")
Back to Journals » Cancer Management and Research » Volume 6
Novel drugs that target the estrogen-related receptor alpha: their therapeutic potential in breast cancer
Authors May F
Received 17 January 2014
Accepted for publication 3 March 2014
Published 23 May 2014 Volume 2014:6 Pages 225—252
DOI https://doi.org/10.2147/CMAR.S35024
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Felicity EB May
Northern Institute for Cancer Research and Department of Pathology, Faculty of Medical Sciences, University of Newcastle upon Tyne, Newcastle upon Tyne, UK
Abstract: The incidence of breast cancer continues to rise: 1.7 million women were diagnosed with and 521,000 women died from breast cancer in 2012. This review considers first current treatment options: surgery; radiotherapy; and systemic endocrine, anti-biological, and cytotoxic therapies. Clinical management includes prevention, early detection by screening, treatment with curative intent, management of chronic disease, and palliative control of advanced breast cancer. Next, the potential of novel drugs that target DNA repair, growth factor dependence, intracellular and intercellular signal transduction, and cell cycle are considered. Estrogen-related receptor alpha has attracted attention as a therapeutic target in triple-negative breast cancers with de novo resistance to, and in breast cancers with acquired resistance to, endocrine therapies such as antiestrogens and aromatase inhibitors. Estrogen-related receptor alpha is an orphan receptor and transcription factor. Its activity is regulated by coregulator proteins and posttranslational modification. It is an energy sensor that controls adaptation to energy demand and may facilitate glycolytic metabolism and mitochondrial oxidative respiration in breast cancer cells. Estrogen-related receptor alpha increases breast cancer cell migration, proliferation, and tumor development. It is expressed at high levels in estrogen receptor-negative tumors, and is proposed to activate estrogen-responsive genes in endocrine-resistant tumors. The structures and functions of the ligand-binding domains of estrogen receptor alpha and estrogen-related receptor alpha, their ability to bind estrogens, phytoestrogens, and synthetic ligands, and the effects of ligand agonists, antagonists, and inverse agonists on biological activity, are evaluated. Synthetic ligands of estrogen-related receptor alpha have activity in preclinical models of metabolic disorders, diabetes, osteoporosis, and oncology. The clinical settings in which these novel drugs might have utility in the management of advanced breast cancer, and biomarkers for stratification of patients likely to benefit, are discussed. Finally, the potential side effects of the novel drugs on metabolism, osteoporosis, osteo-metastasis, and cachexia are considered.
Keywords: estrogen receptor alpha, inverse or reverse agonist, coactivators, ligand-binding domain, TFF1, HER2, antiestrogen-resistance
Introduction to breast cancer
Incidence, mortality, and survival
Breast cancer is the most common cancer in women and the greatest cause of death from cancer in females. In 2012, 1.7 million women were diagnosed with breast cancer worldwide; men have a 100-fold lower risk of developing breast cancer than women. Across the world, over 522,000 women died as a direct result of their breast cancer (GLOBOCAN 2012 IACR).1
Breast cancer is considered very much a cancer of the western developed world; countries of Western Europe and North America, as well as Australia and New Zealand, report the highest rates of new diagnoses (Figure 1A). The incidence is sevenfold higher in richer than in more deprived nations. The prevailing view is that lifestyle drives this higher rate of diagnosis; second-generation migrants from low-incidence countries develop breast cancer with the incidence of their host nation. Belgium has the ignominious claim to the highest incidence; it has an age-standardized rate of diagnosis of over 110 cases per 100,000 women per annum (Figure 1B). Of the other 12 top countries, nine are Western European, but the Bahamas, Barbados, and the United States of America also make the rank. The 12 lowest-incidence countries are mainly in sub-Saharan Africa, South Asia, and the Far East, and are all characterized by poverty.
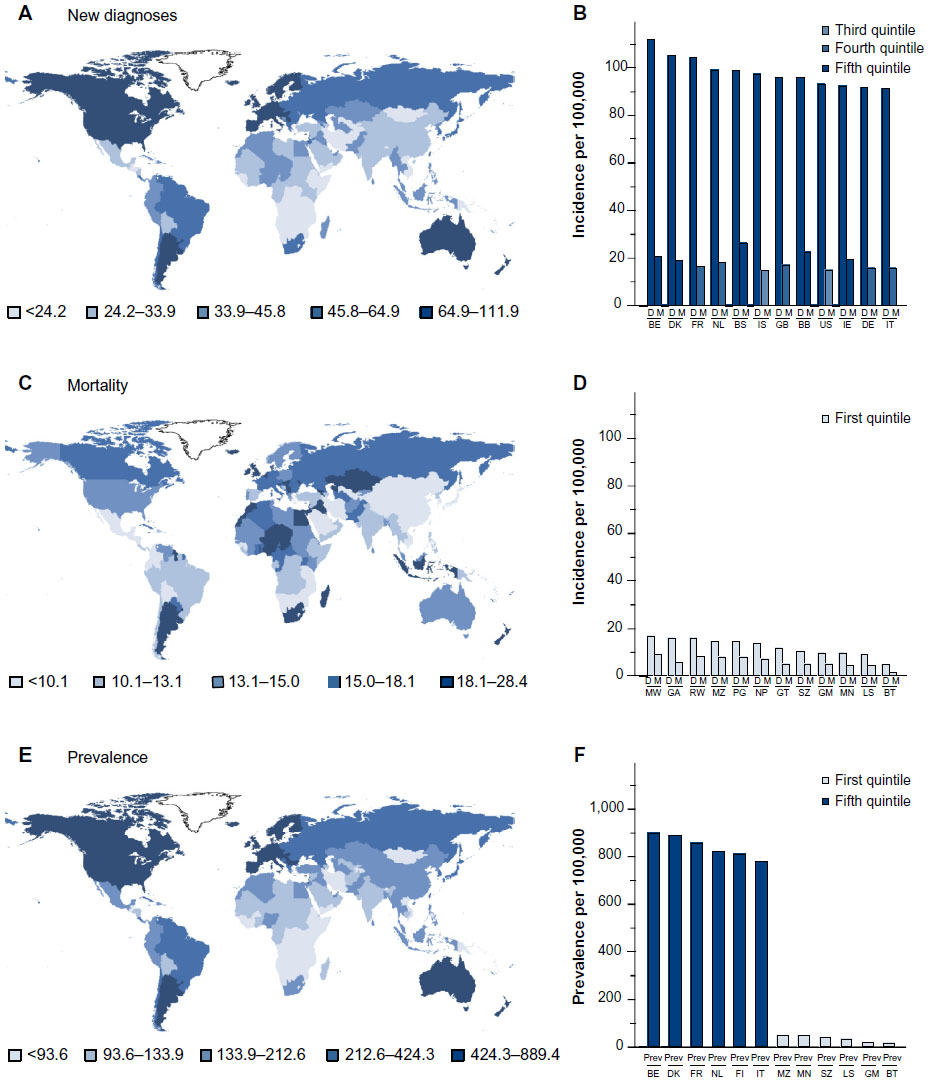 | Figure 1 Worldwide incidence of breast cancer. |
The distribution of countries with the highest age-standardized mortality per 100,000 women throughout the world is very different (Figure 1C). Belgium has the highest mortality followed by the Republic of Ireland of the 12 countries with the highest rate of diagnosis, but they are outranked by poorer countries: Fiji, the Bahamas, Nigeria, and Pakistan. Mortality is relatively low in most of the lowest-incidence countries, but the likelihood that an individual will die from their breast cancer is much higher in low-incidence countries than in high-incidence countries (Figure 1B and D). For instance, a woman diagnosed with breast cancer in Belgium, the UK, France, or Iceland has an 82%, 82%, 84%, or 85% chance, respectively, of surviving her cancer and of dying of other causes. A woman diagnosed with breast cancer in Nepal, Mozambique, Papua New Guinea, or Comoros has only a 47%, 45%, 45%, or 44% chance, respectively, of surviving her disease. The reasons for the differential survival are multiple and include cultural influences, stage of presentation, and standards of health care.
Preferred sites of metastasis
Patients are unlikely to die of primary early breast cancer; the breast is a nonessential organ. Patients die from metastases to essential organs. Metastasis by localized spread is to the overlying skin and underlying muscles and ribs. Distant metastasis via the lymphatic drainage, vascular system, or neural network allows for the formation of secondary tumors in preferred organs notably bone, lung, liver, and brain.2 It is these secondary tumors that cause most of the terrible suffering and pain associated with advanced breast cancer. The success of antibody therapies in some patients has resulted in an increase in deaths from brain metastases because the drugs that target, successfully the cancer cells throughout the rest of the body are unable to cross the blood–brain barrier and the malignant cells within the brain are able to thrive.3 Intrathecal administration of the antibodies provides an attractive solution.4
Risk factors
The strongest risk factor for breast cancer is inheritance of an inactivating mutation in one of the familial breast cancer genes: BRCA1, BRCA2, CHEK2, p53, and ATM, which together contribute to around 5% of breast cancer cases.5 The next highest risk factor is age: three-quarters of breast cancer cases present in postmenopausal women, less than 5% in women less than 40 years of age and breast cancer is rare in women less than 30 years of age.5 Nevertheless, large numbers of premenopausal women develop breast cancer and it is the highest cause of death in women between 34 and 54 years of age. Exposure to estrogens explains the majority of the other known risk factors for breast cancer. Early menarche, late menopause, nulliparity, absence of lactation, hormone replacement therapy, oral contraception, and treatment with the synthetic estrogen diethylstilbestrol are all associated with an increased risk of breast cancer. High circulating insulin-like growth factor-1 (IGF-1) and low serum insulin-like growth factor binding protein 3, as well as alcohol intake, are thought to increase risk, while exercise is thought to reduce risk.6–8
Obesity, the scourge of our times, is associated with a 1.6-fold increased risk of breast cancer in postmenopausal women.6 Given the high basal incidence of postmenopausal breast cancer and the ongoing surge in obesity, predictions are that rates will rise at an alarming rate in the coming decades. The reasons why people with more body fat are more likely to develop breast cancer are debated, but it is incontrovertible that adipose cells express aromatase, which converts androgens into estrogens, and 17β-hydroxysteroid dehydrogenase, which converts estrone into the more active 17β-estradiol. As adipose tissue accumulates in the body, local and circulating concentrations of estrone and 17β-estradiol increase.6 Exposure to high local and circulating concentrations of IGFs secreted by adipose tissue may contribute to the increased risk of breast cancer in obese individuals.6,9,10 In addition, high concentrations of circulating insulin in obese individuals who develop hyperinsulinemia and metabolic syndrome may contribute.6
Recent rise
A recent study reported a huge rise in presentation of young women with advanced, estrogen receptor-positive breast cancer.11 Rates of presentation had risen 2% per year for women less than 40 years of age and 3% per year for women less than 34 years of age. No explanation was proffered for the increases observed, but they are cause for alarm and may be related to changes in lifestyle factors such as obesity.
Brief overview of current treatment options and emerging therapies
Efforts to counteract or lessen the impact of breast cancer can be divided into five main phases: the “holy grail” of prevention, earlier detection, cure, management as a chronic disease, and palliation of symptoms (Figure 2).
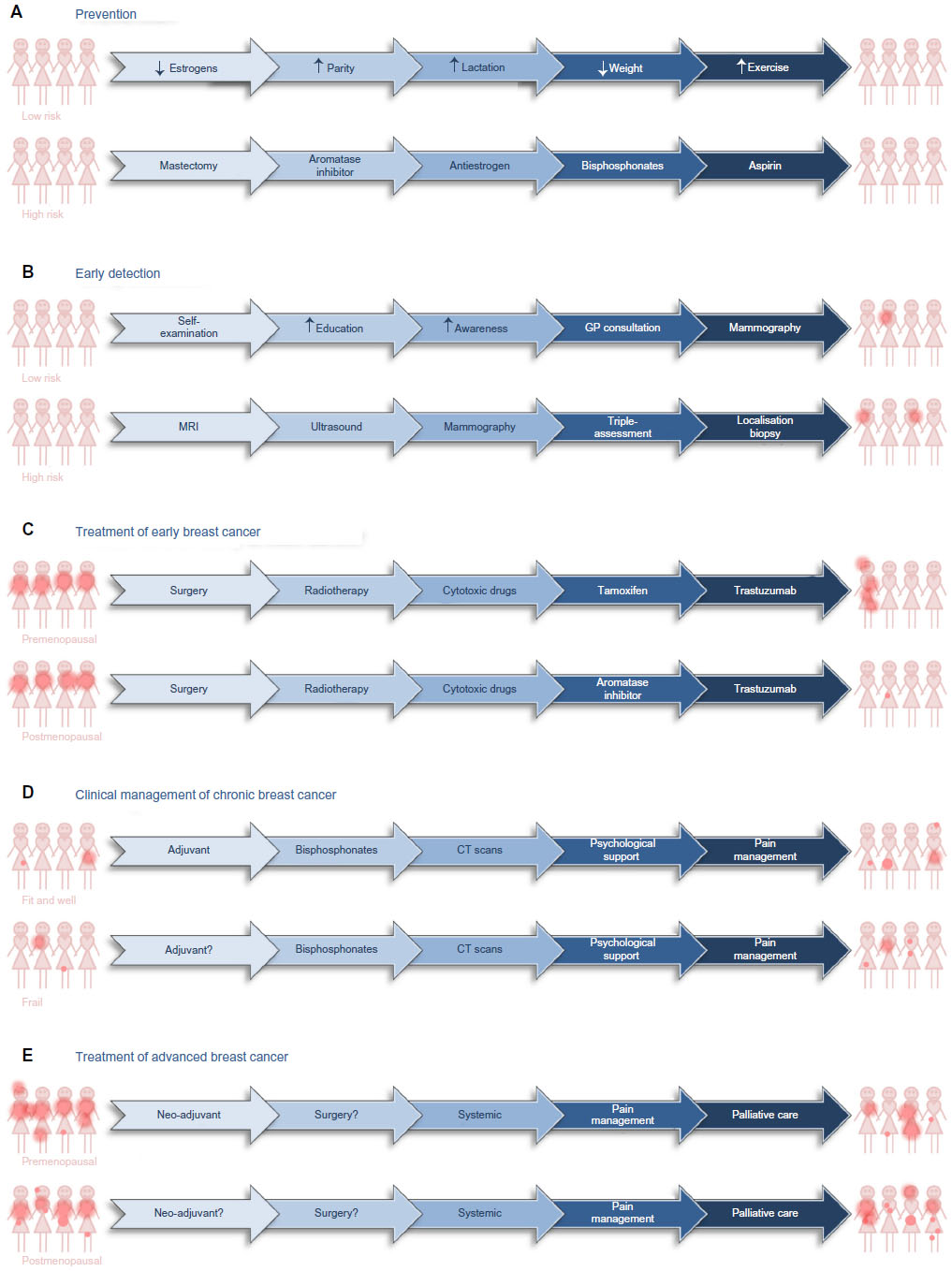 | Figure 2 Treatment options for prevention, early detection, and clinical management of patients diagnosed with breast cancer. |
Prevention
Prevention of any cancer is the ultimate aim (Figure 2A). For individuals who have an inherited predisposition to develop breast cancer because they have inherited a mutated copy of one of the susceptibility genes, radical intervention is appropriate. Surgical removal of the susceptible breast tissue at an early age is one option. Alternatively, individuals may receive long-term systemic therapy with antiestrogens, an aromatase inhibitor, aspirin, bisphosphonates, or metformin, all of which have proven ability to reduce the development of breast cancer in these women.12,13
For others, education to alter lifestyle choices that predispose towards cancer is paramount. Reduction in obesity could have a huge impact on the number of people who will develop breast cancer. Reduction in, restricted duration of, or altered composition of medications such as oral contraceptives and hormone replacement therapy to reduce the length of time that breast tissue is exposed to estrogens and the concentration of estrogens to which it is exposed has the potential to reduce the incidence of breast cancer. General physical and mental well-being, exercise, and reduced alcohol intake may reduce risk.7,8,14
Early detection
If breast cancer cannot be prevented, the next most effective strategy is early detection; the rationale being that a cancer detected early is a curable cancer. Screening is most cost effective in individuals at high risk of developing cancer (Figure 2B). Women in the Western world who have inherited a mutated copy of one of the genes that predisposes to breast cancer are offered annual magnetic resonance imaging from an early age. Magnetic resonance imaging is more effective than mammography in younger breast tissue. Breast tissue in younger women tends to be denser than breast tissue in older women, because breast tissue in older women is more likely to contain a higher proportion of adipose tissue. Dense breast tissue is visible on a mammogram and difficult to distinguish from a breast cancer. Women in the Western world who have been exposed to high levels of therapeutic or industrial radiation are also screened from an early age. It is debatable whether clinically obese individuals should be screened earlier than the general population.
In the low-risk population, younger women tend to rely on self-awareness and self-examination, which are of questionable benefit. Older women are often offered mammographic screening. Three-yearly screening is offered now as standard in the UK for women between 47 and 73 years; in New Zealand for women between 45 and 69 years, and, in Denmark for women between 50 and 69 years of age. Detection of a potential abnormality is followed by triple assessment: further mammographic or ultrasound imaging; clinical examination; and cytological or histological examination of a fine-needle aspirate, core biopsy, or vacuum-assisted mammotome biopsy. The benefits of universal breast screening remain controversial.15,16 Some argue that more women should be screened, while others contend that the lives saved are at an unacceptable human and financial cost. Current estimates suggest that a cancer will be detected in 6.81%, a life saved in 0.43%, and a cancer detected and treated that never would have caused any morbidity in 1.29% of women screened.16 Baum contends that the benefit of universal screening in terms of reduction in death from breast cancer is outweighed by cardiopulmonary and other adverse effects of treatment.17 Others argue that for fit, healthy, slim women, the risk of radiation outweighs the benefits of potential early detection.18
Treatment of early breast cancer
Breast cancers that are detected early may be treated with curative intent (Figure 2C). Research in the 1980s confirmed that wide local excision accompanied by appropriate lymph node clearance was as effective as a simple or radical mastectomy.19,20 More recently, detection of and histological evaluation of the sentinel node has reduced the number of patients who undergo unnecessary lymph node clearance with associated resultant debilitating lymphedema and neuropathic pain. As a result, surgeons now cure breast cancer with much lower associated morbidity than previously.
In contrast to many other major solid tumors, such as lung, gastric, or pancreatic adenocarcinomas, patients with breast cancer often relapse with disseminated disease over 10 years after their initial diagnosis. For this reason, and because of its proven benefit, almost all women diagnosed with early breast cancer will be offered some form of adjuvant therapy, which is therapy given after surgical resection in the absence of demonstrable residual disease. Adjuvant therapy may be radiotherapy to the breast and axilla or systemic cytotoxic, endocrine, or anti-human epidermal growth factor receptor 2 (HER2) therapy. Interestingly, obese patients or patients who gain weight during adjuvant therapy are less likely to benefit.21 A disadvantage of adjuvant treatment is that it is not known which patients will or will not benefit from their treatment because they do not have measurable disease. A consequence is that outwith clinical trials, patients who experience severe side effects from their drugs when they do not feel ill from their potential occult disease are less likely to comply. It is estimated that over 40% of women prescribed adjuvant endocrine therapy outside of clinical trials do not comply.22,23 There is a clear need to develop strategies with which to identify women who do require and who will benefit from different adjuvant therapies.
Management of chronic breast cancer
The combination of early detection, improved clinical management, and general increased longevity has resulted in there being over 6,250,000 survivors of breast cancer worldwide whose breast cancer was diagnosed within the past 5 years (GLOBOCAN, 2012;1 Figure 1E). The worldwide distributions of age-standardized incidence of diagnoses and of prevalence of breast cancer survivors are similar, because countries with high rates of diagnosis tend to have better survival and countries with low rates of diagnosis tend to have worse survival (Figure 1B, D and F). Many of these breast cancer survivors will die from diseases other than their breast cancer and can be considered to have chronic breast cancer. Many will receive adjuvant radiotherapy or systemic cytotoxic, endocrine, or anti-HER2 adjuvant therapy (Figure 2D). Other strategies include bisphosphonates, zoledronic acid, or alendronic acid, to strengthen the bones and reduce the formation of osteo-metastases. Some will receive adjuvant endocrine therapy for 10 years.24 These patients require long-term surveillance, which may include computed tomography scans. Some will relapse and will benefit from multiple successive interventions designed to keep their disease at bay. Patients who relapse during adjuvant therapy will have developed disease that is resistant to the drugs that they took during their adjuvant therapy. A goal for the pharmaceutical industry is to develop strategies and new drugs to reduce or delay the onset of resistance and for the medical oncologist to keep one step ahead of the evolving disease. Patients with significant comorbidities or advanced disease may be offered primary endocrine therapy. Some of these women will benefit from this systemic therapy for the remainder of their lives, but others will relapse with resistant disease and require alternative intervention or palliative care.
Treatment of advanced breast cancer
For patients who present with advanced disease or who relapse with advanced disease, therapy will be largely palliative (Figure 2E). Patients present with advanced disease for different reasons: they may have particularly aggressive disease; there may be cultural taboos; or they may live in a country with inadequate health care or be too poor to seek help. Neoadjuvant therapy may be necessary to downsize the primary tumor prior to surgery. Palliative treatment will aim to reduce tumor burden and slow the progression of the disease. Strategies can include surgery, radiotherapy, cytotoxic or anti-biological therapies, and pain management.
Current systemic therapies
Systemic therapy has important roles in breast cancer management to treat advanced disease, to treat frail patients, as adjuvant therapy for early operable disease, and as neoadjuvant therapy to downsize inoperable tumors or to downsize operable tumors and enable breast conservation. Therapy decisions are based upon the age of and menopausal status of the patient; existence of comorbidities; clinical stage; prognostic indicators such as tumor size, histological grade, and axillary lymph node involvement; and the expression of the estrogen and progesterone receptors and HER2 in the primary tumor cells.
All four of the major classes of cytotoxic drug have a role. Therapy will usually include a topoisomerase inhibitor, doxorubicin or epirubicin, combined with the antimetabolite 5-fluorouracil and the alkylating agent cyclophosphamide. A taxane to disrupt microtubule formation, docetaxel, may be given in combination or sequentially to the former drugs and has utility in treatment of anthracycline-resistant disease. Capecitabine, an alternative antimetabolite, is often given as second-line therapy and the alternative antimicrotubule agent vinorelbine as third-line therapy. Platinum complexes such as carboplatin or cisplatin may be active.
Patients whose tumor cells express estrogen or progesterone receptor are eligible for endocrine therapy. Endocrine therapy prevents ovarian synthesis of estrogens either with the luteinising hormone-releasing hormone (LHRH) analog goserelin in premenopausal women or by oophorectomy in pre- or perimenopausal women. Conversion of androgens into estrogens may be inhibited with aromatase inhibitors such as letrozole, anastrozole, or exemestane in postmenopausal women. Alternatively, the interaction between estrogens and their nuclear receptor may be inhibited with antiestrogens such as tamoxifen, raloxifene, or fulvestrant in all breast cancer patients.
Tumor cell expression of membrane-bound HER2 or amplification of the HER2 gene constitutes eligibility for anti-HER2 therapies. The antibodies trastuzumab and pertuzumab inhibit dimerization of HER2 with other members of its receptor family. Small molecule inhibitors of the activation by phosphorylation of HER2, such as lapatinib, may be used in combination with trastuzumab or in patients who develop trastuzumab-resistant disease. Recently, ado-trastuzumab emtansine (T-DM1), a drug in which trastuzumab is conjugated to the cytotoxin mertansine, has been approved for treatment of advanced breast cancer patients whose disease has progressed after treatment with trastuzumab.25,26
Emerging systemic therapies
Enormous effort has been expended to develop drugs against novel targets and many have been evaluated in clinical trials, either as single agents or in combination with established regimens. Among the most popular are agents that potentiate the DNA damage induced by cytotoxic drugs or mutations in genes that encode enzymes in the DNA damage response of malignant cells.27 Cytotoxic drugs cause substitution with nucleotide analogs, which is reversed by base excision repair; formation of DNA adducts, that are removed by nucleotide excision repair; DNA double-strand breaks that are repaired by nonhomologous end-joining; stalled replication forks due to single-strand breaks which are restored by homologous recombination; or interstrand crosslinks that are excised by interstrand crosslink repair. The rationale behind the development of drugs that inhibit DNA repair is that DNA repair antagonizes cytotoxic drugs and that inhibition of DNA repair enzymes will potentiate the drugs. Agents that inhibit DNA-dependent protein kinase are designed to potentiate drugs that induce DNA double-strand breaks and interstrand crosslinks. Inhibitors of poly(adenosine diphosphate-ribose) polymerase prevent single-strand repair and therefore induce double-strand breaks and subsequent cell death in cells in which enzymes such as BRCA1, BRCA2, or ATM (ataxia telangiectasia mutated) are defective.27
Other agents are designed to prevent the dependence of malignant cells on diverse growth factors and their receptors. At the forefront are agents that target members of the human epidermal growth factor receptor family (HER): epidermal growth factor receptor (EGFR), HER2, HER3 and HER4.28–30 The dependence of many tumor cells on the IGFs31–34 led to the development of drugs that sequester the ligands or inhibit their receptors.6,35 The fibroblast growth factor receptor has received attention. The potency of the agents is low, but promising results have been obtained in patients with amplified FGFR1 analogous to the treatment of patients with amplified HER2.36 Inhibitors of the scatter factor receptor MET are being considered.
Substantial effort has focused on two main intracellular signal-transduction pathways: PI3K–Akt–mTor37 and Ras–Raf–MAPK.38 Many specific or pan-PI3K inhibitors have entered clinical trials, as have inhibitors of Akt, mTor1, and mTor2.39 Mutations of Ras and Raf are relatively infrequent in breast cancer and their inhibitors have received less attention than in other cancers, but MEK inhibitors have shown some success.40 Given the ubiquitous importance of these signal-transduction molecules in the response of all cells to many different extracellular signals, it is questionable whether these agents will have sufficient specificity. Alternatively, the possibility that some breast cancers are dependent on other steroid hormones has led investigators to test antiandrogens such as bicalutamide and enzalutamide.41
Drugs have been developed to target cyclins and cyclin-dependent kinases but tend to lack tumor cell specificity.42 Recent interest has investigated specific characteristics of cancer stem cells,43 the rationale being that elimination of such cells is central to the destruction of the tumor because they control cell renewal and resistance to therapy. Interest prevails in strategies that encompass the interactions between the malignant cells and their microenvironment. For instance, drugs that target chemokines such as CXCL12 and its receptor CXCR4 have been investigated.44 There is considerable interest in the potential to target the interactions between cells via, for instance, β-catenin, or with the extracellular matrix via integrins, or the consequence of integrin signal transduction.45 Another strategy is to potentiate and extend tumor hypoxia.46
Introduction to the estrogen-related receptor alpha
Estrogens
Steroid hormones are small hydrophobic molecules that are transported in the blood bound to sex hormone binding globulin and are able to diffuse in and out of cells. Estrogen target tissues include breast, endometrium, bone, brain, liver, and heart. Derived from cholesterol, estrogens share a common four-ring structure and have important roles in sex determination, fertility, pregnancy, immune response, bone formation, and in the cardiovascular system. Cholesterol is converted into progestins, then into androgens and, finally, estrogens in a series of enzymatic reactions.6 Synthesized predominantly in the ovaries in premenopausal women, the principal site of estrogen synthesis in older women and in men is in peripheral tissues, notably adipose tissue.6
There are three estrogens, which are named for the number of hydroxyl groups: estrone, estradiol, and estriol. Estrone and estradiol are produced by aromatization of androstenedione and testosterone, respectively. Estriol is synthesized in the liver and placenta. Estriol is considered the major estrogen in pregnant women, estradiol in premenopausal women, and estrone in postmenopausal women. Concentrations of estrone and estradiol increase with obesity in postmenopausal women and in men.6,9 The surge in estrogen concentrations at puberty contributes to the development of secondary sexual characteristics including the female breast. Cyclical changes in concentration during the menstrual cycle cause cyclical changes in breast size and tenderness. Reduction in estrogen concentrations on cessation of lactation or menopause lead to breast involution.
The role of the estrogen receptor
The existence of a high-affinity receptor for estrogen was recognized initially by Jensen.47 Each steroid hormone family has its own protein receptor. These receptors are found in the steroid target cells and are ligand-dependent transcription factors, inactive until bound by their ligand (Figure 3A). The activated transcription factors coordinate formation of complexes of coactivator or corepressor proteins on the chromatin of their target genes. The composition of the transcription complexes, which depends on the receptor, the availability of coactivator and corepressor proteins, and on the DNA that surrounds the interaction site in the responsive gene, determines whether transcription is activated or repressed.
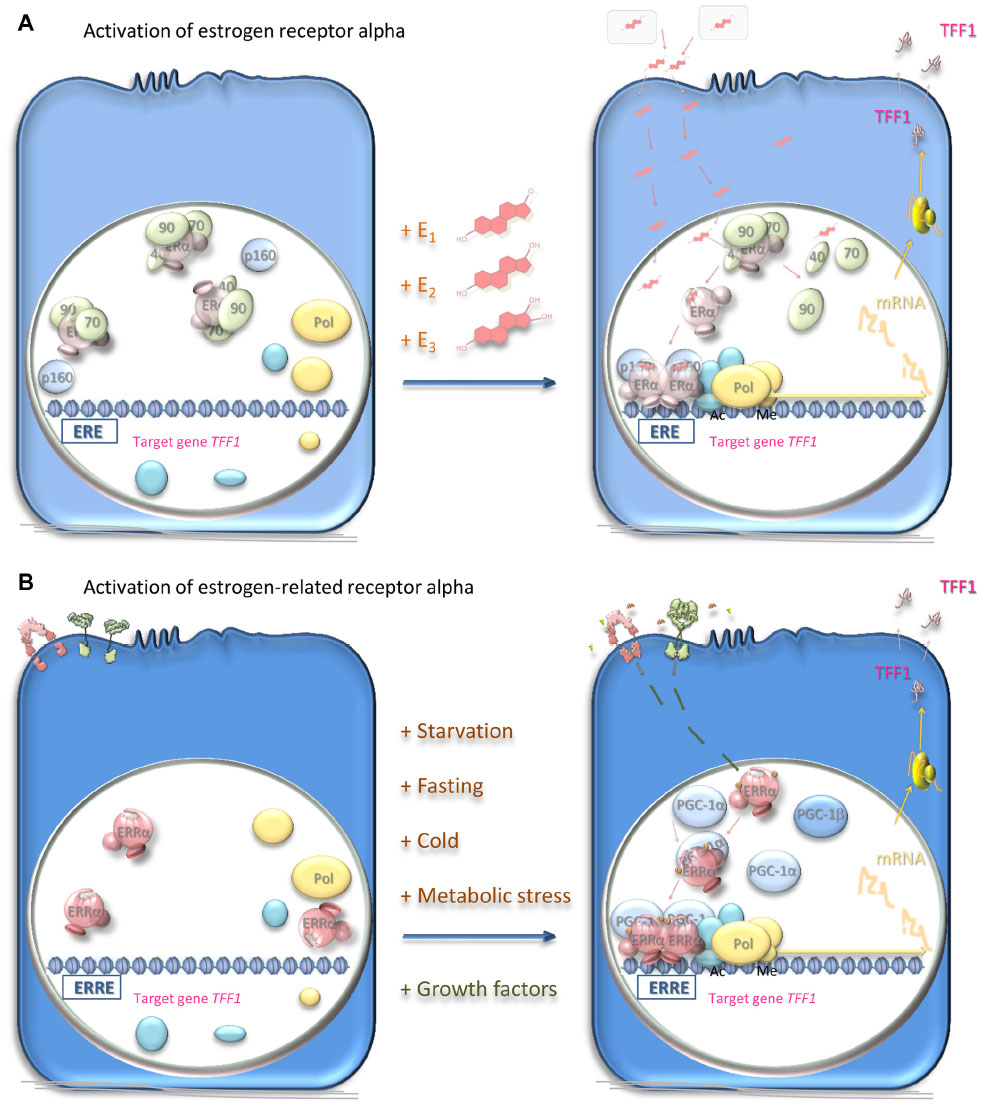 | Figure 3 Mechanisms of activation of estrogen receptor alpha and estrogen-related receptor alpha. |
The estrogen receptor is a 66 kDa protein of 595 amino acid residues sequestered in estrogen target cells in its inactive form in a complex with proteins including heat shock proteins 70 and 90, cyclophilin 40, FKBP51, and FKBP52 (Figure 3A). Estrogens diffuse into the target cells and bind with high affinity to their receptor, which dissociates from its sequestered complex. Estradiol has the highest affinity for the estrogen receptor, followed by estriol and then estrone.48 Interaction with the ligand leads to dimerization of the receptor and stabilizes a conformation that creates a surface on the receptor with which transcriptional coregulators interact. The dimeric receptor binds to estrogen response elements (EREs) in the promoters of estrogen-responsive genes.49,50 Coactivators, for instance, members of the p160 steroid receptor coactivator (SRC) family (SRC-1, SRC-2, and SRC-3), bind through one of three LXXLL motifs that form amphipathic alpha-helices.51,52 The p160 SRC proteins interact, in turn, with the histone acetyltransferases, cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB)-binding protein (CBP), and p300, which acetylate, and with coactivator-associated arginine methyltransferase 1 (CARM1) and protein arginine N-methyltransferase 1 (PRMT1), which methylate, histones within the nucleosomes. An RNA helicase A (RHA), and an ATP-dependent chromatin remodeling complex, SWItch/Sucrose NonFermentable (SW1/SNF), are recruited. The resultant coactivator complex modifies the nucleosomes and alters the surrounding chromatin to allow access to the activating transcription factor proteins, TATA-binding protein, and RNA polymerase II machinery, and transcription ensues. Posttranslational modifications may affect the activity of the estrogen receptor.52,53 Alternatively, corepressors, such as ligand-dependent corepressor (LCoR) and receptor-interacting protein 140 (RIP140), are recruited and attract deacetylases and demethylases to inhibit transcription.
Domain organization of estrogen receptors
Nuclear steroid receptors comprise five separate domains (Figure 4A). From the amino-terminus, the hormone-independent transcription activation domain comprises also a nuclear localization signal. It is followed by the DNA-binding domain, which contains a second nuclear localization signal. The DNA-binding domain is separated by a short hinge region from the ligand-binding domain, which is the largest domain and contains a dimerization domain and a transcription repression domain. The second transcription activator domain, which is ligand dependent, is at the carboxy-terminus. There are two estrogen receptors. The first, cloned in 1985 from estrogen-responsive breast cancer cells,54,55 is expressed in classic estrogen target cells and tissues and is responsible for the standard estrogen responses listed above. It is this receptor that is measured as an important prognostic and predictive biomarker in hormone-dependent breast cancer. The second, which is a 59.2 kDa receptor of 530 amino acids, identified in 1996,56 was called estrogen receptor beta; the former was renamed estrogen receptor alpha at this time to distinguish between the two. Estrogen receptor beta is reported to be expressed more widely than oestrogen receptor alpha and its function is less well understood. Conservation between the two paralogs is variable: low in the domains that interact with transcription activators and in the hinge region, at 25-30%; intermediate in the ligand-binding domain, at 60%; and highest, at 90%, in the DNA-binding domain (Figure 4C).
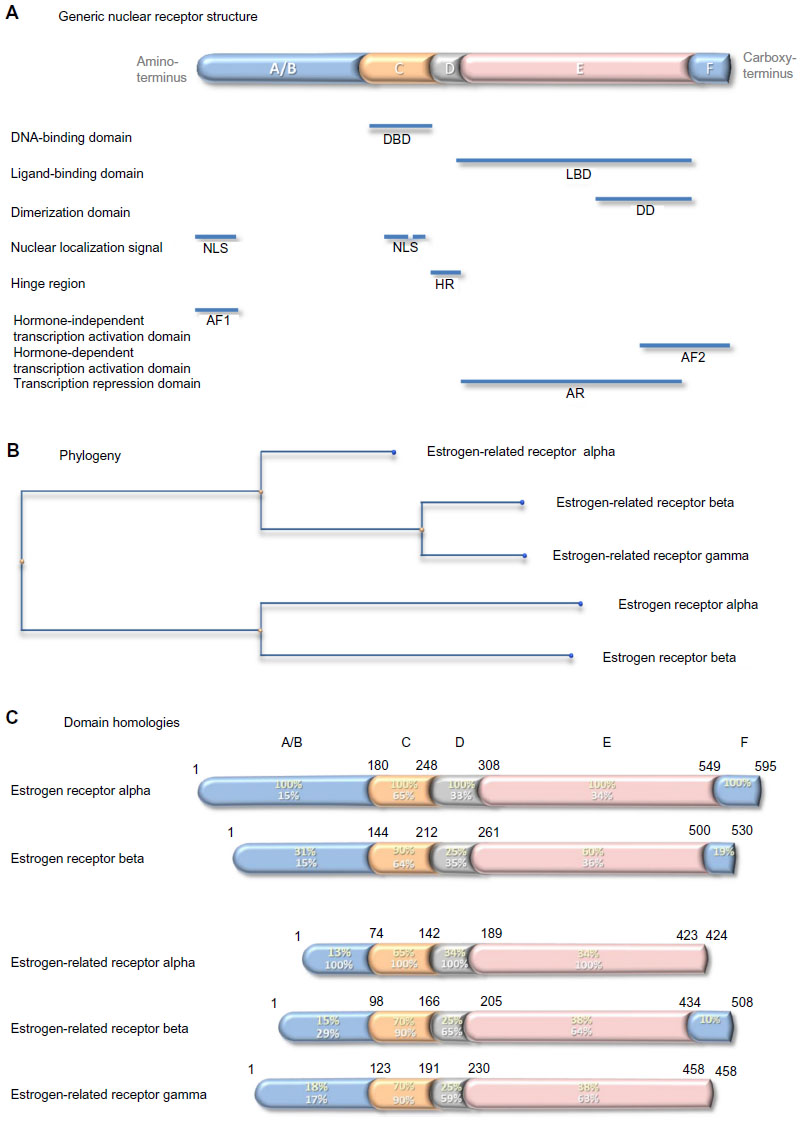 | Figure 4 Generic nuclear receptor structure, and estrogen receptor family phylogeny and shared homologies. |
Estrogen-related receptor alpha
In 1988, 8 years before discovery of estrogen receptor beta, two other members of the estrogen receptor family were discovered by Giguère et al:57 a 45.5 kDa, 423 amino acid residue protein named estrogen-related receptor alpha; and a 56.2 kDa, 508 amino acid residue protein named estrogen-related receptor beta. Later, the estrogen-related receptor gamma of 51.3 kDa, 458 amino acid residue protein was identified.58 Evolutionarily, estrogen-related receptor beta and gamma are closer to each other than to estrogen-related receptor alpha (Figure 4B). Comparison of the primary sequences in the different receptor domains shows relatively strong conservation of around 65% in the DNA-binding domains and less, around 35%, in the ligand-binding domains. Conservation is lower outwith these domains, and the estrogen-related receptors alpha and gamma lack an F domain (Figure 4C).
The estrogen-related receptor alpha interacts with coregulator proteins and binds to specific DNA sequences of its target gene promoters, primarily as a homodimer (Figure 3B). The peroxisome proliferator-activated receptor (PPAR) gamma coactivator (PGC)-1 family (PGC-1α, PGC-1β, and PPRC-1) and the p160 SRC proteins interact with this estrogen-related receptor alpha coactivator surface via LXXLL motifs.51 The most notable difference between estrogen-related receptors and estrogen receptors is that the former function as aporeceptors when they are not bound to ligand.
Estrogen receptor family DNA-binding domains
Arguably the domain most central to the specific functions of the estrogen receptor family is the conserved DNA-binding domain that recognizes sequences in the responsive genes and dictates with which genes the receptor will interact. This relatively short sequence of 70 amino acid residues contains two zinc-binding elements. In each, a zinc ion is ligated tetrahedrally by four cysteine residues (Figure 5). This class II zinc-binding motif comprises, from the amino-terminus, a zinc finger, an alpha helix, the second zinc finger, and the second alpha helix. The first alpha helix is the recognition helix that fits into the major groove of the double-stranded DNA. The structure of the estrogen receptor alpha DNA-binding domain in complex with DNA illustrates that the receptor dimer locates primarily on one face of the DNA helix (Figure 5C).59 The side chains of the residues make specific contacts with four DNA bases. The three residues responsible for the DNA interface are referred to sometimes as the proximal box. One of these residues, alanine in the estrogen-related receptors, is a glycine in the two estrogen receptors. The second region of intermolecular interaction is responsible for the dimerization interface and is referred to sometimes as the distal box. This region contains uniquely in estrogen-related receptor alpha a single conservative substitution of a serine in place of a threonine.
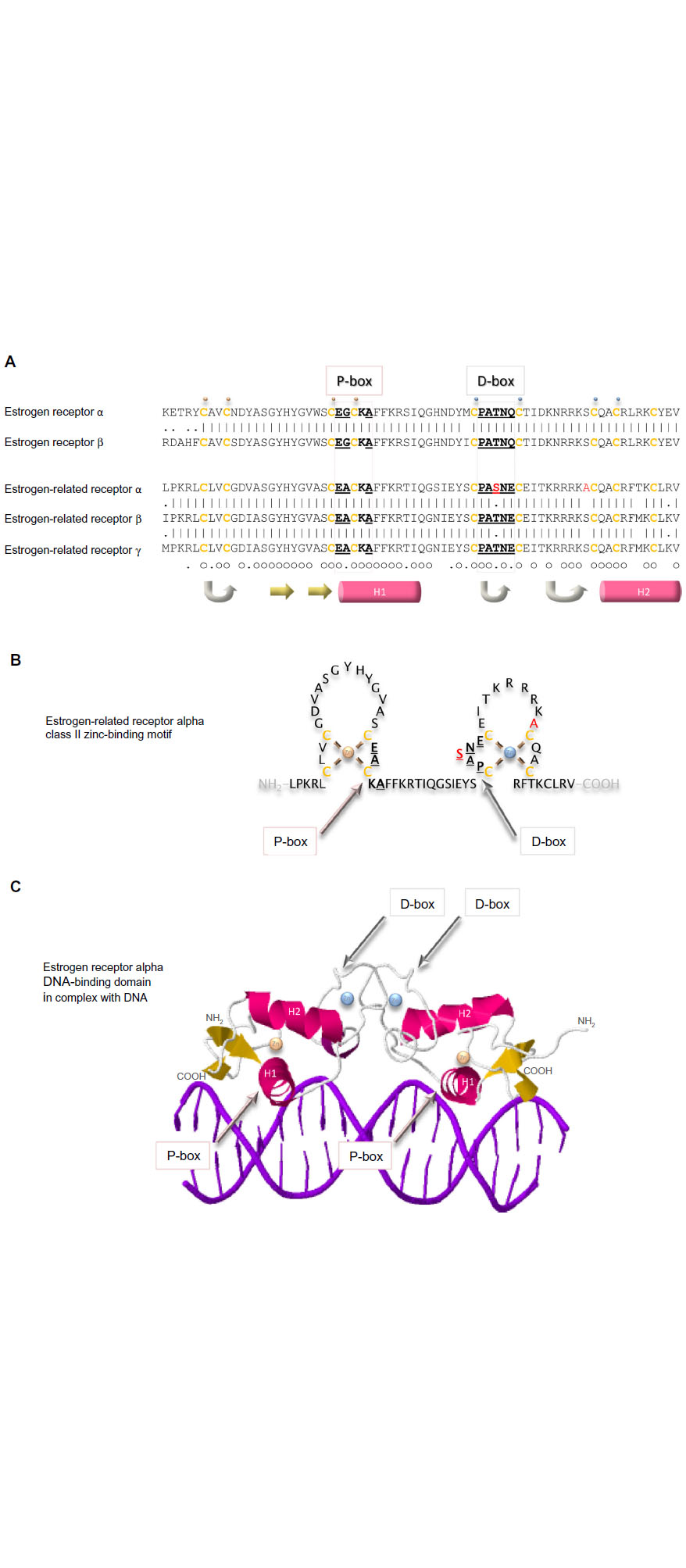 | Figure 5 DNA-binding domains of the estrogen receptors. |
Members of the estrogen receptor family bind to specific conserved DNA recognition sequences in the DNA of their responsive genes (Figure 6). Often, but not always, located proximal to the promoters, they may be up to 100 kb from the promoter of the responsive gene. The canonical estrogen response element or ERE 5′-AGGTCA-3′ is followed by three bases of indeterminate sequence and then by the inverse sequence 5′-TGACCT-3′.60–64 The palindromic nature of the perfect ERE is predictable given the perfect dimeric structure formed by interaction of the two receptor DNA-binding domains (Figure 5C). Reduction in the length of the palindromic sequence by a base on either side reduces the affinity of the interaction with the receptor dimer. The third base pair of the ERE half-site, G–C, provides binding energy, and the fourth base pair, T–A, makes a positive contact with the receptor. Most EREs identified differ from the canonical sequence by at least one base pair.63
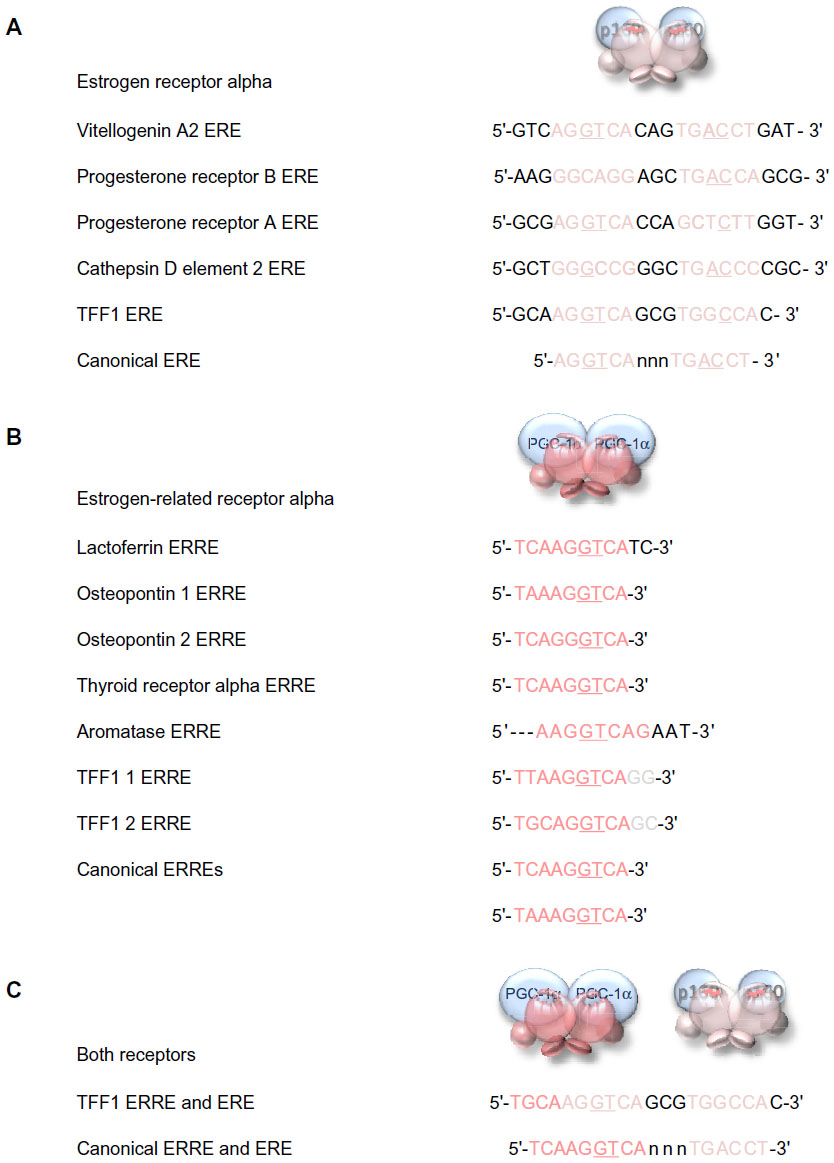 | Figure 6 DNA response elements recognized by estrogen receptor alpha and estrogen-related receptor alpha. |
The estrogen-related receptor alpha affects the transcription of known estrogen-responsive genes, such as those that encode lactoferrin, osteopontin, thyroid receptor alpha, aromatase (CYP19), and TFF165,66 via interaction with the EREs in these genes.67–71 Detailed analysis suggests that estrogen-related receptor alpha binds particularly well if the sequence is preceded by 5′-TAA-3′ or 5′-TCA-3′, and it is suggested that the estrogen-related receptor alpha response element (ERRE) is 5′-TA/CA AGGTCA-3′. The presence of a combined ERRE and ERE will ensure regulation of a gene by both receptors. It is noteworthy that the TFF1 promoter contains an imperfect ERRE and an imperfect ERE (Figure 6).
Estrogen receptor family ligand-binding domains
The ligand-binding domains of the estrogen receptor family are composed almost entirely of 12 helices, of which eleven are arranged in three antiparallel layers.72 Helices 5, 6, 9, and 10 comprise the central core layer,which is sandwiched between helices 1–4 on one face and helices 7, 8, and 11 on the other (Figure 7A). In the estrogen receptors, this wedge-shaped molecular scaffold creates a sizeable hydrophobic cavity at its narrower end into which estrogens slip and interact with high affinity. The remaining secondary structural elements, a small two-stranded antiparallel β-sheet and helix 12, flank the main three-layered motif on either side of the hydrophobic pocket (Figure 7A). After interaction with estrogens, helix 12 is positioned as a lid over the ligand-binding pocket to secure the ligand in position and posit the hydrophobic side chains of helix 12 toward the steroid (Figure 7B).72 This conformation creates a surface on the receptor that includes the charged residues, Lys362 at the end of helix 3 and Asp538, Glu542, and Asp545 from helix 12, that were identified by mutation analysis to be important for transcription activation (Figure 7A).73 Subsequent analysis of the structure of the ligand-binding domain of estrogen receptor alpha bound to the synthetic estrogen diethylstilbestrol and a coactivator peptide with an LXXLL motif identified more fully the coactivator recruitment surface.74 The interaction surface comprises a hydrophobic cleft formed with residues from helices 3, 4, 5, and 12 and the turn between helices 3 and 4 (Figure 7C). Interaction of the amphipathic alpha-helical coactivator peptide buries approximately 1,000 Å2 of the hydrophobic interaction surface. The majority of the residues involved in the interaction are hydrophobic. In addition, the main chain conformation of the coactivator peptide is stabilized by charged capping interactions at either end of the peptide helix with Lys362 from helix 3 and Glu542 from helix 12 of the receptor (Figure 7C).
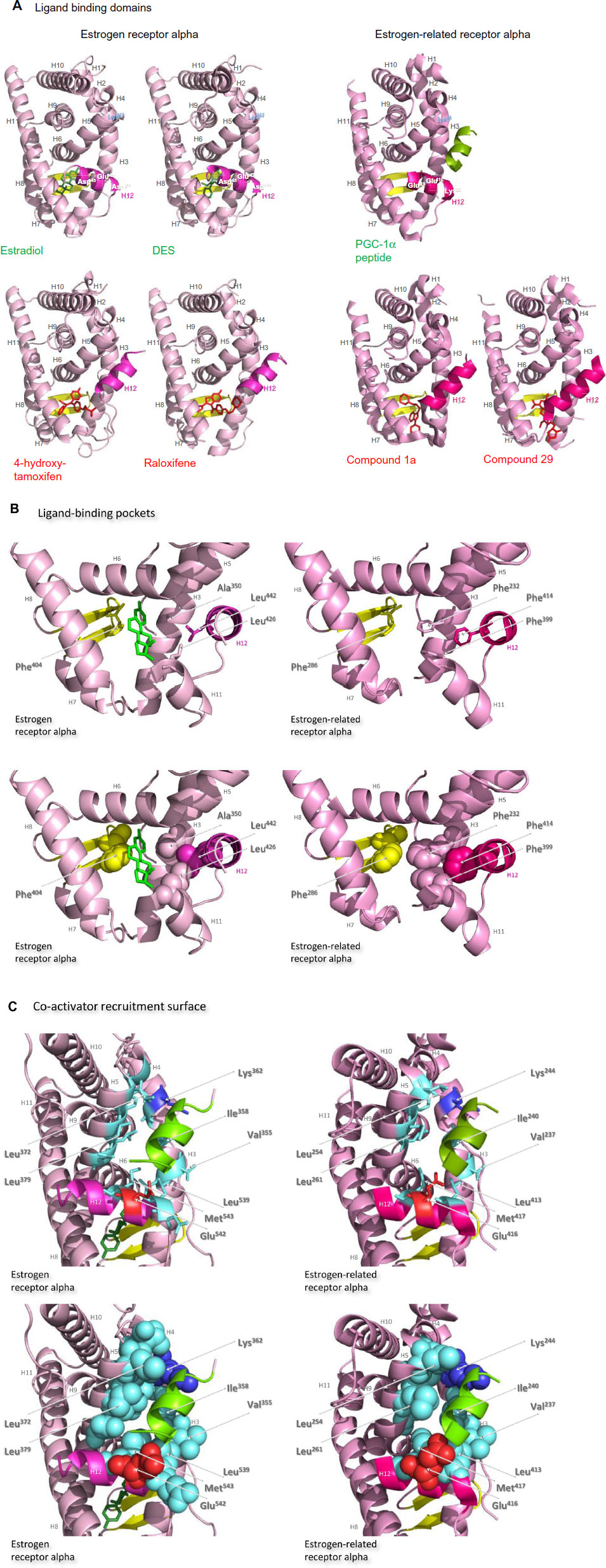 | Figure 7 Structures of the ligand-binding domains of estrogen receptor alpha and estrogen-related receptor alpha in complex with their ligands. |
After interaction of antiestrogens such as the active metabolite of tamoxifen, 4-hydroxytamoxifen, or raloxifene with the ligand-binding domain of estrogen receptor alpha, a portion of the ligand remains outside the ligand-binding pocket (Figure 7A).72,74 The extruded ligand prevents alignment of helix 12 over the ligand-binding pocket and hence formation of a complete coactivator recruitment surface. Instead, helix 12 is positioned over the hydrophobic cleft between helices 3, 4, and 5, in which position it precludes completely interaction of coactivators with this surface of the receptor.
The secondary and tertiary structures of the ligand-binding domains of estrogen-related receptors are extremely similar to those of the estrogen receptors, but subtle differences are proposed to allow them to function as aporeceptors and explain the failure to identify their natural ligands. Notably, in the structure of the estrogen-related aporeceptor alpha, helix 12 is positioned across the ligand-binding domain (Figure 7A).75 Thus the four charged residues, Lys244 in helix 3 and Lys412, Glu416, and Glu419 in helix 12, equivalent to those thought originally to be critical for activation of transcription in estrogen receptor alpha, are on the same face of the receptor in the absence of ligand. The crystal structure of estrogen-related receptor alpha includes the coactivator peptide of PGC-1α bound to the receptor coactivator recruitment surface; it is not known if the presence of this peptide facilitates stabilization of the active conformation of estrogen-related receptor alpha in the crystals, or if the structure of the aporeceptor ligand-binding pocket would be more open in the absence of the PGC-1α peptide. The coactivator recruitment surface of estrogen-related receptor alpha formed from helices 3, 4, 5, and 12 is similar to that of estrogen receptor alpha. The PGC-1α peptide is anchored by canonical charge clamp interactions with Lys244 from helix 3 and Glu416 from helix 12, and many of the conserved hydrophobic residues interact with the coactivator peptides in both structures (Figure 7C).74,75
The estrogen-related receptor alpha ligand-binding pocket is delineated by 22 amino acid residues, most of which have hydrophobic side chains. It is occluded by bulky hydrophobic side chains, in particular that of the phenylalanine Phe232 from helix 3,75 which is an alanine in the other four receptors, to create a cavity of only 100 Å3, which is substantially smaller than those of the estrogen receptor alpha (490 Å3) or beta (390 Å3) (Figure 7B). Removal of this hydrophobic side chain abolishes activity of the aporeceptor. It is proposed that the presence in the ligand-binding pocket of Phe232 and other bulky hydrophobic side chains, notably Phe286 from the small beta sheet, Phe399 from helix 11, and Phe414 from helix 12, recapitulates the interactions provided by hydrophobic steroids in the estrogen receptors and allows the aporeceptor to hold an active conformation able to interact with coactivator proteins.75 Disruption of the interactions between Phe232, Phe286, Phe399, and Phe414 destabilizes the active conformation of the estrogen-related aporeceptor alpha.
Interestingly, estrogen-related receptor alpha retains the charged Glu235 and Arg276, of which the equivalent residues in estrogen receptor alpha form a hydrogen bond network with the hydroxyl group on carbon 3 of the A ring of all three estrogens. Estrogen-related receptor alpha retains also the polar His298 that forms a hydrogen bond with the hydroxyl group on carbon 17 of the D ring of estradiol and estriol. It is suggested, however, that insertion of an estrogen into the ligand-binding pocket of estrogen-related receptor alpha would cause such serious steric clashes, notably with the side chains of Phe232 and Phe399, that even the A ring would not be accommodated (Figure 7B).75
The occlusion of the estrogen-related receptor alpha ligand-binding pocket by bulky hydrophobic side chains indicates that the introduction of a molecule with more than four or five carbon atoms would necessitate a conformational change that would displace helix 12 from the coactivator surface.75 The estrogen-related receptor alpha does not bind estrogens or 4-hydroxytamoxifen, but does interact with the synthetic estrogen diethylstilbestrol to prevent the receptor interaction with SRC-1. Antagonist effects of diethylstilboestrol on estrogen-related receptor alpha activity have been reported by some but not all authors.71,76 Modeling indicates that diethylstilbestrol would only be accommodated in the estrogen-related receptor alpha ligand-binding pocket if the side chains of Phe232 from helix 3 and Phe399 from helix 11 were to assume different conformations, and if Phe414 was removed from the hydrophobic cavity by displacement of helix 12.29,30 That these conformational changes would disrupt the favorable cluster of phenylalanines Phe232, Phe286, Phe399, and Phe414, might indicate that the affinity of diethylstilboestrol for estrogen-related receptor alpha would be weak. Substitution of Phe232 with an alanine residue, which is found in the equivalent position in the other four members of the estrogen receptor family, allows 4-hydroxytamoxifen to bind estrogen-related receptor alpha with a relatively high affinity of 4×10–8 M.77
Despite the steric constraints described above, several phytoestrogens: the flavone 6,3′,4′-trihydroxyflavone and the isoflavones genistein, daidzein, and biochanin A, have been reported to be agonists for estrogen-related receptor alpha activity.76 Phytoestrogens are produced by plants, have bactericidal and fungicidal activity, and represent the major natural exogenous sources of estrogenic compounds. The results indicate that it is possible for a ligand to interact with estrogen-related receptor alpha to augment its activity.
Physiological functions of estrogen-related receptor alpha
Discovery of estrogen-related receptor alpha immediately prompted questions of its physiological function: whether it overlapped with that of the estrogen receptor and if the receptor had a role in breast cancer. Estrogen-related receptor alpha is expressed in the later stages of embryonic development and is abundant in heart, skeletal muscle, and the nervous system. The physiological role of estrogen-related receptor alpha, and of estrogen-related receptor gamma, is to act as an energy sensor to control cellular adaptation to energy demand and stress. To this end, estrogen-related receptor alpha is expressed at high levels in tissues with high energy demands, such as muscle and brown adipose tissue. Cells that do not express active estrogen-related receptor alpha cannot produce sufficient energy in times of peak demand.
Role of estrogen-related receptor alpha in metabolism
In adipose tissue, estrogen-related receptor alpha increases the differentiation of mesenchymal stem cells into adipocytes and hence enhances fat deposition. Further, estrogen-related receptor alpha has a role in the regulation of energy metabolism in adipocytes. It increases lipid uptake, fatty acid beta-oxidation, the tricarboxylic acid cycle, oxidative phosphorylation, and mitochondrial biogenesis and function. Effects of estrogen-related receptor alpha on metabolism extend to other tissues with high energy requirements, notably cardiomyocytes and macrophages. The importance of estrogen-related receptor alpha in metabolic regulation is emphasized by the demonstration that esrra-null mice have impaired fat absorption and metabolism and are relatively resistant to fat-induced obesity.78 These lean mice are unable to adapt to cold environments and develop cardiac contractile dysfunction. The cardiac hypertrophy induced by stress in esrra-null mice is caused by reduced ATP synthesis and reduced phosphocreatine storage.79
Role of estrogen-related receptor alpha in osteogenesis
Estrogen-related receptor alpha influences the differentiation of myocytes, T-cells, intestinal epithelial cells, and osteoblasts. A report indicated that estrogen-related receptor alpha has a role in bone development and metabolism during embryogenesis.80 Its messenger RNA is expressed in murine bone cells during bone formation by endochondral and intramembranous ossification and in primary human osteoblasts. Estrogen-related receptor alpha was found to affect transcription of an osteopontin gene promoter; osteopontin is an important constituent of the mineralized extracellular matrix of bones.80 In essra-null mice, absence of estrogen-related receptor alpha increased modestly osteoblast differentiation and cancellous bone mineral density, as well as mesenchymal cell differentiation into osteoblasts.81 Further, estrogen-related receptor alpha was shown to decrease differentiation of human mesenchymal stem cells into osteoblasts, osteopontin expression, and mineral deposition, but to increase adipocyte differentiation.81 In a different strain of essra-null mice, female bones aged less compared to those of wild-type mice even after estrogen depletion and their marrow mesenchymal stem cells showed greater ability to differentiate into osteoblasts ex vivo.82 Thus estrogen-related receptor alpha has a pivotal role in determination of mesenchymal stem cell fate and is implicated in inhibition of mineralization by osteoblasts.80–82
Genes induced by estrogen-related receptor alpha
Genomic studies indicate that estrogen-related receptor alpha regulates large numbers of genes involved in energy metabolism. Estrogen-related receptors interact with the promoters of most mitochondrial and cellular genes that encode enzymes involved in the glycolytic pathway, the tricarboxylic acid cycle, and oxidative phosphorylation, and in nucleic acid, amino acid, lipid, and pyruvate synthesis. Estrogen-related receptor alpha is involved in the transcriptional regulation of genes required for mitochondrial biogenesis, the tricarboxylic acid cycle, oxidative phosphorylation, fatty acid oxidation, and lipid metabolism.83–85 For instance, estrogen-related receptor alpha induces expression of NRF1, GAPα, and PPARα.86 The nuclear receptor coactivators PGC-1α, PGC-1β, and PPRC-1 are implicated in the regulation of these genes and in the autoregulation of the expression of estrogen-related receptor alpha. It has been suggested that the metabolic effects of estrogen-related receptor alpha are controlled by PGC-1α.85 PGC-1α is expressed at low basal levels but is induced by fasting and other metabolic stresses.83 PGC-1β, a related coactivator, has similar functions, but its expression may not be regulated as acutely by variations in energy demand.84
Activation of estrogen-related receptor alpha
If the estrogen-related receptor alpha is fully functional in the absence of ligand, does this mean that it is constitutively active? Current thoughts are that its activity is regulated in two main ways (Figure 3B). First, its activation is limited by the intracellular concentrations of its coregulators. Rather than being regulated by interaction with a classic ligand with resultant stabilization of an active receptor conformation, the magnitude of estrogen-related receptor alpha activity is thought to be dependent largely on the presence of transcriptional coactivators of transcription such as PGC-1α, PGC-1β and PPARβ, SRC-3 and PPGC183,87,88 or corepressors of transcription such as RIP140 and nuclear receptor corepressor 1 (NCoR1).89,90 Whether or not the coactivators induce or stabilize the active conformation of estrogen-related receptor alpha is unknown. Certainly, the coregulators are essential for most estrogen-related receptor alpha activity and have been termed surrogate ligands. Induction of the expression of the coregulators by external metabolic stress activates estrogen-related receptor alpha.
Secondly, receptor activity is controlled by posttranslational modification (Figure 3B), namely by phosphorylation initiated by the interaction of growth factors such as the IGFs and epidermal growth factors (EGFs) with their cognate receptors and consequent signal transduction. Recruitment of estrogen-related receptor alpha to the TFF1 promoter and resultant transcription are increased in the presence of EGF, possibly via phosphorylation of the DNA-binding domain.91 Activation of HER2 increases the transcriptional activity of estrogen-related receptor alpha by phosphorylation at multiple residues, including in the carboxy-terminus.92 In vitro analyses of the ability of estrogen-related receptor alpha to induce transcription from the TFF1 gene in breast cancer cells demonstrated that the induction is increased by activation of growth factor receptors including EGF receptor, HER2 and the type I IGF receptor.93 In addition, phosphorylation-dependent, amino-terminal SUMOylation reduces transcriptional activity of estrogen-related receptor alpha94 and acetylation by p300 coactivator-associated factor (PCAF) of four lysine resides in its DNA-binding domain modulates its activity.95
Role of the estrogen-related receptor alpha in breast cancer
Preclinical studies
The estrogen-related receptor alpha receptor has been promulgated as a prospective target for breast cancer therapy based in part on preclinical analyses. Knockdown of estrogen-related receptor alpha mRNA in estrogen-responsive and estrogen-unresponsive breast cancer cells decreased their migratory potential, possibly by induction of WNT11 and vascular endothelial growth factor expression.96 Similarly, reduction of estrogen-related receptor alpha mRNA with miR-137 decreased the migratory and proliferative capacity of breast cancer cells.97 While proliferation in vitro was unaffected, proliferation of xenografts of estrogen-unresponsive breast cancer cells was reduced after knockdown of estrogen-related receptor alpha mRNA.98 Further, genetic deletion of esrra delayed tumor development in a mouse model of HER2-induced mammary tumorigenesis.99 The ability of estrogen-related receptor alpha to increase orthotopic tumor growth may be explained by its promotion of angiogenesis by induction of vascular endothelial growth factor.100 In apparent contradiction, estrogen-related receptor alpha expression decreased the growth of osteolytic metastases in a mouse model of breast cancer, possibly via reduced osteolysis as a result of the induction of osteoprotegerin, which inhibits osteoclastogenesis.100
Tumor metabolism
The importance of estrogen-related receptor alpha in the control of cellular energy metabolism suggests that it may be involved in breast cancer cell metabolism.79 Estrogen-related receptor alpha could facilitate glycolytic metabolism by tumor cells. Its relatively high expression in tumors with poorer prognosis, some of which manifest increased glucose uptake, which is associated with a more aggressive phenotype, supports this conjecture. In breast cancer cells, estrogen-related receptor alpha increases the expression of critical enzymes of the glycolytic pathway.98,101 Estrogen-related receptor alpha expression may also favor mitochondrial oxidative respiration in breast cancer cells through its regulation of genes involved in mitochondrial oxidative function.86 Breast cancer cells that had been selected for their ability to metastasize to the brain, expressed estrogen-related receptor alpha, PGC-1α, PGC-1β, and estrogen-related receptor alpha-regulated genes involved in the tricarboxylic acid cycle, glycolysis, and oxidative phosphorylation pathways at higher levels than unselected cells.102 Activation of estrogen-related receptor alpha by overexpression of PGC-1α in breast cancer cells increases oxidative metabolism.103 The induction by estrogen-related receptor alpha of genes involved in lipid, amino acid, and nucleic acid synthesis, in glutaminolysis, and in regulation of the pentose phosphate pathway implicates the receptor in anabolic biosynthesis.98,101 Further, estrogen-related receptor alpha may protect breast cancer cells against oxidative damage and production of reactive oxygen species by induction of detoxifying enzymes such as glutathione S-transferase MU-1 (GSTM1), liver glutaminase (GLS2), and superoxide dismutase 2 (SOD2).98,101
Translational studies
A decade ago, two clinical studies reported that estrogen-related receptor alpha is involved in breast cancer progression.104,105 Estrogen-related receptor alpha expression is inversely correlated with estrogen receptor and progesterone receptor expression but is associated positively with expression of HER2.104 The authors suggested that estrogen-related receptor alpha might be able to substitute for estrogen receptor if expression of the latter is lost, especially in HER2-positive or tamoxifen-resistant tumors.104 Estrogen-related receptor alpha is detected in around 55% of human breast cancers by immunohistochemistry and its expression is associated with increased risk of recurrence and poor prognosis.105 Estrogen-related receptor alpha mRNA levels are similar or higher than estrogen receptor mRNA levels in approximately one-quarter of breast tumors and are highest in tumors in which functional estrogen receptor is absent.105 In this context, estrogen-related receptor alpha response elements often overlap those of estrogen receptor alpha, and both receptors affect the in vitro transcription of genes that encode lactoferrin, osteopontin, aromatase, and TFF1.67,68,70,71 Conversely, estrogen-related receptor alpha is reported to induce transcription of the HER2 gene, whereas the estrogen receptor represses the transcription of HER2 in the presence of estrogen.99 Estrogen-related receptor alpha is not an independent biomarker of prognosis of breast cancer patients, but the concerted expression of a cohort of 86 genes that are regulated by the receptor is associated with poor prognosis.98
Critical analysis of the potential for targeting the estrogen-related receptor alpha in breast cancer management
There has been considerable interest in the therapeutic potential of the activation or suppression of estrogen-related receptor alpha activity. The important role of estrogen-related receptor alpha in mitochondrial biogenesis and oxidative metabolism suggests that its activation could be an effective therapeutic intervention in diabetes or metabolic diseases. Although more controversial, the possibility of targeting estrogen-related receptor alpha activity in the treatment of osteoporosis has attracted attention. Importantly, in the context of this review, interest in the inhibition of estrogen-related receptor alpha in medical oncology has focused on the treatment of breast cancer.
Phenex Pharmaceuticals AG (Ludwigshafen, Germany) was interested in the potential to treat cancer, osteoporosis, obesity, lipid and cardiovascular disorders, or conditions that affect fertility and reproductive health. GlaxoSmithKline (Brentford, UK) investigated estrogen-related receptor alpha activation for the treatment of metabolic disease. Merck & Co., Inc. (Whitehouse Station, NJ, USA) considered its inhibition in the context of breast cancer and metabolic disorders. Novartis (Basel, Switzerland) invested in the possibility of targeting breast cancer, metabolic diseases, and osteoporosis. Johnson and Johnson (New Brunswick, NJ, USA) considered the treatment of obesity and insulin-insensitive diabetes. The last two companies have published structures of their synthetic compounds bound to the receptor.106,107 The SRI International (Menlo Park, CA, USA) has a compound in clinical trial. Notably, medicinal chemists at the Guangzhou International Business Incubator (Guangzhou, People’s Republic of China) have reported the synthesis of several series of biologically active ligands of the estrogen-related receptor alpha.108
Potential drugs
Natural ligands of the estrogen-related receptor alpha have not been identified, and it is generally accepted that the aporeceptor is active. The crystal structure of Kallen et al indicates that the aporeceptor has an active conformation in the absence of bound ligand, that the ligand-binding pocket is too small to accommodate an estrogen-like ligand, and that introduction of such a ligand would disrupt the active conformation (Figure 7).75 The crystals of estrogen-related receptor alpha included a coactivator peptide from PGC-1α, and it is possible that the presence of this peptide and its interaction with the receptor induces or favors adoption of the active conformation by the receptor. The corollary is that it is possible that, in the absence of the PGC-1α coactivator peptide, helix 12 of the receptor may be more mobile than is suggested by the crystal structure, and the ligand-binding site more open and flexible and able to accommodate a ligand larger than might be predicted from examination of the crystal structure.
Agonists
Given the failure to identify natural ligands for the estrogen-related receptor alpha and widespread acceptance that the receptor is fully active in its unliganded form, it was surprising to discover that natural phytoestrogens had agonist activity for estrogen-related receptor alpha. In 2003, Suetsugi et al identified potential agonists76 by virtual ligand screening of a homology model of the estrogen-related receptor alpha ligand-binding domain based on the crystal structure of estrogen-related receptor gamma ligand-binding domain.77 Four ligands, three isoflavones: genistein, daidzein, and biochanin A, and one flavone: 6,3,4-trihydroxyflavone, all of which are present in legumes and various herbs, were identified (Figure 8).76 The virtual screening indicated that estradiol, the synthetic estrogen diethylstilbestrol, and 4-hydroxytamoxifen do not bind. The authors demonstrated that genistein, daidzein, biochanin A, and 6,3,4-trihydroxyflavone increased estrogen-related receptor alpha transcriptional activity.76
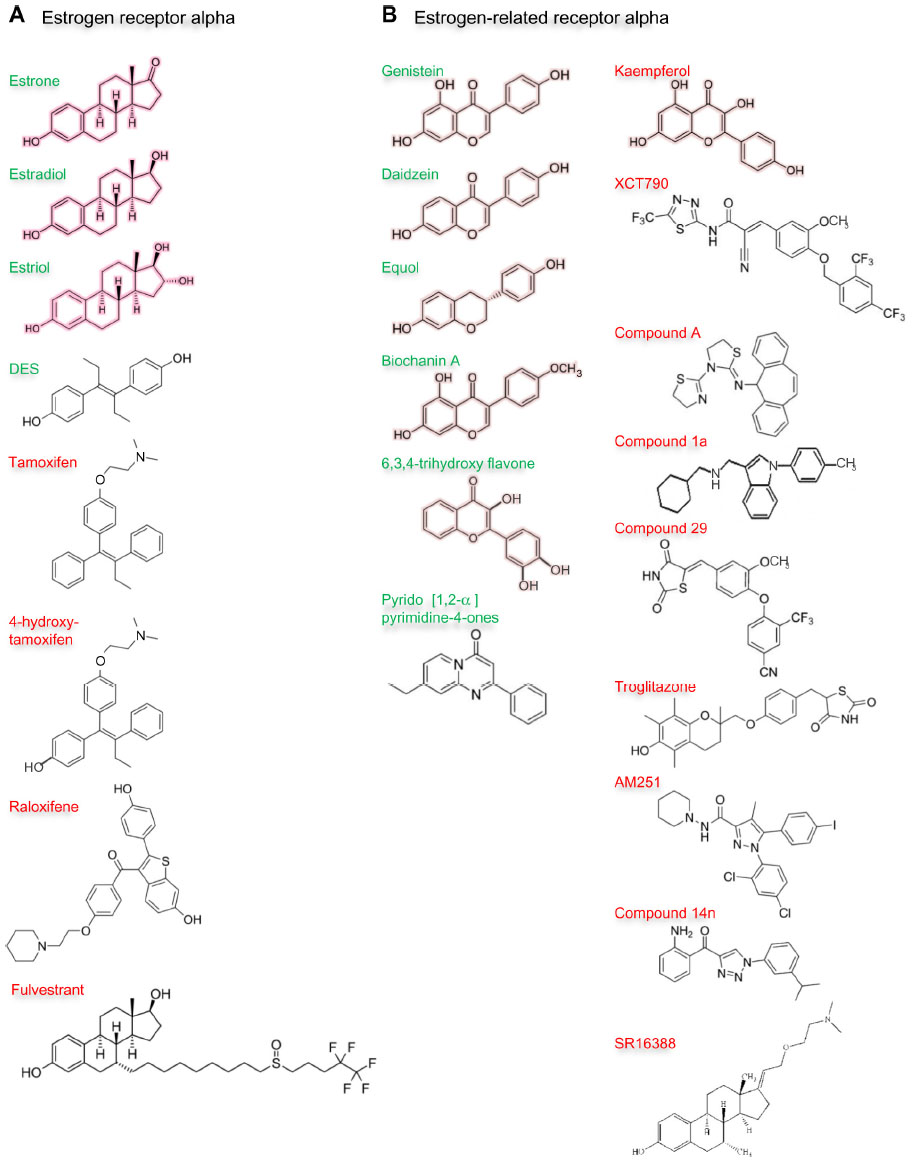 | Figure 8 Ligands of estrogen receptor alpha and estrogen-related receptor alpha. |
Later, scientists at GlaxoSmithKline failed to identify agonists through random screening of their compound collection. Subsequently, they synthesized molecules predicted to interact with the ligand-binding domain by structure-guided design based on the 2004 crystal structure of the estrogen-related receptor alpha,75 but were unable to demonstrate agonist activity.109 Subsequently, Peng et al synthesized a series of pyrido[1,2-α]pyrimidin-4-ones with a view to producing more potent agonists of the estrogen-related receptor alpha and confirmed the compounds’ ability to increase the transcriptional activity of the receptor (Figure 8).110 The compounds improved the uptake of glucose and fatty acids by muscle cells and have potential in the treatment of metabolic disease.110 Taken together, these data indicate that there are ligands whose interaction with the estrogen-related receptor alpha stabilizes the active conformation of the receptor and increases its transcriptional activity. These molecules or their derivatives have potential clinical utility for the treatment of metabolic diseases, including metabolic syndrome and diabetes.
Inverse agonists
The demonstration that two organochlorine pesticides, toxaphene and chlordane, antagonize estrogen-related receptor alpha activity established the principle that the activity of the aporeceptor may be inhibited by a ligand.111 The dietary flavonol kaempferol, which is present in tea, brassicas, legumes, and some fruits, prevents interaction of estrogen-related receptor alpha with DNA response elements and hence its effects on transcription (Figure 8).112 Such compounds are called reverse agonists or inverse agonists. Several groups have synthesized inverse agonists of estrogen-related receptor alpha and sought to demonstrate their therapeutic potential in preclinical models.
In 2004, Busch et al reported the first synthetic inverse agonists of estrogen-related receptor alpha.113 Their search was initiated by a high-throughput screen of their compound library followed by optimization of their lead structure to develop a potent, selective, and orally-available compound, (2E)-3-(4-([2,4-bis(trifluoromethyl)benzyl]oxy)-3-methoxyphenyl)-2-cyano-N-[5-(trifluoromethyl)-1,3,4-thiadiazol-2-yl]acrylamide, or XCT790 (Figure 8).113 Several other groups have studied the effects of XCT790 on the biological activity of estrogen-related receptor alpha and established that XCT790 induces ubiquitin-dependent proteasomal degradation of estrogen-related receptor alpha.114 XCT790 treatment was found to increase glucose uptake and mitochondrial production of reactive oxygen species and decrease mitochondrial mass and membrane potential.115–118 The proliferation of estrogen-responsive and estrogen-unresponsive breast cancer cells is inhibited by XCT790.119 XCT790 prevents the induction in vitro by estrogen-related receptor alpha of the expression of genes whose expression in tumors is associated with poor patient prognosis.93
Chisamore et al searched for inverse agonists by examination of the ability of compounds to antagonize the interaction between estrogen-related receptor alpha ligand-binding domain and the coactivator interaction domain of PGC-1α.120 They identified a novel tricyclic antagonist, N-[(2Z)-3-(4,5-dihydro-1,3-thiazol-2-yl)-1,3-thiazolidin-2-ylidene]-5H dibenzo[a,d][7]annulen-5-amine, or compound A, that binds estrogen-related receptor alpha with high affinity (Figure 8). The authors developed a homology model of estrogen-related receptor alpha in an antagonist conformation to explain the specificity of compound A. Subsequently, Chisamore et al demonstrated that compound A decreased expression of estrogen-related receptor alpha-responsive genes that encode TFF1, osteopontin, and aromatase.121 Estrogen-related receptor alpha protein degradation is induced by compound A through the ubiquitin 26S proteasome pathway.121 Compound A was shown to inhibit the proliferation of estrogen-responsive and estrogen-unresponsive breast cancer cells and reduce their growth in xenograft models.122
The first structure of a cocrystal of a synthetic inverse agonist in complex with the estrogen-related receptor alpha ligand-binding domain was reported in 2007 by Kallen et al.106 The authors produced cyclohexylmethyl-(1-p-tolyl-1H-indol-3-ylmethyl)-amine, or compound 1a (Figure 8), by evolution of a lead compound identified through high-throughput screening. In the complex with compound 1a, part of the ligand fits into the ligand-binding pocket of estrogen-related receptor alpha and the aromatic side chains of Phe232 from helix 3, Phe399 from helix 11, and Phe414 from helix 12 do not cluster (Figure 7A). The remainder of compound 1a protrudes from the ligand-binding pocket. The last turn of helix 11 is unwound and the amino-terminus of helix 3 is in a different position than in the structure of the aporeceptor in complex with the coactivator peptide from PGC-1α. Helix 12 of the estrogen-related receptor alpha is not aligned in its agonist position but lies in the groove occupied by the coactivator peptide from PGC-1α in the cocrystal with the aporeceptor. This alignment of helix 12 is similar to the alignment of helix 12 in the crystal structures of estrogen receptor alpha with the antiestrogens 4-hydroxytamoxifen and raloxifene. Interaction of coactivator LXXLL motif peptides with the coactivator recruitment surface is prevented, which may well account for the inverse agonistic effects of compound 1a.106
More recently, Patch et al used a high-throughput binding assay to identify unbiased ligands for estrogen-related receptor alpha.107 Compounds were tested for their ability to inhibit interaction with a coactivator peptide of SRC-2. Subsequent optimization produced 4-(4-[[(5R)-2,4-dioxo-1,3-thiazolidin-5-yl]methyl]- 2-methoxyphenoxy)-3-(trifluoromethyl)benzonitrile, or compound 29, as the most potent ligand (Figure 8). A cocrystal structure of the ligand-binding domain of estrogen-related receptor alpha with compound 29 revealed a covalent interaction between the ligand and the receptor that is known to be reversible.107 Helix 12 is aligned in a similar position to that found in the cocrystal with compound 1a adjacent to the hydrophobic cleft formed between helices 3, 4, and 5, which means that helix 12 prevents interaction of coactivators with this receptor coactivator recruitment surface (Figure 7A). Interestingly, Patch et al demonstrated that compound 29 was functionally active in metabolic animal models and proposed its development as a novel antidiabetic agent. Oral administration of compound 29 stabilized insulin and circulating triglyceride levels and improved insulin sensitivity but did not affect body weight in diet-induced murine models of obesity and in an overt diabetic rat model.107
Wang et al found that that the thiazolidinedione (RS)-5-(4-[(6-hydroxy-2,5,7,8-tetramethylchroman-2-yl)methoxy]benzyl)thiazolidine-2,4-dione, or troglitazone (Figure 8), which was marketed originally as an antidiabetic agent but withdrawn due to liver toxicity, is an effective inverse agonist of estrogen-related receptor alpha.123 The troglitazone-receptor complex does not interact with the coactivators PGC-1α and PGC-1β, and troglitazone inhibits mitochondrial function. Mitochondrial mass was reduced and the expression of superoxide dismutases suppressed, which elevated reactive oxygen species production. The increase in reactive oxygen species in turn induced expression of the cell cycle inhibitor p21.123 Investigation of a pyrazole derivative, the cannabinoid 1 receptor inverse antagonist biarylpyrazole compound 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide, or AM251 (Figure 8), in cannabinoid 1 receptor-null cells identified estrogen-related receptor alpha as an alternative receptor.124 Interaction between AM251 and estrogen-related receptor alpha inhibited its transcriptional activity and induced its proteolytic degradation. AM251 displaced diethylstilboestrol from the ligand-binding domain of recombinant estrogen-related receptor alpha. Subsequent studies confirmed that AM251 effects are mediated by proteasomal degradation of nuclear estrogen-related receptor alpha.125 Downregulation of estrogen-related receptor alpha by AM251 or small interfering RNA led to increased mitochondria biogenesis and reduced mitochondrial membrane potential. These results indicate that the clinical analog of AM251, rimonabant, which is prescribed to decrease food intake, could exert its inhibitory effects against obesity, at least in part, through estrogen-related receptor alpha.
Most recently, Xu et al reported synthesis of a series of 1-phenyl-4-benzoyl-1H-1,2,3-triazoles as novel suppressors of estrogen-related receptor alpha transcriptional activation.108 The most promising compound, 2-aminophenyl-(1-(3-isopropylphenyl)-1H-1,2,3-triazol-4-yl)methanone, or compound 14n (Figure 8), reduced with an IC50 of 0.021 μM the transcriptional activation by estrogen-related receptor alpha as assessed in a cell-based reporter gene assay and decreased breast cancer cell migration and proliferation. Preliminary pharmacokinetic studies suggested that compound 14n has a good pharmacokinetic profile, with an oral bioavailability of 71.8%.108
The synthetic inverse inhibitor of estrogen-related receptor alpha that has advanced furthest towards clinical application is (E)-3-Hydroxy-7α-methyl-21-[2′-(N,N,-dimethylamino)ethoxy]-19-norpregna-1,3,5(10),17(20)-tetraene, or SR16388 or TAS-108 (Figure 8).126,127 This novel steroidal compound was developed by SRI International for the treatment of breast, prostate, ovarian, and colon cancer in the hope that it would inhibit the effects of estrogen-related receptor alpha on energy metabolism of tumor cells by inhibition of hypoxia-inducible factor 1-alpha and hence prevent tumor cell survival in hypoxic conditions by prevention of angiogenesis and induction of apoptosis.126,127 SR16388 also inhibits estrogen receptor alpha. Malignant cell growth in xenograft models is inhibited with an IC50 of around 0.2 μM and the inhibition is potentiated in the presence of microtubulin inhibitors such as paclitaxel or vincristine.127 Promulgated for treatment of tamoxifen-resistant breast cancer and bicalutamide-resistant prostate cancer, SR16388 has completed Phase I trials, as well as Phase II trials for different tumor types. Benefits demonstrated in these studies include: antitumor efficacy in breast cancer patients who have relapsed while on treatment with an antiestrogen or an aromatase inhibitor; similar reduction in bone mineral density compared with aromatase inhibitors; no thickening of the endometrium; and a good overall safety profile. A Phase III trial of SR16388 in combination with a microtubulin inhibitor has been proposed.
Inhibitors of protein interactions
The second most popular approach by which to inhibit the activity of estrogen-related receptor alpha is to prevent interaction between the receptor and the coactivators and corepressors that interact with the coactivator surface that lies between helices 3, 4, 5, and 12 in the active conformation of the receptor (Figure 7A and C).75 There are no prototype drugs reported, but the importance of these interactions in modulating the activity of the receptor is recognized.90,98,128 The success of the nutlins that disrupt interaction between p53 and MDM2 demonstrates the feasibility of a strategy to disrupt interactions between two important intracellular proteins.
Repression of expression by microRNA
A third possibility that has been mooted is to exploit the regulation of estrogen-related receptor alpha expression by the microRNA miR-137.97 This microRNA reduced expression of estrogen-related receptor alpha in breast cancer cells and consequently reduced breast cancer cell proliferation and migration.97 Therapeutic intervention with inhibitory RNAs is in its infancy, but the existence of this regulatory control of estrogen-related receptor alpha provides an alternative strategy for therapeutic intervention.
Phosphorylation
Another possible intervention with which to inhibit estrogen-related receptor alpha activity would be a drug designed to inhibit the activity of the intracellular signal transduction proteins responsible for activation of the receptor by phosphorylation. Inhibitors that target the IGF and EGF pathways6,30 are likely to affect the transcriptional activity of the estrogen-related receptor alpha.93 Examples of these drugs are figitumumab and lapatinib.30,35 An advantage of such a therapeutic approach would be that other tumor promotion signal transduction induced through the growth factor pathway would be prevented.
Clinical setting
Drugs that target estrogen-related receptor alpha are most likely to be introduced initially for the treatment of patients with advanced breast cancer, most probably with palliative intent (Figure 2E). There is evidence that the estrogen-related receptor alpha may be an effective target in estrogen-dependent and estrogen-independent tumors. The dearth of effective noncytotoxic drugs for the treatment of patients with triple-negative breast tumors that lack expression of estrogen receptor, progesterone receptor, and HER2 means that these patients are more likely to be offered such novel therapy. In addition, there is evidence from early clinical trials of SR16388 that estrogen-related receptor alpha inhibitors are effective in patients who have relapsed after treatment with tamoxifen or an aromatase inhibitor. Such patients might benefit from estrogen-related receptor alpha inhibitors.
Evidence of clinical benefit from drugs that target estrogen-related receptor alpha in the palliative setting would indicate that they should be considered for adjuvant therapy in patients diagnosed with early triple-negative breast cancers (Figure 2C). It would be necessary to consider if drugs that target estrogen-related receptor alpha activity should be given in combination with cytotoxic therapies or as single agents. The decision might be influenced by patient prognosis.
Patient stratification
Increasingly, promulgation of new drugs is accompanied by consideration of which patients are most likely to receive clinical benefit. While pharmaceutical companies might wish that their novel drug could benefit, for example, all breast cancer patients, they must recognize that it is better to demonstrate activity in a subgroup of patients than no significant effect in all patients. Analogous to the estrogen receptor alpha, patients in whom the estrogen-related receptor alpha is expressed at high levels in tumor cells might be more likely to receive benefit. Presence of high concentrations of the coactivators PGC-1α and PGC-1β may also indicate that the patient is more likely to benefit from drugs that target the dependence of tumor cells on the estrogen-related receptor alpha. It is possible that assessment of the metabolic reliance of the tumor cells might select those most likely to benefit. Measurement of genes whose expression is particularly closely dependent on the activity of the estrogen-related receptor alpha, for instance, those that encode aromatase or TFF1, or enzymes in the glycolytic pathway or enzymes involved in oxidative phosphorylation, might predict the dependence of the tumor cells on its transcriptional pathway.
Estrogen receptor alpha might be a biomarker that predicts lack of response to drugs that target estrogen-related receptor alpha activity because the presence of estrogen receptor alpha and the progesterone receptor indicate the importance of the estrogen-responsive transcriptional pathway in the tumor cells. In addition, based on the evidence that the estrogen-related receptor alpha may inhibit estrogen receptor alpha activity, inhibition of the former might promote the activity of the latter and enhance the progression of the estrogen-dependent breast disease.70,129,130,131 Other evidence suggests that inhibition of estrogen-related receptor alpha might potentiate the effects of fulvestrant.114
Potential side effects
Consideration of the therapeutic potential of the estrogen-related receptor alpha encompasses its effectiveness in target malignant cells and its effects in other cells and other tissues. Clearly, toxicity studies and early clinical trials will investigate these effects and potential side effects, and dose limitation work will determine the optimum therapeutic threshold.
Osteoporosis
The effects of interventions that target exclusively the estrogen-related receptor alpha are unknown. It is thought that estrogen-related receptor alpha increases adipocyte formation at the expense of osteoblast formation and activity.81 This bone inhibitory role is supported by the demonstration that, in estrogen-related receptor alpha-null mice, the bone mineral density does not reduce with age.82 The protective effect prevails during estrogen deficiency. The true effects of estrogen-related receptor alpha on osteogenesis in vivo in humans are particularly important given the widespread incidence of osteoporosis in many breast cancer patients, the drug-limiting side effects of pure antiestrogens and aromatase inhibitors on bone density, and the potential complications of treatment in patients who may have received bisphosphonates or their replacements. SRI International has reported that early trial data indicate that the effect of SR16388, which inhibits both estrogen receptor alpha and estrogen-related receptor alpha, on bone mineral density is comparable to that of aromatase inhibitors.
Metastasis
Most relevant studies of estrogen-related receptor alpha activity suggest that it promotes reduction of bone formation.81,82 The bone is a favored site of breast cancer cell metastasis. The possibility that an inhibitor of estrogen-related receptor alpha activity might strengthen bone structure suggests that such a drug might, as a consequence, militate against the establishment of clinically significant metastatic deposits of breast cancer secondary tumors in bones. Such an activity would be desirable in the treatment of advanced breast cancer and could augment the effects of bisphosphonates. Evidence from a preclinical mouse model of bone metastasis contradicts this supposition; overexpression of estrogen-related receptor alpha in the tumor cells reduced formation of osteolytic lesions. The corollary would be that inhibition of estrogen-related receptor alpha activity might promote formation of bone metastases.100
Metabolism
Many of the inverse agonists of the estrogen-related receptor alpha have been produced with the aim of managing metabolic diseases. Consideration must be given to the potential of detrimental side effects of oncological intervention via estrogen-related receptor alpha on metabolic control in normal tissues with high energy requirements, such as skeletal muscle, heart, and kidney. There is concern that the decrease in PGC-1α and PGC-1β in the skeletal muscle of patients with diabetes and obesity would mean that inhibition of estrogen-related receptor alpha activity could be particularly detrimental in diabetic or obese breast cancer patients.132
Another consideration is that esrra-null mice exhibit cardiac hypertrophy.79 It would be important to consider potential detrimental effects of drugs that target estrogen-related receptor activity on patients with underlying heart conditions, including cardiomyopathy. Combined treatment with drugs associated with heart damage, such as anthracyclines and trastuzumab, might be contraindicated.
Cachexia
A major concern is that estrogen-related receptor alpha inhibition would be catastrophic for patients with established cachexia or would exacerbate its development. Poorly understood, cachexia is a major cause of morbidity and contributor to mortality in patients with advanced breast cancer. Cachexic patients have extensive muscle atrophy and fatigue, as well as severe weight loss associated with impaired metabolic function and nutrient sensing ability. The possibility that this condition might be worsened or hastened in breast cancer patients by drugs that inhibit estrogen-related receptor alpha activity is a major concern and should be a focus of any early clinical trials that involve dose escalation of the drugs.
Conclusion
Estrogen-related receptor alpha is an attractive target in breast cancer patients, in particular, in those for whom there are limited therapeutic options in either the palliative or adjuvant clinical setting. Several compounds that have inverse agonist activity for the receptor have been developed, and some have been shown to have high affinity and good oral availability and to interact with the ligand-binding domain to destabilize the active conformation and inhibit the transcriptional activity of the receptor. Preclinical studies suggest that inhibitors of its activity prevent breast cancer cell migration in vitro and proliferation in vivo in mouse models. The possibility that they might enhance metastasis to the bone warrants further investigation. The pivotal role of estrogen-related receptor alpha in energy metabolism suggests that its inhibition might target the high metabolism characteristic of many tumor cells.
Being an attractive target is a long way from introduction into routine clinical practice. Important areas of investigation include identification of valid biomarkers of dependence upon estrogen-related receptor alpha transcriptional activity and potential response to therapeutic intervention. Successful identification of such biomarkers would ensure effective patient stratification and that only those with a high probability of benefit would receive treatment. The important roles of estrogen-related receptor alpha in energy metabolism and bone formation mean that inhibition of its effects in tissues with high energy requirements and in bone must be considered. Further, effects of inhibition of estrogen-related receptor alpha activity in those with metabolic disorders or diabetes, many of whom will develop breast cancer,6 will require careful investigation.
Acknowledgments
The author is grateful to the Breast Cancer Campaign and Cancer Research UK for their support of our work in some of the areas covered in this review. The author is also indebted to colleagues for invaluable discussions and for their important contributions to this review.
Disclosure
The author reports no conflicts of interest in this work.
References
GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. [homepage on the Internet]. World Health Organization. Available from: http://globocan.iarc.fr/Default.aspx. Accessed January 20, 2014 | |
Cummings MC, Simpson PT, Reid LE, et al. Metastatic progression of breast cancer: insights from 50 years of autopsies. J Pathol. 2014;232(1): 23–31. | |
Gori S, Rimondini S, De Angelis V, et al. Central nervous system metastases in HER-2-positive metastatic breast cancer patients treated with trastuzumab: incidence, survival, and risk factors. Oncologist. 2007;12(7):766–773. | |
Zagouri F, Sergentanis TN, Bartsch R, et al. Intrathecal administration of trastuzumab for the treatment of meningeal carcinomatosis in HER2-positive metastatic breast cancer: a systematic review and pooled analysis. Breast Cancer Res Treat. 2013;139(1):13–22. | |
Collaborative Group on Hormonal Factors in Breast Cancer. Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet. 2001;358(9291): 1389–1399. | |
Westley RL, May FEB. A twenty-first century cancer epidemic caused by obesity: the involvement of insulin, diabetes, and insulin-like growth factors. Int J Endocrinol. 2013;632461. | |
Allen NE, Beral V, Casabonne D, et al; Million Women Study Collaborators. Moderate alcohol intake and cancer incidence in women. J Natl Cancer Inst. 2009;101(5):296–305. | |
McCullough LE, Eng SM, Bradshaw PT, et al. Fat or fit: the joint effects of physical activity, weight gain, and body size on breast cancer risk. Cancer. 2012;118(19):4860–4868. | |
Schneider G, Kirschner MA, Berkowitz R, Ertel NH. Increased estrogen production in obese men. J Clin Endocr Metab. 1979;48(4):633–638. | |
Lamar CA, Dorgan JF, Longcope C, Stanczyk FZ, Falk RT, Stephenson HE Jr. Serum sex hormones and breast cancer risk factors in postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2003; 12(4):380–383. | |
Johnson RH, Chien FL, Bleyer A. Incidence of breast cancer with distant involvement among women in the United States, 1976 to 2009. JAMA. 2013;309(8):800–805. | |
Sestak I, Cuzick J. Preventive therapy for breast cancer. Curr Oncol Rep. 2012;14(6):568–573. | |
Cuzick J, Sestak I, Bonanni B, et al; SERM Chemoprevention of Breast Cancer Overview Group. Selective oestrogen receptor modulators in prevention of breast cancer: an updated meta-analysis of individual participant data. Lancet. 2013;381(9880):1827–1834. | |
Williams PT. Breast cancer mortality vs exercise and breast size in runners and walkers. Plos One. 2013;8(12):e80616. | |
Jørgensen KJ1, Gøtzsche PC. Overdiagnosis in publicly organised mammography screening programmes: systematic review of incidence trends. BMJ. 2009;339. | |
Independent UK Panel on Breast Cancer Screening. The benefits and harms of breast cancer screening: an independent review. Lancet. 2012;380(9855):1778–1786. | |
Baum M. Harms from breast cancer screening outweigh benefits if death caused by treatment is included. BMJ. 2013;346;f385. | |
Heyes GJ, Mill AJ, Charles MW. Mammography-oncogenecity at low doses. J Radiol Prot. 2009;29(2A):A123–A132. | |
Effects of adjuvant tamoxifen and of cytotoxic therapy on mortality in early breast cancer. An overview of 61 randomized trials among 28,896 women. Early Breast Cancer Trialists’ Collaborative Group. N Engl J Med. 1988;319(26):1681–1692. | |
Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group. Lancet. 1998;351(9114):1451–1467. | |
Sestak I, Distler W, Forbes JF, Dowsett M, Howell A, Cuzick J. Effect of body mass index on recurrences in tamoxifen and anastrozole treated women: an exploratory analysis from the ATAC trial. J Clin Oncol. 2010;28(21):3411–3415. | |
Hershman DL, Kushi LH, Shao T, et al. Early discontinuation and nonadherence to adjuvant hormonal therapy in a cohort of 8,769 early-stage breast cancer patients. J Clin Oncol. 2010;28(27): 4120–4128. | |
van Herk-Sukel MPP, van de Poll-Franse LV, Voogd AC, Nieuwenhuijzen GAP, Coebergh JWW, Herings RMC. Half of breast cancer patients discontinue tamoxifen and any endocrine treatment before the end of the recommended treatment period of 5 years: a population-based analysis. Breast Cancer Res Treat. 2010;122(3):843–851. | |
Davies C, Pan H, Godwin J, et al; Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) Collaborative Group. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381(9869):805–816. | |
Miller KD, Dieras V, Harbeck N, et al. Phase IIa Trial of Trastuzumab Emtansine With Pertuzumab for Patients With Human Epidermal Growth Factor Receptor 2-Positive, Locally Advanced, or Metastatic Breast Cancer. J Clin Oncol. Epub 2014 April 14. | |
Welslau M, Dieras V, Sohn JH, et al. Patient-reported outcomes from EMILIA, a randomized phase 3 study of trastuzumab emtansine (T-DM1) versus capecitabine and lapatinib in human epidermal growth factor receptor 2-positive locally advanced or metastatic breast cancer. Cancer. 2014;120(5):642–651. | |
Rios J, Puhalla S. PARP inhibitors in breast cancer: BRCA and beyond. Oncology. 2011;25(11):1014–1025. | |
Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2012;9(1):16–32. | |
Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. 2012;12(8):553–563. | |
Tebbutt N, Pedersen MW, Johns TG. Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer. 2013;13(9):663–673. | |
Clemmons DR. Modifying IGF1 activity: an approach to treat endocrine disorders, atherosclerosis and cancer. Nature Rev Drug Discov. 2007;6(10):821–833. | |
Stewart AJ, Johnson MD, May FEB, Westley BR. Role of insulin-like growth factors and the type I insulin-like growth factor receptor in the estrogen-stimulated proliferation of human breast cancer cells. J Biol Chem. 1990;265(34):21172–21178. | |
Davison Z, de Blacquière GE, Westley BR, May FEB. Insulin-like growth factor-dependent proliferation and survival of triple-negative breast cancer cells: implications for therapy. Neoplasia. 2011;13(6):504–515. | |
Westley BR, Clayton SJ, Daws MR, Molloy CA, May FEB. Interactions between the oestrogen and insulin-like growth factor signalling pathways in the control of breast epithelial cell proliferation. Biochem Soc Symp. 1998;(63):35–44. | |
Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12(3):159–169. | |
Zardavas D, Baselga J, Piccart M. Emerging targeted agents in metastatic breast cancer. Nat Rev Clin Oncol. 2013;10(4):191–210. | |
Zardavas D, Fumagalli D, Loi S. Phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin pathway inhibition: a breakthrough in the management of luminal (ER+/HER2-) breast cancers? Curr Opin Oncol. 2012;24(6):623–634. | |
Hatzivassiliou G, Haling JR, Chen HF, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501(7466):232–236. | |
Bendell JC, Rodon J, Burris HA, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282–290. | |
Ibrahim YH, García-García C, Serra V, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2(11):1036–1047. | |
Gucalp A, Tolaney S, Isakoff SJ, et al; Translational Breast Cancer Research Consortium (TBCRC 011). Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic breast cancer. Clin Cancer Res. 2013;19(19):5505–5512. | |
Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11(8): 558–572. | |
Pandya K, Meeke K, Clementz AG, et al. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br J Cancer. 2011;105(6): 796–806. | |
Huang EH, Singh B, Cristofanilli M, et al. A CXCR4 antagonist CTCE-9908 inhibits primary tumor growth and metastasis of breast cancer. J Surg Res. 2009;155(2):231–236. | |
Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1): 9–22. | |
Wong CCL, Zhang HF, Gilkes DM, et al. Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J Mol Med (Berl). 2012;90(7):803–815. | |
Jacobson HI, Gupta GN, Fernandez C, Hennix S, Jensen EV. Determination of tritium in biological material. Arch Biochem Biophys. 1960;86:89–93. | |
Blair RM, Fang H, Branham WS, et al. The estrogen receptor relative binding affinities of 188 natural and xenochemicals: structural diversity of ligands. Toxicol Sci. 2000;54(1):138–153. | |
May FEB, Westley BR. Identification and characterization of estrogen-regulated Rnas in human-breast cancer-cells. J Biol Chem. 1988;263(26):12901–12908. | |
Wright PK, May FEB, Darby S, Saif R, Lennard TWJ, Westley BR. Estrogen regulates vesicle trafficking gene expression in EFF-3, EFM-19 and MCF-7 breast cancer cells. Int J Clin Exp Pathol. 2009;2(5):463–475. | |
Xu JM, Wu RC, O’Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer. 2009;9(9):615–630. | |
Manavathi B, Dey O, Gajulapalli VNR, Bhatia RS, Bugide S, Kumar R. Derailed estrogen signaling and breast cancer: an authentic couple. Endocr Rev. 2013;34(1):1–32. | |
Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L. Cracking the estrogen receptor’s posttranslational code in breast tumors. Endocr Rev. 2011;32(5):597–622. | |
Walter P, Green S, Greene G, et al. Cloning of the human estrogen-receptor cDNA. Proc Natl Acad Sci U S A. 1985;82(23):7889–7893. | |
Green S, Walter P, Greene G, et al. Cloning of the human estrogen-receptor cDNA. J Steroid Biochem. 1986;24(1):77–83. | |
Kuiper GGJM, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel estrogen receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A. 1996;93(12):5925–5930. | |
Giguère V, Yang N, Segui P, Evans RM. Identification of a new class of steroid-hormone receptors. Nature. 1988;331(6151):91–94. | |
Chen F, Zhang Q, McDonald T, et al. Identification of two hERR2-related novel nuclear receptors utilizing bioinformatics and inverse PCR. Gene. 1999;228(1–2):101–109. | |
Schwabe JWR, Chapman L, Finch JT, Rhodes D. The crystal-structure of the estrogen-receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell. 1993;75(3): 567–578. | |
Klein-Hitpass L, Schorpp M, Wagner U, Ryffel GU. An estrogen-responsive element derived from the 5′ flanking region of the xenopus vitellogenin A2 gene functions in transfected human-cells. Cell. 1986;46(7):1053–1061. | |
May FEB, Smith DJ, Westley BR. The human cathepsin D-encoding gene is transcribed from an estrogen-regulated and a constitutive start point. Gene. 1993;134(2):277–282. | |
Berry M, Nunez AM, Chambon P. Estrogen-responsive element of the human pS2 gene is an imperfectly palindromic sequence. Proc Natl Acad Sci U S A. 1989;86(4):1218–1222. | |
May FEB, Westley BR. Estrogen regulated messenger RNAs in human breast cancer cells. Biomed Pharmacother. 1995;49(9):400–414. | |
Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29(14):2905–2919. | |
May FEB, Westley BR. Cloning of estrogen-regulated messenger RNA sequences from human breast cancer cells. Cancer Res. 1986;46(12): 6034–6040. | |
Prest SJ, May FEB, Westley BR. The estrogen-regulated protein, TFF1, stimulates migration of human breast cancer cells. FASEB J. 2002;16(2):592–594. | |
Yang NY, Shigeta H, Shi HP, Teng CT. Estrogen-related receptor, hERR1, modulates estrogen receptor-mediated response of human lactoferrin gene promoter. J Biol Chem. 1996;271(10):5795–5804. | |
Vanacker JM, Delmarre C, Guo XJ, Laudet V. Activation of the osteopontin promoter by the orphan nuclear receptor estrogen receptor related alpha. Cell Growth Differ. 1998;9(12):1007–1014. | |
Vanacker JM, Bonnelye E, Delmarre C, Laudet V. Activation of the thyroid hormone receptor alpha gene promoter by the orphan nuclear receptor ERR alpha. Oncogene. 1998;17(19):2429–2435. | |
Yang C, Zhou DJ, Chen S. Modulation of aromatase expression in the breast tissue by ERR alpha-1 orphan receptor. Cancer Res. 1998;58(24):5695–5700. | |
Lu DS, Kiriyama Y, Lee KY, Giguère V. Transcriptional regulation of the estrogen-inducible pS2 breast cancer marker gene by the ERR family of orphan nuclear receptors. Cancer Res. 2001;61(18):6755–6761. | |
Brzozowski AM, Pike ACW, Dauter Z, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389(6652): 753–758. | |
Danielian PS, White R, Lees JA, Parker MG. Identification of a conserved region required for hormone dependent transcriptional activation by steroid hormone receptors. EMBO J. 1992;11(3):1025–1033. | |
Shiau AK, Barstad D, Loria PM, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95(7):927–937. | |
Kallen J, Schlaeppi JM, Bitsch F, et al. Evidence for ligand-independent transcriptional activation of the human estrogen-related receptor alpha (ERRalpha): crystal structure of ERRalpha ligand binding domain in complex with peroxisome proliferator-activated receptor coactivator-1alpha. J Biol Chem. 2004;279(47):49330–49337. | |
Suetsugi M, Su L, Karlsberg K, Yuan YC, Chen S. Flavone and isoflavone phytoestrogens are agonists of estrogen-related receptors. Mol Cancer Res. 2003;1(13):981–991. | |
Coward P, Lee D, Hull MV, Lehmann JM. 4-Hydroxytamoxifen binds to and deactivates the estrogen-related receptor gamma. Proc Natl Acad Sci U S A. 2001;98(15):8880–8884. | |
Luo JM, Sladek R, Carrier J, Bader JA, Richard D, Giguère V. Reduced fat mass in mice lacking orphan nuclear receptor estrogen-related receptor alpha. Mol Cell Biol. 2003;23(22):7947–7956. | |
Huss JM, Imahashi KI, Dufour CR, et al. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6(1):25–37. | |
Bonnelye E, Vanacker JM, Dittmar T, et al. The ERR-1 orphan receptor is a transcriptional activator expressed during bone development. Mol Endocrinol. 1997;11(7):905–916. | |
Delhon I, Gutzwiller S, Morvan F, et al. Absence of estrogen receptor-related-alpha increases osteoblastic differentiation and cancellous bone mineral density. Endocrinology. 2009;150(10):4463–4472. | |
Teyssier C, Gallet M, Rabier B, et al. Absence of ERR alpha in female mice confers resistance to bone loss induced by age or estrogen-deficiency. PLoS One. 2009;4(11). | |
Puigserver P, Wu ZD, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–839. | |
Yoon JC, Puigserver P, Chen GX, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413(6852):131–138. | |
Huss JM, Torra IP, Staels B, Giguère V, Kelly DP. Estrogen-related receptor alpha directs peroxisome proliferator-activated receptor at signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol. 2004;24(20):9079–9091. | |
Mootha VK, Handschin C, Arlow D, et al. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci U S A. 2004;101(17):6570–6575. | |
Kressler D, Schreiber SN, Knutti D, Kralli A. The PGC-1-related protein PERC is a selective coactivator of estrogen receptor alpha. J Biol Chem. 2002;277(16):13918–13925. | |
Lin JD, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 beta (PGC-1 beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277(3):1645–1648. | |
Pérez-Schindler J, Summermatter S, Salatino S, et al. The corepressor NCoR1 antagonizes PGC-1α and estrogen-related receptor α in the regulation of skeletal muscle function and oxidative metabolism. Mol Cell Biol. 2012;32(24):4913–4924. | |
Castet A, Herledan A, Bonnet S, Jalaguier S, Vanacker JM, Cavailles V. Receptor-interacting protein 140 differentially regulates estrogen receptor-related receptor transactivation depending on target genes. Mol Endocrinol. 2006;20(5):1035–1047. | |
Barry JB, Giguère V. Epidermal growth factor-induced signaling in breast cancer cells resufts in selective target gene activation by orphan nuclear receptor estrogen-related receptor alpha. Cancer Res. 2005;65(14):6120–6129. | |
Ariazi EA, Kraus RJ, Farrell ML, Jordan VC, Mertz JE. Estrogen-related receptor alpha 1 transcriptional activities are regulated in part via the ErbB2/HER2 signalling pathway. Mol Cancer Res. 2007;5(1): 71–85. | |
Chang CY, Kazmin D, Jasper JS, Kunder R, Zuercher WJ, McDonnell DP. The metabolic regulator ERRα, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell. 2011;20(4):500–510. | |
Tremblay AM, Wilson BJ, Yang XJ, Giguère V. Phosphorylation-dependent sumoylation regulates estrogen-related receptor-alpha and -gamma transcriptional activity through a synergy control motif. Mol Endocrinol. 2008;22(3):570–584. | |
Wilson BJ, Tremblay AM, Deblois G, Sylvain-Drolet G, Giguère V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor alpha. Mol Endocrinol. 2010;24(7): 1349–1358. | |
Dwyer MA, Joseph JD, Wade HE, et al. WNT11 expression is induced by estrogen-related receptor alpha and beta-catenin and acts in an autocrine manner to increase cancer cell migration. Cancer Res. 2010;70(22):9298–9308. | |
Zhao YY, Li YP, Lou GY, et al. MiR-137 targets estrogen-related receptor alpha and impairs the proliferative and migratory capacity of breast cancer cells. PLoS One. 2012;7(6). | |
Stein RA, Chang CY, Kazmin DA, et al. Estrogen-related receptor alpha is critical for the growth of estrogen receptor-negative breast cancer. Cancer Res. 2008;68(21):8805–8812. | |
Deblois G, Chahrour G, Perry MC, Sylvain-Drolet G, Muller WJ, Giguère V. Transcriptional control of the ERBB2 amplicon by ERRalpha and PGC-1 beta promotes mammary gland tumorigenesis. Cancer Res. 2010;70(24):10277–10287. | |
Fradet A, Sorel H, Bouazza L, et al. Dual function of ERRα in breast cancer and bone metastasis formation: implication of VEGF and osteoprotegerin. Cancer Res. 2011;71(17):5728–5738. | |
Deblois G, Hall JA, Perry MC, et al. Genome-wide identification of direct target genes implicates estrogen-related receptor alpha as a determinant of breast cancer heterogeneity. Cancer Res. 2009;69(15): 6149–6157. | |
Chen EI, Hewel J, Krueger JS, et al. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007;67(4): 1472–1486. | |
Tiraby C, Hazen BC, Gantner ML, Kralli A. Estrogen-related receptor gamma promotes mesenchymal-to-epithelial transition and suppresses breast tumor growth. Cancer Res. 2011;71(7): 2518–2528. | |
Ariazi EA, Clark GM, Mertz JE. Estrogen-related receptor alpha and estrogen-related receptor gamma associate with unfavorable and favorable biomarkers, respectively, in human breast cancer. Cancer Res. 2002;62(22):6510–6518. | |
Suzuki T, Miki Y, Moriya T, et al. Estrogen-related receptor a in human breast carcinoma as a potent prognostic factor. Cancer Res. 2004;64(13):4670–4676. | |
Kallen J, Lattmann R, Beerli R, et al. Crystal structure of human estrogen-related receptor alpha in complex with a synthetic inverse agonist reveals its novel molecular mechanism. J Biol Chem. 2007;282(32):23231–23239. | |
Patch RJ, Searle LL, Kim AJ, et al. Identification of diaryl ether-based ligands for estrogen-related receptor α as potential antidiabetic agents. J Med Chem. 2011;54(3):788–808. | |
Xu S, Zhuang X, Pan X, et al. 1-Phenyl-4-benzoyl-1H-1,2,3-triazoles as orally bioavailable transcriptional function suppressors of estrogen-related receptor α. J Med Chem. 2013;56(11):4631–4640. | |
Hyatt SM, Lockamy EL, Stein RA, et al. On the intractability of estrogen-related receptor alpha as a target for activation by small molecules. J Med Chem. 2007;50(26):6722–6724. | |
Peng L, Gao X, Duan L, Ren X, Wu D, Ding K. Identification of pyrido[1,2-α]pyrimidine-4-ones as new molecules improving the transcriptional functions of estrogen-related receptor α. J Med Chem. 2011;54(21):7729–7733. | |
Yang C, Chen S. Two organochlorine pesticides, toxaphene and chlordane, are antagonists for estrogen-related receptor alpha-1 orphan receptor. Cancer Res. 1999;59(18):4519–4524. | |
Wang JJ, Fang F, Huang ZY, Wang YF, Wong CW. Kaempferol is an estrogen-related receptor alpha and gamma inverse agonist. FEBS Lett. 2009;583(4):643–647. | |
Busch BB, Stevens WC, Martin R, et al. Identification of a selective inverse agonist for the orphan nuclear receptor estrogen-related receptor alpha. J Med Chem. 2004;47(23):5593–5596. | |
Lanvin O, Bianco S, Kersual N, Chalbos D, Vanacker JM. Potentiation of ICI182,780 (Fulvestrant)-induced estrogen receptor-alpha degradation by the estrogen receptor-related receptor-alpha inverse agonist XCT790. J Biol Chem. 2007;282(39):28328–28334. | |
Wang J, Wang Y, Wong C. Oestrogen-related receptor alpha inverse agonist XCT-790 arrests A549 lung cancer cell population growth by inducing mitochondrial reactive oxygen species production. Cell Prolif. 2010;43(2):103–113. | |
Chen LH, Wong CW. Estrogen-related receptor alpha inverse agonist enhances basal glucose uptake in myotubes through reactive oxygen species. Biol Pharm Bull. 2009;32(7):1199–1203. | |
Nie YH, Wong CW. Suppressing the activity of ERR alpha in 3T3-L1 adipocytes reduces mitochondrial biogenesis but enhances glycolysis and basal glucose uptake. J Cell Mol Med. 2009;13(9B):3051–3060. | |
Wu F, Wang JJ, Wang YF, Kwok TT, Kong SK, Wong CW. Estrogen-related receptor alpha (ERRalpha) inverse agonist XCT-790 induces cell death in chemotherapeutic resistant cancer cells. Chem Biol Interact. 2009;181(2):236–242. | |
Bianco S, Lanvin O, Tribollet V, Macari C, North S, Vanacker JM. Modulating estrogen receptor-related receptor-alpha activity inhibits cell proliferation. J Biol Chem. 2009;284(35):23286–23292. | |
Chisamore MJ, Mosley RT, Cai SJ, et al. Identification of small molecule estrogen-related receptor alpha-specific antagonists and homology modeling to predict the molecular determinants as the basis for selectivity over ERR beta and ERR gamma. Drug Dev Res. 2008;69(4):203–218. | |
Chisamore MJ, Cunningham ME, Flores O, Wilkinson HA, Chen JD. Characterization of a novel small molecule subtype specific estrogen-related receptor alpha antagonist in MCF-7 breast cancer cells. Plos One. 2009;4(5). | |
Chisamore MJ, Wilkinson HA, Flores O, Chen JD. Estrogen-related receptor-alpha antagonist inhibits both estrogen receptor-positive and estrogen receptor-negative breast tumor growth in mouse xenografts. Mol Cancer Ther. 2009;8(3):672–681. | |
Wang YF, Fang F, Wong CW. Troglitazone is an estrogen-related receptor alpha and gamma inverse agonist. Biochem Pharmacol. 2010;80(1):80–85. | |
Fiori JL, Sanghvi M, O’Connell MP, Krzysik-Walker SM, Moaddel R, Bernier M. The cannabinoid receptor inverse agonist AM251 regulates the expression of the EGF receptor and its ligands via destabilization of oestrogen-related receptor alpha protein. Br J Pharmacol. 2011;164(3): 1026–1040. | |
Krzysik-Walker SM, González-Mariscal I, Scheibye-Knudsen M, Indig FE, Bernier M. The biarylpyrazole compound AM251 alters mitochondrial physiology via proteolytic degradation of ERR alpha. Mol Pharmacol. 2013;83(1):157–166. | |
Duellman SJ, Calaoagan JM, Sato BG, et al. A novel steroidal inhibitor of estrogen-related receptor alpha (ERR alpha). Biochem Pharmacol. 2010;80(6):819–826. | |
Chao WR, Amin K, Shi YH, et al. SR16388: a steroidal antiangiogenic agent with potent inhibitory effect on tumor growth in vivo. Angiogenesis. 2011;14(1):1–16. | |
Gaillard S, Grasfeder LL, Haeffele CL, et al. Receptor-selective coactivators as tools to define the biology of specific receptor-coactivator pairs. Mol Cell. 2006;24(5):797–803. | |
Zhang ZP, Teng CT. Estrogen receptor alpha and estrogen receptor-related receptor alpha 1 compete for binding and coactivator. Mol Cell Endocrinol. 2001;172(1–2):223–233. | |
Kraus RJ, Ariazi EA, Farrell ML, Mertz JE. Estrogen-related receptor alpha 1 actively antagonizes estrogen receptor-regulated transcription in MCF-7 mammary cells. J Biol Chem. 2002;277(27):24826–24834. | |
Watanabe A, Kinoshita Y, Hosokawa K, Mori T, Yamaguchi T, Honjo H. Function of estrogen-related receptor alpha in human endometrial cancer. J Clin Endocrinol Metab. 2006;91(4):1573–1577. | |
Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1 alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.