")
Back to Journals » Journal of Experimental Pharmacology » Volume 13
Novel Experimental Therapies for Treatment of Pulmonary Arterial Hypertension
Authors Zolty R
Received 2 January 2021
Accepted for publication 7 July 2021
Published 17 August 2021 Volume 2021:13 Pages 817—857
DOI https://doi.org/10.2147/JEP.S236743
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Abdel Abdel-Rahman
Ronald Zolty
Pulmonary Hypertension Program, University of Nebraska Medical Center, Lied Transplant Center, Omaha, NE, USA
Correspondence: Ronald Zolty 982265 Nebraska Medical Center, Room 8712, Omaha, NE, 68198-2265 Tel +1 4025595591
Fax +1 4025598355
Email [email protected]
Abstract: Pulmonary arterial hypertension (PAH) is a progressive and devastating disease characterized by pulmonary artery vasoconstriction and vascular remodeling leading to vascular rarefaction with elevation of pulmonary arterial pressures and pulmonary vascular resistance. Often PAH will cause death from right heart failure. Current PAH-targeted therapies improve functional capacity, pulmonary hemodynamics and reduce hospitalization. Nevertheless, today PAH still remains incurable and is often refractory to medical therapy, underscoring the need for further research. Over the last three decades, PAH has evolved from a disease of unknown pathogenesis devoid of effective therapy to a condition whose cellular, genetic and molecular underpinnings are unfolding. This article provides an update on current knowledge and summarizes the progression in recent advances in pharmacological therapy in PAH.
Keywords: pulmonary hypertension, investigational drugs, vascular remodelling, animal models, clinical trials, new drug targets, toxicity
Introduction
Pulmonary hypertension (PH) is a vascular disease characterized by progressive increase in pulmonary pressures, ultimately leading to death from right heart failure.1
In 2018, the sixth World Symposium on Pulmonary Hypertension has recommended considering a new hemodynamic definition of PH to be mean pulmonary artery pressure at rest of greater than 20 mmHg.2
PH may occur as a consequence of pulmonary vascular disease, chronic left heart or lung disease, pulmonary embolism, or other etiologies.3 The World Symposium on PH has led to an internationally accepted classification of PH in five groups: Group 1 comprises pulmonary arterial hypertension (PAH), Group 2 comprises PH associated with left heart disease, Group 3 consists of PH associated with lung disease and/or hypoxia, Group 4 includes chronic thromboembolic PH, and ultimately, Group 5 constitutes of PH due to various causes including hemolytic anemia, sarcoidosis, histiocytosis X, glycogen storage disease, and renal diseases.3
Group 1 PAH can also be classified into many subcategories including idiopathic PAH (IPAH), heritable PAH (HPAH), and PAH associated with other diseases, such as connective tissue diseases, liver cirrhosis, congenital heart malformations, HIV infection, and schistosomiasis.3 All types of PAH share common pathological changes, such as pulmonary artery endothelial cell (PAEC) dysfunction and proliferation; pulmonary artery smooth muscle cell (PASMC) proliferation, migration, and contraction; inflammation; and fibroblast proliferation, activation, and migration.
Vascular lesions found in the lungs of PAH patients are responsible for the increase in pulmonary vascular resistance (PVR). The proliferative and fibrotic PAH lesions mainly concern small lung vessels. Precapillary lesions are typically located in muscular arteries with diameters less than 500 um diameter and arterioles.
The main histopathological characteristic of PAH is vascular wall remodeling, which comprises intima proliferation, medial and adventitial layer hypertrophy, as well as extracellular matrix deposition.4
In severe cases of PAH, plexiform lesions are found. Plexiform lesions, glomeruloid-like structures, are typically located at branching points of muscular arteries and consist of a network of vascular channels lined by endothelial cells and a core of myofibroblastic or less differentiated cells.5
The pathobiology of PAH is complex, involving cross-talk between several signaling pathways that comprises metabolic shift (Warburg effect), humoral modulation, impaired angiogenesis with loss of distal vessels, growth factors (eg, PDGF), and chronic inflammation.6–17
PAEC dysfunction is considered to be a critical initiating factor in the pathobiology of PAH.18 PAEC dysfunction observed in PAH includes an imbalance in the secretion of vasodilators and anti-mitogenic factors such as nitric oxide and prostacyclin and vasoconstrictors and pro-mitogenic molecules such as endothelin.19
Endothelial dysfunction also leads to the production of factors affecting PASMCs, thrombotic mediators and inflammatory cytokines.19 As a result, there is migration and proliferation of PASMCs into the small precapillary pulmonary arterioles, which normally lack a smooth muscle layer. Abnormal presence of myofibroblasts is also seen.20 The proliferation of PASMCs and myofibroblasts will lead to luminal narrowing and diminished ability for the vessel to dilate appropriately, ultimately causing elevation of the pulmonary arterial pressures and PVR.
Additionally, PAECs display alteration in energy production with a shift to anaerobic glycolysis, lactate synthesis and reduced oxidative phosphorylation.21 In PASMCs, there is evidence of proliferation and apoptosis resistance due to genetically22 controlled mechanisms, metabolic reprogramming,23 calcium mishandling,24 and abnormal mitochondrial function.25 Mitochondrial dysfunction creates a Warburg effect that will stimulate proliferation of vascular fibroblasts.26 Extracellular matrix (ECM) remodeling promotes PAH by reducing pulmonary arterial compliance. Finally, the expanding body of evidence has associated PAH with a substantial inflammatory process with infiltration of T- and B-lymphocytes as well as elevation of circulating inflammatory cytokines27 [Figure 1].
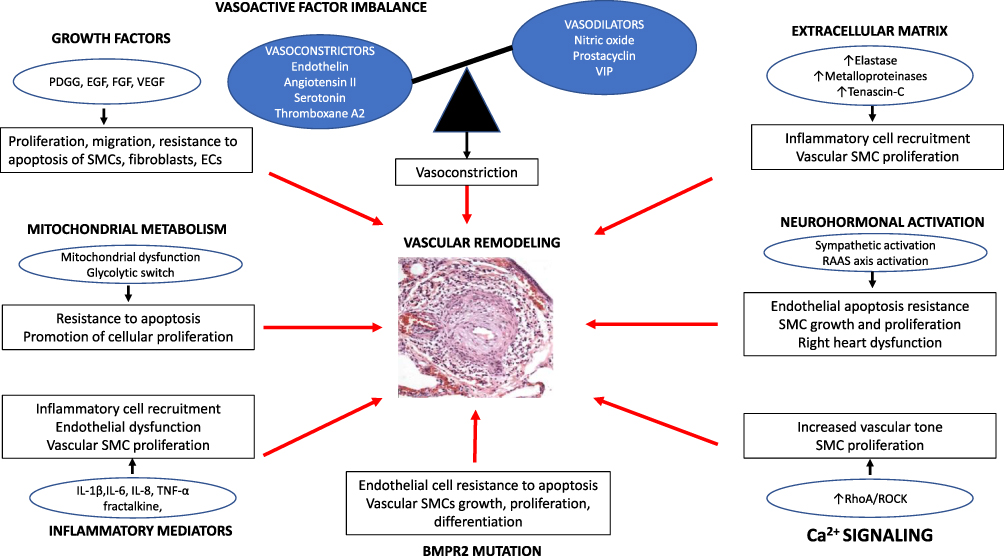 |
Figure 1 Physiopathological mechanisms of pulmonary arterial hypertension. Abbreviations: ↑, increase in; ECs, endothelial cells; EGF, epidermal growth factor; FGF, fibroblast growth factor IL, interleukin; PDGF, platelet-derived growth factor RAAS, renin–angiotensi–-aldosterone system; SMCs, smooth muscle cells; VEGF, vascular endothelial growth factor; VIP, vasoactive intestinal peptide. |
Available current FDA-approved pharmacotherapies to treat PAH fall into three main therapeutic groups: prostacyclins [cyclic adenosine monophosphate (cAMP) pathway], phosphodiesterase (PDE) 5-inhibitors and soluble guanylate cyclase stimulators [nitric oxide (NO)-cyclic guanosine monophosphate (cGMP) pathway], and endothelin receptor antagonists [endothelin-1 (ET-1) pathway]. Used as single agent or in combination, these different agents have significantly improved functional status, quality of life, hemodynamics and PAH hospitalizations.28 In the case of intravenous epoprostenol survival, these responses are usually partial and often temporary.29 Also, these therapeutics do not target the fundamental key factors involved in the pathogenesis of the disease as they act essentially as vasodilator agents.
There is an imperative necessity, therefore, for the development of novel therapies that target key molecular pathways and will potentially cure this devastating disease.
This article presents current knowledge and summarizes the progression and recent advances in pharmacological therapy in PAH (Table 1).
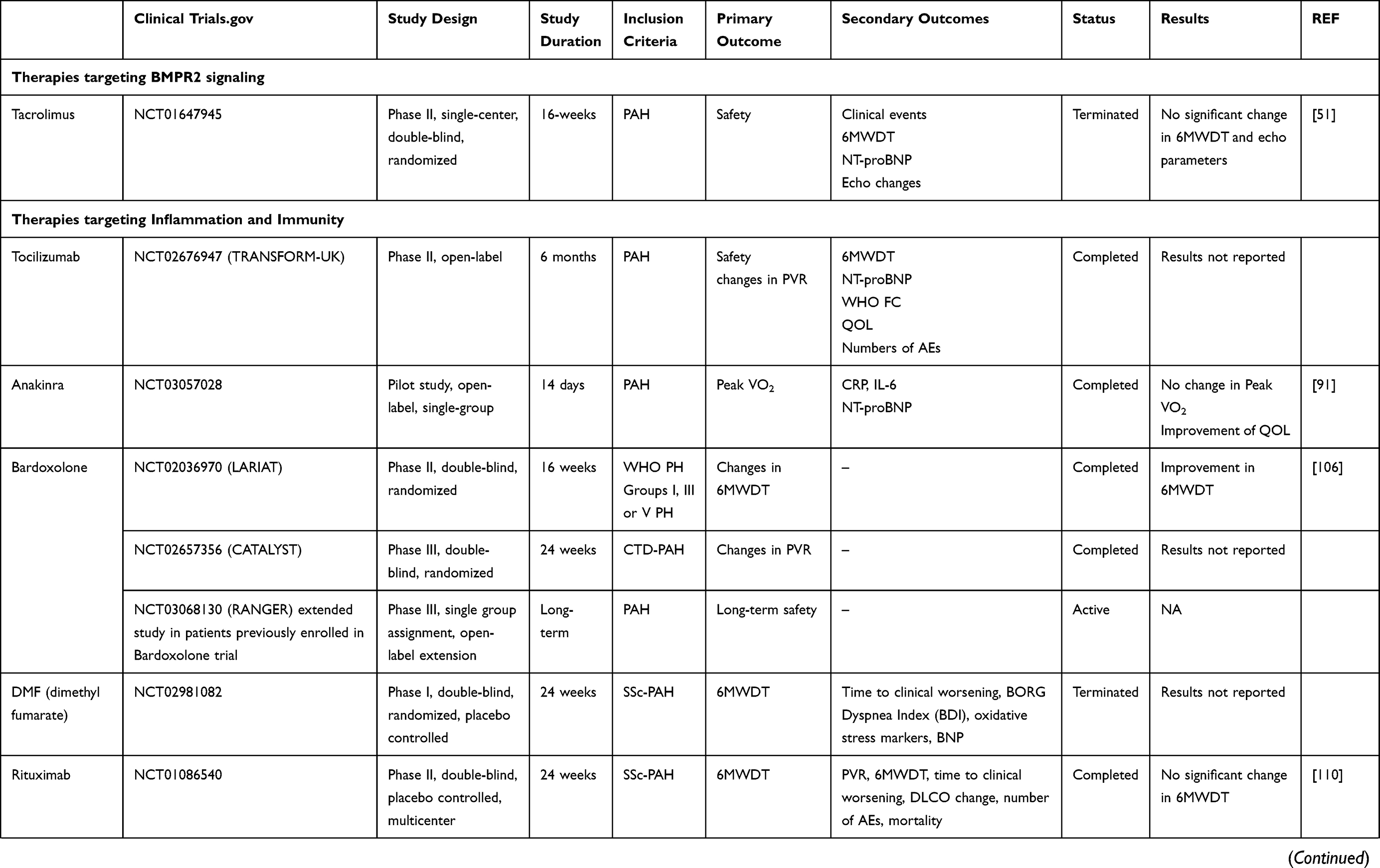 | 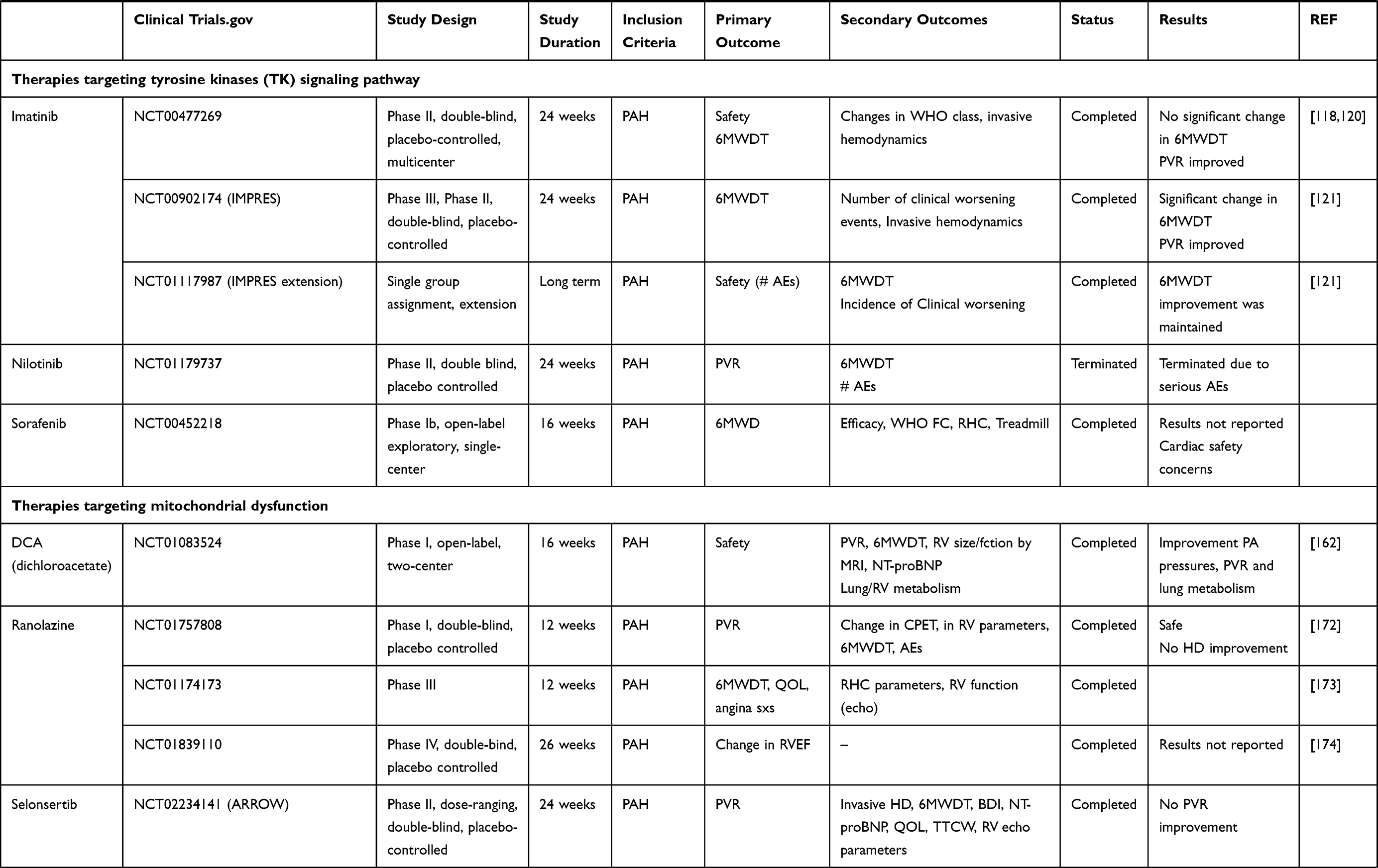 | 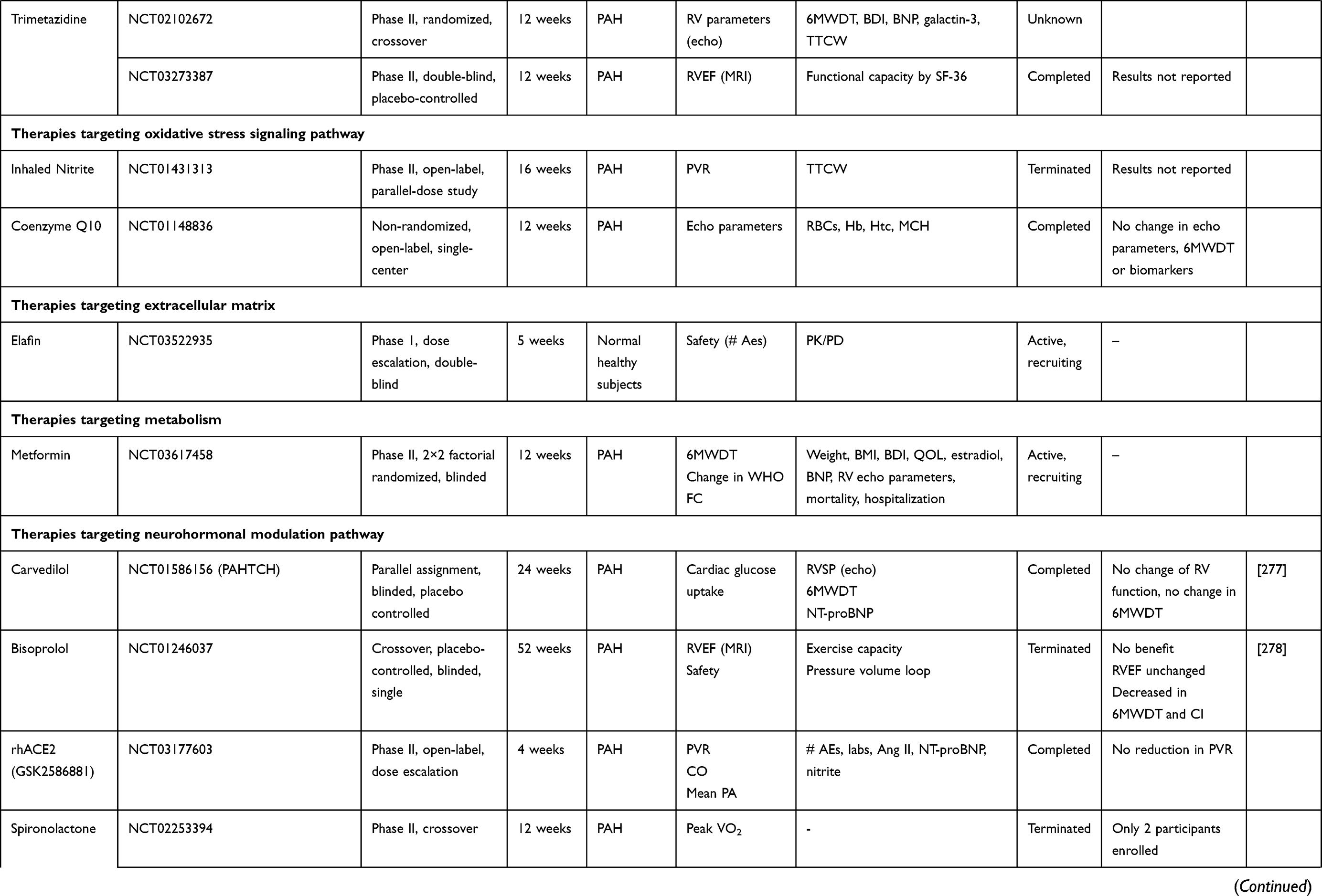 | 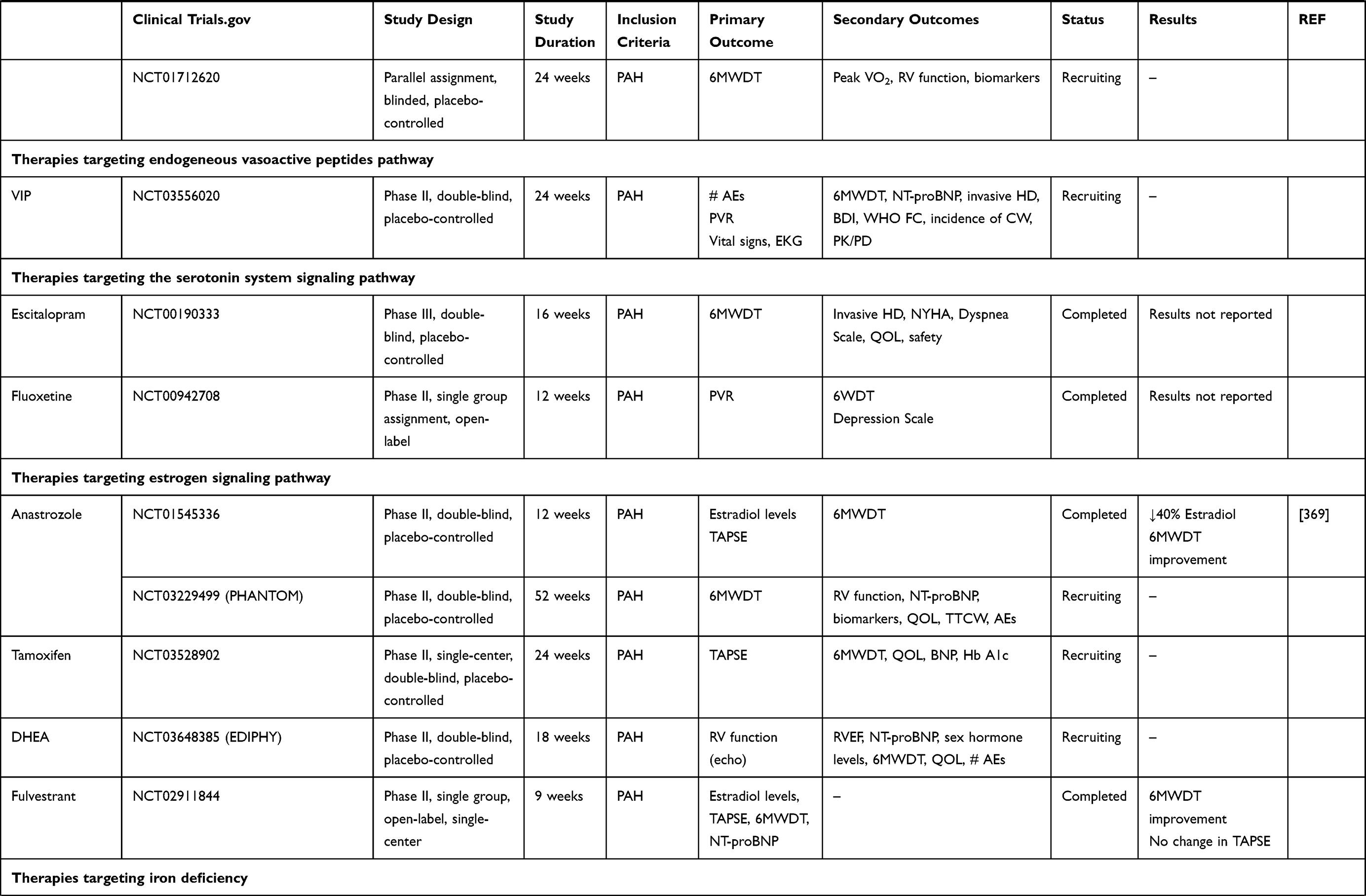 |
Table 1 Summary of Clinical Trials Data of New Potential Drugs in the Treatment of PAH |
Bone Morphogenetic Protein Receptor-2 (BMPR2) Signaling
Aberrant bone morphogenetic protein (BMP) and, more specifically dysfunctional bone morphogenetic protein receptor-2 (BMPR2) is considered to be one of the cornerstones in the PAH pathogenesis.
The BMPR2 is a membrane receptor that belongs to the TGF-β superfamily. This receptor was initially described as being involved in the regulation of cell growth and differentiation of cartilages and bones.30,31 More recently, it was demonstrated that the BMPR2 receptor is involved in the regulation of growth and apoptosis of other cells such as PAECS and PASMCs.
BMPR2 mutations are responsible for a decrease in the receptor expression and function, leading to an abnormal proliferation of PASMCs.32,33 This decrease may promote PAECs apoptosis resistance, development of vascular remodeling and pulmonary hypertension.34 Thus, BMPR2 signaling appears to have a protective function by promoting the survival of PAECs, inhibiting PASMCs proliferation and triggering anti-inflammatory response.35–37
BMPR2 ligands, the bone morphogenetic proteins (BMPs), such as BMP9 and BMP10, are cytokines regulating growth, differentiation, and apoptosis of PAECs and PASMCs. BMPs play a role in cell differentiation during embryogenesis and in the maintenance and repair of adult tissues.38–40 Initially BMPs, such as BMP9 and BMP 10, bind and bring BMPR2 and ALK1 together, resulting in phosphorylation of cytoplasmic protein called Smads (Smad1, Smad5 Smad8). These Smad proteins then bind with Smad4 in the cytosol. This new complex is translocated into the nucleus and modulates the expression of target genes43,44 (Figure 2).
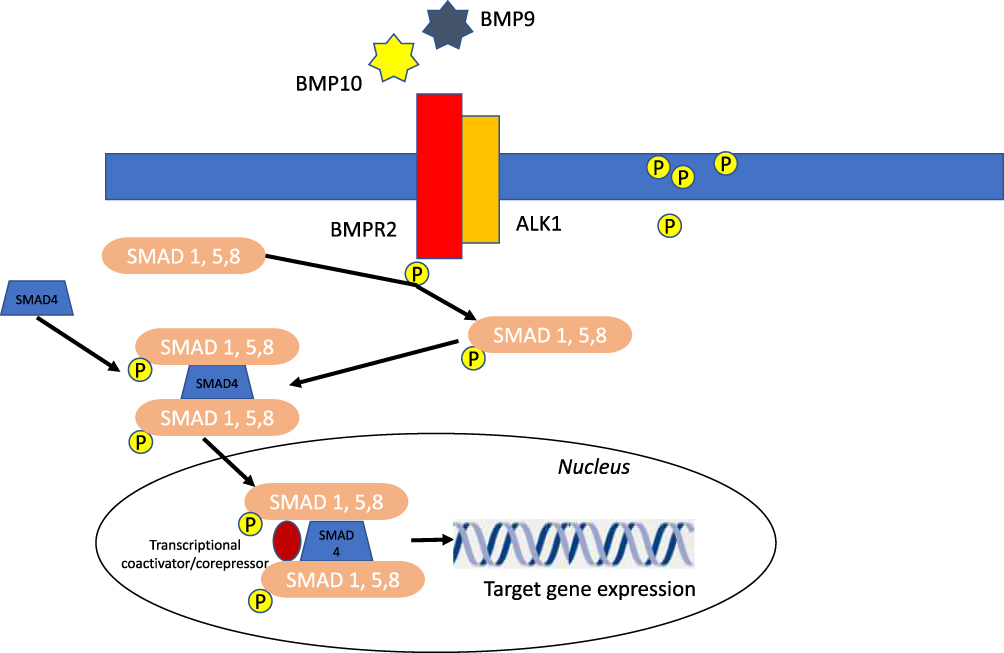 |
Figure 2 Schematic of BMP signaling pathway signaling implicated in PAH pathogenesis. BMP9 and BMP10 present in the circulation initiate signaling by binding and bringing together BMPR2 and ALKI. BMPR2 phosphorylated ALK1 which then propagate the signal through phosphorylation of SMAD 1/5/8. Subsequently, SMAD 4 forms a complex with SMAD 1/5/8, which translocates to the nucleus regulating the expression of target genes. Abbreviations: ASK1, apoptosis signal regulating kinase 1; P, phosphate. |
The importance of the BMPR2 signaling pathway in the PAH pathobiology is confirmed by the observation that >80% of subjects with heritable PAH and approximately 20% of the subjects with idiopathic PAH have a gene mutation leading to BMPR2 dysfunction.41,42 Also, subjects with no BMPR2 mutation and IPAH or PAH associated with other conditions such as connective tissue disease or HIV have reduced pulmonary vascular expression of BMPR2.43 Estrogen also can diminish BMPR2 expression, potentially explaining the female predisposition of developing the disease compared to males.44
Studies from PH animal models have confirmed the involvement of BMPR2 mutation in the development of PH. West et al45 demonstrated the development of pulmonary vascular remodeling in mice expressing a dominant negative form of BMPR2. Specifically in their PASMCs. These mice developed increased pulmonary artery pressures, pulmonary arterial muscularization and right ventricular hypertrophy (RVH). Also, Hong et al46 showed that deletion of the BMPR2 gene in PAECs predisposed mice to develop PH.
Even though the exact mechanism of how BMPR2 dysfunction leads to vascular remodeling remains unexplained, the BMPR2 signaling seems to play a critical role in thwarting PAECs and PASMCs proliferation from several pro-mitogenic factors such as VEGF, bFDF, hEGF, platelet-derived growth factor (PDGF),37,47 and prevents PAECs from apoptosis.34
Based on all these observations and studies, regulation of the BMPR2 signaling is thought to be an important potential target for the treatment of PAH.
Therapies Targeting BMPR2 Signaling Pathway
Tacrolimus (FK506)
Tacrolimus (FK506) is an immunosuppressive drug used in allogeneic organ transplant.48 It was shown to be a potent BMPR2 activator that reversed endothelial dysfunction in PAH patient cells and experimental PH.49 Tacrolimus reverses pulmonary vascular remodeling, lessens right ventricular hypertrophy (RVH) and reduces right ventricular systolic pressure (RVSP) in a mouse model of chronic hypoxia.49
Compassionate use of low-dose Tacrolimus for 12 months in three patients with end-stage PAH increased their exercise capacity with amelioration of their functional class (FC), 6-minute walk distance (6MWD), N-terminal pro-brain natriuretic peptide (NT-proBNP) levels, and augmentation of BMPR2 expression in the peripheral blood mononuclear cells (PBMCs).50
A phase IIa, placebo-controlled, double-blind, randomized, 16-week, single-center trial in 14 subjects with PAH, World Health Organization (WHO) functional class II/III symptoms was conducted using three Tacrolimus target levels (<2, 2–3 and 3–5 ng·mL-1) (NCT01647945).51 Tacrolimus was generally well tolerated. While some subjects demonstrated a marked increase in BMPR2 expression, amelioration in 6MWD, and echocardiographic parameters, these changes were not significant.51
Chloroquine and Hydroxychloroquine
Chloroquine and hydroxychloroquine have shown favorable effects in preclinical animal PH models. The mechanism of action of chloroquine comprises inhibition of lysosomal degradation of BMPR2 as well as inhibition of autophagy pathways. Today¸ chloroquine is being used to treat malaria, as well as rheumatologic disease such as systemic lupus erythematosus (SLE).52
In preclinical studies, administration of chloroquine in monocrotaline (MCT)-exposed male Sprague Dawley rats was shown to prevent the elevation of pulmonary arterial pressures and RVH, to increase cardiac output, to inhibit the development of pulmonary artery remodeling, and to block the progression of established PH.51 In rats cultured PASMCs, the administration of chloroquine was also shown to augment levels of BMPR2 expression, stimulate apoptosis and diminish PASMCs proliferation.53
The efficacy and safety of chloroquine has not been assessed in PAH clinical trials. The doses used in animal studies might be too elevated for humans.
Sotatercept
Disturbances in the bone morphogenetic protein receptor type II (BMPR2) and the transforming growth factor (TGF)-β pathways lead to cellular proliferation and vascular remodeling and cellular proliferation as seen in PAH. Sotatercept, a first-in-class, is a ligand trap with high selectivity for multiple proteins within the TGF-β superfamily, including activins and GDFs that can restore BMPR2 signaling and pulmonary vascular homeostasis. A Phase 2, multicenter trial with 106 PAH subjects showed that sotatercept in combination with background therapies for 24 weeks significantly reduces PVR and PA pressures, improves 6MWD, lessens NT-proBNP, and enhances WHO-FC compared with placebo.54
These results led to the STELLAR study, a Phase 3, randomized, double-blind, controlled trial that will evaluate the efficacy and safety of sotatercept 0.7 mg/kg SC every 3 weeks plus background therapy compared with placebo in 284 PAH patients. The primary end point of the trial is change from baseline in 6MWD. Secondary outcomes include hemodynamic improvements, time to death or clinical worsening, FC and QOL score changes (NCT04576988).
Inflammation and Immunity
Numerous PH experimental preclinical data and human studies have linked a malfunction of the inflammatory/immune system to the development of PAH.55 First, PAH is associated with systemic autoimmune diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and scleroderma.56–58 Second, perivascular inflammation has been frequently observed in subjects with idiopathic PAH59 and PAH associated with systemic autoimmune diseases.57,60 In lung sections of these PAH patients, perivascular lesions are characterized by the infiltration of immune cells of varying levels,27 including B and T lymphocytes,61–65 macrophages,67 neutrophils,66–68 dendritic cells,65,69 and mast cells.65,69 Third, higher levels of chemokines and cytokines such as tumor necrosis factor (TNF)-α, Interleukin-1 (IL-1), and Interleukin-6 (IL-6) are measured in PAH subjects.27,70 These cytokines and chemokines include TNF-α, interleukin (IL)-1β, IL-6, IL-8, MCP-1 (monocyte chemoattractant protein-1), CCL5/RANTES, fractalkine, and MCP-1 (monocyte chemoattractant protein-1). Some of these cytokines and chemokines are associated with a poor prognosis and might be used as markers of disease progression. Also, some, such as IL-1β and TNF-α, have been linked to the pulmonary peri-vascular deposition of extracellular proteins such as fibronectin.71,72 Others, such as IL-6, have been associated with the proliferation of PASMCs.73 Fourth, in the MCT and chronic hypoxia rats models, anti-inflammatory agents have been shown to prevent and inhibit the development of PH.74,75 Fifth, human studies have shown that the combination of immunosuppression and vasodilators may ameliorate pulmonary pressures, exercise capacity and prognosis of PAH subjects with certain systemic autoimmune diseases.76,77 Finally, the absence of regulatory T cells (Treg), also known as suppressor T cells, the dysregulation of B cells, and the presence of endothelial autoantibodies have also been incriminated in the pathobiology of PAH. The lack of Tregs in athymic rats submitted to SU5416 and chronic hypoxia led to peri-vascular accumulation of B cells and development of anti-PAEC antibodies as well as severe PH.78 On the other hand, the presence of Treg in euthymic rats was shown to inhibit the deposition of B-cells, development of anti-PAEC antibodies and elevation of PA pressures.78
The safety and efficiency of several agents modulating and regulating the immune system are currently being evaluated in clinical studies.
Therapies Targeting Inflammation and Immunity
Interleukin-6 (IL-6)
There is substantial evidence from the literature implicating elevated activity of the cytokine IL-6 in the development of PAH. PASMCs produce IL-6 as a pro-inflammatory cytokine.
In preclinical studies, the overexpression of IL-6 in mice was associated with elevated pulmonary pressures, RVH, and severe luminal obliteration of small pre-capillary arteries with infiltration of mast cells and lymphocytes.79
Hypoxia-induced PH transgenic IL6-deficient mice exhibit less inflammation and less pulmonary vascular remodeling compared to the wild-type mice.80 IL-6 receptor (IL-6 R) was also shown to be up-regulated in PASMCs from PH experimental animal models leading to the development of pulmonary vascular remodeling with PAECs and PASMCs proliferation.81 The administration of an IL-6R specific antagonist in experimental PH-induced rodent models prevented the development of PH.81
In human studies, IL-6 was shown to be augmented in serum and lungs of subjects with IPAH and PAH associated with connective tissue diseases (PAH-CTD). In PAH subjects, elevation of circulating IL-6 correlates with an increased risk of mortality.82,83 Similar to the experimental animal models, IL-6R expression is upregulated in PASMCs from subjects with PAH.
Tocilizumab
Tocilizumab, also known as atlizumab, is an immunosuppressive drug given for the treatment of rheumatoid arthritis (RA) and systemic juvenile idiopathic arthritis. It is a humanized monoclonal antibody against the interleukin-6 receptor (IL-6R).
The significant improvement of several PAH-CTD subjects treated with tocilizimab84–86 led to the Phase II TRANSFORM-UK clinical trial (Therapeutic Open-Label Study of Tocilizumab in the Treatment of Pulmonary Arterial Hypertension) (NCT02676947). This study was a 6-month open-label trial with tocilizumab in patients with group 1 PAH excluding those with SLE, RA, or CTD. Endpoints included safety change in exercise capacity, PVR and quality of life. Although the full data have not yet been reported, the drug was administered to 23 patients with a good safety profile. A larger, therapeutic study is expected.
Interleukin-1 (IL-1)
Serum levels of IL-1, which also promotes IL-6 synthesis, are augmented in PAH patients.83
In an inflammatory model of MCT-induced PH, increased IL-mRNA are expressed in the lung tissue87 In the same animal model, the development of PH is prevented with administration of IL-1 receptor antagonist.88 Furthermore, when IL-1 is administered to the R899X transgenic mouse with decreased BMPR2 expression, the rodent will develop significant pulmonary arterial remodeling with severely elevated pulmonary pressures.89
All these results are indicative of a strong association between IL-1 and inflammation in the PAH pathogenesis.
Anakinra
Anakinra is a recombinant of the IL-1 receptor antagonist and is approved for the treatment of RA.90
In an open-label study of 6 IPAH subjects, anakinra was shown to have a safe profile. (NCT03057028). In this small study, the six patients also had moderate functional impairment and right ventricular dysfunction at baseline. After 2 weeks of treatment, a significant reduction of hsCRP with a trend reduction of IL-6 was observed. There was also substantial improvement in symptoms as assessed by the Minnesota Living with Heart Failure Questionnaire.91 However, there was no significant change in circulating NT-proBNP, peak oxygen consumption, or RV systolic function compared to baseline.
This study indicates that anakinra in PAH appears safe with patients’ symptomatology improvement. A longer and larger study is necessary to establish the efficacy of this agent in the treatment of group 1 PAH.
Tumor Necrotic Factor-α (TNF-α)
Systemic circulating TNF-α levels were shown to be more elevated in PAH patients compared to normal control subjects.83
In preclinical studies, transgenic mice overexpressing TNF-α will lead to the development of PH.93 Also, in MCT-induced PH (MCT-PH) in dogs and rats, the lung expression of TNF-α is significantly augmented.94,95
TNF-α was shown to selectively reduce BMPR2 transcription and translation in PASMCs,93 stimulate the pro-proliferative NOTCH2 pathway, and simultaneously inhibit the anti-proliferative NOTCH3 axis, leading to excessive PASMC proliferation.92
In animal models, the administration of anti- TNF-α agent was shown to improve the disease progression and to reestablish the normal BMP/NOTCH pathway.
These results confirmed the role of TNF-α in the pathogenesis of PAH, justifying the need of anti- TNF-α approaches in the treatment of PAH.96
Etanercept
Etanercept is a TNF-α inhibitor, which is approved to treat autoimmune diseases such as rheumatoid arthritis, inflammatory bowel disease, juvenile idiopathic arthritis, psoriatic arthritis, plaque psoriasis, and ankylosing spondylitis.97–100
Etanercept was shown to prevent and reverse MCT-PH in rodents92 and in endotoxemic pigs.101 Etanercept in SU-5416 hypoxia rats restores BMPR2 expression, reduces NOTCH2 signaling activity, and normalizes activated SMAD function.58 This leads to RVSP reduction, improvement of disease progression, RVH, and vascular remodeling.96
No study is currently investigating the safety and efficacy effect of etanercept in PAH patients. The above findings suggest that a clinical trial should be considered in the future.
Nuclear Factor κB
Many of the inflammatory and immune pathways are under the control of the transcription nuclear factor κB. Nuclear factor κB stimulation leads to the upregulation of genes encoding IL-6, IL-1β, TNF, and other important immunologic signals.102
Multiple lines of evidence from rodent models have shown that nuclear factor κB blockade can prevent the development of PH.103 IPAH subjects have activated nuclear factor κB in their lung tissue,104 indicating a potential role for this pathway in human disease.
Bardoxolone
Bardoxolone methyl blocks nuclear factor κB by stimulating the regulatory transcription nuclear factor (erythroid-derived 2)–related factor 2,105 activates Nrf2, and targets mitochondrial dysfunction.
In the phase II LARIAT trial (NCT 02036970) with 22 PAH subjects, administration of bardoxolone methyl vs placebo for 16 weeks showed a significant improvement in 6MWDT in the bardoxolone methyl group.106
A 24 week Phase III study with PAH-CTD patients (CATALYST) (NCT02657356), followed by a long-term open-label extension follow-up study (NCT03068130), with bardoxolone methyl vs placebo, to evaluate the change from baseline in 6MWD has been recently completed. Results have not yet been released.
Dimethyl Fumarate (DMF)
Dimethyl fumarate (DMF) activates the transcription factor nuclear factor erythroid-derived 2-related factor 2 (Nrf2) pathway and has been approved by the US Food and Drug Administration (FDA) as a treatment option for adults with relapsing multiple sclerosis.
DMF is considered to be an immunomodulatory agent, causing a shift in T helper cells (Th) and reducing inflammatory cytokine production, such as IL-6.
DMF improves pathological hemodynamics in PH-induced hypoxic mice models.107 Not only does DMF prevent the development of PH and RVH in hypoxic and hypoxia/SU5416-treated mice, it also reverses PH in a chronic hypoxia mice model.107 DMF blocks pro-inflammatory pathways, including NFκB, and STAT3 with evidence of less lung tissue infiltration by immune cells and macrophages. Also, DMF was shown to inhibit HIF1α expression108 and provide additional anti-PH effects.
A randomized, multicenter, double-blinded, placebo-controlled trial with DMF in 34 Systemic Sclerosis-PAH (SSc-PAH) patients is currently ongoing (NCT02981082). The primary outcome of this clinical trial is improvement in 6MWD. This study will also assess the safety profile of nuclear factor κB manipulation in patients with SSc-PAH.
Rituximab
Rituximab is a chimeric monoclonal antibody against the protein CD20, which is primarily found on the surface of immune system B cells.109
Following several case reports of disease regression in CTD-PAH with Rituximab, a randomized, Phase II, placebo-controlled, double-blind, NIH-funded ASC01 trial (NCT01086540) investigated the efficacy and safety of Rituximab on disease progression in patients with SSc-PAH with change in 6MWDT at 24 weeks as primary endpoint. Secondary endpoints included RV function changes evaluated by cardiac MRI. At week 24, 6MWWD trended towards improvement. Changes in PVR were highly variable but on average improved.110
Growth Factors and Tyrosine Kinases (TK) Signaling Pathway
Pathological proliferation of PAECs and PASMCs in PAH, as well as abnormal increase in the production of growth factors such as vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF), have caused a paradigm shift in the treatment strategies of the disease as some researchers have associated the pathogenesis of PAH to a neoplastic process. These growth factors may act as powerful mitogens and chemo-attractants for SMCs, ECs and fibroblasts and may also cause resistance to apoptosis.111
PDGF can be synthesized in PAECs, PASMCs and macrophages and was shown to stimulate proliferation and migration of fibroblasts and PASMCs.111 In PASMC and PAECs from lungs of PAH subjects, levels of PDGF and its receptor, PDGFR, were found to be elevated.112 PDGF acts through the stimulation of two PDGF receptors (PDGFR-α and PDGFR-β), leading to fibroblasts and PASMCs’ proliferation.
Increased PDGF signaling has been strongly implicated in playing an important role in the pathogenesis of PAH, including the efficacy of the PDGF pathway inhibitors in PH animal models. Animal model data highlight that inhibiting the PDGF pathway may prevent/reverse the cellular proliferation of the intimal lining of the pulmonary arteries, a key characteristic of PAH.113
Tyrosine kinases (TKs) are enzymes that activate specific proteins by transferring a phosphate group from ATP to their tyrosine residues. Receptor tyrosine kinases (RTK) are composed of N-terminal extracellular ligand-binding domains and C-terminal intracellular tyrosine kinase domains which, upon ligand binding, catalyze phosphorylation of tyrosine residues of these RTKs, thereby altering their activity. PDGF acting via RTKs triggers the activation of major signaling pathways. RTKs have been associated with the pathogenesis of PAH.114
Different agents have been explored to block PDGFR and TK signaling pathway (Figure 3).
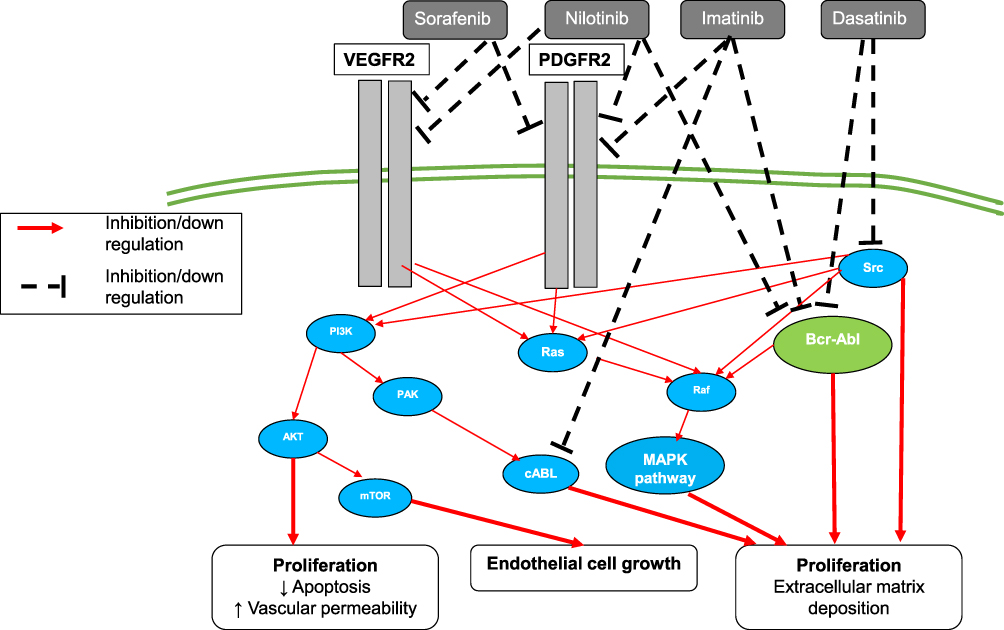 |
Figure 3 Growth factors and tyrosine kinases (TK) signaling pathway. Abbreviations: ↑, increase in; ↓, decrease in; PDGFR2, platelet-derived growth factor receptor type 2; VEGFR2, vascular endothelial growth factor type 2; BCR, breakpoint cluster region; ABL, abelson 1 kinase. |
Therapies Targeting Growth Factors and Tyrosine Kinases (TK) Signaling Pathway
Imatinib
Imatinib is an oral chemotherapy agent used to treat chronic myeloid leukemia (CML),115 gastrointestinal stromal tumors (GISTs)116 and a number of other malignancies. Imatinib functions as a specific inhibitor of a number of tyrosine kinase enzymes. Imatinib inhibits PDGF-induced arterial remodeling and SMCs proliferation. In rat preclinical experiments, imatinib hinders PASMCs proliferation.117
Following reports of improvement in several PAH patients with Imatinib,118,119 a phase II, multicenter trial evaluated the efficacy, safety, and tolerability of imatinib in 59 PAH subjects (NCT00477269). The study demonstrated that this agent was well tolerated. There was no significant change in 6MWDT, which was the primary end point. However, there was a significant decrease in PVR and an increase in cardiac output in imatinib-treated patients versus placebo.119,120 Post hoc studies of this clinical trial showed improvement of 6MWD in a subgroup of patients with a PVR >1000 dyne·s·cm (−5),120 indicating that imatinib might be efficient in severe PAH patients. These results led to the IMPRES trial (Imatinib in PAH, a Randomized Efficacy Study) (NCT00902174), which was a Phase III, 24-week, randomized, placebo-controlled, double-blind, multicenter clinical study evaluating the efficacy and safety of imatinib in 202 symptomatic PAH patients with PVR ≥ 800 dyne·s·cm (−5) on ≥2 specific PAH background therapies. At the completion of the study, the 6MWDT in the active treated group improved by 32 meters compared to the placebo arm (P= 0.002), an effect maintained in the open-extension study. Also, the PVR significantly diminished by 379 dyne·s·cm (−5).121 Moreover, treatment with imatinib was associated with significant improvement of the RV function compared to placebo in the echocardiographic IMPRES sub-study.122 Nevertheless, discontinuations and serious adverse events were more common in the imatinib arm than in the placebo arm. Subdural hematoma occurred in eight patients who received both imatinib and anticoagulation.121 The seriousness and the numbers of the adverse effects of imatinib arm, despite the strong efficacy of the drug, precluded the FDA approval of imatinib for the treatment of severe PAH patients.
Seralutinib
Although the studies of imatinib in PAH demonstrated significant hemodynamic and 6MWD improvements in PAH subjects, its poor tolerability and serious side effects suggested that while targeting the PDGFR and TK signaling pathway is significant as a potential therapeutic for PAH, a drug with an improved therapeutic window would be more optimal.
Based on this rationale, seralutinib was developed as a highly potent, small-molecule PDGFR kinase inhibitor that is formulated to be delivered via inhalation to limit systemic exposure and thereby improves tolerability and side effect profile of this class of drugs.
A current Phase II randomized trial with seralutinib is currently undergoing to determine the effect of seralutinib on improving pulmonary hemodynamics in subjects with PAH who are FC II and III with PVR improvement as primary endpoint. The secondary objective of this trial is to determine the effect of seralutinib on improving exercise capacity in this population. (NCT04456998)
Sorafenib
Sorafenib is multi-kinase inhibitor drug approved for the treatment of advanced renal cell carcinoma,123 hepatocellular carcinoma,124 FLT3-ITD-positive AML,125 and radioactive iodine resistant advanced thyroid carcinoma.126
This tyrosine-kinase inhibitor also exerts activity against angiogenesis growth factor receptors PDGFR and VEGFR as well as against Raf-1 kinase, a regulator of endothelial apoptosis.127
Sorafenib demonstrated anti-pulmonary arterial remodeling effects and hemodynamic improvement in MCT induced PH rodent.114,128 Sorafenib was shown to have positive hemodynamic effects in subjects with refractory PAH as well.129
A small exploratory phase Ib, single-arm open-label study, revealed that 16 weeks of sorafenib administration improved 6MWD significantly in 12 very limited PAH patients despite continuous administration of intravenous prostacyclin analog130 (NCT00452218). Potential cardiovascular adverse effects with sorafenib have been described in several reports.
Nilotinib
Nilotinib, a second-generation TK inhibitor is structurally related to imatinib.17 It is 10–30-fold more potent than imatinib in inhibiting BCR-ABL tyrosine kinase activity and proliferation of BCR-ABL expressing cells.131–134
Nilotinib is indicated for the treatment of both initial CML and CML resistant to imatinib as well as for the treatment of gastrointestinal stromal tumors.135
A Phase II multicenter, double-blind, randomized, 24-week, placebo controlled study was performed in PAH subjects to assess safety, tolerability and PK of nilotinib (NCT01179737). The trial was discontinued due to serious adverse reactions in the nilotinib arm. To be noted that nilotinib also carries a black box warning in the United States for possible heart complications.
Dasatinib
In contrast to imatinib, sorafenib, and nilotinib, dasatinib another second-generation BCR-ABL TK inhibitor indicated to treat positive CML and Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL)136 was shown in several reports to increase the risk of developing PAH.137,138
In October 2011, the US Food and Drug Administration (FDA) issued a safety announcement warning health care professionals of the elevated risk of developing PAH in patients receiving dasatinib. The drug is considered to induce lung vascular toxicity and predispose the development of PAH.
Thus, not all the TK inhibitors can be considered similar and may have potential opposite effects on PAH. Better comprehension of the different TK inhibitor properties and outcomes is needed.
RhoA/Rho-Kinase Inhibitors Signaling Pathway
Rho-kinase is part of a family of enzymes that are involved in regulating various cellular responses, such as cellular growth, contraction, gene expression migration, differentiation, (7) and, more specifically, smooth muscle tone.139
Intracellular signaling of the RhoA/Rho-kinase Inhibitor signaling pathway is increasingly appreciated as an important signaling pathway in the pathogenesis of PAH because of its vasoconstriction and its pro-proliferative effects of the pulmonary artery wall cells.140,141
Rho-A is a small GTP-binding protein that acts via its downstream effector Rho-kinase. This pathway couples membrane receptor/G protein signaling with phosphorylation of intracellular proteins. Rho-kinases contribute to agonist-induced vascular contraction via Ca2+ sensitization of PASMCs. Rho-kinase might also contribute to endothelial dysfunction through negative regulation of eNOS and phosphatidylinositol 3-kinase (PI3K) activity causing reduced endothelial NO bioavailability.
RhoA/Rho-kinase signaling pathway has been linked to various key upstream mediators (eg, endothelin, thromboxane, angiotensin II and serotonin)142 (Figure 4).
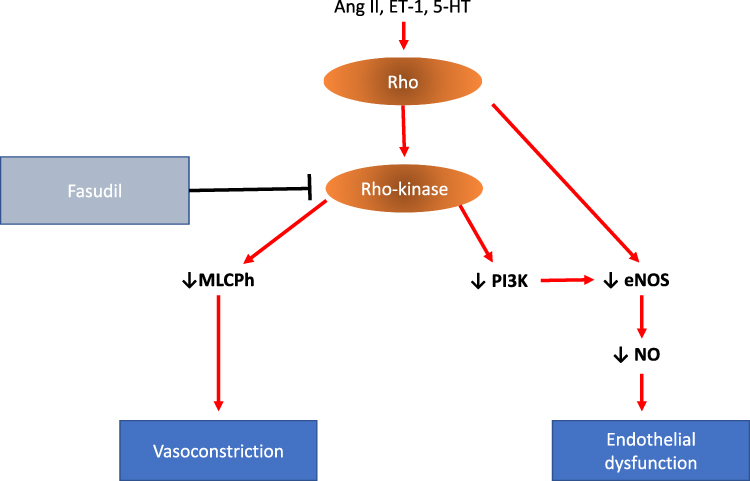 |
Figure 4 RhoA/Rho-kinase signaling pathway. Wthin PAEcs and PASMCs, Rho-Rho kinase can be activated by several pathological mediators, including angiotensin II (Ang II), endothelin-1 (ET-1) and 5-hydroxytryptamine (5-HT). Rho-Rho kinase activation leads to inhibition of myosin light chain phosphatase (MLCPh) and subsequently vasoconstriction. Rho-Rho kinase also contributes to endothelial dysfunction through negative regulation of endothelial nitric oxide synthase (eNOS) and phosphatidylinositol 3-kinase (PI3K) activity causing reduced NO bioavailability. Abbreviations: ↓, decrease in; 5-HT, 5-hydroxytryptamine; Ang II, angiotensin II; ET-1, endothelin-1; eNOS, endothelial nitric oxide synthase; MLCPh, myosin light-chain phosphatase; NO, nitric oxide; PI3K, phosphatidylinositol 3-kinase. |
Moreover, there is evidence that chronic hypoxia activates RhoA in PASMCs and PAEC.143–146
The importance of RhoA/Rho-kinase signaling in preclinical hypoxic PH studies is highlighted by the experiments of Fagan et al, in which the RhoA/Rho-kinase inhibitor Y267632, but not nifedipine, caused marked vasodilatation in hypoxic induced PH mice.140
In preclinical hypoxia-PH rodent studies, the RhoA/Rho signaling pathway showed vascular remodeling and vasoconstriction with sustained augmentation of PVR.140,142
Likewise, in humans, the RhoA/Rho kinase activity has been shown to be augmented in the PASMCs and PAECs of idiopathic PAH patients.147
Therapies Targeting Rho/ROCK Signaling Pathway: Rho-Kinase Inhibitors
Fasudil
Fasudil, a Rho-kinase inhibitor, has been studied in clinical trials as an antianginal148 and for the treatment of cerebral vasospasm.149
In studies with MCT-induced PH rats, administration of Fasudil demonstrated improvement of PA pressures, arterial vascular remodeling and BNP levels compared to sildenafil and bosentan.150 Clinically, fasudil administered intravenously has also shown significant improvement of hemodynamic parameters in subjects with severe PAH.151
A randomized, placebo-controlled, double-blind trial with 20 PAH subjects who received oral fasudil extended release demonstrated a significant increase in the cardiac index after 30 months of fasudil administration; nevertheless, no 6MWD amelioration was seen.152 Preliminary results of another trial with fasudil demonstrated reverse pulmonary vascular remodeling effect.153 Inhaled fasudil was also shown to lessen PVR significantly in a small PAH subject study.154
The positive experimental work with Rho-kinase inhibitors in both preclinical and clinical studies suggests the need for larger clinical trials to study their efficacy and safety in PAH.
Mitochondrial Dysfunction
Growing evidence has demonstrated the role of mitochondrial dysfunction in the pathogenesis of PAH. Mitochondria control the redox-related enzymes, the proteins of the electron transport chain, the regulators of proton gradient and apoptosis as well as calcium homeostasis and mitophagy. Mitochondria are the major source of energy production and are central to cellular metabolism. The metabolic pathways of mitochondria comprise fatty acid oxidation, glucose oxidation, and glutaminolysis.155–158 Mitochondrial dysfunction in PAH is associated with increased production of acid lactic production and uncoupled glycolysis at the cost of pyruvate production and glucose oxidation.
Mitochondrial dysfunction might lead to pulmonary artery remodeling and therefore is linked to the underlying pathobiology contributing to the disease.159
Pulmonary vessels have a mitochondrial-metabolic phenotype comparable to that observed in cancer and described by Warburg.160 This Warburg effect stimulates apoptosis resistance and proliferation of the pulmonary vessel cells.10
The Warburg effect mimics a pseudohypoxic state in which the transcription factor hypoxia inducible factor 1α (HIF-1α) is being upregulated. HIF-1α controls energy metabolism, vasomotor tone and angiogenesis. The HIF‐1α upregulation takes place in spite of normal pO2 and appropriately is called pseudohypoxia.
Increased HIF-1α is also caused by decreased levels of superoxide dismutase (SOD2) activity. The decrease in SOD2 activity found in PH will cause a reduction in the production of hydrogen peroxide (H2O2), generating this pseudo-hypoxia “milieu”. The upregulation of HIF‐1α inhibits the production of pyruvate dehydrogenase (PDH), leading to the PASMCs proliferation, apoptosis resistance and inflammation, which are key factors of the pathobiology of the disease and, finally, to the reduction of the acetyl-CoA entry into the Krebs cycle.161
Therapies Targeting Mitochondrial Dysfunction
Dichloroacetate (DCA)
The inhibitor dichloroacetate (DCA) was shown to activate pyruvate dehydrogenase, restore glucose oxidation, and prevent as well as reverse the development of PH in preclinical rodent studies.162–164
The effect of DCA to reverse the glycolytic shift, increase energy and ameliorate RV function in preclinical studies has led to an open-label, Phase I study to investigate DCA in subjects with PAH FC III–IV (NCT01083524).
In this 4-month, open-label study conducted in Canada and the UK, DCA was given to twenty PAH subjects already on standard PAH treatment and had a positive effect on symptoms. The treatment reduced PA pressures and PVR, improved lung metabolism as assessed by positron emission tomography (FDG-PET) scan, and ameliorated exercise functionality with a large variation of individual response.165
Selonsertib
Selonsertib (GS-4997), an inhibitor of apoptosis ASK1 (signal regulating kinase 1)166 that also targets mitochondrial dysfunction, was evaluated in a Phase II study to determine the effect of selonsertib on PVR in 151 subjects with PAH over a period of 24 weeks (NCT02234141). Selonsertib did not significantly improve PVR compared to placebo.
Ranolazine
Ranolazine is a piperazine derivative that inhibits persistent or late inward sodium current (INa) in heart muscle167 and promotes glucose oxidation in mitochondria. Ranolazine is currently approved for the treatment of chronic angina.168,169
In a rodent model of RVH, ranolazine was reported to successfully reverse metabolic dysfunction and improve cardiac output and exercise capacity.170,171
In a safety Phase I clinical trial (NCT01757808) 12 PAH patients were given ranolazine and were followed for 12 weeks. Ranolazine was found safe without significantly impacting hemodynamics (NCT01757808).172
Ranolazine was investigated in a 3-month phase III study with 11 PAH subjects (NCT01174173). In this study, ranolazine was shown to ameliorate RV function (p = 0.037), reduce RV size (p = 0.015), and enhance WHO FC (p = 0.0013) with a trend toward increase in exercise time on bike echocardiogram (p = 0.06). Nevertheless, no significant hemodynamics by right heart catheterization was observed.173
Another multicenter study (NCT01839110) looking at the effect of ranolazine in stable PAH patients with RV dysfunction (RVEF <45%) has been completed, but results have not yet been published.174
Trimetazidine
Trimetazidine is another piperazine derivative that also promotes glucose oxidation in mitochondria. Preclinical data have demonstrated that fatty acid oxidation inhibition by trimetazidine reverts the PH adverse phenotype by restoring mitochondrial function, decreases cellular proliferation and restores apoptosis susceptibility in PASMCs.
Preclinical rodent studies have demonstrated improvement of PA pressures and remodeling in animal treated with trimetazidine.175
A Phase II trial study is currently investigating the effect of trimetazidine on right ventricular function change in 25 PAH patients (NCT02102672).
Oxidative Stress
Abundant evidence suggests that oxidative stress (OS) is highly implicated in the pathobiology of PAH.
Increased levels of oxidative stress have been demonstrated in serum and lungs of animal PH models as well as PAH subjects.176–179 Furthermore, the levels of oxidative stress increase concomitantly with the gravity of the disease, signifying that oxidative stress markers could potentially be used as markers of disease severity and evolution.180–182
Increased production of oxidant species and/or decreased production of antioxidants characterize oxidative stress. Oxidative stress is associated with augmented production of reactive oxygen species (ROS) and reactive nitrogen species (RNS), diminished NO bioavailability and decreased glutathione peroxidase, superoxide dismutase (SOD), and catalase activity.176
ROS are created as a result of oxidation–reduction reactions and include free radical molecules such as superoxide (O2−), hydroxyl radical (OH−), lipid peroxyl and non-free radical species like hydrogen peroxide (H2O2). RNS like (ONOO−) are produced by the reaction between NO and O2−.183 Cellular ROS are generated as by-products of mitochondrial respiration, NADPH oxidase, eNOS,184 and xanthine oxidase.185
OS plays a crucial role in the pathobiology of PH176,186–188 as it causes vessel wall thickening by stimulating the activity of transforming growth factor-β1 (TGF-β1), VEGF, fibroblast growth factor-2 (FGF-2),189 and PDGF.190 Additionally, OS can trigger vasoconstriction by increasing the production of endothelin-1191,192 and thromboxane A2,193 and by decreasing prostacyclin levels.194,195 Also, OS was shown to upregulate the transcription of the factors HIF-1α and HIF-2α,196,197 factors that are also involved in the development of PAH.198,199 Finally, augmented OS causes inflammation and cell damage via oxidation of proteins, lipids and DNA.184,186,200–203
Preclinical rodent models have demonstrated that increased OS contributes to the pathogenesis and the development of PH.
Extracellular superoxide dismutase (SOD3) is essential for removing extracellular superoxide anions and is highly expressed in the lungs. SOD3 appears to play a major role in protecting against the development of PH through decreasing O2− and increasing H2O2 and NO bioavailability. SOD3 knockout mice and SOD3 loss of function gene mutation in rats resulted in the development of PH, RVH and vascular remodeling in response to hypoxia or MCT.204
Therapies Targeting Oxidative Stress
In spite of a wealth of data demonstrating the involvement of the oxidative stress in PH, and several cases reports of antioxidant therapies improving PH in animal models, these agents have been essentially ineffective in treating PAH subjects.
Dihydroartemisinin (DHA)
Dihydroartemisinin (DHA) is an anti-inflammatory, anti-malaria, and anti-tumor agent. DHA blocks PAECs proliferation and diminishes OS by augmenting SOD expression and decreasing ROS in MCT-PH rats.205
No clinical studies have yet been performed to investigate the effects of DHA in PAH subjects.
Fenofibrate
The PPARα agonist fenofibrate is another anti-oxidative stress therapy. In MCT-PH rats, fenofibrate reduces RVH, OS, ROS, and NADPH oxidase (NOX-1) activity.206 No clinical trial has yet investigated the effects of fenofibrate in PAH patients.
Trapidil
Trapidil is a potent vasodilator, which was shown to improve the effects of OS in PH in augmenting lipid and glutathione peroxidation as well as decrease RV dilatation in MCT-PH experimental animals.207
No clinical studies have been conducted to investigate the effects of trapidil on PAH subjects.
Nitrite
NO2− is a reactive nitrogen species (RNS) created by the oxidation of NO. In animal models of PH, nebulized nitrite caused reduction in mean PAP and regression of pulmonary remodeling.208 Also, administration of inorganic nitrate, which is metabolized into nitrite and NO, was shown to improve the severity of PH in mice.209
However, a phase II trial single center evaluating the effect of inhaled nitrite in 48 PAH subjects undergoing RHC did not significantly improve PVR (NCT01431313).
Coenzyme Q
Coenzyme Q (CoQ) is an antioxidant via the redox cycle. CoQ participates in oxidative phosphorylation and plays a critical role in biochemical generation of ATP.
In a prospective randomized double-blind study with 15 PAH patients evaluating the use of the coenzyme Q10 vs placebo for 12 weeks, there was a small enhancement in right ventricular hemodynamics with no significant improvement in 6MWD or biomarker levels (NCT01148836).210
Allopurinol
Xanthine oxidase (XO) generates oxygen species. XO catalyzes the oxidation of hypoxanthine to xanthine and can further catalyze the oxidation of xanthine to uric acid with the creation of four superoxide anions.211 Thus, XO plays a key role as a regulator of the cellular OS.212
Under conditions of tissue hypoxia in an experimental model,213 the breakdown of ATP to AMP to hypoxanthine provides substrate to XO. Subsequently, XO uses oxygen rather than NAD as an oxidant. As a result, XO produces superoxide and hydrogen peroxide (H2O2) rather than NADH.214,215 Increased vascular O2•− production due to XO affects negatively endothelial function by impairing nitric oxide (NO) signaling216 and triggering pulmonary arterial remodeling.217
Rodent animal studies have tested the effect of allopurinol in PH.
Elevated levels of serum and lung xanthine oxidase activity as well as increased levels of phosphatidylcholine hydroperoxide, a marker of oxidative stress have been demonstrated in rats exposed to chronic hypoxia217,221 Administration of allopurinol to these hypoxic rats decreased PCOOH levels, improved pulmonary vascular remodeling, and diminished RVH.217,218
In PAH clinical studies, an abnormal role for the xanthine oxidase pathway was also demonstrated. In a study with 99 IPAH subjects, there was a strong association between the right atrial elevation and the level of uric acid.219 In another retrospective study with 29 PAH patients, uric acid levels correlated with New York Heart Association class. Also, elevated uric acid levels correlated with lower 6MWD and a worse survival.220 Additionally, xanthine oxidase activity is increased in subjects with PAH compared to normal controls.221 Moreover, expression of the antioxidant enzyme SOD2 is reduced in PAH lungs.186,222
Despite the potential benefits of treating PAH patients with allopurinol, no clinical study has investigated the potential benefits of allopurinol in PAH subjects.
Extracellular Matrix (ECM)
The extracellular matrix (ECM) plays a critical role in controlling cell shape and cell signaling, in maintaining cell–cell communication, and in regulating cell differentiation. Vascular stiffness is directly associated with the proportion of the different ECM components regulating PASMCs contractility and proliferation.223–225 Elastin and collagen are the main structural ECM constituents. A variety of growth factors and cytokines regulate the production of the various components of the ECM. Turnover of the ECM is controlled by the balance between proteolytic enzymes such as serine elastases and matrix metalloproteinases (MMPs). The MMPs are matrix-degrading enzymes implicated in ECM turnover and in PASMCs migration and proliferation. It is, therefore, not surprising that perturbations in the regulation of the ECM would lead to pathogenic pulmonary vascular remodeling and PAH.
Remodeling of the extracellular matrix (ECM) with augmented collagen deposition and cross-linkage as well as increase in the elastic laminae breakdown are characteristics of PAH.19
Among the MMPs, MMP-2, and MMP-9 are the major MMPs in the pulmonary vessels. They are implicated in the PASMC stimulation and neointimal development characterizing the vascular remodeling seen in PAH.226,227 MMPs are regulated by the tissue inhibitors of metalloproteinase (TIMP) TIMP-1 and TIMP-2. The imbalance of MMPs/TIMPs will lead to matrix abnormality and remodeling.228 Also, pulmonary arterial stiffness stimulates proliferation of PASMCs through mechano-activation of several signaling pathways, including transcriptional cofactors YAP/TAZ, transforming growth factor-β, Toll-like receptor, and NF-κB.229
Preclinical studies have revealed that transgenic expression of MMP-9 is augmented in the monocrotaline-PH rodent model. The data further point out that MMP-9 contributes to the fibrosis and remodeling of pulmonary vessels in this experimental model. Inhibition of MMP-9 should be considered as possible PAH treatment.230
Therapies Targeting the Extracellular Matrix
MMP Inhibitors
Preclinical research has revealed potential therapeutic benefits with MMP inhibitors in the monocrotaline-PH rodent preclinical studies.231,232
A study demonstrated that intratracheal instillation of human TIMP-1 gene in the lungs of rodents subjected to monocrotaline improved pulmonary vascular remodeling, RVH, and muscularization of small pulmonary vessels.233
Several pharmacological compounds have been shown to improve PH via regulation of MMP/TIMPs.234
Lercanidipine, a vasoselective dihydropyridine calcium channel blocker, showed improvement in PAH subjects by reducing serum MMP-9 levels and OS with no modification of proMMP-2 activity or TIMP-1 level.235,236
The calcium channel blocker, amlodipine, administered to the MCT-PH rat was shown to inhibit MMP-2 activity, platelet activation, PAECs damage, and PASMCs proliferation.237
The endothelin receptor antagonist, bosentan, was also shown to significantly diminish MMP-2, TIMP-1, and NO synthase activity in monocrotaline-induced PH rodent.238
Periostat, the only MMP inhibitor currently approved by the FDA, is only indicated for periodontal disease. The probable cause for its low use in clinical practice is its numerous undesirable adverse reactions and poor oral bioavailability.
Serine Elastase Inhibitor: Elafin
The neutrophil elastase, which is be secreted by PASMCs, plays a fundamental role in the adverse remodeling of the ECM of the pulmonary vessels in PAH.
Transgenic mice overexpressing elafin, a serine elastase inhibitor when subjected to hypoxia, showed diminished serine elastase and MMP activity compared to controls. Elafin transgenic mice demonstrated improved PH pressures, reduced muscularization and conservation of the peripheral small pulmonary arteries compared to the non-transgenic mice.239
Although the clinical use of elastase inhibitors has been limited by hepatotoxicity, the endogenous elastase inhibitor. Elafin, remains a promising therapeutic possibility. Elafin, in addition to blocking the action of elastase, enhances BMPR2-mediated signaling, inhibits nuclear factor κB and diminishes the innate immune system.240 Administration of elafin in animal models reverses PH.232 As such, clinical exploration of elafin as a PAH therapy is underway, beginning with a safety and tolerance trial in healthy volunteers (NCT03522935).
Metabolism
Peroxisome Proliferator-Activated Receptor-γ (PPAR-γ)
Emerging evidence has demonstrated that the Peroxisome proliferator-activated receptor-γ (PPAR-γ) pathway dysregulation is involved in PAH development.241–244 PPAR-γ, a nuclear transcription factor, is known to modulate various cellular and physiological processes including cell differentiation, lipid metabolism, inflammation, and tumorigenesis.
In healthy PAECs and PASMCs, PPAR-γ is profusely expressed, but its expression declines significantly in the PAECs and PASMCs of PH affected patients and animals.242
Moreover, the elimination of PPAR-γ from PAECs and PASMCs produces PH and activation of PPAR-γ reduces the PH development.245 PPARγ plays a key role in regulating the BMP2/BMPR2 and transforming growth factor–β1 (TGFβ1) pathways in vascular SMC, regulating several miRNAs, and cell proliferation, as well as the glucose metabolism.246,247
PPAR-γ acts downstream of BMP-RII. BMP2/BMPR2 activates PPARγ in PASMC,16 and mRNA expression of both BMP2248 and PPARγ242 is decreased in lung tissue from IPAH patients.
Thus, PPAR-γ has been proposed to be an important therapeutic target for PAH.
Therapies Targeting PPAR-γ
A class of drug substances that are known to be potent agonists of PPAR-γ are thiazolidinediones. Rosiglitazone and pioglitazone, for example, are two thiazolidinedione-based orally active drugs used in type 2 diabetes. These drugs improve glycemic control and enhance insulin sensitivity in diabetic patients by selectively activating PPAR-γ in the liver, skeletal muscle, and adipose tissue.
Rosiglitazone
Rosiglitazone has been shown to decrease PASMC proliferation by modulating cell growth and apoptosis.37,249 Rosiglitazone reduces mPAP in both MCT- and chronic hypoxia-induced PH animal models.250,251 Unlike other anti-PH drugs, which reduce only the pulmonary pressures but have little or no direct effect on vascular remodeling, rosiglitazone appears to reduce pulmonary pressures and pulmonary vascular remodeling in PH animals.249
At the molecular level, rosiglitazone stimulates adiponectin, which suppresses PDGF with subsequent inhibition of PASMC proliferation.252 In chronically hypoxic rats, rosiglitazone increases PPAR‐γ levels and decreases endothelin (ET1) and VEGF levels.253 In hypoxic mice, rosiglitazone reduces PH by improving oxidative signaling by decreasing superoxide generation and PDGF activation.251
Pioglitazone
In monocrotaline-PH rats, pioglitazone was shown to improve pulmonary pressures, as well as peripheral pulmonary vessel muscularization and wall thickness. Furthermore, RVH and fibrosis are also attenuated.254
In SU‐5416 hypoxia rats, pioglitazone decreases PA pressures, reduces pulmonary vascular remodeling and improves RV function with reduction of RV fibrosis.255 Mitochondrial function and organization from right ventricular cells also improve. Pioglitazone appears to stimulate multiple genes involved in the FAO pathway, with subsequent utilization enhancement of fatty acid. Finally, pioglitazone was shown to reduce the elevated levels of miR‐197 and miR‐146b in SU‐5416 of the RV cardiomyocytes.255
Although the preclinical data are promising, the administration of pioglitazone and rosiglitazone for PAH patients should be considered carefully as these drugs can cause fluid retention and HF.256
No clinical studies have been conducted to investigate the effects of rosiglitazone and pioglitazone in PAH subjects.
Metformin
In experimental studies, metformin, a biguanide agent used in type 2 diabetes mellitus,257 has demonstrated favorable effects on the prostacyclin, endothelin and nitric oxide signaling pathways. Metformin, via AMPK activation, inhibits the proliferation of murine PASMCs induced by ET-1.258 AMPK activation also causes an increase in NO synthesis via stimulation of eNOS.259 Furthermore, Metformin increases the tyrosine nitration of prostacyclin synthase.260 Additionally, Metformin was shown to block the estrogen pathway via inhibition of aromatase transcription,261 and to decrease PASMCs stimulation by blocking MAPK (mitogen‐activated protein kinase)262 and by decreasing Rho kinase activity.262 In MCT rats, administration of metformin reduces pulmonary vascular remodeling, decreases pulmonary pressures and RVH.262
Metformin in PAH subjects is presently being tested in a phase II clinical study (NCT03617458). The primary end points include safety and measure of OS biomarkers. Secondary end points include BMPR2 expression in mononuclear cells, RVEF and RV volume measures using MRI, and 6MWD.
Neurohormonal Modulation
In PAH, both sympathetic nervous (SNS) and renin–angiotensin–aldosterone (RAA) systems are activated. Even though agents targeting the sympathetic or RAA signaling pathways have been shown to improve PH in animal models, the efficiency of such treatments in humans has been ambiguous.
Sympathetic Nervous System Activation
A large body of evidence has confirmed the contribution of the sympathetic nervous system in the pathogenesis PAH.263,264
In one study with 60 group 1 PAH subjects, circulating norepinephrine levels inversely correlated with cardiac output.265 In another study with 32 PAH patients, circulating norepinephrine levels strongly correlated with PVR and 5-year mortality rate.266
Similarly to the left ventricle in HFrEF, the right ventricle in PAH, undergoes maladaptation with downregulation and desensitization of α-1 and β-1 adrenergic receptors.267,268
Therapies Targeting Sympathetic Nervous System Activation
Preclinical animal studies have demonstrated that β‐blockade improves pulmonary arterial remodeling. For example, arotinolol, an α- and β- adrenergic receptor blocker, reduces pulmonary arterial pressures in MCT rats.269 Propranolol and nebivolol were both shown to stimulate NO activity by blocking the protein kinase C.270,271 Nebivolol also demonstrated improvement of the pulmonary vascular remodeling in PH-rodent models.272
Carvedilol demonstrated improvement in survival as well as matrix deposition, RVH, and Nrf2 pathway in MCT-treated animals.273,274
Administration of bisoprolol to MCT-rats demonstrated an increase in RV contractility and cardiac output as well as a decrease in the degree of inflammation and fibrosis.275
A few small clinical trials have investigated the efficiency and safety of β‐blockade in PAH.
In an open‐label trial with 6 PAH subjects, the use of carvedilol increased RV systolic function by cardiac MRI.276
In a double‐blind, placebo‐controlled, randomized study with 30 PAH subjects (NCT01586156), carvedilol was shown to increase RV function at 3 but not 6 months and did not improve exercise capacity or cardiac output. In this study, carvedilol was also shown to increased β‐adrenergic receptor density on white blood cells.277 In another placebo-controlled, crossover, randomized, single-center study with 18 IPAH patients (NCT01246037), no benefit with bisoprolol was seen, with even a drop in cardiac index, along with a trend towards a decreased in 6MWD. RVEF also remained unchanged.278
Renin–Angiotensin–Aldosterone System (RAAS)
Activation of the RAAS in PAH is well recognized.279–282 Even if benefits from ACE inhibitors and angiotensin II receptor antagonists have been well demonstrated in HFrEF, their positive effect cannot be extrapolated to the field of pulmonary hypertension. Clinical trials with ACE-inhibitors in PAH have demonstrated no benefit.283,284
Although AT-1 receptor antagonism has demonstrated benefit in the MCT rat model of PH, this drug class has not been trialed in humans with PAH.279
ACE2 is a homologue of ACE that is insensitive to ACE inhibitors. It converts angiotensin I and angiotensin II to angiotensin-(1–7), angiotensin-(1–9), and angiotensin-(1–5). These vascular peptides have both cardiac and vascular protecting effects. Ang-(1–7) activates the Mas receptor (Mas1), which is present on ECs and has vasodilatory, anti-inflammatory and anti-fibrotic effects,285,286 functionally antagonizing the effects of AT1 receptor stimulation.287–289
Low ACE2 levels and antibodies against ACE2 have been measured in the blood of PAH subjects and may potentially play a role in the pathogenesis of PAH.290
Therapies Targeting Renin–Angiotensin–Aldosterone System
A small open-label study with 5 PAH subjects demonstrated that one infusion of rhACE2 (purified intravenous formulation of soluble recombinant human ACE 2) was associated with improvement of PA pressures and reduction of inflammatory and OS biomarkers.291 However, another open-label, dose-escalation study with a single dose of rhACE2 was not able to confirm the significant improvement of the PVR (NCT03177603).
Aldosterone Antagonist
Aldosterone is a mineralocorticoid hormone that plays a central role in the regulation of the circulatory homeostasis including blood pressure, sodium level, and potassium retention. Aldosterone is implicated in the development of myocardial fibrosis and stimulation of the adrenergic autonomous system.292 Mineralocorticoid receptors are located in numerous organs including the pulmonary vasculature.
Current clinical indications of mineralocorticoid-receptor antagonists include treatment of HFrEF293 and refractory ascites in cirrhotic patients.294
In preclinical studies, aldosterone levels are elevated in the serum and lungs of PH-MCT rodents.296 Similarly, in PAH patients, serum aldosterone levels are increased and their levels correlate with PVR.295
In PH-induced rodents, elevated serum aldosterone was shown to reduce eNOS in PAECs,296 promote maladaptive vascular remodeling including fibrosis,297 and stimulate PASMC proliferation in the pulmonary vasculature.298 Additionally, aldosterone negatively regulates endothelin type B receptor.296
Clinical studies in PAH subjects with a mineralocorticoid receptor antagonist such as spironolactone have demonstrated conflicting results.
A single-center, 16-week randomized double-blinded crossover clinical trial was performed to assess the effect of spironolactone (50 mg daily) in 42 patients with PAH. At the end of week 8, the treatment arm was switched. The treatment with spironolactone was safe but no change in the levels of amino-terminal propeptide of procollagen type III (PIIINP), MMP-9, TIMP-1, and MMP-9/TIMP-1 ratio at weeks 8 and 16 was seen compared to baseline and placebo arm. No change in walk distance was noted at weeks 8 and 16.299
A post-hoc study from ARIES 1 and ARIES 2 clinical trials indicated some positive effects in the addition of spironolatone to ambrisantan.300 Subsequently, a prospective study examined in 30 subjects with PAH, the effect of spironolactone when added to ambrisentan on exercise capacity (NCT02253394). Unfortunately, due to low enrollment, the study was terminated early.
Another randomized, double‐blind, multicenter, placebo‐controlled study (NCT01712620) is currently investigating the long-term effects of spironolactone in 70 subjects with PAH.
Vasoactive Factors
Vasoconstriction is one of the vascular changes that have been identified in PAH, implicating an imbalance between vasodilator and vasoconstrictor factors. Several vasoactive factors that may affect pulmonary vascular tone, including natriuretic peptides, adrenomedullin, vasoactive intestinal peptide (VIP) and relaxin, have been studied in PAH.
Natriuretic Peptides
Natriuretic peptides (atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and c-type natriuretic peptide) activate soluble guanylate cyclase, and increase intracellular cGMP production via stimulation of soluble guanylate cyclase, with subsequent vasodilation and PASMCs anti-proliferation effect. Also, the natriuretic peptides relax vascular SMCs, inhibit salt and fluid retention, block PASMCs proliferation and endothelin-1 synthesis, and antagonize many of the actions of the renin–angiotensin–aldosterone system. Circulating levels of ANP and BNP correlate directly with the degree of RV failure. ANP and BNP bind to natriuretic peptide receptor-A and B.301–303
All three natriuretic peptides have been shown to inhibit hypertrophy of cardiac myocytes in vitro.304 Mice with transgenic overexpression of ANP have smaller RVs than their non-transgenic littermates.305
ANP and BNP are potent pulmonary vasodilators306–310 and have been shown to blunt hypoxia-induced PH and RVH in preclinical rodent animal models311,312 Also, the natriuretic peptides may play an important role in limiting RVH during the development of PAH.
One mechanism by which BNP limits its anti-PAH effects may be by its profound inhibitory effect on TGF-β-induced pro-inflammatory, fibrotic, and proliferative effects on cardiac fibroblasts.313
Nesiritide
Nesiritide is a recombinant form of BNP and is FDA approved for the management of acute decompensated HF. Nesiritide ameliorates hemodynamic variables, such as the pulmonary capillary wedge pressure, in patients hospitalized for HF management.314–316
The infusion of IV nesiritide was shown to ameliorate pulmonary pressures and wedge pressures in 10 subjects with PH secondary to LV dysfunction (post-capillary PH). However, the drug did not demonstrate hemodynamic improvement of the other 10 patients with PAH (pre-capillary PH).317
Vasoactive Intestinal Peptide (VIP)
Vasoactive intestinal peptide (VIP), is produced by PAECs and is a potent systemic vasodilator. VIP decreases PA pressure and PVR in MCT-PH rabbits318 and in healthy human subjects.319 VIP inhibits PASMCs proliferation320 and platelet activation.321 Also, PAH has been associated with low levels of VIP in the serum and PAH lungs.322
In a preliminary case series, eight patients with PAH treated with inhaled VIP showed marked clinical and hemodynamic improvement.322 However, a double-blinded, phase II, randomized trial with 56 PAH receiving inhaled VIP was negative.323 The reasons for these discrepant findings are unclear and may be due to the dosing or delivery system.
Another randomized, multicenter, double-blind phase II clinical trial investigating PB1046, a VIP analogue, is currently being conducted (NCT 03556020).
Adrenomedullin
Adrenomedullin vasodilates pulmonary vessels, increases pulmonary blood flow, and is synthesized in the normal lung by several cell populations. High levels of mRNA for adrenomedullin and its receptor in the lungs suggest a regulatory role for the peptide in the pulmonary circulation.324
Plasma levels of adrenomedullin are elevated in preclinical PH animal models as well as in PAH patients.325–327 Plasma levels of adrenomedullin have been correlated with CVP, PVR and mean PA pressure, suggesting that levels of adrenomedullin increase in proportion to the extent of PAH.328
Administration of adrenomedullin improves PA pressures as well as medial thickening of pulmonary arteries in MCT-rats.329 Similarly, the infusion of adrenomedullin to PH subjects was shown to improve PVR.330–332 Nonetheless, larger and longer studies in human PAH with adrenomedullin are needed to examine its efficacy and long-term effects.
Relaxin
The anti-fibrotic, anti-inflammatory and vasodilatory parturition natural peptide hormone, relaxin,333 was shown to cause positive hemodynamic effects in an experimental PH rat model.334 Clinically, relaxin in PAH subjects is elevated compared to normal controls.335 No clinical trial with relaxin in PAH subjects is currently ongoing despite its potential therapeutic effect.
Serotonin System
Serotonin (5-hydroxytryptamine or 5-HT) is a vasoconstrictor that promotes PASMC hyperplasia hypertrophy and proliferation.336
Serotonin is a potent growth factor released from the endothelium.337 It has a paracrine effect on neighboring cells and diffuse to the vascular SMC layer.337 In the normal vasculature, serotonin is largely stored in circulating platelets in the bloodstream.338 This sequestration helps to maintain low normal plasma levels of 5-HT.338 Increased 5 HT levels in the plasma and low platelet storage are observed in PAH.339 5-HT is mitogenic.340 Also, the 5-HT pathway elicits endothelial-dependent and PASMCs vasoconstriction.341 The 5 HT-induced growth and proliferative effect appear to be dependent on the RhoA/ROCK pathways.342,343
Elevated plasma serotonin levels and reduced content of serotonin in platelet have been reported in patients with IPAH.344
Use of appetite suppressant medications such as dexfenfluramine, fenfluramine, aminorex, and benfluorex, which increase platelet serotonin release and inhibit its re-uptake, is associated with an increased incidence of PAH.345
Data have described elevated expression of the 5HT transporter (5-HTT) in the PASMCs.346,347 Mutation in the 5HTT and/or 5-HT receptors in the platelets and lung tissues from PAH patients has also been demonstrated.346
5HT2A/2B Receptor Antagonists
Terguride
Terguride is a serotonin (5-HT2A and 5-HT2B) receptor blocker.348–350
In the experimental MCT-PH rodent model, the administration of terguride was shown to inhibit the development of PH and RVH.351 Nonetheless, a clinical phase II, placebo-controlled, randomized trial revealed no hemodynamic or clinical beneficial effects. Also, significant side effects were reported in the terguride group.352
Selective Serotonin Reuptake Inhibitors (SSRIs)
The SSRI, escitalopram was tested in PAH subjects in a clinical trial, but results were never released (NCT00190333). Another phase II, 3-month, open-label trial examining the tolerability and safety of fluoxetine was performed with PAH subjects, but results were never published (NCT 00942708).
Estrogens Signaling Pathway
Regulation of the estrogen signaling pathway may also lead to novel potential PAH therapies.353 Indeed, the estrogen metabolism pathway and its interaction with other PAH signaling pathways appear to be PAH disease modifiers. The focus of the estrogen signaling pathways in the understanding of PAH pathogenesis is based on the prevalence of the disease affecting women more than men with a female/male ratio of 3–4:1.354–357 Furthermore, estrogen was shown to downregulate the expression of BMPR2 in the pulmonary vasculature.358
Data have found that 17β-estradiol (E2), the most common female sex hormone and its metabolite, 16α-hydroxyestrone, are important factors in the development of PAH.353,359 Higher circulating E2 levels were measured in PAH-men compared to normal controls in a case–control study.360 E2 is mostly synthesized through aromatisation of androgens by the enzyme aromatase (encoded by the CYP19A1 gene).
In hypoxia-induced PH rodents, estrogen enhances RV function and administration of 17b-estradiol improves exercise endurance, probably by inhibition of RV myocytes apoptosis and anti-inflammation mechanism.361 Furthermore, estrogen also improves the right ventricle by protecting mitochondrial organization, structure, and oxidative function.362
It is puzzling that while women are more susceptible to develop PAH than men, they exhibit better RV function and survival than men with PAH.363,364 It is also interesting to note that women with PAH seem to respond better to specific PAH treatment than their male counterparts.363
Finally, PAH postmenopausal women and PAH-men were found to have more elevated circulating levels of estrogen with lower levels of dehydroepiandrosterone sulfate (DHEA-S) than female and male controls.360,365 Elevated levels of estrogen are associated with worse 6-minute walk distances while elevated levels of DHEA-S were linked with lower right atrial pressure and PVR.
Therapies Targeting the Estrogen Signaling Pathway
Anastrozole
Anastrozole is an antiestrogen agent used in women in the treatment and prevention of estrogen receptor-positive breast cancer. Anastrozole blocks the enzyme aromatase, which catalyzes the conversion of androgens to estrogens.366,367
In experimental PH models, the positive effects of anastrozole are seen in female rodents but not in males.
Female mice exposed to chronic hypoxia and then treated with anastrozole have diminished vessel remodeling, less elevated RV systolic pressure (RVSP), and less hypertrophy.368 Their counterpart hypoxic PH-male, on the other hand, does not exhibit any beneficial effect from the administration of anastrozole.368
In another protocol with SU-5416 hypoxia rats, and again only in females, anastrozole, decreases the amount of remodeled vessels.368 The difference in gender response might be explained by lesser amounts of aromatase activity in PASMCs in males than in females. Indeed, males have less aromatase activity in the SMCs of their pulmonary arteries than the female rodents.368
The use of the combination of anastrozole and fulvestrant, another selective estrogen receptor modulator, to obtain complete estrogen blockade formation was shown to inhibit pulmonary arterial remodeling and lessen the pulmonary pressures in the BMPR2 R899X transgenic female rodent model.358
Clinically, anastrozole was shown to significantly decrease 17b-estradiol levels by 40% and improve the 6MWD by 26 meters in a 12-weeks randomized clinical trial with 18 PAH patients (NCT01545336).369 There was, however, no change in quality of life or RV function.369 Based on these results, PHANTOM (Pulmonary Hypertension and Anastrozole Trial) a phase II, NIH-funded, randomized, double-blind, placebo-controlled multicenter trial, is currently assessing safety and 6MWDT change in 84 PAH patients with anastrozole administered for 12 months (NCT03229499).
Tamoxifen
The effect of tamoxifen, another selective estrogen receptor inhibitor,370 is also being studied. In this randomized, double-blind, single-center, placebo-controlled trial, tolerance and impact of tamoxifen on functionality, biomarkers, echocardiographic parameters, 6MWDT, and quality of life for 24 weeks in 24 PAH patients will be evaluated (NCT03528902).
Several other NIH-funded trials targeting the estrogen and androgens signaling pathways in PAH, including studies with fulvestrant (NCT02911844), tamoxifen (NCT03528902), and DHEA (NCT03648385)(NIH HL141268) are currently ongoing. Results are not yet available.
Iron Deficiency
In PAH, a large body of evidence has demonstrated the importance of iron metabolism. In several PAH registries, iron deficiency has been shown to be common,371–374 is being associated with diminished functional capacity372,373 and survival.375 Furthermore, iron deficiency in PAH is associated with disease severity including higher pulmonary pressures, diminished cardiac output and worse functional class.340 Positive effects of iron administration in PAH patients have been shown in two small clinical trials,376,377 even though another study has demonstrated that a low iron level might be protective.378
A multicenter phase II trial with 40 PAH subjects examined the effect of intravenous Ferinject® (ferric carboxymaltose) on functional capacity and PVR (NCT01447628). Results are not yet published. Another 120week, open-label trial also studied the effects of oral ferrous sulfate in 40 PAH (NCT01446848). No results for this trial have been reported either.
Vitamin D Deficiency
Recent data have established the consequences of vitamin D deficiency in PAH. In a 68 PAH patients study compared to 100 controls, vitamin D deficiency was shown to be highly prevalent. Also, 25-hydroxy vitamin D levels appear to be a potential predictor of adverse outcomes in PAH.379 In this study, 70% of the PAH studied subjects had severe vitamin D deficiency with secondary hyperparathyroidism. PAH subjects with total 25-hydroxy vitamin D plasma above the median (7.17 ng/mL) exhibited better 6MWDT, exercise capacity, functional class, RV systolic function and survival. Lower levels of bioavailable 25-hydroxy vitamin D were linked to lower exercise capacity, more advanced functional class, and higher risk of mortality.379
In a rat model with PH, administration of 1,25(OH)2D3, the bioactive metabolite of vitamin D, was shown to restore normal lung function by modifying the function of miR-204 through the transforming growth factor (Tgfbr2), TGFβ-1 and Smad pathways.380
Restoration of vitamin D levels in the PAH population represents a very achievable goal. Whether symptoms, prognosis and survival of PAH subjects improve after restoring vitamin D levels remains unclear. In a uncontrolled small cohort of PAH subjects, restoring vitamin D levels for 3 months was shown to ameliorate 6MWD and right-ventricular size without improving the pulmonary pressures or the functional class.381 Thus, a large randomized study is warranted to evaluate the long-term effects of vitamin D substitution in the PAH population.
DNA Damage
Reports have shown that in patients taking etoposide, an anticancer drug causing DNA damage and PARP-1 activation may develop PAH.382 Poly [ADP-ribose] polymerase 1 (PARP-1), member of the PARP family, detects DNA damage and contributes to the repair of DNA by modifying the structure of the chromatin and by interacting with multiple DNA repair factors.383
As discussed previously, PAH is characterized by sustained inflammation and elevated circulating cytokines such as TNF-α and IL-6,6 both of which are known to promote DNA damage.384–386
Increased DNA damage in the distal pulmonary arteries in PAH has been demonstrated in comparison with control patients. Also, PARP1 activity was shown to be augmented in PASMCs from subjects with PAH.387 In PAH, PARP-1 appears to promote the activation of a pro-proliferative and anti-apoptotic program, via miR-204 downregulation and subsequent NFAT and HIF-1 (hypoxia-inducible factor 1-α) upregulation in PASMCs.387
It is possible that PARP-1 activation is a common denominator for diverse conditions that cause DNA damage in PAH, making it an attractive therapeutic target.
Therapies Targeting DNA Damage
Olaparib
PARP-1 inhibition is associated with a complete reversal of miR-204 downregulation, limiting NFAT activation, and decrease in PAH-PASMC proliferation.387 In preclinical studies, inhibition of PARP-1 reduces PH severity in MCT and SU‐5416 hypoxia rodents.387
Olaparib, an orally available PARP1 inhibitor approved for the treatment of BRCA (breast cancer gene)-related breast cancer, is being investigated in a small clinical trial (NCT03782818). Twenty PAH subjects are getting Olaparib for 24 weeks, with PVR change, as primary end point.
FoxO1 Signaling Pathway
FoxO1 is a transcription factor that belongs to the forkhead O family and plays a critical role in biological processes, including inflammation, metabolism, proliferation, and vascular homeostasis.388,389 FoxO1 regulates PASMC stimulation via its pro-apoptotic properties.390
In MCT and SU-5416 hypoxia rats as well as in PAH patient, FoxO1 is downregulated and inactivated.390,391 This change will cause PASMCs stimulation leading to the development of PH.390
Therapies Targeting FoxO1 Signaling Pathway
Paclitaxel
Paclitaxel is one of the most potent and effective anti-neoplastic agents and is widely used in a variety of tumors including breast, ovarian, and lung cancers.392 Paclitaxel activates cellular apoptotic program by inhibiting microtubule depolymerization and arresting cycling cells in M (mitotic) phase.393
Paclitaxel was shown to stimulate FOXO1. Activated FOXO1 reduces cellular proliferation and promotes apoptosis by modifying the expression of numerous cell-cycle modulators and by stimulating the BMPR2 pathway.390,394
In both MCT and SU-5416 hypoxic rats, administration of paclitaxel activates BMPR2 signaling and induces PASMCs apoptosis resulting in lesser PH, increase in RV systolic function and decrease in RV dilatation and hypertrophy.390,395
Paclitaxel is currently not being tested in clinical trials, probably due to its potential numerous adverse effects.396 Use of an inhaled form of the drug should, nevertheless, be considered as it was shown to be effective in animal models with limited systemic effects.390
Emerging Therapies
Over the last decade, vast research efforts have led to the development of inventive therapeutic approaches in the field of PAH. Undeniably, recent preclinical studies indicate that immunotherapy, gene therapy, microRNA-based therapy and epigenetic medicines may offer new therapeutic perspectives in the area of PAH.
Immunotherapy
Therapeutic vaccine is a new approach for the treatment of oncologic and cardiovascular diseases. Vaccines generate antibodies against specific target molecules and may offer a new safe and effective way to treat PAH.
Activation of the endothelin system and more specifically of the endothelin-1 receptor type A (ETAR) have been implicated in the pathogenesis of PAH.397 ETRQβ-002 is a vaccine developed against the second extracellular loop (ECL2) of ETAR and was administered to MCT-rats and Sugen/hypoxia-exposed mice. The ETRQβ-002 vaccine was shown to significantly decrease RV systolic pressures and improve pathological remodeling of pulmonary arterioles in both the MCT-rats and Sugen/hypoxia-induced mice, two and 3 months after the vaccine injection.398 The results of this positive study may provide a novel approach for the treatment of PAH.
Gene Therapy
Abnormal or malfunctioning genes due to mutation or abnormal gene expression may promote the development of PAH by stimulating PASMCs and PAECs proliferation, causing apoptosis resistance and by altering the integrity of PAECs.399
Heritable PAH (hPAH) represents approximately 6–10% of all PAH. Genetic studies have identified more than 450 mutations heterozygous germline mutations in the BMPR2 gene.400
Gene therapy is a technique that focuses on the utilization of the therapeutic delivery of nucleic acids into a patient’s cells to treat disease.401 Thus, delivery of targeted-gene such as BMPR2 may treat, block the disease progression or even prevent the development of PAH. The delivery of the adenoviral vector comprising the BMPR2 gene was shown to improve PH, RVH and pulmonary vessel remodeling in the chronic hypoxia-PH rodent model.402
The use of the adenovirus vectors remains limited due to its strong inflammatory reaction associated with its gene transfer.403 Advances in gene delivery technology have led to the development of new vectors with limited inflammatory reaction called adeno-associated virus (AAV). The use of such vectors is currently being used in experimental PH models.404
MicroRNAs (miRNAs)
MicroRNAs (miRNAs) are small, non-coding endogenous RNA molecules consisting of 17–25 nucleotides.405,406 A single miRNA can regulate hundreds of genes or proteins and conversely multiple miRNAs can regulate one protein. miRNAs target both mRNA degradation and suppression of protein translation on the basis of sequence complementarity between miRNA and its target.
Currently, many miRNA have been discovered and found to have a critical role in the pathogenesis of many disease, including PAH.407
Several miRNAs expressed in human lungs with PAH have been found to be altered compared with normal controls. While miR-138, miR-145 and miR-210 are upregulated in PASMCs of PAH subjects,408–410 miR-124, miR-204, and miR-206 are downregulated.411–413 Also miR-204 down-regulation appears to be strongly associated with PAH severity and seems to correlate with PASMCs proliferation and PAECs apoptosis resistance levels.412
Also, intratracheal nebulization of miR-204 was shown to improve pulmonary vascular remodeling and RVH in a MCT-PH rat experimental model.412
Epigenetics
Growing data indicate that epigenetics plays a crucial role in the development of PAH. Epigenetics is the study of heritable phenotype changes that do not involve alterations in the DNA sequence.414 Examples of mechanisms that produce such changes are DNA methylation and histone modification, each of which alters how genes are expressed without altering the underlying DNA sequence. Gene expression can be controlled through the action of repressor proteins that attach to silencer regions of the DNA.
The discovery of epigenetic components to PAH may offer new potential therapeutic targets for the treatment of PAH.
DNA Methyltransferase (DNMT) Inhibitors
DNA methylation is the modification of DNA nucleotides by addition of a methyl group. Because DNA methylation affects cellular function through successive generations of cells without changing the underlying DNA sequence, demethylating agents are considered a type of epigenetic therapy. Demethylating agents block the activity of DNA methyltransferase (DNA methyltransferase inhibitors/DNMT inhibitors). Currently, two members of this class, azacitidine and decitabine, are FDA-approved for use in the United States in myelodysplastic syndrome and are being investigated for use in a number of tumors.415
The activity and expression of the mitochondrial superoxide dismutase-2 (SOD2) have been shown to be significantly diminished in PAH subjects and PH rodent experimental models. No mutation was found in the SOD2 gene in PAH patient and rat’s PH experimental models. However, study of the SOD2 gene has revealed selective hypermethylation of the SOD2 promoter region in fawn-hooded rats, known to develop PH spontaneously.3 Administration of the DNMT inhibitor methyltransferase 5-aza-2′-deoxycytidine was shown to re-establish SOD2 expression, block cell proliferation and apoptosis resistance in the fawn-hooded rats’ PASMCs.156 These results indicate that targeting DNA methylation can be an appealing therapeutic possibility to restore SOD2 expression in PAH.
No clinical study has yet been performed to investigate the effects of 5-aza-2′-deoxycytidine in PAH subjects.
Histone Deacetylase (HDAC) Inhibitors
Histone deacetylases (HDAC) are enzymes that eliminate the acetyl group from histone proteins, altering the chromatin condensing process and approachability of transcription factors. HDAC inhibitors represent a new class of cytostatic agents that inhibit the proliferation of tumor cells in culture and in vivo by inducing cell differentiation, cell cycle arrest, and/or apoptosis. HDAC inhibitors exert their anti-tumor effects via the induction of expression changes of oncogenes or tumor suppressor, through modulating the acetylation/deacetylation of histones and/or non-histone proteins such as transcription factors.416
Preclinical experiments demonstrated that HDAC1, and HDAC5 are upregulated in PAH patients as well as experimental animal models of PH.417 Both valproic acid, a class I HDAC inhibitor, and suberoylanilide hydroxamic acid (vorinostat), an inhibitor of class I, II, and IV HDACs, were shown to lessen the development and at least to partially reverse rat hypoxia-induced PH.417
Likewise, the activity of the transcription factor myocyte enhancer factor 2 (MEF2) was demonstrated to be significantly decreased in PAECs of PAH subjects. MEF2 regulates the expression of miRNA-424 and miRNA-523, two miRNAs involved in the maintenance of pulmonary vascular homeostasis418 as well as other number of transcriptional targets involved in pulmonary vascular homeostasis, such as microRNAs 424 and 503, connexins 37 and 40, and Krüppel-like Factors 2 and 4, which were found to be significantly diminished in PAH PAECs. The impaired MEF2 activity in PAH PAECs was found to be linked to excessive accumulation of HDAC4 and HDAC5 that block MEF2 function. The selective inhibition by MC1568 of class IIa HDACs led to restoration of MEF2 activity in PAECs by inducing degradation of HDAC4 and HDAC5 in both the MCT and the Sugen animal models with subsequent significant decrease in cell proliferation in the pulmonary vasculature.419
Overall, these studies validate the therapeutic potential of targeting histone acetylation using HDAC inhibitors to inhibit vascular remodeling in PAH.
Bromodomain and Extra-Terminal Motif (BET) Inhibitors
Epigenetic mechanisms include the placing (writing) or removal (erasing) of histone alterations that permit the chromatin to change to an open, activable chromatin state necessary for transcription. A third, less studied epigenetic pathway includes the reading of these specific histone marks once placed. The BETs (bromodomain and extra-terminal-containing protein family), which includes BRD2, BRD3, BRD4 and the testis-restricted BRDT, are epigenetic “reader” regulator proteins. They bind to specific acetylated lysine residues on histone tails and modulate gene transcription involved in many cell functions, such as cell proliferation and apoptosis.420 The BETs facilitate the assembly of transcription complexes including transcription factors like RNA Polymerase II. BRD4 is the most characterized studied member of the BET family. Compared to controls, BRD4 was shown to be upregulated in lungs, distal PAs, and PASMCs of PAH patients. BRD4 expression in PAH is microRNA-204 dependent.421
BRD4 inhibition, by JQ1, was shown to increase p21 expression, and trigger cell cycle arrest. Moreover, in the same study, blocking BRD4 inhibition by JQ1, demonstrated a decrease in the expression of three major oncogenes, which are overexpressed in PAH, survivin, the nuclear factor of activated T cells, and the B-cell lymphoma 2. Inhibition of these oncogenes leads to diminished PAH-PASMC proliferation. BRD4 inhibition also significantly improved hemodynamic measurements by significantly decreasing mean PA pressures and by increasing cardiac output in the Sugen/hypoxia-induced PAH rat model. Finally, BRD4 inhibition by JQ1 was shown to decrease pulmonary artery wall area by 40%.421
These promising preclinical results led to the ongoing early Phase 1 pilot study assessing apabetalone in the PAH population (NCT03655704). Apabetalone (development code RVX-208) is an orally available small molecule that targets bromodomain and extra terminal domain (BET) proteins, and in particular BRD4. The overall hypothesis of this pilot study is that BRD4 inhibition with apabetalone is a safe and effective therapy for PAH to assess the feasibility of a Phase 2 clinical trial.
Conclusions
The hallmark of PAH is a lethal and progressive hyperproliferative vascular remodeling process that involves all cell types of the pulmonary artery. Initially thought to be a disease primarily characterized by vasoconstriction, it is now recognized that the pathobiology of PAH is much more complex and includes cellular apoptosis resistance, genetic abnormalities, altered microRNA function, increased extracellular matrix deposition with enhanced interaction interplay of inflammation, metabolic derangements, and mitochondrial dysfunction, leading to pulmonary artery remodeling and elevated pulmonary vascular resistance.
Although major advances in our understanding of the mechanism of disease development and treatment have been achieved, substantial gaps in our knowledge remain.
Current approved PAH therapies do not cure the disease and appear to have limited effects on the underlying pathogenesis.
While multiple novel signaling pathways in the pathogenesis of PAH have been discovered over the last three decades, most clinical trial investigating new therapeutic agents targeting these pathways so far have been disappointing. Hence, the importance and necessity to improve comprehension of the PAH pathobiology and to discover new molecules capable of disrupting the disease and its development as well as improving outcomes of PAH.
Disclosure
Dr Zolty is a consultant at Actelion, Bayer, United Therapeutics, and Alnylam. The author report no other conflicts of interest in this work.
References
1. Rubin LJ. Pulmonary arterial hypertension. Proc Am Thorac Soc. 2006;3(1):111–115. PMID: 16493158. doi:10.1513/pats.200510-112JH
2. Galiè N, McLaughlin VV, Rubin LJ, et al. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur Respir J. 2019;53(1):1802148. PMID: 30552088. doi:10.1183/13993003.02148-2018
3. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. PMID: 30545968. doi:10.1183/13993003.01913-2018
4. Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99(7):675–691. PMID: 17008597. doi:10.1161/01.RES.0000243584.45145.3f
5. Stacher E, Graham BB, Hunt JM, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(3):261–272. PMID: 22679007. doi:10.1164/rccm.201201-0164OC
6. Tuder RM, Archer SL, Dorfmüller P, et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D4–D12. PMID: 24355640. doi:10.1016/j.jacc.2013.10.025
7. Humbert M, Montani D, Perros F, et al. Endothelial cell dysfunction and cross talk between endothelium and smooth muscle cells in pulmonary arterial hypertension. Vascul Pharmacol. 2008;49:113–118. PMID: 18606248. doi:10.1016/j.vph.2008.06.003
8. Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Investig. 2012;122:4306–4313. PMID: 23202738. doi:10.1172/JCI60658
9. Cohen-Kaminsky S, Hautefort A, Price L, et al. Inflammation in pulmonary hypertension: what we know and what we could logically and safely target first. Drug Discov Today. 2014;19:1251–1256. PMID: 24747559. doi:10.1016/j.drudis.2014.04.007
10. Ryan J, Dasgupta A, Huston J, et al. Mitochondrial dynamics in pulmonary arterial hypertension. J Mol Med. 2015;93:229–242. PMID: 25672499. doi:10.1007/s00109-015-1263-5
11. Potus F, Ruffenach G, Dahou A, et al. Downregulation of MicroRNA-126 contributes to the failing right ventricle in pulmonary arterial hypertension. Circulation. 2015;132:932–943. PMID: 26162916. doi:10.1161/CIRCULATIONAHA.115.016382
12. Potus F, Malenfant S, Graydon C, et al. Impaired angiogenesis and peripheral muscle microcirculation loss contribute to exercise intolerance in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;190:318–328. PMID: 2497762. doi:10.1164/rccm.201402-0383OC
13. Ranchoux B, Antigny F, Rucker-Martin C, et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–1018. PMID: 25593290. doi:10.1161/CIRCULATIONAHA.114.008750
14. Kherbeck N, Tamby MC, Bussone G, et al. The role of inflammation and autoimmunity in the pathophysiology of pulmonary arterial hypertension. Clin Rev Allergy Immunol. 2013;44:31–38. PMID: 21394427. doi:10.1007/s12016-011-8265-z
15. Malenfant S, Neyron AS, Paulin R, et al. Signal transduction in the development of pulmonary arterial hypertension. Pulm Circ. 2013;3:278–293. PMID: 24015329. doi:10.4103/2045-8932.114752
16. Perros F, Humbert M, Cohen-Kaminsky S. Pulmonary arterial hypertension: a flavor of autoimmunity. Méd Sci. 2013;29:607–616. PMID: 23859515. doi:10.1051/medsci/2013296013.12
17. Montani D, Günther S, Dorfmüller P, et al. Pulmonary arterial hypertension. Orphanet J Rare Dis. 2013;8:97. PMID: 23829793. doi:10.1186/1750-1172-8-97
18. Toshner M, Tajsic T, Morrell NW. Pulmonary hypertension: advances in pathogenesis and treatment. Br Med Bull. 2010;94:21–32. PMID: 20447940. doi:10.1093/bmb/ldq012
19. Morrell NW, Adnot S, Archer SL, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1Suppl):S20–31. PMID: 19555855. doi:10.1016/j.jacc.2009.04.018
20. Wagenvoort CA, Wagenvoort N. Primary pulmonary hypertension: a pathologic study of the lung vessels in 156 clinically diagnosed cases. Circulation. 1970;42:1163–1184. doi:10.1161/01.CIR.42.6.1163
21. Caruso P, Dunmore BJ, Schlosser K, et al. Identification of MicroRNA‐124 as a major regulator of enhanced endothelial cell glycolysis in pulmonary arterial hypertension via PTBP1 (polypyrimidine tract binding protein) and pyruvate kinase M2. Circulation. 2017;136:2451–2467. PMID: 28971999. doi:10.1161/CIRCULATIONAHA.117.028034
22. Lane KB, Machado RD, Pauciulo MW, et al.; Consortium IP. Heterozygous germline mutations in BMPR2, encoding a TGF‐beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. PMID: 10973254. doi:10.1038/79226
23. Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation. 2010;121:2045–2066. PMID: 20458021. doi:10.1161/CIRCULATIONAHA.108.847707
24. Landsberg JW, Yuan JX. Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol Sci. 2004;19:44–50. PMID: 15016901. doi:10.1152/nips.01457.2003
25. Archer SL. Pyruvate kinase and Warburg metabolism in pulmonary arterial hypertension: uncoupled glycolysis and the cancer‐like phenotype of pulmonary arterial hypertension. Circulation. 2017;136:2486–2490. PMID: 29255124. doi:10.1161/CIRCULATIONAHA.117.031655
26. Zhang H, Wang D, Li M, et al. Metabolic and proliferative state of vascular adventitial fibroblasts in pulmonary hypertension is regulated through a MicroRNA‐124/PTBP1 (polypyrimidine tract binding protein 1)/pyruvate kinase muscle axis. Circulation. 2017;136:2468–2485. PMID: 28972001 F. doi:10.1161/CIRCULATIONAHA.117.028069
27. Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res. 2014;115:165–175. PMID: 24951765. doi:10.1161/CIRCRESAHA.113.301141
28. Galiè N, Humbert M, Vachiery JL, et al.; ESC Scientific Document Group. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37:67–119. PMID: 26320113. doi:10.1093/eurheartj/ehv317
29. Humbert M, Sitbon O, Chaouat A, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122(2):156–163. PMID: 20585011. doi:10.1161/CIRCULATIONAHA.109.911818
30. Fujii M, Takeda K, Imamura T, et al. Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol Biol Cell. 1999;10(11):3801–3813. PMID: 10564272. doi:10.1091/mbc.10.11.3801
31. Miyazono K, Maeda S, Imamura T. BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 2005;16(3):251–263. PMID: 15871923. doi:10.1016/j.cytogfr.2005.01.009
32. Machado RD, Eickelberg O, Elliott CG, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1Suppl):S32–42. PMID: 19555857. doi:10.1016/j.jacc.2009.04.015
33. Valdimarsdottir G, Goumans MJ, Rosendahl A, et al. Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation. 2002;106(17):2263–2270. PMID: 12390958. doi:10.1161/01.cir.0000033830.36431.46
34. Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, et al. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. 2006;98(2):209–217. PMID: 16357305. doi:10.1161/01.RES.0000200180.01710.e6
35. Soon E, Crosby A, Southwood M, et al. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production. A gateway to pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192(7):859–872. PMID: 26073741. doi:10.1164/rccm.201408-1509OC18
36. de Jesus Perez VA, Alastalo T-P, Wu JC, et al. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt-beta-catenin and Wnt-RhoA-Rac1 pathways. J Cell Biol. 2009;184(1):83–99. PMID: 19139264. doi:10.1083/jcb.200806049
37. Hansmann G, de Jesus Perez VA, Alastalo TP, et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118:1846–1857. PMID: 18382765. doi:10.1172/jci32503
38. Kawabata M, Imamura T, Miyazono K. Signal transduction by bone morphogenetic proteins. Cytokine Growth Factor Rev. 1998;9(1):49–61. PMID: 9720756. doi:10.1016/s1359-6101(97)00036-1
39. Massagué J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14(6):627–644. PMID: 10733523.
40. Attisano L, Wrana J. Signal transduction by the TGF-beta superfamily. Science. 2002;296(5573):1646–1647. PMID: 12040180. doi:10.1126/science.1071809
41. Deng Z, Morse JH, Slager SL, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67(3):737–744. PMID: 10903931. doi:10.1086/303059
42. Thomson JR, Machado RD, Pauciulo MW, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet. 2000;37:741–745. PMID: 11015450. doi:10.1136/jmg.37.10.741
43. Atkinson C, Stewart S, Upton PD, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105(14):1672–1678. PMID: 11940546. doi:10.1161/01.CIR.0000012754.72951.3D
44. Austin ED, Hamid R, Hemnes AR, et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differ. 2012;3(1):6. PMID: 22348410. doi:10.1186/2042-6410-3-6
45. West J, Fagan K, Steudel W, et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res. 2004;94(8):1109–1114. PMID: 15031260. doi:10.1161/01.RES.0000126047.82846.20
46. Hong KH, Lee YJ, Lee E, et al. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation. 2008;118(7):722–730. PMID: 18663089. doi:10.1161/CIRCULATIONAHA.107.736801
47. Morrell NW, Yang X, Upton PD, et al. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation. 2001;104:790–795. PMID: 11502704. doi:10.1161/hc3201.094152
48. Fung JJ. Tacrolimus and transplantation: a decade in review. Transplantation. 2004;77(9):S41–S43. PMID: 15201685. doi:10.1097/01.tp.0000126926.61434.a5
49. Spiekerkoetter E, Tian X, Cai J, et al. FK506 activates BMPR-II, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. 2013;123:3600–3613. PMID: 23867624. doi:10.1172/JCI65592
50. Spiekerkoetter E, Sung YK, Sudheendra D, et al. Low-dose FK506 (tacrolimus) in end-stage pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192(2):254–257. PMID: 26177174. doi:10.1164/rccm.201411-2061LE
51. Spiekerkoetter E, Sung YK, Sudheendra D, et al. Randomised placebo controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur Respir J. 2017;50:1602449. PMID: 28893866. doi:10.1183/13993003.02449-2016
52. Al‐Bari MA. Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother. 2015;70:1608–1621. PMID: 25693996. doi:10.1093/jac/dkv018
53. Long L, Yang X, Southwood M, et al. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type ii receptor degradation. Circ Res. 2013;112(8):1159–1170. PMID: 23446737. doi:10.1161/CIRCRESAHA.111.300483
54. Humbert M, McLaughlin V, Gibbs JSR, et al.; PULSAR Trial Investigators. Sotatercept for the treatment of pulmonary arterial hypertension. N Engl J Med. 2021;384(13):1204–1215. PMID: 33789009. doi:10.1056/NEJMoa2024277
55. Goldenberg NM, Steinberg BE. Inflammation drives pulmonary arterial hypertension. Anesthesiology. 2019;130(5):820–821. PMID: 30994559. doi:10.1097/ALN.0000000000002561
56. Mouthon L, Guillevin L, Humbert M. Pulmonary arterial hypertension: an autoimmune disease? Eur Respir J. 2005;26:986–988. PMID: 16319325. doi:10.1183/09031936.05.00112105
57. Nicolls MR, Taraseviciene-Stewart L, Rai PR, et al. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26:1110–1118. PMID: 16319344. doi:10.1183/09031936.05.00045705
58. Rabinovitch M. Autoimmune disease and unexplained pulmonary hypertension. Circulation. 1992;85:380–381. PMID: 1728476. doi:10.1161/01.cir.85.1.380
59. Perros F, Dorfmuller P, Montani D, et al. Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:311–321. PMID: 22108206. doi:10.1164/rccm.201105-0927OC
60. Dorfmuller P, Perros F, Balabanian K, et al. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358–363. PMID: 12952274. doi:10.1183/09031936.03.00038903
61. Blum LK, Cao RRL, Sweatt AJ, et al. Circulating plasmablasts are elevated and produce pathogenic anti-endothelial cell autoantibodies in idiopathic pulmonary arterial hypertension. Eur J Immunol. 2018;48:874–884. PMID: 29369345. doi:10.1002/eji.201747460
62. de Bourcy CFA, Dekker CL, Davis MM, et al. Dynamics of the human antibody repertoire after B cell depletion in systemic sclerosis. Sci Immunol. 2017;2(15):eaan8289. PMID: 28963118. doi:10.1126/sciimmunol.aan8289
63. Huertas A, Phan C, Bordenave J, et al. Regulatory T cell dysfunction in idiopathic, heritable and connective tissue-associated pulmonary arterial hypertension. Chest. 2016;149:1482–1493. PMID: 26836928. doi:10.1016/j.chest.2016.01.004
64. Hautefort A, Girerd B, Montani D, et al. T-helper 17 cell polarization in pulmonary arterial hypertension. Chest. 2015;147:1610–1620. PMID: 25429518. doi:10.1378/chest.14-1678
65. Savai R, Pullamsetti SS, Kolbe J, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:897–908. PMID: 22955318. doi:10.1164/rccm.201202-0335OC
66. Klinke A, Berghausen E, Friedrichs K, et al. Myeloperoxidase aggravates pulmonary arterial hypertension by activation of vascular Rho-kinase. JCI Insight. 2018;3(11):e97530. PMID: 29875311. doi:10.1172/jci.insight.97530
67. Harbaum L, Baaske KM, Simon M, et al. Exploratory analysis of the neutrophil to lymphocyte ratio in patients with pulmonary arterial hypertension. BMC Pulm Med. 2017;17(1):72. PMID: 28446163. doi:10.1186/s12890-017-0407-5
68. Taylor S, Dirir O, Zamanian RT, et al. The role of neutrophils and neutrophil elastase in pulmonary arterial hypertension. Front Med (Lausanne). 2018;5:217. PMID: 30131961. doi:10.3389/fmed.2018.00217
69. Marsh LM, Jandl K, Grünig G, et al. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2018;51(1):1701214. PMID: 29371380. doi:10.1183/13993003.01214-2017
70. Cracowski JL, Chabot F, Labarere J, et al. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur Respir J. 2014;43:915–917. PMID: 24232704. doi:10.1183/16000617.0084-2016387
71. Molossi S, Clausell N, Rabinovitch M. Reciprocal induction of tumor necrosis factor-alpha and interleukin-1 beta activity mediates fibronectin synthesis in coronary artery smooth muscle cells. J Cell Physiol. 1995;163:19–29. PMID: 7896895. doi:10.1002/jcp.1041630104
72. Jones PL, Cowan KN, Rabinovitch M. Tenascin-C, proliferation and subendothelial fibronectin in progressive pulmonary vascular disease. Am J Pathol. 1997;150:1349–1360. PMID: 9094991.
73. Courboulin A, Tremblay VL, Barrier M, et al. Krüppel-like factor 5 contributes to pulmonary artery smooth muscle proliferation and resistance to apoptosis in human pulmonary arterial hypertension. Respir Res. 2011;12:128. PMID: 21951574F. doi:10.1186/1465-9921-12-128
74. Frid MG, Brunetti JA, Burke DL, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–669. PMID: 16436679. doi:10.2353/ajpath.2006.050599
75. Ito T, Okada T, Miyashita H, et al. Interleukin-10 expression mediated by an adeno-associated virus vector prevents monocrotaline-induced pulmonary arterial hypertension in rats. Circ Res. 2007;101:734–741. PMID: 17673672. doi:10.1161/CIRCRESAHA.107.153023
76. Miyamichi-Yamamoto S, Fukumoto Y, Sugimura K, et al. Intensive immunosuppressive therapy improves pulmonary hemodynamics and long-term prognosis in patients with pulmonary arterial hypertension associated with connective tissue disease. Circ J. 2011;75:2668–2674. PMID: 21873802. doi:10.1253/circj.cj-11-0473
77. Kommireddy S, Bhyravavajhala S, Kurimeti K, et al. Pulmonary arterial hypertension in systemic lupus erythematosus may benefit by addition of immunosuppression to vasodilator therapy: an observational study. Rheumatology (Oxford). 2015;54:1673–1679. PMID: 25929760. doi:10.1093/rheumatology/kev097
78. Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, et al. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med. 2007;175:1280–1289. PMID: 17413127. doi:10.1164/rccm.200608-1189OC
79. Steiner MK, Syrkina OL, Kolliputi N, et al. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104:236–244. PMID: 19074475. doi:10.1161/CIRCRESAHA.108.182014
80. Savale L, Tu L, Rideau D, et al. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir Res. 2009;10(1):6. PMID: 19173740. doi:10.1186/1465-9921-10-6
81. Tamura Y, Phan C, Tu L, et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J Clin Investig. 2018;128:1956–1970. PMID: 29629897. doi:10.1172/JCI96462
82. Humbert M, Monti G, Brenot F, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–1631. PMID: 7735624. doi:10.1164/ajrccm.151.5.7735624
83. Soon E, Holmes AM, Treacy CM, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–927. PMID: 20713898. doi:10.1161/CIRCULATIONAHA.109.933762
84. Taniguchi K, Shimazaki C, Fujimoto Y, et al. Tocilizumab is effective for pulmonary hypertension associated with multicentric Castleman’s disease. Int J Hematol. 2009;90(1):99–102. PMID: 19554396. doi:10.1007/s12185-009-0346-x
85. Arita Y, Sakata Y, Sudo T, et al. The efficacy of tocilizumab in a patient with pulmonary arterial hypertension associated with Castleman’s disease. Heart Vessels. 2010;25(5):444–447. PMID: 20676969. doi:10.1007/s00380-009-1215-5
86. Kadavath S, Zapantis E, Zolty R, et al. A novel therapeutic approach in pulmonary arterial hypertension as a complication of adult-onset Still’s disease: targeting IL-6. Int J Rheum Dis. 2014;17(3):336–340. PMID: 24581387. doi:10.1111/1756-185X.12324
87. Voelkel NF, Tuder RM, Bridges J, Arend WP. Interleukin‐1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am J Respir Cell Mol Biol. 1994;11:664–675. PMID: 7946395. doi:10.1165/ajrcmb.11.6.7946395
88. Voelkel NF, Tuder R. Interleukin-1 receptor antagonist inhibits pulmonary hypertension induced by inflammation. Ann N Y Acad Sci. 1994;725:104–109. PMID: 8030981. doi:10.1111/j.1749-6632.1994.tb39794.x
89. Pickworth J, Rothman A, Iremonger J, et al. Differential IL‐1 signaling induced by BMPR2 deficiency drives pulmonary vascular remodeling. Pulm Circ. 2017;7:768–776. PMID: 28828907. doi:10.1177/2045893217729096
90. Mertens M, Singh JA. Anakinra for rheumatoid arthritis: a systematic review. J Rheumatol. 2009;36:1118–1125. PMID: 19447938. doi:10.3899/jrheum.090074
91. Trankle CR, Canada JM, Kadariya D, et al. Interleukin‐1 (IL‐1) blockade reduces inflammation in pulmonary arterial hypertension and right ventricular failure: a single‐arm, open‐label, phase IB/II pilot study. Am J Respir Crit Care Med. 2019;199(3):381–384. PMID: 30418047. doi:10.1164/rccm.201809-1631LE
92. Zhang LL, Lu J, Li MT, et al. Preventive and remedial application of etanercept attenuate monocrotaline‐induced pulmonary arterial hypertension. Int J Rheum Dis. 2016;19:192–198. PMID: 24612527. doi:10.1111/1756-185X.12304
93. Fujita M, Shannon JM, Irvin CG, et al. Overexpression of tumor necrosis factor-alpha produces an increase in lung volumes and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2001;280(1):L39–49. PMID: 11133493. doi:10.1152/ajplung.2001.280.1.L39
94. Li XQ, Wang HM, Yang CG, et al. Fluoxetine inhibited extracellular matrix of pulmonary artery and inflammation of lungs in monocrotaline-treated rats. Acta Pharmacol Sin. 2011;32(2):217–222. PMID: 21217769. doi:10.1038/aps.2010.187
95. Chen D, Zhou D, Qian J, et al. Atorvastatin prevents dehydromonocrotaline-induced pulmonary hypertension in beagles. Exp Lung Res. 2012;38(7):333–343. PMID: 22888849. doi:10.3109/01902148.2012.702852
96. Hurst LA, Dunmore BJ, Long L, et al. TNFα drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat Commun. 2017;8:14079. PMID: 28084316. doi:10.1038/ncomms14079
97. Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor: FC fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340:253–259. PMID: 9920948. doi:10.1056/NEJM199901283400401
98. Moreland LW, Schiff MH, Baumgartner SW, et al. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann Intern Med. 1999;130:478–486. PMID: 10075615. doi:10.7326/0003-4819-130-6-199903160-00004
99. Ford AC, Sandborn WJ, Khan KJ, et al. Efficacy of biological therapies in inflammatory bowel disease: systematic review and meta‐analysis. Am J Gastroenterol. 2011;106:644–659. PMID: 21407183. doi:10.1038/ajg.2011.73
100. Mease PJ. Tumor necrosis factor (TNF) in psoriatic arthritis: pathophysiology and treatment with TNF inhibitors. Ann Rheum Dis. 2002;61:298–304. PMID: 11874829. doi:10.1136/ard.61.4.298
101. Mutschler D, Wikström G, Lind L, et al. Etanercept reduces late endotoxin-induced pulmonary hypertension in the pig. J Interferon Cytokine Res. 2006;26(9):661–667. PMID: 16978070. doi:10.1089/jir.2006.26.661
102. Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell. 2017;168:37–57. PMID: 28086098. doi:10.1016/j.cell.2016.12.012
103. Sawada H, Mitani Y, Maruyama J, et al. A nuclear factor-kappaB inhibitor pyrrolidine dithiocarbamate ameliorates pulmonary hypertension in rats. Chest. 2007;132:1265–1274. PMID: 17934115. doi:10.1378/chest.06-2243
104. Price LC, Caramori G, Perros F, et al. Nuclear factor κ-B is activated in the pulmonary vessels of patients with end-stage idiopathic pulmonary arterial hypertension. PLoS One. 2013;8(10):e75415. PMID: 24124488. doi:10.1371/journal.pone.0075415
105. Chin MP, Bakris GL, Block GA, et al. Bardoxolone methyl improves kidney function in patients with chronic kidney disease stage 4 and type 2 diabetes: post-hoc analyses from bardoxolone methyl evaluation in patients with chronic kidney disease and type 2 diabetes study. Am J Nephrol. 2018;47:40–47. PMID: 29402767. doi:10.1159/000486398
106. Oudiz R, Meyer C, Chin M, et al. Initial data report from “LARIAT”: a phase 2 study of bardoxolone methyl in PAH patients on stable background therapy. Chest. 2015;148:639 A. doi:10.1378/chest.2345856
107. Grzegorzewska AP, Seta F, Han R, et al. Dimethyl fumarate ameliorates pulmonary arterial hypertension and lung fibrosis by targeting multiple pathways. Sci Rep. 2017;7:41605. PMID: 28150703. doi:10.1038/srep41605
108. Zhao G, Liu Y, Fang J, et al. Dimethyl fumarate inhibits the expression and function of hypoxia-inducible factor-1α (HIF-1α). Biochem Biophys Res Commun. 2014;448:303–307. PMID: 24569076. doi:10.1016/j.bbrc.2014.02.062
109. Bosch X, Ramos-Casals M, Khamashta MA. Drugs Targeting B-Cells in Autoimmune Diseases. Springer Science & Business Media; 2013:1–4.
110. Nicolls M, Badesch D, Chung L, et al.; ASC01 Investigators. Safety and efficacy of B-cell depletion with rituximab for the treatment of systemic sclerosis-associated pulmonary arterial hypertension in a multi-center NIH Clinical Trial. Arthritis Rheumatol. 2019;71(S10):Abstract 0867.
111. Hassoun PM, Mouthon L, Barbera JA, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54:S10–19. PMID: 19555853. doi:10.1016/j.jacc.2009.04.006
112. Perros F, Montani D, Dorfmuller P, et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:81–88. PMID: 18420966. doi:10.1164/rccm.200707-1037OC
113. Medarametla V, Festin S, Sugarragchaa C, et al. PK10453, a nonselective platelet-derived growth factor receptor inhibitor, prevents the progression of pulmonary arterial hypertension. Pulm Circ. 2014;4(1):82–102. PMID: 25006424. doi:10.1086/674881
114. Klein M, Schermuly RT, Ellinghaus P, et al. Combined tyrosine and serine/threonine kinase inhibition by sorafenib prevents progression of experimental pulmonary hypertension and myocardial remodeling. Circulation. 2008;118(20):2081–2090. PMID: 18955668. doi:10.1161/CIRCULATIONAHA.108.779751
115. Redner RL. Why doesn’t imatinib cure chronic myeloid leukemia? Oncologist. 2010;15(2):182–186. PMID: 20124443. doi:10.1634/theoncologist.2009-0297
116. Dagher R, Cohen M, Williams G. Approval summary: imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8:3034–3038. PMID: 12374669.
117. Schermuly RT, Dony E, Ghofrani HA, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115(10):2811–2821. PMID: 16200212. doi:10.1172/JCI24838
118. Patterson KC, Weissmann A, Ahmadi T, et al. Imatinib mesylate in the treatment of refractory idiopathic pulmonary arterial hypertension. Ann Intern Med. 2006;145(2):152–153. PMID: 16847299. doi:10.7326/0003-4819-145-2-200607180-00020
119. ten Freyhaus H, Dumitrescu D, Berghausen E, et al. Imatinib mesylate for the treatment of pulmonary arterial hypertension. Expert Opin Investig Drugs. 2012;21(1):119–134. PMID: 22074410. doi:10.1517/13543784.2012.632408
120. Ghofrani HA, Morrell NW, Hoeper MM, et al. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med. 2010;182(9):1171–1177. PMID: 20581169. doi:10.1164/rccm.201001-0123OC
121. Hoeper MM, Barst RJ, Bourge RC, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127(10):1128–1138. PMID: 23403476. doi:10.1161/CIRCULATIONAHA.112.000765
122. Shah AM, Campbell P, Rocha GQ, et al. Effect of imatinib as add-on therapy on echocardiographic measures of right ventricular function in patients with significant pulmonary arterial hypertension. Eur Heart J. 2015;36(10):623–632. PMID: 24566799. doi:10.1093/eurheartj/ehu035
123. Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–134. PMID: 17215530. doi:10.1056/NEJMoa060655
124. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. PMID: 18650514. doi:10.1056/NEJMoa0708857
125. Man CH, Fung TK, Ho C, et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood. 2012;119(22):5133–5143. PMID: 22368270. doi:10.1182/blood-2011-06-363960
126. Worden F. Treatment strategies for radioactive iodine refractory differentiated thyroid cancer. Ther Adv Med Oncol. 2014;6(6):267–279. PMID: 25364392. doi:10.1177/1758834014548188
127. Ranieri G, Gadaleta-Caldarola G, Goffredo V, et al. Sorafenib (BAY 43-9006) in hepatocellular carcinoma patients: from discovery to clinical development. Curr Med Chem. 2012;19(7):938–944. PMID: 22214462. doi:10.2174/092986712799320736
128. Moreno-Vinasco L, Gomberg-Maitland M, Maitland ML, et al. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol Genomics. 2008;33(2):278–291. PMID: 18303084. doi:10.1152/physiolgenomics.00169.2007
129. Kimura G, Kataoka M, Inami T, et al. Sorafenib as a potential strategy for refractory pulmonary arterial hypertension. Pulm Pharmacol Ther. 2017;44:46–49. PMID: 28315488. doi:10.1016/j.pupt.2017.03.009
130. Gomberg-Maitland M, Maitland ML, Barst RJ, et al. A dosing/cross-development study of the multikinase inhibitor sorafenib in patients with pulmonary arterial hypertension. Clin Pharmacol Ther. 2010;87(3):303–310. PMID: 20010555. doi:10.1038/clpt.2009.217
131. Manley P, Cowan-Jacob S, Mestan J. Advances in the structural biology, design and clinical development of Bcr-Abl kinase inhibitors for the treatment of chronic myeloid leukaemia. Biochim Biophys Acta. 2005;1754(1–2):3–13. PMID: 16172030. doi:10.1016/j.bbapap.2005.07.040
132. Jabbour E, Cortes J, Kantarjian H. Nilotinib for the treatment of chronic myeloid leukemia: an evidence-based review. Core Evid. 2010;4:207–213. PMID: 20694077. doi:10.2147/ce.s6003
133. Olivieri A, Manzione L. Dasatinib: a new step in molecular target therapy. Ann Oncol. 2007;18(Suppl 6):vi42–6. PMID: 17591830. doi:10.1093/annonc/mdm223
134. Breccia M, Alimena G. Nilotinib: a second-generation tyrosine kinase inhibitor for chronic myeloid leukemia. Leuk Res. 2010;34(2):129–134. PMID: 19783301. doi:10.1016/j.leukres.2009.08.031
135. Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–2551. PMID: 16775235. doi:10.1056/NEJMoa055104
136. Keating GM. Dasatinib: a review in chronic myeloid leukaemia and Ph+ acute lymphoblastic leukaemia. Drugs. 2017;77(1):85–96. PMID: 28032244. doi:10.1007/s40265-016-0677-x
137. Dumitrescu D, Seck C, ten Freyhaus H, et al. Fully reversible pulmonary arterial hypertension associated with dasatinib treatment for chronic myeloid leukaemia. Eur Respir J. 2011;38(1):218–220. PMID: 21719499. doi:10.1183/09031936.00154210
138. Montani D, Bergot E, Günther S, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125(17):2128–2137. PMID: 22451584. doi:10.1161/CIRCULATIONAHA.111.079921
139. Wang Y, Zheng XR, Riddick N, et al. ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ Res. 2009;104(4):531–540. PMID: 19131646. doi:10.1161/CIRCRESAHA.108.188524
140. Fagan KA, Oka M, Bauer NR, et al. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol. 2004;287:L656–L664. PMID: 14977625. doi:10.1152/ajplung.00090.2003
141. Loirand G, Guérin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res. 2006;98(3):322–334. PMID: 16484628. doi:10.1161/01.RES.0000201960.04223.3c
142. Nagaoka T, Morio Y, Casanova N, et al. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2004;287(4):L665–72. PMID: 12959926. doi:10.1152/ajplung.00050.2003
143. Walker J, Undem C, Yun X, Lade J, Jiang H, Shimoda LA. Role of Rho kinase and Na+/H+ exchange in hypoxia-induced pulmonary arterial smooth muscle cell proliferation and migration. Physiol Rep. 2016;4(6):e12702. PMID: 27009277. doi:10.14814/phy2.12702
144. Madden JA, Ray DE, Keller PA, Kleinman JG. Ion exchange activity in pulmonary artery smooth muscle cells: the response to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2001;280(2):L264–71. PMID: 11159005. doi:10.1152/ajplung.2001.280.2.L264
145. McMurtry IF, Bauer NR, Fagan KA, Nagaoka T, Gebb SA, Oka M. Hypoxia and Rho/Rho-kinase signaling. Lung development versus hypoxic pulmonary hypertension. Adv Exp Med Biol. 2003;543:127–137. PMID: 14713118.
146. Wojciak-Stothard B, Tsang LY, Haworth SG. Rac and Rho play opposing roles in the regulation of hypoxia/reoxygenation-induced permeability changes in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;288:L749–L760. PMID: 15591411. doi:10.1152/ajplung.00361.2004
147. Guilluy C, Eddahibi S, Agard C, et al. RhoA and Rho kinase activation in human pulmonary hypertension: role of 5-HT signaling. Am J Respir Crit Care Med. 2009;179(12):1151–1158. PMID: 19299501. doi:10.1164/rccm.200805-691OC
148. Shimokawa H, Hiramori K, Iinuma H, et al. Anti-anginal effect of fasudil, a Rho-kinase inhibitor, in patients with stable effort angina: a multicenter study. J Cardiovasc Pharmacol. 2002;40(5):751–761. PMID: 12409984. doi:10.1097/00005344-200211000-00013
149. Tachibana E, Harada T, Shibuya M, et al. Intra-arterial infusion of fasudil hydrochloride for treating vasospasm following subarachnoid haemorrhage. Acta Neurochir (Wien). 1999;141(1):13–19. PMID: 10071681. doi:10.1007/s007010050260
150. Somlyo AP, Somlyo AV. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol. 2000;522(Pt 2):177–185. PMID: 10639096. doi:10.1111/j.1469-7793.2000.t01-2-00177.x
151. Fukumoto Y, Matoba T, Ito A, et al. Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart. 2005;91(3):391–392. PMID: 15710736. doi:10.1136/hrt.2003.029470
152. Fukumoto Y, Yamada N, Matsubara H, et al. Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ J. 2013;77(10):2619–2625. PMID: 23912836. doi:10.1253/circj.cj-13-0443
153. Liu AJ, Ling F, Wang D, et al. Fasudil inhibits platelet-derived growth factor-induced human pulmonary artery smooth muscle cell proliferation by up-regulation of p27kip1 via the ERK signal pathway. Chin Med J (Engl). 2011;124(19):3098–3104. PMID: 22040563.
154. Fujita H, Fukumoto Y, Saji K, et al. Acute vasodilator effects of inhaled fasudil, a specific Rho-kinase inhibitor, in patients with pulmonary arterial hypertension. Heart Vessels. 2010;25(2):144–149. PMID: 20339976. doi:10.1007/s00380-009-1176-8
155. Archer SL, Fang YH, Ryan JJ, Piao L. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulm Circ. 2013;3(1):144–152. PMID: 23662191. doi:10.4103/2045-8932.109960
156. Archer SL, Marsboom G, Kim GH, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010;121(24):2661–2671. PMID: 20529999F. doi:10.1161/CIRCULATIONAHA.109.916098
157. Sutendra G, Michelakis ED. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014;19:
158. Ryan JJ, Archer SL. Emerging concepts in the molecular basis of pulmonary arterial hypertension: part I: metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation. 2015;131:
159. Wilkins MR. Pulmonary hypertension: the science behind the disease spectrum. Eur Respir Rev. 2012;21:19–26. PMID: 22379170. doi:10.1183/09059180.00008411
160. Warburg O. On the origin of cancer cells. Science. 1956;123:
161. Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res. 2014;115:148–164. PMID: 24951764. doi:10.1161/CIRCRESAHA.115.301130.
162. Michelakis ED, McMurtry MS, Wu XC, et al. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation. 2002;105:244–250. PMID: 11790708. doi:10.1161/hc0202.101974
163. McMurtry MS, Bonnet S, Wu X, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95:830–840. PMID: 15375007. doi:10.1161/01.RES.0000145360.16770.9f
164. Piao L, Sidhu VK, Fang YH, et al. FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: therapeutic benefits of dichloroacetate. J Mol Med (Berl). 2013;91:333–346. PMID: 23247844. doi:10.1007/s00109-012-0982-0
165. Michelakis ED, Gurtu V, Webster L, et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med. 2017;9(413):
166. Nelson CH, Etchevers K, Yi S, et al. Pharmacokinetics, safety, and tolerability of selonsertib, an apoptosis signal-regulating kinase 1 (ASK1) inhibitor, following first-in-human single and multiple ascending doses in healthy subjects. Clin Pharmacokinet. 2020;59(9):1109–1117. PMID: 32333325. doi:10.1007/s40262-020-00878-y
167. Hawwa N, Menon V. Ranolazine: clinical applications and therapeutic basis. Am J Cardiovasc Drugs. 2013;13(1):5–16. PMID: 23335347. doi:10.1007/s40256-012-0003-2
168. Chaitman BR, Skettino SL, Parker JO, et al. Anti-ischemic effects and long-term survival during ranolazine monotherapy in patients with chronic severe angina. J Am Coll Cardiol. 2004;43:1375–1382. PMID: 15093870. doi:10.1016/j.jacc.2003.11.045
169. Chaitman BR, Pepine CJ, Parker JO, et al. Effects of ranolazine with atenolol, amlodipine, or diltiazem on exercise tolerance and angina frequency in patients with severe chronic angina: a randomized controlled trial. JAMA. 2004;291:309–316. PMID: 14734593. doi:10.1001/jama.291.3.309
170. Hasenfuss G, Maier LS. Mechanism of action of the new anti-ischemia drug ranolazine. Clin Res Cardiol. 2008;97:222–226. PMID: 18046526. doi:10.1007/s00392-007-0612-y
171. Fang YH, Piao L, Hong Z, et al. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. J Mol Med. 2012;90:31–43. PMID: 21874543. doi:10.1007/s00109-011-0804-9
172. Gomberg-Maitland M, Schilz R, Mediratta A, et al. Phase I safety study of ranolazine in pulmonary arterial hypertension. Pulm Circ. 2015;5:691–700. PMID: 26697176. doi:10.1086/683813
173. Khan SS, Cuttica MJ, Beussink-Nelson L, et al. Effects of ranolazine on exercise capacity, right ventricular indices, and hemodynamic characteristics in pulmonary arterial hypertension: a pilot study. Pulm Circ. 2015;5(3):547–556. PMID: 26401256. doi:10.1086/682427
174. Han Y, Forfia PR, Vaidya A, et al. Rationale and design of the ranolazine PH-RV study: a multicentred randomised and placebo-controlled study of ranolazine to improve RV function in patients with non-group 2 pulmonary hypertension. Open Heart. 2018;5(1):e000736. PMID: 29531764. doi:10.1136/openhrt-2017-000736
175. Sutendra G, Bonnet S, Rochefort G, et al. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med. 2010;2(44):44–58. PMID: 20702857. doi:10.1126/scitranslmed.3001327
176. Aggarwal S, Gross CM, Sharma S, et al. Reactive oxygen species in pulmonary vascular remodeling. Compr Physiol. 2013;3:1011–1034. PMID: 23897679. doi:10.1002/cphy.c120024
177. Fulton D, Li X, Bordan Z, et al. Reactive oxygen and nitrogen species in the development of pulmonary hypertension. Antioxidants (Basel). 2017;6(3):54. PMID: 28684719. doi:10.3390/antiox6030054
178. Jernigan NL, Naik JS, Weise-Cross L, et al. Contribution of reactive oxygen species to the pathogenesis of pulmonary arterial hypertension. PLoS One. 2017;12(6):e0180455. PMID: 28666030. doi:10.1371/journal.pone.0180455
179. Zuo L, Rose BA, Roberts WJ, et al. Molecular characterization of reactive oxygen species in systemic and pulmonary hypertension. Am J Hypertens. 2014;27(5):643–650. PMID: 24552887. doi:10.1093/ajh/hpt292
180. Anwar A, Ruffenach G, Mahajan A, et al. Novel biomarkers for pulmonary arterial hypertension. Respir Res. 2016;17(1):88. PMID: 27439993. doi:10.1186/s12931-016-0396-6
181. Reis GS, Augusto VS, Silveira APC, et al. Oxidative-stress biomarkers in patients with pulmonary hypertension. Pulm Circ. 2013;3(4):856–861. PMID: 25006401. doi:10.1086/674764
182. Zhang S, Yang T, Xu X, et al. Oxidative stress and nitric oxide signaling related biomarkers in patients with pulmonary hypertension: a case control study. BMC Pulm Med. 2015;15:50. PMID: 25934483. doi:10.1186/s12890-015-0045-8
183. Beckman JS, Beckman TW, Chen J, et al. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87(4):1620–1624. PMID: 2154753. doi:10.1073/pnas.87.4.1620
184. Chen CA, Wang TY, Varadharaj S, et al. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468(7327):1115–1118. PMID: 21179168. doi:10.1038/nature09599
185. Elahi MM, Kong YX, Matata BM. Oxidative stress as a mediator of cardiovascular disease. Oxid Med Cell Longev. 2009;2(5):259–269. PMID: 20716913. doi:10.4161/oxim.2.5.9441
186. Bowers R, Cool C, Murphy RC, et al. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med. 2004;169:764–769. PMID: 14701708. doi:10.1164/rccm.200301-147OC
187. Demarco VG, Whaley-Connell AT, Sowers JR, et al. Contribution of oxidative stress to pulmonary arterial hypertension. World J Cardiol. 2010;2:316–324. PMID: 21160609. doi:10.4330/wjc.v2.i10.316
188. Dorfmüller P, Chaumais M-C, Giannakouli M, et al. Increased oxidative stress and severe arterial remodeling induced by permanent high-flow challenge in experimental pulmonary hypertension. Respir Res. 2011;12:119. PMID: 21906276. doi:10.1186/1465-9921-12-119
189. Black SM, DeVol JM, Wedgwood S. Regulation of fibroblast growth factor-2 expression in pulmonary arterial smooth muscle cells involves increased reactive oxygen species generation. Am J Physiol Cell Physiol. 2008;294:C345–C354. PMID: 17942638. doi:10.1152/ajpcell.00216.2007
190. Montisano DF, Mann T, Spragg RG. H2O2 increases expression of pulmonary artery endothelial cell platelet-derived growth factor mRNA. J Appl Physiol. 1992;73:2255–2262. PMID: 1490932. doi:10.1152/jappl.1992.73.6.2255
191. Wedgwood S, Dettman RW, Black SM. ET-1 stimulates pulmonary arterial smooth muscle cell proliferation via induction of reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1058–1067. PMID: 11597896. doi:10.1152/ajplung.2001.281.5.L1058
192. Cheng TH, Shih NL, Chen SY, et al. Reactive oxygen species mediate cyclic strain-induced endothelin-1 gene expression via Ras/Raf/extracellular signal-regulated kinase pathway in endothelial cells. J Mol Cell Cardiol. 2001;33:1805–1814. PMID: 11603923. doi:10.1006/jmcc.2001.1444
193. Tate RM, Morris HG, Schroeder WR, Repine JE. Oxygen metabolites stimulate thromboxane production and vasoconstriction in isolated saline-perfused rabbit lungs. J Clin Investig. 1984;74:608–613. PMID: 6547730. doi:10.1172/JCI111458
194. Lee DS, McCallum EA, Olson DM. Effects of reactive oxygen species on prostacyclin production in perinatal rat lung cells. J Appl Physiol. 1989;66:1321–1327. PMID: 2651389. doi:10.1152/jappl.1989.66.3.1321
195. Brito R, Castillo G, González J, et al. Oxidative stress in hypertension: mechanisms and therapeutic opportunities. Exp Clin Endocrinol Diabetes. 2015;123:325–335. PMID: 25918881. doi:10.1055/s-0035-1548765
196. Block K, Gorin Y, Hoover P, et al. NAD(P)H oxidases regulate HIF-2alpha protein expression. J Biol Chem. 2007;282:8019–8026. PMID: 17200123. doi:10.1074/jbc.M611569200
197. Chandel NS, McClintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia A mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. PMID: 10833514. doi:10.1074/jbc.M001914200
198. Gale DP, Harten SK, Reid CDL, et al. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2α mutation. Blood. 2008;112:919–921. PMID: 18650473. doi:10.1182/blood-2008-04-153718
199. Brusselmans K, Compernolle V, Tjwa M, et al. Heterozygous deficiency of hypoxia-inducible factor–2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Investig. 2003;111:1519–1527. PMID: 12750401. doi:10.1172/JCI15496
200. Cracowski JL, Cracowski C, Bessard G, et al. Increased lipid peroxidation in patients with pulmonary hypertension. Am J Respir Crit Care Med. 2001;164:1038–1042. PMID: 11587993. doi:10.1164/ajrccm.164.6.2104033
201. Wong C-M, Bansal G, Pavlickova L, et al. Reactive oxygen species and antioxidants in pulmonary hypertension. Antioxid Redox Signal. 2013;18:1789–1796. PMID: 22657091. doi:10.1089/ars.2012.4568
202. Tabima DM, Frizzell S, Gladwin MT. Reactive oxygen and nitrogen species in pulmonary hypertension. Free Radic Biol Med. 2012;52:1970–1986. PMID: 22401856. doi:10.1016/j.freeradbiomed.2012.02.041
203. Crosswhite P, Sun Z. Nitric oxide, oxidative stress and inflammation in pulmonary arterial hypertension. J Hypertens. 2010;28:201–212. PMID: 20051913. doi:10.1097/HJH.0b013e328332bcdb
204. Xu D, Guo H, Xu X, et al. Exacerbated pulmonary arterial hypertension and right ventricular hypertrophy in animals with loss of function of extracellular superoxide dismutase. Hypertens. 2011;58:303–309. PMID: 21730301. doi:10.1161/HYPERTENSIONAHA.110.166819
205. Yu H, Liu J, Dong Y, et al. Anti-hypoxic effect of dihydroartemisinin on pulmonary artery endothelial cells. Biochem Biophys Res Commun. 2018;506:840–846. PMID: 30391003. doi:10.1016/j.bbrc.2018.10.176
206. Galhotra P, Prabhakar P, Meghwani H, et al. Beneficial effects of fenofibrate in pulmonary hypertension in rats. Mol Cell Biochem. 2018;449:185–194. PMID: 29761247. doi:10.1007/s11010-018-3355-3
207. Türck P, Lacerda DS, Carraro CC, et al. Trapidil improves hemodynamic, echocardiographic and redox state parameters of right ventricle in monocrotaline-induced pulmonary arterial hypertension model. Biomed Pharmacother. 2018;103:182–190. PMID: 29653363. doi:10.1016/j.biopha.2018.04.001
208. Zuckerbraun BS, Shiva S, Ifedigbo E, et al. Nitrite potently inhibits hypoxic and inflammatory pulmonary arterial hypertension and smooth muscle proliferation via xanthine oxidoreductase-dependent nitric oxide generation. Circulation. 2010;121:98–109. PMID: 20026772. doi:10.1161/CIRCULATIONAHA.109.891077
209. Baliga RS, Milsom AB, Ghosh SM, et al. Dietary nitrate ameliorates pulmonary hypertension: cytoprotective role for endothelial nitric oxide synthase and xanthine oxidoreductase. Circulation. 2012;125:2922–2932. PMID: 22572914. doi:10.1161/CIRCULATIONAHA.112.100586
210. Sharp J, Farha S, Park MM, et al. Coenzyme Q supplementation in pulmonary arterial hypertension. Redox Biol. 2014;2:884–891. PMID: 25180165. doi:10.1016/j.redox.2014.06.010
211. Archer SL, Nelson DP, Weir EK. Detection of activated O2 species in vitro and in rat lungs by chemiluminescence. J Appl Physiol. 1989;67:1912–1921. PMID: 2532194. doi:10.1152/jappl.1989.67.5.1912
212. Pacher P, Nivorozhkin A, Szabó C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006;58:87–114. PMID: 16507884. doi:10.1124/pr.58.1.6
213. Poss WB, Huecksteadt TP, Panus PC, et al. Regulation of xanthine dehydrogenase and xanthine oxidase activity by hypoxia. Am J Physiol Lung Cell Mol Physiol. 1996;270:L941–L946. PMID: 8764218. doi:10.1152/ajplung.1996.270.6.L941
214. Engerson TD, McKelvey TG, Rhyne DB, et al. Conversion of xanthine dehydrogenase to oxidase in ischemic rat tissues. J Clin Invest. 1987;79:1564–1570. PMID: 3294898. doi:10.1172/JCI112990
215. Pritsos CA. Cellular distribution, metabolism and regulation of the xanthine oxidoreductase enzyme system. Chem Biol Interact. 2000;129:195–208. PMID: 11154741. doi:10.1016/s0009-2797(00)00203-9
216. Houston M, Estevez A, Chumley P, et al. Binding of xanthine oxidase to vascular endothelium. Kinetic characterization and oxidative impairment of nitric oxide-dependent signaling. J Biol Chem. 1999;274:4985–4994. PMID: 9988743. doi:10.1074/jbc.274.8.4985
217. Hoshikawa Y, Ono S, Suzuki S, et al. Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J Appl Physiol. 2001;90:1299–1306. PMID: 11247927. doi:10.1152/jappl.2001.90.4.1299
218. Jankov RP, Kantores C, Pan J, Belik J. Contribution of xanthine oxidase‐derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am J Physiol Lung Cell Mol Physiol. 2008;294:L233–L245. PMID: 18083771. doi:10.1152/ajplung.00166.2007
219. Voelkel MA, Wynne KM, Badesch DB, et al. Hyperuricemia in severe pulmonary hypertension. Chest. 2000;117:19–24. PMID: 10631193. doi:10.1378/chest.117.1.19
220. Bendayan D, Shitrit D, Ygla M, et al. Hyperuricemia as a prognostic factor in pulmonary arterial hypertension. Respir Med. 2003;97:130–133. PMID: 12587962. doi:10.1053/rmed.2003.1440
221. Spiekermann S, Schenk K, Hoeper MM. Increased xanthine oxidase activity in idiopathic pulmonary arterial hypertension. Eur Respir J. 2009;34(1):276. PMID: 19567609. doi:10.1183/09031936.00013309
222. Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn‐SOD (SOD1), Mn‐SOD (SOD2), and EC‐SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–349. PMID: 12126755. doi:10.1016/s0891-5849(02)00905-x
223. Eble JA, Niland S. The extracellular matrix of blood vessels. Curr Pharm Des. 2009;15:1385–1400. PMID: 19355976. doi:10.2174/138161209787846757
224. Lammers SR, Kao PH, Qi HJ, et al. Changes in the structure-function relationship of elastin and its impact on the proximal pulmonary arterial mechanics of hypertensive calves. Am J Physiol Heart Circ Physiol. 2008;295:H1451–9. PMID: 18660454. doi:10.1152/ajpheart.00127.2008
225. Frisdal E, Gest V, Vieillard-Baron A, et al. Gelatinase expression in pulmonary arteries during experimental pulmonary hypertension. Eur Respir J. 2001;18:838–845. PMID: 11757635. doi:10.1183/09031936.01.00084601
226. Ambalavanan N, Nicola T, Li P, et al. Role of matrix metalloproteinase-2 in newborn mouse lungs under hypoxic conditions. Pediatr Res. 2008;63:26–32. PMID: 18043506. doi:10.1203/PDR.0b013e31815b690d
227. Novotná J, Herget J. Possible role of matrix metalloproteinases in reconstruction of peripheral pulmonary arteries induced by hypoxia. Physiol Res. 2002;51:323–334. PMID: 12449429.
228. Lepetit H, Eddahibi S, Fadel E, et al. Smooth muscle cell matrix metalloproteinases in idiopathic pulmonary arterial hypertension. Eur Respir J. 2005;25:834–842. PMID: 15863640. doi:10.1183/09031936.05.00072504
229. Thenappan T, Chan SY, Weir EK. Role of extracellular matrix in the pathogenesis of pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2018;315(5):H1322–H1331. PMID: 30141981. doi:10.1152/ajpheart.00136.2018
230. George J, D’Armiento J. Transgenic expression of human matrix metalloproteinase-9 augments monocrotaline-induced pulmonary arterial hypertension in mice. J Hypertens. 2011;29(2):299–308. PMID: 21063214. doi:10.1097/HJH.0b013e328340a0e4
231. Cowan KN, Jones PL, Rabinovitch M. Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J Clin Invest. 2000;105(1):21–34. PMID: 10619858. doi:10.1172/JCI6539
232. Cowan KN, Heilbut A, Humpl T, et al. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. 2000;6(6):698–702. PMID: 10835689. doi:10.1038/76282
233. Vieillard-Baron A, Frisdal E, Raffestin B, et al. Inhibition of matrix metalloproteinases by lung TIMP-1 gene transfer limits monocrotaline-induced pulmonary vascular remodeling in rats. Hum Gene Ther. 2003;14(9):861–869. PMID: 12828857. doi:10.1089/104303403765701150
234. Pullamsetti S, Krick S, Yilmaz H, et al. Inhaled tolafentrine reverses pulmonary vascular remodeling via inhibition of smooth muscle cell migration. Respir Res. 2005;6(128):1–14. PMID: 16262900. doi:10.1186/1465-9921-6-128
235. Martinez MLL, Lopes LF, Coelho EB, et al. Lercanidipine reduces matrix metalloproteinase-9 activity in patients with hypertension. J Cardiovasc Pharmacol. 2006;47(1):117–122. PMID: 16262900. doi:10.1097/01.fjc.0000196241.96759.71
236. Tayebjee MH, Nadar S, Blann AD, et al. Matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 in hypertension and their relationship to cardiovascular risk and treatment: a substudy of the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT). Am J Hypertens. 2004;17(9):764–769. PMID: 15363817. doi:10.1016/j.amjhyper.2004.05.019
237. Mawatari E, Hongo M, Sakai A, et al. Amlodipine prevents monocrotaline-induced pulmonary arterial hypertension and prolongs survival in rats independent of blood pressure lowering. Clin Exp Pharmacol Physiol. 2007;34(7):594–600. PMID: 17581214. doi:10.1111/j.1440-1681.2007.04618.x.
238. Koo HS, Kim KC, Hong YM. Gene expressions of nitric oxide synthase and matrix metalloproteinase-2 in monocrotaline induced pulmonary hypertension in rats after bosentan treatment. Korean Circ J. 2011;41(2):83–90. PMID: 21430993. doi:10.4070/kcj.2011.41.2.83
239. Zaidi SHE, You X-M, Ciura S, et al. Overexpression of the serine elastase inhibitor elafin protects transgenic mice from hypoxic pulmonary hypertension. Circulation. 2002;105(4):516–521. PMID: 11815437. doi:10.1161/hc0402.102866
240. Nickel NP, Spiekerkoetter E, Gu M, et al. Elafin reverses pulmonary hypertension via caveolin-1-dependent bone morphogenetic protein signaling. Am J Respir Crit Care Med. 2015;191:1273–1286. PMID: 25853696. doi:10.1164/rccm.201412-2291OC
241. Sutliff RL, Kang BY, Hart CM. PPARgamma as a potential therapeutic target in pulmonary hypertension. Ther Adv Respir Dis. 2010;4:143–160. PMID: 20530063. doi:10.1177/1753465809369619
242. Ameshima S, Golpon H, Cool CD, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res. 2003;92:1162–1169. PMID: 12714563. doi:10.1161/01.RES.0000073585.50092.14
243. Hansmann G, Zamanian RT. PPARgamma activation: a potential treatment for pulmonary hypertension. Sci Transl Med. 2009;1:12–14. PMID: 20371457. doi:10.1126/scitranslmed.3000267
244. Tian J, Smith A, Nechtman J, et al. Effect of PPARgamma inhibition on pulmonary endothelial cell gene expression: gene profiling in pulmonary hypertension. Physiol Genomics. 2009;40:48–60. PMID: 19825830. doi:10.1152/physiolgenomics.00094.2009
245. Lu X, Murphy TC, Nanes MS, Hart CM. PPAR{gamma} regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-{kappa}B. Am J Physiol Lung Cell Mol Physiol. 2010;299:L559–566. PMID: 20622120. doi:10.1152/ajplung.00090.2010
246. Calvier L, Chouvarine P, Legchenko E, Hansmann G. Transforming growth factor β1- and bone morphogenetic protein 2/PPARγ-regulated miRNAs in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2017;196:1227–1228. PMID: 28653861. doi:10.1164/rccm.201705-0923LE
247. Calvier L, Chouvarine P, Legchenko E, et al. PPARγ links BMP2 and TGFβ1 pathways in vascular smooth muscle cells, regulating cell proliferation and glucose metabolism. Cell Metab. 2017;25:1118–1134.e7. PMID: 28467929. doi:10.1016/j.cmet.2017.03.011
248. Geraci MW, Moore M, Gesell T, et al. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: a gene microarray analysis. Circ Res. 2001;88:555–562. PMID: 11282888. doi:10.1161/01.res.88.6.555
249. Green DE, Murphy TC, Kang BY, et al. PPARgamma ligands attenuate hypoxia-induced proliferation in human pulmonary artery smooth muscle cells through modulation of MicroRNA-21. PLoS One. 2015;10(7):e0133391. PMID: 26208095. doi:10.1371/journal.pone.0133391
250. Matsuda Y, Hoshikawa Y, Ameshima S, et al. Effects of peroxisome proliferator-activated receptor gamma ligands on monocrotaline-induced pulmonary hypertension in rats. Nihon Kokyuki Gakkai Zasshi. 2005;43:283–288. PMID: 15969209.
251. Nisbet RE, Bland JM, Kleinhenz DJ, et al. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am J Respir Cell Mol Biol. 2010;42:482–490. PMID: 19520921. doi:10.1165/rcmb.2008-0132OC
252. Hansmann G, Wagner RA, Schellong S, et al. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator‐activated receptor‐gamma activation. Circulation. 2007;115:1275–1284. PMID: 17339547. doi:10.1161/CIRCULATIONAHA.106.663120
253. Kim EK, Lee JH, Oh YM, et al. Rosiglitazone attenuates hypoxia‐induced pulmonary arterial hypertension in rats. Respirology. 2010;15:659–668. PMID: 20546541. doi:10.1111/j.1440-1843.2010.01756.x.
254. Behringer A, Trappiel M, Berghausen EM, et al. Pioglitazone alleviates cardiac and vascular remodelling and improves survival in monocrotaline induced pulmonary arterial hypertension. Arch Pharmacol. 2016;389(4):369–379. PMID: 26742933. doi:10.1007/s00210-015-1205-3
255. Legchenko E, Chouvarine P, Borchert P, et al. Pparγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med. 2018;10(438):eaao0303. PMID: 29695452. doi:10.1126/scitranslmed.aao0303
256. Mannucci E, Monami M, Di Bari M, et al. Cardiac safety profile of rosiglitazone: a comprehensive meta‐analysis of randomized clinical trials. Int J Cardiol. 2010;143:135–140. PMID: 19328563. doi:10.1016/j.ijcard.2009.01.064
257. Rojas LB, Gomes MB. Metformin: an old but still the best treatment for type 2 diabetes. Diabetol Metab Syndr. 2013;5:6. PMID: 23415113. doi:10.1186/1758-5996-5-6
258. Wu Y, Liu L, Zhang Y, et al. Activation of AMPK inhibits pulmonary arterial smooth muscle cells proliferation. Exp Lung Res. 2014;40:251–258. PMID: 24809794. doi:10.3109/01902148.2014.913092.
259. Davis BJ, Xie Z, Viollet B, Zou MH. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55:496–505. PMID: 16443786. doi:10.2337/diabetes.55.02.06.db05-1064
260. Zou MH, Kirkpatrick SS, Davis BJ, et al. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem. 2004;279:43940–43951. PMID: 15265871. doi:10.1074/jbc.M404421200
261. Dean A, Nilsen M, Loughlin L, et al. Metformin reverses development of pulmonary hypertension via aromatase inhibition. Hypertension. 2016;68(2):446–454. PMID: 27296990. doi:10.1161/HYPERTENSIONAHA.116.07353
262. Agard C, Rolli‐Derkinderen M, Dumas‐de‐La‐Roque E, et al. Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br J Pharmacol. 2009;158:1285–1294. PMID: 19814724. doi:10.1111/j.1476-5381.2009.00445.x
263. Maron BA, Leopold JA. Emerging concepts in the molecular basis of pulmonary arterial hypertension: part II: neurohormonal signaling contributes to the pulmonary vascular and right ventricular pathophenotype of pulmonary arterial hypertension. Circulation. 2015;131:2079–2091. PMID: 26056345. doi:10.1161/CIRCULATIONAHA.114.006980
264. Vaillancourt M, Chia P, Sarji S, et al. Autonomic nervous system involvement in pulmonary arterial hypertension. Respir Res. 2017;18(1):201. PMID: 29202826. doi:10.1186/s12931-017-0679-6
265. Nagaya N, Nishikimi T, Uematsu M, et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation. 2000;102:865–870. PMID: 10952954. doi:10.1161/01.cir.102.8.865
266. Nootens M, Kaufmann E, Rector T, et al. Neurohormonal activation in patients with right ventricular failure from pulmonary hypertension: relation to hemodynamic variables and endothelin levels. J Am Coll Cardiol. 1995;26:1581–1585. PMID: 7594089. doi:10.1016/0735-1097(95)00399-1
267. Bristow MR, Minobe W, Rasmussen R, et al. Beta-adrenergic neuroeffector abnormalities in the failing human heart are produced by local rather than systemic mechanisms. J Clin Investig. 1992;89:803–815. PMID: 1311717. doi:10.1172/JCI115659
268. Piao L, Fang YH, Parikh KS, et al. GRK2-mediated inhibition of adrenergic and dopaminergic signaling in right ventricular hypertrophy: therapeutic implications in pulmonary hypertension. Circulation. 2012;126:2859–2869. PMID: 23124027. doi:10.1161/CIRCULATIONAHA.112.109868
269. Ishikawa M, Sato N, Asai K, et al. Effects of a pure alpha/beta‐adrenergic receptor blocker on monocrotaline‐induced pulmonary arterial hypertension with right ventricular hypertrophy in rats. Circ J. 2009;73:2337–2341. PMID: 19822980. doi:10.1253/circj.cj-09-0213
270. Ghosh S, Gupta M, Xu W, et al. Phosphorylation inactivation of endothelial nitric oxide synthesis in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2016;310:L1199–L1205. PMID: 27130529. doi:10.1152/ajplung.00092.2016
271. Perros F, Ranchoux B, Izikki M, et al. Nebivolol for improving endothelial dysfunction, pulmonary vascular remodeling, and right heart function in pulmonary hypertension. J Am Coll Cardiol. 2015;65:668–680. PMID: 25677428. doi:10.1016/j.jacc.2014.11.050
272. Perros F, De man FS, Bogaard HJ, et al. Use of β‐blockers in pulmonary hypertension. Circ Heart Fail. 2017;10(4):e003703. PMID: 28364092. doi:10.1161/CIRCHEARTFAILURE.116.003703
273. Bogaard HJ, Natarajan R, Mizuno S, et al. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med. 2010;182:652–660. PMID: 20508210. doi:10.1164/rccm.201003-0335OC
274. Drake JI, Gomez-Arroyo J, Dumur CI, et al. Chronic carvedilol treatment partially reverses the right ventricular failure transcriptional profile in experimental pulmonary hypertension. Physiol Genom. 2013;45:449–461. PMID: 23632417. doi:10.1152/physiolgenomics.00166.2012
275. de Man FS, Handoko ML, van Ballegoij JJ, et al. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail. 2012;5:97–105. PMID: 22157723. doi:10.1161/CIRCHEARTFAILURE.111.964494
276. Grinnan D, Bogaard HJ, Grizzard J, et al. Treatment of group I pulmonary arterial hypertension with carvedilol is safe. Am J Respir Crit Care Med. 2014;189:1562–1564. PMID: 24930531. doi:10.1164/rccm.201311-2025LE
277. Farha S, Saygin D, Park MM, et al. Pulmonary arterial hypertension treatment with carvedilol for heart failure: a randomized controlled trial. JCI Insight. 2017;2(16):e95240. PMID: 28814664. doi:10.1172/jci.insight.95240
278. van Campen JS, de Boer K, van de Veerdonk MC, et al. Bisoprolol in idiopathic pulmonary arterial hypertension: an explorative study. Eur Respir J. 2016;48:787–796. PMID: 27390285. doi:10.1183/13993003.00090-2016
279. de Man FS, Tu L, Handoko ML, et al. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:780–789. PMID: 22859525. doi:10.1164/rccm.201203-0411OC
280. Morrell NW, Danilov SM, Satyan KB, et al. Right ventricular angiotensin converting enzyme activity and expression is increased during hypoxic pulmonary hypertension. Cardiovasc Res. 1997;34:393–403. PMID: 9205554. doi:10.1016/s0008-6363(97)00049-7
281. Morrell NW, Atochina EN, Morris KG, et al. Angiotensin converting enzyme expression is increased in small pulmonary arteries of rats with hypoxia-induced pulmonary hypertension. J Clin Invest. 1995;96:1823–1833. PMID: 7560074. doi:10.1172/JCI118228
282. Morrell NW, Morris KG, Stenmark KR. Role of angiotensin-converting enzyme and angiotensin II in development of hypoxic pulmonary hypertension. Am J Physiol. 1995;269:H1186–94. PMID: 7485548. doi:10.1152/ajpheart.1995.269.4.H1186
283. Alpert MA, Pressly TA, Mukerji V, et al. Short- and long-term hemodynamic effects of captopril in patients with pulmonary hypertension and selected connective tissue disease. Chest. 1992;102:1407–1412. PMID: 1424860. doi:10.1378/chest.102.5.1407
284. Ikram H, Maslowski AH, Nicholls MG, et al. Haemodynamic and hormonal effects of captopril in primary pulmonary hypertension. Br Heart J. 1982;48:541–545. PMID: 6756445. doi:10.1136/hrt.48.6.541
285. Lambert DW, Hooper NM, Turner AJ. Angiotensin-converting enzyme 2 and new insights into the renin-angiotensin system. Biochem Pharmacol. 2008;75:781–786. PMID: 17897633. doi:10.1016/j.bcp.2007.08.012
286. Tallant EA, Lu X, Weiss RB, et al. Bovine aortic endothelial cells contain an angiotensin-(1–7) receptor. Hypertension. 1997;29:388–393. PMID: 9039132. doi:10.1161/01.hyp.29.1.388
287. Ferrario CM, Chappell MC, Tallant EA, et al. Counterregulatory actions of angiotensin-(1–7). Hypertension. 1997;30:535–541. PMID: 9322978. doi:10.1161/01.hyp.30.3.535
288. Brosnihan KB, Li P, Ferrario CM. Angiotensin-(1–7) dilates canine coronary arteries through kinins and nitric oxide. Hypertension. 1996;27:523–528. PMID: 8613197. doi:10.1161/01.hyp.27.3.523
289. Nakamoto H, Ferrario CM, Fuller SB, et al. Angiotensin-(1–7) and nitric oxide interaction in renovascular hypertension. Hypertension. 1995;25:796–802. PMID: 7536715. doi:10.1161/01.hyp.25.4.796
290. Tan WSD, Liao W, Zhou S, et al. Targeting the renin-angiotensin system as novel therapeutic strategy for pulmonary diseases. Curr Opin Pharmacol. 2018;40:9–17. PMID: 29288933. doi:10.1016/j.coph.2017.12.002
291. Hemnes AR, Rathinasabapathy A, Austin EA, et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur Respir J. 2018;51(6):
292. Maron BA, Leopold JA. Aldosterone receptor antagonists: effective but often forgotten. Circulation. 2010;121:934–939. PMID: 20177008. doi:10.1161/CIRCULATIONAHA.109.895235
293. Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized aldactone evaluation study investigators. N Engl J Med. 1999;341:709–717. PMID: 10471456. doi:10.1056/NEJM199909023411001
294. Moore KP, Aithal GP. Guidelines on the management of ascites in cirrhosis. Gut. 2006;55(suppl6):vi1–vi12. PMID: 16966752. doi:10.1136/gut.2006.099580
295. Maron BA, Opotowsky AR, Landzberg MJ, et al. Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: a pilot study. Eur J Heart Fail. 2013;15:277–283. PMID: 23111998. doi:10.1093/eurjhf/hfs173
296. Maron BA, Zhang YY, White K, et al. Aldosterone inactivates the endothelin‐B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation. 2012;126:963–974. PMID: 22787113. doi:10.1161/CIRCULATIONAHA.112.094722
297. Maron BA, Oldham WM, Chan SY, et al. Upregulation of steroidogenic acute regulatory protein by hypoxia stimulates aldosterone synthesis in pulmonary artery endothelial cells to promote pulmonary vascular fibrosis. Circulation. 2014;130:168–179. PMID: 25001622. doi:10.1161/CIRCULATIONAHA.113.007690
298. Aghamohammadzadeh R, Zhang YY, Stephens TE, et al. Up‐regulation of the mammalian target of rapamycin complex 1 subunit raptor by aldosterone induces abnormal pulmonary artery smooth muscle cell survival patterns to promote pulmonary arterial hypertension. FASEB J. 2016;30(7):2511–2527. PMID: 27006450. doi:10.1096/fj.201500042
299. Safdar Z, Frost A, Basant A, et al. Spironolactone in pulmonary arterial hypertension: results of a cross-over study. Pulm Circ. 2020;10(2):2045894019898030. PMCID: PMC7219009. PMID: 32426108. doi:10.1177/2045894019898030
300. Maron BA, Waxman AB, Opotowsky AR, et al. Effectiveness of spironolactone plus ambrisentan for treatment of pulmonary arterial hypertension (from the [ARIES] Study 1 and 2 Trials). Am J Cardiol. 2013;112(5):720–725. PMID: 23751938. doi:10.1016/j.amjcard.2013.04.051
301. Hobbs AJ. Soluble guanylate cyclase: the forgotten sibling. Trends Pharmacol Sci. 1997;18(12):484–491. PMID: 9458697. doi:10.1016/s0165-6147(97)01137-1
302. Casserly B, Klinger JR. Brain natriuretic peptide in pulmonary arterial hypertension: biomarker and potential therapeutic agent. Drug Des Devel Ther. 2009;3:269–287. PMID: 20054445. doi:10.2147/dddt.s4805
303. Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev. 2006;27(1):47–72. PMID: 16291870. doi:10.1210/er.2005-0014
304. Rosenkranz AC, Woods RL, Dusting GJ, Ritchie RH. Antihypertrophic actions of the natriuretic peptides in adult rat cardiomyocytes: importance of cyclic GMP. Cardiovasc Res. 2003;57(2):515–522. PMID: 12566124. doi:10.1016/s0008-6363(02)00667-3
305. Klinger JR, Petit RD, Curtin LA, et al. Cardiopulmonary responses to chronic hypoxia in transgenic mice that overexpress ANP. J Appl Physiol. 1993;75(1):198–205. PMID: 7690745. doi:10.1152/jappl.1993.75.1.198.
306. Hill NS, Klinger JR, Warburton RR, Pietras L, Wrenn DS. Brain natriuretic peptide: possible role in the modulation of hypoxic pulmonary hypertension. Am J Physiol. 1994;266(3 Pt 1):L308–15. PMID: 8166300. doi:10.1152/ajplung.1994.266.3.L308
307. Angus RM, McCallum MJ, Hulks G, et al. Bronchodilator, cardiovascular, and cyclic guanylyl monophosphate response to high-dose infused atrial natriuretic peptide in asthma. Am Rev Respir Dis. 1993;147(5):1122–1125. PMID: 8387254. doi:10.1164/ajrccm/147.5.1122
308. Baliga RS, Zhao L, Madhani M, et al. Synergy between natriuretic peptides and phosphodiesterase 5 inhibitors ameliorates pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178(8):861–869. PMID: 18689467. doi:10.1164/rccm.200801-121OC
309. Klinger JR, Houtchens J, Thaker S, et al. Acute cardiopulmonary hemodynamic effects of brain natriuretic peptide in patients with pulmonary arterial hypertension. Chest. 2005;128(6Suppl):618S–619S. PMID: 16373873. doi:10.1378/chest.128.6_suppl.618S-a
310. Klinger JR, Thaker S, Houtchens J, et al. Pulmonary hemodynamic responses to brain natriuretic peptide and sildenafil in patients with pulmonary arterial hypertension. Chest. 2006;129(2):417–425. PMID: 16478861. doi:10.1378/chest.129.2.417
311. Klinger JR, Warburton RR, Pietras L, Hill NS. Brain natriuretic peptide inhibits hypoxic pulmonary hypertension in rats. J Appl Physiol. 1998;84(5):1646–1652. PMID: 9572812. doi:10.1152/jappl.1998.84.5.1646
312. Jin H, Yang RH, Chen YF, Jackson RM, Oparil S. Atrial natriuretic peptide attenuates the development of pulmonary hypertension in rats adapted to chronic hypoxia. J Clin Invest. 1990;85(1):115–120. PMID: 2136863. doi:10.1172/JCI114400.
313. Kapoun AM, Liang F, O’Young G, et al. B-type natriuretic peptide exerts broad functional opposition to transforming growth factor-beta in primary human cardiac fibroblasts: fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ Res. 2004;94(4):453–461. PMID: 14726474. doi:10.1161/01.RES.0000117070.86556.9F
314. VMAC Investigators. (Vasodilatation in the Management of Acute CHF). Intravenous nesiritide vs. nitroglycerin for treatment of congestive heart failure. A randomized controlled trial. JAMA. 2002;287(12):1531–1540. PMID: 11911755. doi:10.1001/jama.287.12.1531
315. Nesiritide Study Group. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heartfailure. N Engl J Med. 2000;343(4):246–253. PMID: 10911006. doi:10.1056/NEJM200007273430403
316. Mills RM, LeJemtel TH, Horton DP, et al. Sustained hemodynamic effects of an infusion of nesiritide (human B-type natriuretic peptide) in heart failure: a randomized, double-blind, placebo-controlled clinical trial. Natrecor Study Group. J Am Coll Cardiol. 1999;34:155–162. PMID: 10400005. doi:10.1016/s0735-1097(99)00184-9
317. Michaels AD, Chatterjee K, De Marco T. Effects of intravenous nesiritide on pulmonary vascular hemodynamics in pulmonary hypertension. J Card Fail. 2005;11(6):425–431. PMID: 16105633. doi:10.1016/j.cardfail.2005.01.009
318. Gunaydin S, Imai Y, Takanashi T, et al. The effects of vasoactive intestinal peptide on monocrotaline induced pulmonary hypertensive rabbits following cardiopulmonary bypass: a comparative study with isoproterenol and nitroglycerine. Cardiovasc Surg. 2002;10(2):138–145. PMID: 11888743. doi:10.1016/s0967-2109(01)00126-0
319. Söderman C, Eriksson LS, Juhlin-Dannfelt A, Lundberg JM, Broman L, Holmgren A. Effect of vasoactive intestinal polypeptide (VIP) on pulmonary ventilation-perfusion relationships and central haemodynamics in healthy subjects. Clin Physiol. 1993;13(6):677–685. PMID: 8119061. doi:10.1111/j.1475-097x.1993.tb00482.x
320. Maruno K, Absood A, Said SI. VIP inhibits basal and histamine-stimulated proliferation of human airway smooth muscle cells. Am J Physiol. 1995;268(6 Pt 1):L1047–51. PMID: 7541947. doi:10.1152/ajplung.1995.268.6.L1047
321. Cox CP, Linden J, Said SI. VIP elevates platelet cyclic AMP (cAMP) levels and inhibits in vitro platelet activation induced by platelet-activating factor (PAF). Peptides. 1984;5(2):325–328. PMID: 6206481. doi:10.1016/0196-9781(84)90228-6
322. Petkov V, Mosgoeller W, Ziesche R, et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest. 2003;111(9):1339–1346. PMID: 12727925 PMID: 12727925. doi:10.1172/JCI17500
323. Galie N, Boonstra A, Ewert R. Effects of inhaled aviptadil (Vasoactive Intestinal Peptide) in patients with pulmonary arterial hypertension (PAH). Am J Resp Crit Care Med. 2010;181:A2516.
324. Nicholls MG, Lainchbury JG, Lewis LK, et al. Bioactivity of adrenomedullin and proadrenomedullin N-terminal 20 peptide in man. Peptides. 2001;22(11):1745–1752. PMID: 11754960. doi:10.1016/s0196-9781(01)00508-3
325. Kakishita M, Nishikimi T, Okano Y, et al. Increased plasma levels of adrenomedullin in patients with pulmonary hypertension. Clin Sci (Lond). 1999;96(1):33–39. PMID: 9857104. doi:10.1042/CS19980070
326. Shimukubo T, Sakata J, Kitamura K, Kangawa K, Matsuo H, Eto T. Augmented adrenomedullin concentrations in right ventricle and plasma of experimental pulmonary hypertension. Life Sci. 1995;57(19):1771–1779. PMID: 7475919. doi:10.1016/0024-3205(95)02155-c
327. Yoshibayashi M, Kamiya T, Kitamura K, et al. Plasma levels of adrenomedullin in primary and secondary pulmonary hypertension in patients <20 years of age. Am J Cardiol. 1997;79(11):1556–1558. PMID: 9185657. doi:10.1016/s0002-9149(97)00195-1
328. Nishikimi T, Nagata S, Sasaki T, et al. Plasma concentrations of adrenomedullin correlate with the extent of pulmonary hypertension in patients with mitral stenosis. Heart. 1997;78(4):390–395. PMID: 9404257. doi:10.1136/hrt.78.4.390
329. Yoshihara F, Nishikimi T, Horio T, et al. Eur Chronic infusion of adrenomedullin reduces pulmonary hypertension and lessens right ventricular hypertrophy in rats administered monocrotaline. J Pharmacol. 1998;355(1):33–39. PMID: 9754936. doi:10.1016/s0014-2999(98)00475-0
330. Nagaya N, Nishikimi T, Uematsu M, et al. Haemodynamic and hormonal effects of adrenomedullin in patients with pulmonary hypertension. Heart. 2000;84(6):653–658. PMID: 11083748. doi:10.1136/heart.84.6.653
331. Nagaya N, Miyatake K, Kyotani S, et al. Pulmonary vasodilator response to adrenomedullin in patients with pulmonary hypertension. Hypertens Res. 2003;26(Suppl):S141–6. PMID: 12630825. doi:10.1291/hypres.26.s141
332. Nagaya N, Kyotani S, Uematsu M, et al. Effects of adrenomedullin inhalation on hemodynamics and exercise capacity in patients with idiopathic pulmonary arterial hypertension. Circulation. 2004;109(3):351–356. PMID: 14718403. doi:10.1161/01.CIR.0000109493.05849.14
333. Giordano N, Papakostas P, Lucani B, et al. Serum relaxin in systemic sclerosis. J Rheumatol. 2005;32:2164–2166. PMID: 16265695.
334. Tozzi CA, Poiani GJ, McHugh NA, et al. Recombinant human relaxin reduces hypoxic pulmonary hypertension in the rat. Pulm Pharmacol Ther. 2005;18:346–353. PMID: 15939313. doi:10.1016/j.pupt.2005.01.003
335. Mazurek JA, Horne BD, Kelesidis I, et al. Relaxin levels in pulmonary hypertension: a comparison between pulmonary arterial hypertension and diastolic heart failure-induced pulmonary hypertension. J Heart Lung Transplant. 2013;32(3):371–374. PMID: 23273743. doi:10.1016/j.healun.2012.11.030
336. Lee SL, Simon AR, Wang WW, Fanburg BL. H(2)O(2) signals 5-HT-induced ERK MAP kinase activation and mitogenesis of smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2001;281(3):L646–52. doi:10.1152/ajplung.2001.281.3.L646
337. Eddahibi S, Guignabert C, Barlier-Mur AM, et al. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: critical role for serotonin-induced smooth muscle hyperplasia. Circulation. 2006;113(15):1857–1864. PMID: 16606791. doi:10.1161/CIRCULATIONAHA.105.591321
338. Morrell NW, Archer SL, Stenmark KR, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12Suppl S):13S–24S. PMID: 15194174. doi:10.1016/j.jacc.2004.02.029
339. MacLean MR, Herve P, Eddahibi S, Adnot S. 5-hydroxytryptamine and the pulmonary circulation: receptors, transporters and relevance to pulmonary arterial hypertension. Br J Pharmacol. 2000;131(2):161–168. doi:10.1038/sj.bjp.0703570
340. Morecroft I, Heeley RP, Prentice HM, Kirk A, MacLean MR. 5-hydroxytryptamine receptors mediating contraction in human small muscular pulmonary arteries: importance of the 5-HT1B receptor. Br J Pharmacol. 1999;128(3):730–734. PMID: 10516655. doi:10.1038/sj.bjp.0702841
341. Wilson SM, Mason HS, Ng LC, et al. Role of basal extracellular Ca2+ entry during 5-HT-induced vasoconstriction of canine pulmonary arteries. Br J Pharmacol. 2005;144(2):252–264. PMID: 15655514. doi:10.1038/sj.bjp.0706077
342. Liu Y, Fanburg BL. Serotonin-induced growth of pulmonary artery smooth muscle requires activation of phosphatidylinositol 3-kinase/serine-threonine protein kinase B/mammalian target of rapamycin/p70 ribosomal S6 kinase 1. Am J Respir Cell Mol Biol. 2006;34(2):182–191. PMID: 16195541. doi:10.1165/rcmb.2005-0163OC
343. Mair KM, MacLean MR, Morecroft I, Dempsie Y, Palmer TM. Novel interactions between the 5-HT transporter, 5-HT1B receptors and Rho kinase in vivo and in pulmonary fibroblasts. Br J Pharmacol. 2008;155(4):606–616. PMID: 18695640. doi:10.1038/bjp.2008.310
344. Hervé P, Launay JM, Scrobohaci ML, et al. Increased plasma serotonin in primary pulmonary hypertension. Am J Med. 1995;99(3):249–254. PMID: 7653484. doi:10.1016/s0002-9343(99)80156-9
345. Abenhaim L, Moride Y, Brenot F, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;335(9):609–616. PMID: 8692238. doi:10.1056/NEJM199608293350901
346. Eddahibi S, Humbert M, Fadel E, et al. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest. 2001;108(8):1141–1150. PMID: 11602621. doi:10.1172/JCI12805
347. Fanburg BL, Lee SL. A new role for an old molecule: serotonin as a mitogen. Am J Physiol. 1997;272(5 Pt 1):L795–806. PMID: 9176241. doi:10.1152/ajplung.1997.272.5.L795
348. Kren V, Eich E, Pertz HH. Pergolide, terguride and N,N’-spacer-linked oligomers of both interact with 5-HT2A receptors of rat tail artery. Physiol Res. 2004;53(1):35–43. PMID: 14984312.
349. Millan MJ, Maiofiss L, Cussac D, et al. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. J Pharmacol Exp Ther. 2002;303(2):791–804. PMID: 12388666. doi:10.1124/jpet.102.039867
350. Jähnichen S, Horowski R, Pertz HH. Agonism at 5-HT2B receptors is not a class effect of the ergolines. Eur J Pharmacol. 2005;513(3):225–228. PMID: 15862804. doi:10.1016/j.ejphar.2005.03.010
351. Dumitrascu R, Kulcke C, Königshoff M, et al. Terguride ameliorates monocrotaline-induced pulmonary hypertension in rats. Eur Respir J. 2011;37(5):1104–1118. PMID: 20947677. doi:10.1183/09031936.00126010
352. Ghofrani HA, Al-Hiti H, Vonk-Noordegraaf A, et al. Proof-of-concept study to investigate the efficacy, hemodynamics and tolerability of terguride vs. placebo in subjects with pulmonary arterial hypertension: results of A Double Blind, Randomised, Prospective Phase IIa Study. Am J Resp Crit Care Med. 2012;185:A2496.
353. Lahm T, Tuder RM, Petrache I. Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014;307(1):L7–26. PMID: 24816487. doi:10.1152/ajplung.00337.2013
354. Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216–223. PMID: 3605900. doi:10.7326/0003-4819-107-2-216
355. Frost AE, Badesch DB, Barst RJ, et al. The changing picture of patients with pulmonary arterial hypertension in the United States: how reveal differs from historic and non‐us contemporary registries. Chest. 2011;139:128–137. PMID: 20558556. doi:10.1378/chest.10-0075
356. McGoon MD, Benza RL, Escribano‐Subias P, et al. Pulmonary arterial hypertension: epidemiology and registries. J Am Coll Cardiol. 2013;62:D51–D59. PMID: 24355642. doi:10.1016/j.jacc.2013.10.023
357. Prins KW, Thenappan T. World health organization group i pulmonary hypertension: epidemiology and pathophysiology. Cardiol Clin. 2016;34:363–374. PMID: 27443134. doi:10.1016/j.ccl.2016.04.001
358. Chen X, Austin ED, Talati M, et al. Oestrogen inhibition reverses pulmonary arterial hypertension and associated metabolic defects. Eur Respir J. 2017;50:1602337. PMID: 28775043. doi:10.1183/13993003.02337-2016
359. Foderaro A, Ventetuolo CE. Pulmonary arterial hypertension and the sex hormone paradox. Curr Hypertens Rep. 2016;18(11):84. PMID: 27832457. doi:10.1007/s11906-016-0689-7
360. Ventetuolo CE, Baird GL, Barr RG, et al. Higher estradiol and lower dehydroepiandrosterone-sulfate levels are associated with pulmonary arterial hypertension in men. Am J Respir Crit Care Med. 2016;193:1168–1175. PMID: 26651504. doi:10.1164/rccm.201509-1785OC
361. Frump AL, Goss KN, Vayl A, et al. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol. 2015;308:L873–L890. PMID: 25713318. doi:10.1152/ajplung.00006.2015
362. Liu A, Philip J, Vinnakota KC, et al. Estrogen maintains mitochondrial content and function in the right ventricle of rats with pulmonary hypertension. Physiol Rep. 2017;5(6):e13157. PMID: 28320896. doi:10.14814/phy2.13157
363. Jacobs W, van de Veerdonk MC, Trip P, et al. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest. 2014;145(6):1230–1236. PMID: 24306900. doi:10.1378/chest.13-1291
364. Kawut SM, Al-Naamani N, Agerstrand C, et al. Determinants of right ventricular ejection fraction in pulmonary arterial hypertension. Chest. 2009;135(3):752–759. PMID: 18849396. doi:10.1378/chest.08-1758
365. Baird GL, Archer-Chicko C, Barr RG, et al. Lower DHEA-S levels predict disease and worse outcomes in post-menopausal women with idiopathic, connective tissue disease- and congenital heart disease-associated pulmonary arterial hypertension. Eur Respir J. 2018;51:1800467. PMID: 29954925. doi:10.1183/13993003.00467-2018
366. Simpson ER, Clyne C, Rubin G, et al. Aromatase—a brief overview. Annu Rev Physiol. 2002;64:93–127. PMID: 11826265. doi:10.1146/annurev.physiol.64.081601.142703
367. Burstein HJ, Griggs JJ, Prestrud AA, Temin S. American society of clinical oncology clinical practice guideline update on adjuvant endocrine therapy for women with hormone receptor‐positive breast cancer. J Oncol Pract. 2010;6:243–246. PMID: 21197188. doi:10.1200/JOP.000082
368. Mair KM, Wright AF, Duggan N, et al. Sex‐dependent influence of endogenous estrogen in pulmonary hypertension. Am J Respir Crit Care Med. 2014;190:456–467. PMID: 24956156. doi:10.1164/rccm.201403-0483OC
369. Kawut SM, Archer‐Chicko CL, DeMichele A, et al. Anastrozole in pulmonary arterial hypertension. A randomized, double‐blind, placebo‐controlled trial. Am J Respir Crit Care Med. 2017;195:360–368. PMID: 27602993. doi:10.1164/rccm.201605-1024OC
370. Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov. 2003;2:205–213. PMID: 12612646. doi:10.1038/nrd1031
371. van Empel VP, Lee J, Williams TJ, et al. Iron deficiency in patients with idiopathic pulmonary arterial hypertension. Heart Lung Circ. 2014;23:287–292. PMID: 24094431. doi:10.1016/j.hlc.2013.08.007
372. Rhodes CJ, Wharton J, Howard L, et al. Iron deficiency in pulmonary arterial hypertension: a potential therapeutic target. Eur Respir J. 2011;38:1453–1460. PMID: 21478213. doi:10.1183/09031936.00037711
373. Ruiter G, Lankhorst S, Boonstra A, et al. Iron deficiency is common in idiopathic pulmonary arterial hypertension. Eur Respir J. 2011;37:1386–1391. PMID: 20884742. doi:10.1183/09031936.00100510
374. Soon E, Treacy CM, Toshner MR, et al. Unexplained iron deficiency in idiopathic and heritable pulmonary arterial hypertension. Thorax. 2011;66(4):326–332. PMID: 21297151. doi:10.1136/thx.2010.147272
375. Krasuski RA, Hart SA, Smith B, et al. Association of anemia and long-term survival in patients with pulmonary hypertension. Int J Cardiol. 2011;150(3):291–295. PMID: 20472313. doi:10.1016/j.ijcard.2010.04.038
376. Ruiter G, Manders E, Happe CM, et al. Intravenous iron therapy in patients with idiopathic pulmonary arterial hypertension and iron deficiency. Pulm Circ. 2015;5(3):466–472. PMID: 26401247. doi:10.1086/682217
377. Viethen T, Gerhardt F, Dumitrescu D, et al. Ferric carboxymaltose improves exercise capacity and quality of life in patients with pulmonary arterial hypertension and iron deficiency: a pilot study. Int J Cardiol. 2014;175:233–239. PMID: 24880481. doi:10.1016/j.ijcard.2014.04.233
378. Robinson JC, Graham BB, Rouault TC, et al. The crossroads of iron with hypoxia and cellular metabolism. Implications in the pathobiology of pulmonary hypertension. Am J Respir Cell Mol Biol. 2014;51(6):721–729. PMID: 24988529. doi:10.1165/rcmb.2014-0021TR
379. Callejo M, Mondejar-Parreño G, Esquivel-Ruiz S, et al. Total, bioavailable, and free vitamin D levels and their prognostic value in pulmonary arterial hypertension. J Clin Med. 2020;9(2):448. PMID: 32041235. doi:10.3390/jcm9020448
380. Yu H, Xu M, Dong Y, Liu J. (OH)2D3 attenuates pulmonary arterial hypertension via microRNA-204 mediated Tgfbr2/Smad signaling. Exp Cell Res. 2018;362(2):311–323. PMID: 29196166. doi:10.1016/j.yexcr.2017.11.032.
381. Mirdamadi A, Moshkdar P. Benefits from the correction of vitamin D deficiency in patients with pulmonary hypertension. Caspian J Intern Med. 2016;7(4):253–259. PMID: 27999642.
382. Yeh ET, Tong AT, Lenihan DJ, et al. Cardiovascular complications of cancer therapy: diagnosis, pathogenesis, and management. Circulation. 2004;109:3122–3131. PMID: 15226229. doi:10.1161/01.CIR.0000133187.74800.B9
383. Pascal JM. The comings and goings of PARP-1 in response to DNA damage. DNA Repair (Amst). 2018;71:177–182. PMID: 30177435. doi:10.1016/j.dnarep.2018.08.022
384. Wheelhouse NM, Chan YS, Gillies SE, et al. TNF-alpha induced DNA damage in primary murine hepatocytes. Int J Mol Med. 2003;12:889–894. PMID: 14612962.
385. Fehsel K, Kolb-Bachofen V, Kolb H. Analysis of TNF alpha-induced DNA strand breaks at the single cell level. Am J Pathol. 1991;139:251–254. PMID: 1867316.
386. Gorenne I, Kumar S, Gray K, et al. Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation. 2013;127:386–396. PMID: 23224247. doi:10.1161/CIRCULATIONAHA.112.124404
387. Meloche J, Pflieger A, Vaillancourt M, et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation. 2014;129:786–797. PMID: 24270264. doi:10.1161/CIRCULATIONAHA.113.006167
388. Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2014;117:421–426. PMID: 15137936. doi:10.1016/s0092-8674(04)00452-0
389. Sengupta A, Molkentin JD, Paik JH, et al. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J Biol Chem. 2011;286(9):7468–7478. doi:10.1074/jbc.M110.179242
390. Savai R, Al‐Tamari HM, Sedding D, et al. Pro‐proliferative and inflammatory signaling converge on FoXO1 transcription factor in pulmonary hypertension. Nat Med. 2014;20:1289–1300. PMID: 25344740. doi:10.1038/nm.3695
391. Deng C, Zhong Z, Wu D, et al. Role of FoxO1 and apoptosis in pulmonary vascular remolding in a rat model of chronic thromboembolic pulmonary hypertension. Sci Rep. 2017;7(1):2270. PMID: 28536427. doi:10.1038/s41598-017-02007-5
392. Nehate C, Jain S, Saneja A, et al. Paclitaxel formulations: challenges and novel delivery options. Curr Drug Deliv. 2014;11(6):666–686. PMID: 24909147. doi:10.2174/1567201811666140609154949
393. Veldhoen RA, Banman SL, Hemmerling DR, et al. The chemotherapeutic agent paclitaxel inhibits autophagy through two distinct mechanisms that regulate apoptosis. Oncogene. 2013;32(6):736–746. PMID: 22430212. doi:10.1038/onc.2012.92
394. Goto T, Takano M, Hirata J, Tsuda H. The involvement of FOXO1 in cytotoxic stress and drug-resistance induced by paclitaxel in ovarian cancers. Br J Cancer. 2008;98(6):1068–1075. PMID: 18319717. doi:10.1038/sj.bjc.6604279
395. Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152(6):1237–1251. PMID: 23498934. doi:10.1016/j.cell.2013.02.014
396. Yin Y, Wu X, Yang Z, et al. The potential efficacy of R8-modified paclitaxel loaded liposomes on pulmonary arterial hypertension. Pharm Res. 2013;30:2050–2062. PMID: 23756757. doi:10.1007/s11095-013-1058-8
397. Galié N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res. 2004;61(2):227–237. PMID: 14736539. doi:10.1016/j.cardiores.2003.11.026
398. Dai Y, Chen X, Song X, et al. Immunotherapy of endothelin-1 receptor Type A for pulmonary arterial hypertension. J Am Coll Cardiol. 2019;73(20):2567–2580. PMID: 31118151. doi:10.1016/j.jacc.2019.02.067
399. Fessel JP, Loyd JE, Austin ED. The genetics of pulmonary arterial hypertension in the postBMPR2 era. Pulm Circ. 2011;1(3):305–319. doi:10.4103/2045-8932.87293
400. Kaji EH, Leiden JM. Gene and stem cell therapies. JAMA. 2001;285(5):545–550. doi:10.1001/jama.285.5.545
401. Reynolds AM, Xia W, Holmes MD, et al. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonaryhypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1182–92. doi:10.1152/ajplung.00020.2006
402. Lee CS, Bishop ES, Zhang R, et al. Adenovirus-mediated gene delivery: potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017;4(2):43–63. doi:10.1016/j.gendis.2017.04.001
403. Bisserier M, Pradhan N, Lahouaria H. Current and emerging therapeutic approaches to pulmonary hypertension. Rev Cardiovasc Med. 2020;21(2):163–179. doi:10.31083/j.rcm.2020.02.597
404. Lee R, Feinbaum R, Ambros V. A short history of a short RNA. Cell. 2004;116(Suppl):S89–S92,1 p. following S96. doi:10.1016/s0092-8674(04)00035-2
405. Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of posttranscriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9(2):102–114. doi:10.1038/nrg2290
406. Giannakakis A, Sandaltzopoulos R, Greshock J, et al. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol Ther. 2008;7(2):255–264. PMID: 18059191. doi:10.4161/cbt.7.2.5297
407. Fasanaro P, Greco S, Lorenzi M, et al. An integrated approach for experimental target identification of hypoxia-induced miR-210. J Biol Chem. 2009;284(50):35134–35143. PMID: 19826008. doi:10.1074/jbc.M109.052779
408. Li S, Ran Y, Zhang D, et al. MicroRNA-138 plays a role in hypoxic pulmonary vascular remodelling by targeting Mst1. Biochem J. 2013;452(2):281–291. PMID: 23485012. doi:10.1042/BJ20120680
409. Caruso P, Dempsie Y, Stevens HC, et al. A role for miR-145 in pulmonary arterial hypertension: evidence from mouse models and patient samples. Circ Res. 2012;111(3):290–300. PMID: 22715469. doi:10.1161/CIRCRESAHA.112.267591
410. Gou D, Ramchandran R, Peng X, et al. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am J Physiol Lung Cell Mol Physiol. 2012;303(8):L682–91. PMID: 22886504. doi:10.1152/ajplung.00344.2011
411. Kang K, Peng X, Zhang X, et al. MicroRNA-124 suppresses the transactivation of nuclear factor of activated T cells by targeting multiple genes and inhibits the proliferation of pulmonary artery smooth muscle cells. J Biol Chem. 2013;288(35):25414–25427. PMID: 23853098. doi:10.1074/jbc.M113.460287
412. Courboulin A, Paulin R, Giguère NJ, et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med. 2011;208(3):535–548. PMID: 21321078. doi:10.1084/jem.20101812
413. Jalali S, Ramanathan GK, Parthasarathy PT, et al. Mir-206 regulates pulmonary artery smooth muscle cell proliferation and differentiation. PLoS One. 2012;7(10):e46808. doi:10.1371/journal.pone.0046808
414. Dupont C, Armant DR, Brenner CA. Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med. 2009;27(5):351–357. PMID: 19711245. doi:10.1055/s-0029-1237423
415. Hambach L, Ling KW, Pool J, et al. Hypomethylating drugs convert HA-1 negative solid tumors into targets for stem cell based immunotherapy. Blood. 2009;113(12):2715–2722. PMID: 19096014. doi:10.1182/blood-2008-05-158956
416. Chueh AC, Tse JW, Tögel L, Mariadason JM. Mechanisms of histone deacetylase inhibitor-regulated gene expression in cancer cells. Antioxid Redox Signal. 2015;23(1):66–84. PMC 4492771. PMID 24512308. doi:10.1089/ars.2014.5863
417. Zhao L, Chen CN, Hajji N, et al. Histone deacetylation inhibition in pulmonary hypertension: therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation. 2012;126(4):455–467. PMID: 22711276. doi:10.1161/CIRCULATIONAHA.112.103176
418. Kim J, Kang Y, Kojima Y, et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med. 2013;19(1):74–82. PMID: 23263626. doi:10.1038/nm.3040
419. Kim J, Hwangbo C, Hu X, et al. Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation. 2015;131(2):190–199. PMID: 25336633. doi:10.1161/CIRCULATIONAHA.114.013339
420. Borck PC, Guo LW, Plutzky J. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ Res. 2020;126(9):1190–1208. PMID: 32324495. doi:10.1161/CIRCRESAHA.120.315929
421. Meloche J, Potus F, Vaillancourt M, et al. Bromodomain-containing protein 4: the epigenetic origin of pulmonary arterial hypertension. Circ Res. 2015;117(6):525–535. PMID: 26224795. doi:10.1161/CIRCRESAHA.115.307004
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.