic illustration of the SN38 prodrug, MLP. (B) Morphological characteristics of MLP. (C) Particle size distribution of MLP. (D) Size changes of MLP were examined as a function of time at 4°C for 3 months. (E) Size changes of MLP in different medium were examined for 7 days. |
The morphology of the MLP was observed by transmission electron microscopy. As shown in Figure 1B, the image obviously verified the formation of liposomes with uniform spherical nanostructures. With the addition of moeixitecan, MLP became more stable and maintained a consistent size distribution without significant variation during storage at 4°C for 3 months compared with the blank liposomes (Figure 1D). In order to provide further evidence for in vivo applications, the particle stability in plasma of the liposomes for 7 days was evaluated. MLP showed no significant change in sizes within rat plasma (~150 nm) in 7 days (Figure 1E). In contrast, without moeixitecan, the mixture of lipid could not form stable bilayer liposomes, and formed large aggregates in rat plasma (data were not shown). All of these results proved that moeixitecan entrapped in liposomes formed a stable nanoparticle, and MLP exhibited great size stability either in storage condition (4°C) or physiological environment (in plasma).
In vitro drug release
The in vitro release properties of MLP were investigated in PBS containing 0.2% Tween 80 via dialysis followed by HPLC. Figure 2 presents in vitro release of free SN38 from liposomal formulation and its solution as control. As shown in Figure 2, MLP exhibited a slower and sustained release behavior in comparison with its solution. Cumulative release of SN38 after 72 hrs was 63.55±6.81% from the MLP, whereas 80.57±5.37% for moeixitecan solution within 48 hrs. There was no burst release of SN38 detected from the liposomes, which strongly suggested that moeixitecan was located in lipid bilayers, and thereby enhance drug retention within the liposomes but SN38 can be slowly released. The sustained release of SN38 could be beneficial for cancer treatment as a result of the maintenance of constant SN38 concentration in tumor tissue.36
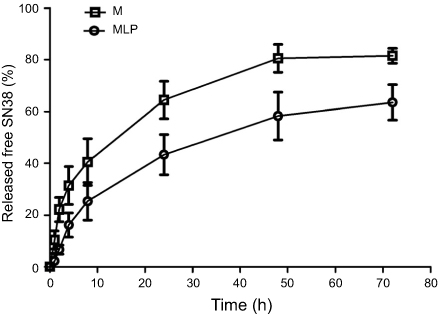 |
Figure 2 SN38 released from moeixitecan (M) solution and MLP at 37°C in PBS (pH 7.4) containing 0.2% Tween 80 at 37°C.Abbreviation: MLP, moeixitecan-loaded liposomal nanoparticle. |
MD simulations of moeixitecan-lipid membranes interaction
To clarify the interaction between moeixitecan and simplified lipid bilayer in more detail, we carried out atomistic MD simulations. We performed all-atom MD simulations of a DPPC bilayer before and after adding 5 mol%, 10 mol%, 15 mol%, 20 mol%, and 25 mol% moeixitecan. The MD simulations can provide a detailed description of the changes in bilayer structure incurred by the incorporation of moeixitecan into a lipid bilayer. Figure 3A and B illustrates the location (depth) and orientation of moeixitecan molecules inside membranes at the end of the simulations. The moeixitecan molecular may integrate itself into the hydrocarbon chain region of the lipid bilayer. To estimate the position of moeixitecan with respect to the lipid molecules, we calculated their mass density profiles across the bilayer. Figure 3C shows the density profiles along the membrane normal for the moeixitecan. At the end of the modeling, the distance between the moeixitecan molecule and the center of membrane was 4 nm. Therefore, these figures show that moeixitecan folding is preferably located inside the fatty acid chains of the phospholipid. The thickness of the membrane increased with increasing concentration of moeixitecan (Figure 3D). Meanwhile, the areas per molecule of lipid bilayer decreased with increasing concentration of moeixitecan (Figure 3E). Lipid membrane thickness and the area per molecule are structural parameters that are needed to accurately determine other bilayer structural parameters and are directly related to lipid–lipid, and lipid–protein interactions in biomembranes.37 Our MD simulations results confirmed the experimental results and provided an accurate picture of moeixitecan’s location and effect for lipid bilayers: moeixitecan molecules fold themselves located inside the hydrocarbon chain of the lipid bilayer inducing an increased acyl chain order of the lipid bilayers and lactone ring stability. Both physical stability of MLP and MD simulations data consistent with each other and showed that moeixitecan has an ordering effect, and increases the stability of bilayer.
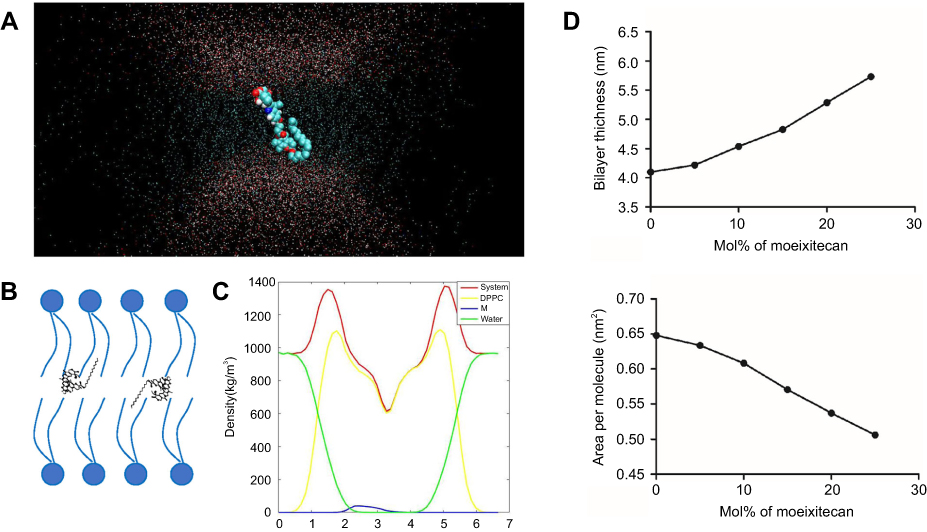 |
Figure 3 Molecular dynamics simulations of dipalmitoyl phosphatidylcholine (DPPC) bilayers with moeixitecan. (A) Snapshots of the systems consisting of moeixitecan molecules in DPPC bilayers. (B) Schematics illustrating the proposed locations of moeixitecan in the lipid membrane. (C) Mass density profiles of moeixitecan and the selected lipid atoms along the bilayer. (D) The thickness of the membrane with increasing concentrations of moeixitecan. (E) The areas per molecule of lipid bilayer with increasing concentrations of moeixitecan. |
In vitro cytotoxicity
The in vitro cytotoxicity was evaluated by CCK8 assay on human colon adenocarcinoma cells (HT-29). The MLP, moeixitecan, and CPT-11 were incubated with HT29 cells for 24 and 48 hrs to compare their cytotoxicity in vitro (Figure 4). The IC50 (concentration inhibiting cell growth to 50% of control) values of CPT-11, MLP, and moeixitecan at 48 hrs were 79,024, 0.8461, and 3.139 μg/mL, respectively. More specifically, the MLP achieved an outstanding cytotoxic effect compared to the clinical anticancer drug CPT-11. Two possible reasons were thought to induce the increased cytotoxicity of moeixitecan and MLP.38 First, the combination of moeixitecan and phospholipids was more effective for cellular uptake, which could contribute to the enhanced cytotoxicity. Second, once be uptaken by cancer cells, the moeixitecan can sufficiently convert to active SN38 molecules by hydrolysis, and reached a relatively high drug concentration. Hence, MLP exhibited potent antitumor activity comparable or even slightly superior in comparison with moeixitecan, both of which were significantly better than CPT-11 in vitro.
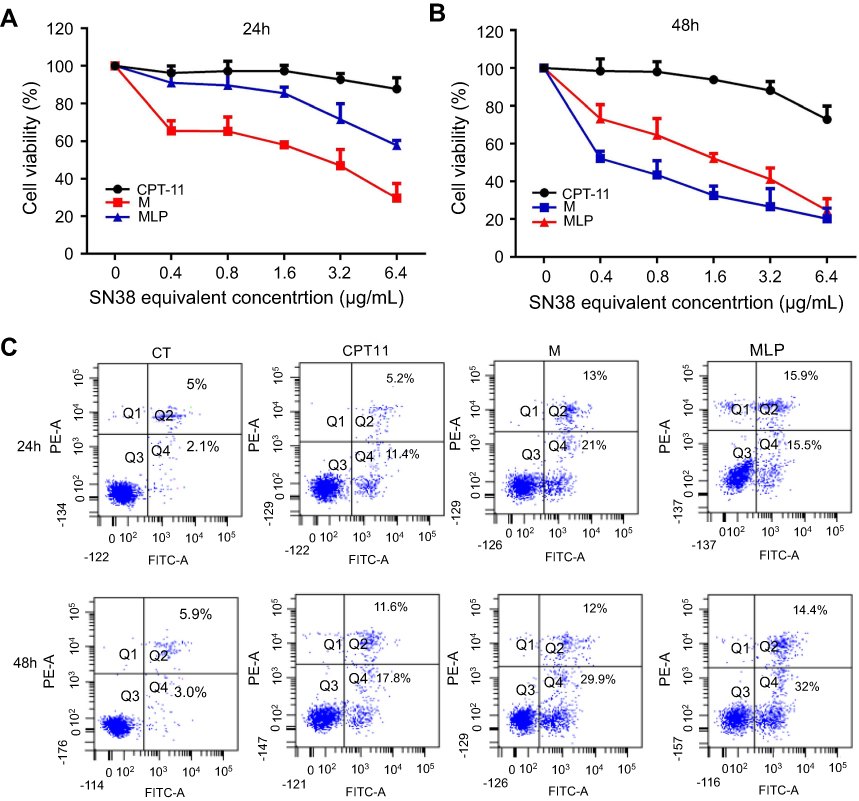 |
Figure 4 (A and B) The cytotoxicity of CPT-11, moeixitecan (M), and moeixitecan-loaded liposomal nanoparticles (MLP) toward HT-29 cells for 24 and 48 hrs. (C) Flow cytometry analysis for apoptosis of HT-29 cells in induced by control, CPT-11, M, and MLP at the same SN38 equivalent concentration for 24 and 48 hrs. |
In vitro cell apoptosis induced by released SN38 molecules
The apoptosis assay was used to elucidate the death mechanism of HT-29 cells incubated with various drug formulations. After treatment for 24 and 48 hrs, the cells were measured using Annexin-V-FITC/PI double staining analyzed by flow cytometry. The motion of phosphatidylserine to the exterior of the cell membrane is an essential event in apoptosis cells which can be specifically detected by fluorescently labeled Annexin V. Indeed, a high level of early and late apoptosis after 24 or 48 hrs of treatment was induced by MLP and moeixitecan, which was comparable to the level induced by CPT-11. After 24 hrs, the percentage of apoptotic HT-29 cells treated with CPT-11, moeixitecan, and MLP were 16.6%, 34%, and 31.4%, respectively (Figure 4C). However, after 48 hrs of incubation, the apoptosis proportion of HT-29 cells incubated with CPT-11, moeixitecan, and MLP increased to 29.4%, 41.6%, and 46.4%, respectively (Figure 4C). Hence, increased cellular apoptosis was observed because of extending treatment time. In summary, the cell apoptosis against cancer cells was consistent with the results of in vitro cytotoxicity assays, suggesting that moeixitecan-loaded NPs can perform better anti-proliferative potency than CPT-11.
In vivo pharmacokinetic studies
To evaluate the MLP in the blood circulation system, the pharmacokinetic study was assessed in Sprague-Dawley rats after intravenous injection of the CPT-11, moeixitecan, and MLP solutions (at the equivalent SN38 dose of 2.5 mg/kg). The concentration-time profiles of CPT-11, moeixitecan, and MLP are shown in Figure 5 and the main pharmacokinetic parameters are provided in Table 3. The maximum SN38 concentrations (Cmax) for MLP were evidently higher when moeixitecan was encapsulated in the liposomal formulations. The Cmax of the rats treated with CPT-11, moeixitecan, and MLP were 56, 137, and 616 ng/mL, respectively. This was further reflected in differences in t1/2 and the values of mean residence time (MRT): t1/2 with CPT-11 at 1.73 hrs, moeixitecan at 2.42 hrs, and MLP at 5.23 hrs; MRT with CPT-11 at 2.44 hrs, moeixitecan at 4.64 hrs, and MLP at 6.70 hrs. Moreover, the AUC of SN38 is higher upon administration of both moeixitecan (774 ng/L h) and MLP (3454 ng/L h) compared to CPT-11 (118 ng/L h). In particular, the AUC value of MLP was 30 times higher than that of CPT-11. These results indicate that the MLP exhibited an increase in net drug exposure and systemic circulation time in the blood.
 |
Table 3 The main pharmacokinetic parameters in SD rats after intravenous injection of CPT-11, moeixitecan (M) ,and moeixitecan-loaded liposomal nanoparticles (MLP) (n=3) |
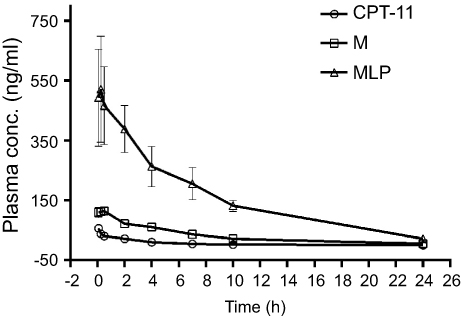 |
Figure 5 In vivo plasma concentration-time profiles of SN38 after intravenous injection of CPT-11, moeixitecan (M), and moeixitecan-loaded liposomal nanoparticles (MLP) in SD rats (n=3). |
Very recently, we have identified a novel NIR dye, iDSPE, which are able to insert into the lipid bilayers of liposomes to evaluate the blood retention and distribution of liposomes conveniently.34 To evaluate the real-time biodistribution and tumor targeting efficiency of liposomes in HT-29 tumor-bearing mice, various formulations were injected into tumor-bearing mice via tail intravenous injection. The results for real-time imaging and biodistribution of ir623 and the iDSPE-embeded MLP (iMLP) were observed in HT-29 tumor-bearing mice by ex/in vivo imaging system (Figure 6). Obviously, after intravenous injection of free ir623, nonspecific distribution of fluorescence all over the body was observed within 2 hrs, and with rapidly decreased fluorescence signals within 48 hrs injection. While iMLP group exhibited detectable NIR fluorescence signals at 24 hrs postinjection, which were mainly concentrated at the tumor sites and retained long term in the tumor (Figure 6A). After living imaged, the mice were sacrificed and tumor tissue as well as other main organs were harvested for the ex vivo fluorescence imaging, also revealing the obvious tumor accumulation of the iMLP (Figure 6B). These results indicated that the MLP showed an excellent tumor-targeting capability and a potential to help extend drug retention in blood correlate well with the pharmacokinetic studies. Therefore, nanoliposomes may be an ideal delivery platform for the passive accumulation of drugs in tumor lesions.
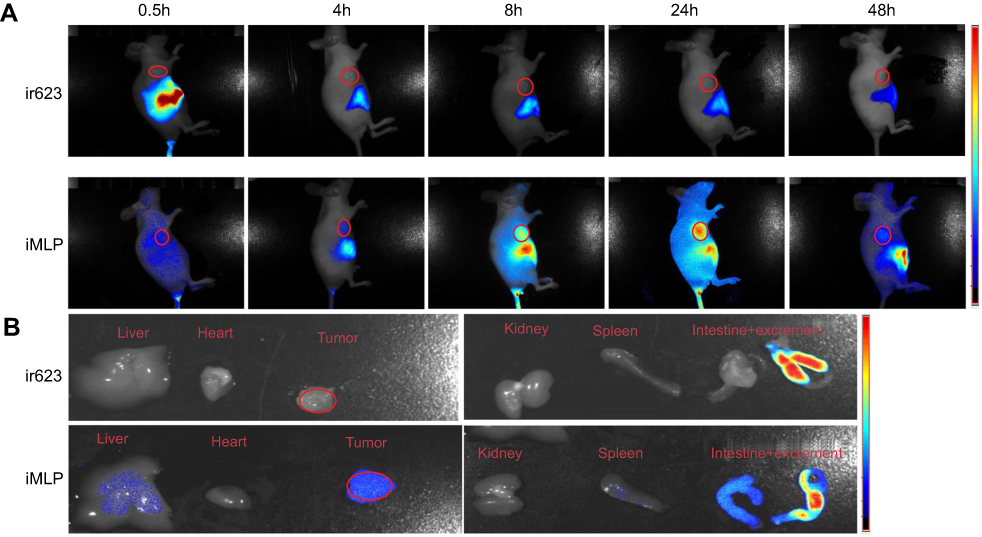 |
Figure 6 Biodistribution of ir623 and iMLP in HT-29 tumor-bearing mice. (A) Representative images of in vivo whole-body imaging of mice at 0.5, 4, 8, 24, and 48 hrs postintravenous injection of ir623 and iMLP. (B) The ex vivo optical images of the tumors and other major organs from the sacrificed mice (n=3).Abbreviation: iMLP, iDSPE-embeded moeixitecan-loaded liposomal nanoparticles. |
In vivo antitumor activity in human colon xenograft tumors
The effectiveness of MLP in suppressing of tumor growth was evaluated in a HT-29 colorectal xenograft model. The group with saline solution and that received CPT-11 served as the control. As shown in Figure 7A, the saline group showed a rapid increase in tumor growth in HT-29 tumor-bearing mice over the whole period of the experiment with tumor volume reaching approximately 1025±139 mm3 at day 15. Meanwhile, the tumor growth was remarkably inhibited after treated with CPT-11, moeixitecan, and MLP as compared to saline solution. When the treatment was finished, the mice were sacrificed and the tumors were weighed. The tumor weights of mice were consistent with the tumor volume results: the overall inhibition rates of CPT-11, moeixitecan, and MLP were 46.06%±6.30%, 54.41%±7.30%, and 66.86%±6.54%, respectively (p˂0.01, Figure 7C and D). Treated with liposomally formulated moeixitecan with a low dose of SN38 (2.5 mg/kg) had an enhanced effect, resulting in 66.86% tumor inhibition compared with CPT-11 treatment (46.06%, p<0.05). Moreover, MLP were even more effective than our previously developed prodrug moeixitecan that only led to 54.41% in the inhibition rates (p<0.05). The increased in vivo antitumor activity of MLP is likely due to the sustained drug release kinetics, improved systemic circulation, and preferential tumor accumulation via the enhanced permeability and retention (EPR) effect. The results are consistent with in vitro cytotoxicity indicating that MLP exerted excellent therapeutic activity better than CPT-11 on the xenograft tumors.
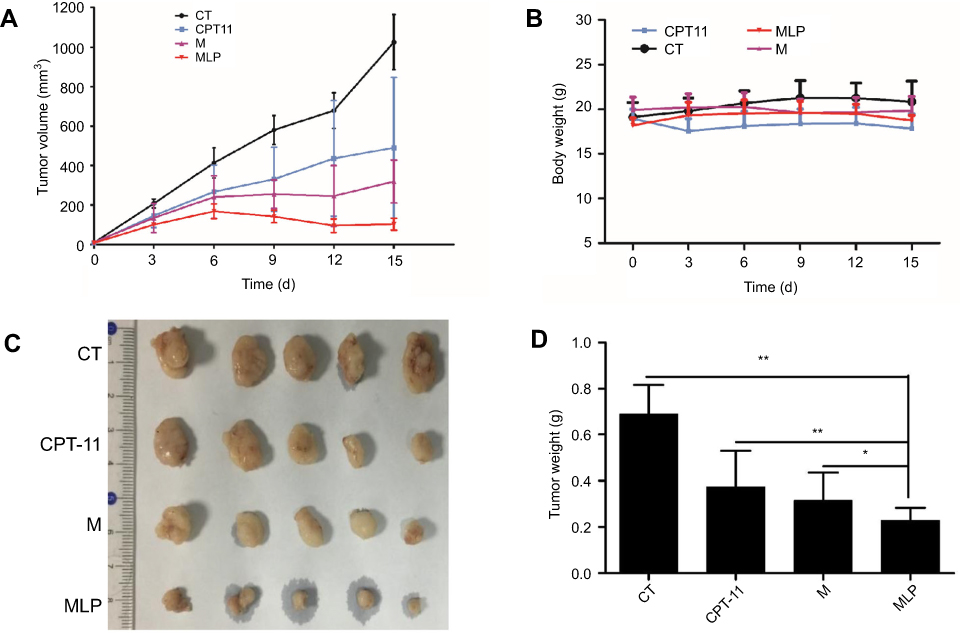 |
Figure 7 Anti-tumor effect on HT-29 tumor-bearing mice treated with moeixitecan-loaded liposomal nanoparticles (MLP). (A) Tumor volume changes as a function of time after intravenous injection of CPT-11, moeixitecan (M), and MLP. (B) Body weight profile of tumor-bearing mice after treatment. (C) Typical excised tumors at the time of sacrifice. (D) Statistical results for tumor weight. |
Safety evaluation
The variations in the body weights of mice in all the treatment group were plotted against time as shown in Figure 7B. Obviously, the body weights of mice did not decrease significantly compared with control group. Afterward, we evaluated the toxicity of different formulations by histological studies on excised tissue sections with H&E staining. As shown in Figure 8A, we did not observe any appreciable abnormality in major organs. Finally, we further evaluated hematological affects by routine peripheral blood examination. The blood cell counts were assessed to investigate the hematological toxicity against MLP. There was no significant variation in the counts of RBC, hemoglobin, or in any other routine peripheral blood tests of CPT-11 treatment groups and MLP treatment group when compared with the control group (Figure 8B). However, the blood cells counts were significantly increased in the moeixitecan treatment groups compared to the control group and the MLP treatment group. The reductive toxicity for MLP might be attributed to liposomal incorporation can encapsulate drug for providing more selective delivery or targeting to the tumor tissue by reduced accumulation in the normal tissues. Thus, these results indicated the MLP, at the test dose for in vivo administration, was relatively safe. Although animal experiments with higher doses of MLP were not explored, raised doses should be expected to improve antitumor activity.
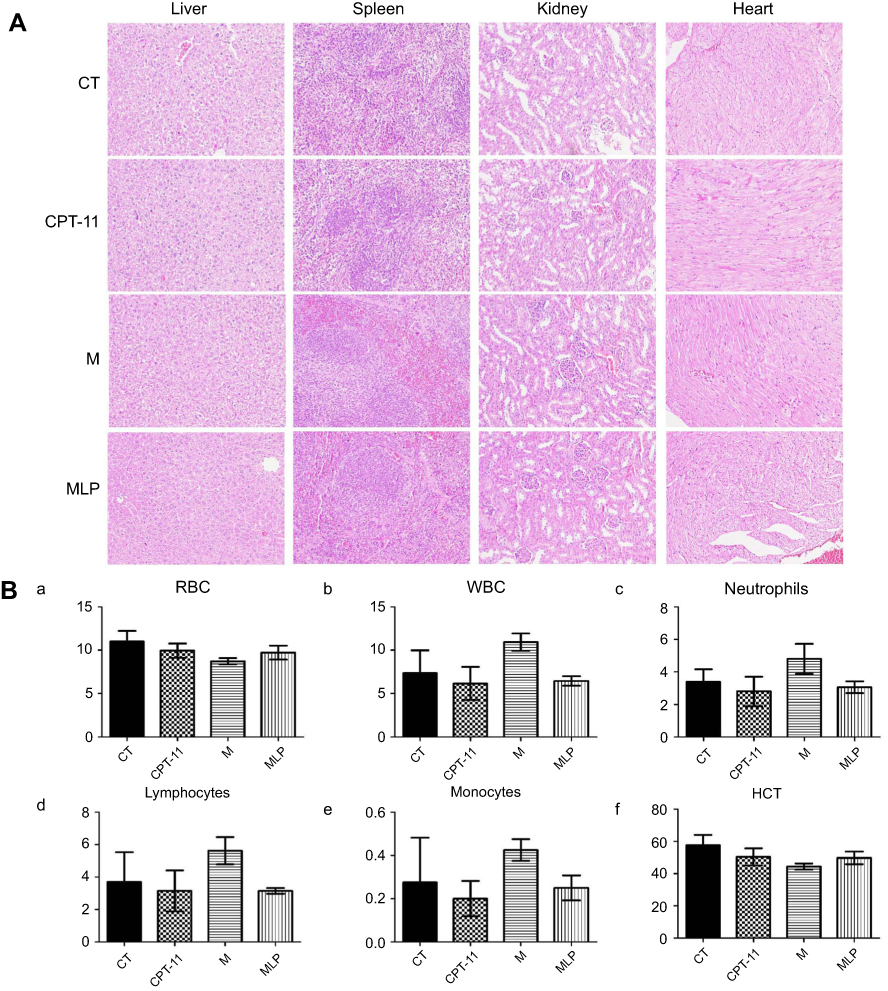 |
Figure 8 Safety evaluation of moeixitecan-loaded liposomal nanoparticles (MLP).Notes: (A) H&E stained images of organ sections separated from mice. Magnification, ×200 (B) blood parameters panel data from mice injected i.v. with saline (control, CT), CPT-11, moeixitecan (M), and moeixitecan-loaded liposomal nanoparticles (MLP). Mean parameters value±SD, n=5. RBC count (a), WBC count (b), neutrophil count (c), lymphocyte count (d), monocyte count (e), and HCT value (f).Abbreviations: WBC, white blood cell; RBC, red blood cell; HCT, hematocrit. |
Conclusion
In summary, we successfully designed stable liposomal formulations containing a lipophilic prodrug of SN38, moeixitecan. The MLP not only overcome various drawbacks of CPT-11 and SN38, but also resolve various limitations of the reported SN38-incorporating drug delivery systems, especially the premature drug release. The moeixitecan was able to incorporate into liposomes with high and stable drug loading, significantly improved solubility, stability, and sustained drug release kinetics. Furthermore, the moeixitecan-loaded liposomes exhibited significant cytotoxicity with selective promotion of cell apoptosis against HT-29 cell lines compared with CPT-11, and showed more therapeutic effect in a colorectal xenograft model than the clinically approved CPT-11 and moeixitecan. Simultaneously, the in vivo pharmacokinetics of the MLP were improved relative to moeixitecan and the toxicities were lower to normal tissues in tumor-bearing mice. Particularly, the results of MD simulations indicated that folding moeixitecan molecules were preferably accumulated in the hydrophobic acyl chain region of the lipid bilayers, which caused packing and ordering of lipids increasing the stability of lipid bilayers and protect stabilization of the lactone form from hydrolysis. Both experimental are in good agreement with the theoretical data. The MD simulations provide an insight into the nature of moeixitecan-lipid membrane interactions at the molecular level and could be used for optimizing the liposomal formulations. Therefore, our results suggest that the moeixitecan-loaded liposomes could be a promising chemotherapeutic drug better than CPT-11 for clinical applications.
Acknowledgments
This research was financially supported by National Natural Science Foundation of China General Program (81671745), and Science and Technology Development Program of Suzhou (ZXY201412).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gerrits CJ, Jonge MJ, Schellens JH, Stoter G, Verweij J. Topoisomerase I inhibitors: the relevance of prolonged exposure for present clinical development. Br J Cancer. 1997;76(7):952–962. doi:10.1038/bjc.1997.491
2. Sriram D, Yogeeswari P, Thirumurugan R, Bal TR. Camptothecin and its analogues: a review on their chemotherapeutic potential. Nat Prod Res. 2005;19(4):393–412. doi:10.1080/14786410412331299005
3. Ramesh M, Ahlawat P, Srinivas NR. Irinotecan and its active metabolite, SN-38: review of bioanalytical methods and recent update from clinical pharmacology perspectives. Biomed Chromatogr. 2010;24(1):104–123. doi:10.1002/bmc.1345
4. Palakurthi S. Challenges in SN38 drug delivery: current success and future directions. Expert Opin Drug Deliv. 2015;12(12):1911–1921. doi:10.1517/17425247.2015.1070142
5. Bala V, Rao S, Boyd BJ, Prestidge CA. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J Control Release. 2013;172(1):48–61. doi:10.1016/j.jconrel.2013.07.022
6. Smith NF, Figg WD, Sparreboom A. Pharmacogenetics of irinotecan metabolism and transport: an update. Toxicol In Vitro. 2006;20(2):163–175. doi:10.1016/j.tiv.2005.06.045
7. Senter PD, Beam KS, Mixan B, Wahl AF. Identification and activities of human carboxylesterases for the activation of CPT-11, a clinically approved anticancer drug. Bioconjug Chem. 2001;12(6):1074–1080.
8. Yoshimatsu K, Yokomizo H, Fujimoto T, et al. Pilot study of simplified low-dose S-1 plus CPT-11 as first-line chemotherapy for patients with advanced colorectal cancer. Anticancer Res. 2007;27(3B):1657–1661.
9. Oostendorp LJ, Stalmeier PF, Pasker-de Jong PC, Van der Graaf WT, Ottevanger PB. Systematic review of benefits and risks of second-line irinotecan monotherapy for advanced colorectal cancer. Anticancer Drugs. 2010;21(8):749–758. doi:10.1097/CAD.0b013e32833c57cf
10. Slatter JG, Su P, Sams JP, Schaaf LJ, Wienkers LC. Bioactivation of the anticancer agent CPT-11 to SN-38 by human hepatic microsomal carboxylesterases and the in vitro assessment of potential drug interactions. Drug Metab Dispos. 1997;25(10):1157–1164.
11. Mathijssen RH, Alphen RJ, Verweij J, et al. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin Cancer Res. 2001;7(8):2182–2194.
12. Charasson V, Haaz MC, Robert J. Determination of drug interactions occurring with the metabolic pathways of irinotecan. Drug Metab Dispos. 2002;30(6):731–733. doi:10.1124/dmd.30.6.731
13. Du Y, Zhang W, He R, et al. Dual 7-ethyl-10-hydroxycamptothecin conjugated phospholipid prodrug assembled liposomes with in vitro anticancer effects. Bioorg Med Chem. 2017;25(12):3247–3258. doi:10.1016/j.bmc.2017.04.025
14. Sapra P, Zhao H, Mehlig M, et al. Novel delivery of SN38 markedly inhibits tumor growth in xenografts, including a camptothecin-11-refractory model. Clin Cancer Res. 2008;14(6):1888–1896. doi:10.1158/1078-0432.CCR-07-4456
15. Zhang H, Wang J, Mao W, et al. Novel SN38 conjugate-forming nanoparticles as anticancer prodrug: in vitro and in vivo studies. J Control Release. 2013;166(2):147–158. doi:10.1016/j.jconrel.2012.12.019
16. Patnaik A, Papadopoulos KP, Tolcher AW, et al. Phase I dose-escalation study of EZN-2208 (PEG-SN38), a novel conjugate of poly(ethylene) glycol and SN38, administered weekly in patients with advanced cancer. Cancer Chemother Pharmacol. 2013;71(6):1499–1506. doi:10.1007/s00280-013-2149-2
17. Wang J, Sun X, Mao W, et al. Tumor redox heterogeneity-responsive prodrug nanocapsules for cancer chemotherapy. Adv Mater. 2013;25(27):3670–3676. doi:10.1002/adma.201300929
18. Wang H, Xie H, Wu J, et al. Structure-based rational design of prodrugs to enable their combination with polymeric nanoparticle delivery platforms for enhanced antitumor efficacy. Angew Chem Int Ed Engl. 2014;53(43):11532–11537. doi:10.1002/anie.201406685
19. Santi DV, Schneider EL, Ashley GW. Macromolecular prodrug that provides the irinotecan (CPT-11) active-metabolite SN-38 with ultralong half-life, low C(max), and low glucuronide formation. J Med Chem. 2014;57(6):2303–2314. doi:10.1021/jm401644v
20. Peng CL, Lai PS, Lin FH, Yueh-Hsiu WS, Shieh MJ. Dual chemotherapy and photodynamic therapy in an HT-29 human colon cancer xenograft model using SN-38-loaded chlorin-core star block copolymer micelles. Biomaterials. 2009;30(21):3614–3625. doi:10.1016/j.biomaterials.2009.03.048
21. Akbarzadeh A, Rezaei-Sadabady R, Davaran S, et al. Liposome: classification, preparation, and applications. Nanoscale Res Lett. 2013;8(1):102. doi:10.1186/1556-276X-8-102
22. Liu Y, Li M, Yang F, Gu N. Magnetic drug delivery systems. Sci Chin Mat. 2017;60(6):471–486. doi:10.1007/s40843-017-9049-0
23. Majumder P, Bhunia S, Chaudhuri A. A lipid-based cell penetrating nano-assembly for RNAi-mediated anti-angiogenic cancer therapy. Chem Commun (Camb). 2018;54(12):1489–1492. doi:10.1039/c7cc08517f
24. Fang YP, Chuang CH, Wu YJ, Lin HC, Lu YC. SN38-loaded <100 nm targeted liposomes for improving poor solubility and minimizing burst release and toxicity: in vitro and in vivo study. Int J Nanomedicine. 2018;13:2789–2802. doi:10.2147/IJN.S158426
25. Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomedicine. 2012;7:49–60. doi:10.2147/IJN.S26766
26. Bala V, Rao S, Li P, Wang S, Prestidge CA. Lipophilic prodrugs of SN38: synthesis and in vitro characterization toward oral chemotherapy. Mol Pharm. 2016;13(1):287–294. doi:10.1021/acs.molpharmaceut.5b00785
27. Fang T, Dong Y, Zhang X, Xie K, Lin L, Wang H. Integrating a novel SN38 prodrug into the PEGylated liposomal system as a robust platform for efficient cancer therapy in solid tumors. Int J Pharm. 2016;512(1):39–48. doi:10.1016/j.ijpharm.2016.08.036
28. Bala V, Rao S, Prestidge CA. Facilitating gastrointestinal solubilisation and enhanced oral absorption of SN38 using a molecularly complexed silica-lipid hybrid delivery system. Eur J Pharm Biopharm. 2016;105:32–39. doi:10.1016/j.ejpb.2016.05.021
29. Wei X, Patil Y, Ohana P, et al. Characterization of pegylated liposomal mitomycin C lipid-based prodrug (Promitil) by high sensitivity differential scanning calorimetry and cryogenic transmission electron microscopy. Mol Pharm. 2017;14(12):4339–4345. doi:10.1021/acs.molpharmaceut.6b00865
30. Drolle E, Kucerka N, Hoopes MI, et al. Effect of melatonin and cholesterol on the structure of DOPC and DPPC membranes. Biochim Biophys Acta. 2013;1828(9):2247–2254. doi:10.1016/j.bbamem.2013.05.015
31. Wilkosz N, Rissanen S, Cyza M, et al. Effect of piroxicam on lipid membranes: drug encapsulation and gastric toxicity aspects. Eur J Pharm Sci. 2017;100:116–125. doi:10.1016/j.ejps.2017.01.007
32. Pazin WM, Olivier DDS, Vilanova N, et al. Interaction of Artepillin C with model membranes. Eur Biophys J. 2017;46(4):383–393. doi:10.1007/s00249-016-1183-5
33. Zhang X, Cao M, Xing J, et al. Identification, characterization, and synthesis of process-related impurities in antiproliferative agent TQ-B3203. J Liq Chromatogr Relat Technol. 2016;39(10):488–496. doi:10.1080/10826076.2016.1196216
34. Zhang X, Cao M, Xing J, et al. TQ-B3203, a potent proliferation inhibitor derived from camptothecin. Med Chem Res. 2017;26(12):3395–3406. doi:10.1007/s00044-017-2032-5
35. Xing J, Liu D, Zhou G, et al. Liposomally formulated phospholipid-conjugated novel near-infrared fluorescence probe for particle size effect on cellular uptake and biodistribution in vivo. Colloids Surf B Biointerfaces. 2018;161:588–596. doi:10.1016/j.colsurfb.2017.11.033
36. Li J, Cheng X, Chen Y, et al. Vitamin E TPGS modified liposomes enhance cellular uptake and targeted delivery of luteolin: an in vivo/in vitro evaluation. Int J Pharm. 2016;512(1):262–272. doi:10.1016/j.ijpharm.2016.08.037
37. Ma J, Domicevica L, Schnell JR, Biggin PC. Position and orientational preferences of drug-like compounds in lipid membranes: a computational and NMR approach. Phys Chem Chem Phys. 2015;17(30):19766–19776. doi:10.1039/c5cp03218k
38. Irby D, Du C, Li F. Lipid-drug conjugate for enhancing drug delivery. Mol Pharm. 2017;14(5):1325–1338. doi:10.1021/acs.molpharmaceut.6b01027
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.