")
Back to Journals » OncoTargets and Therapy » Volume 8
Novel covalent modification of human anaplastic lymphoma kinase (ALK) and potentiation of crizotinib-mediated inhibition of ALK activity by BNP7787
Authors Parker A, Petluru P, Nienaber V, Zhao M, Ayala P, Badger J, Chie-Leon B, Sridhar V, Logan C, Kochat H, Hausheer F
Received 3 September 2014
Accepted for publication 22 October 2014
Published 4 February 2015 Volume 2015:8 Pages 375—383
DOI https://doi.org/10.2147/OTT.S73690
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Aulma R Parker,1 Pavankumar N Petluru,1 Vicki L Nienaber,2 Min Zhao,1 Philippe Y Ayala,1 John Badger,2 Barbara Chie-Leon,2 Vandana Sridhar,2 Cheyenne Logan,2 Harry Kochat,1 Frederick H Hausheer1
1BioNumerik Pharmaceuticals, Inc., San Antonio, TX, USA; 2Zenobia Therapeutics, Inc., La Jolla, CA, USA
Abstract: BNP7787 (Tavocept, disodium 2,2’-dithio-bis-ethanesulfonate) is a novel, investigational, water-soluble disulfide that is well-tolerated and nontoxic. In separate randomized multicenter Phase II and Phase III clinical trials in non-small-cell lung cancer (NSCLC) patients, treatment with BNP7787 in combination with standard chemotherapy resulted in substantial increases in the overall survival of patients with advanced adenocarcinoma of the lung in the first-line treatment setting. We hypothesized that BNP7787 might interact with and modify human anaplastic lymphoma kinase (ALK). At least seven different variants of ALK fusions with the gene encoding the echinoderm microtubule-associated protein-like 4 (EML4) are known to occur in NSCLC. EML4–ALK fusions are thought to account for approximately 3% of NSCLC cases. Herein, we report the covalent modification of the kinase domain of human ALK by a BNP7787-derived mesna moiety and the functional consequences of this modification in ALK assays evaluating kinase activity. The kinase domain of the ALK protein crystallizes as a monomer, and BNP7787-derived mesna-cysteine adducts were observed at Cys 1235 and Cys 1156. The BNP7787-derived mesna adduct at Cys 1156 is located in close proximity to the active site and results in substantial disorder of the P-loop and activation loop (A-loop). Comparison with the P-loop of apo-ALK suggests that the BNP7787-derived mesna adduct at Cys 1156 interferes with the positioning of Phe 1127 into a small pocket now occupied by mesna, resulting in a destabilization of the loop's binding orientation. Additionally, in vitro kinase activity assays indicate that BNP7787 inhibits ALK catalytic activity and potentiates the activity of the ALK-targeted drug crizotinib.
Keywords: adenocarcinoma, ALK, BNP7787, chemo-enhancing, crizotinib, non-small-cell lung cancer, Tavocept
Introduction
BNP7787 (Tavocept, disodium 2,2’-dithio-bis-ethanesulfonate) is an investigational agent that is a novel, water-soluble disulfide that has been evaluated in Phase II and Phase III clinical trials in patients with advanced non-small-cell lung cancer (NSCLC).1 The American Cancer Society estimates that approximately 159,260 deaths from lung cancer will occur in 2014 accounting for approximately 27% of all cancer deaths in the US.2 Worldwide more than 1.59 million deaths per year are attributed to lung cancer.3 Approximately 40–65% of these deaths will be due to primary adenocarcinoma of the lung/bronchus.2 While there have been some advances in chemotherapeutic agents for the treatment of NSCLC, and improved treatments are available for the management of toxic side effects, there have been only modest gains in survival for patients with advanced disease. For example, modest improvements in median duration of survival have been reported with the use of bevacizumab (Avastin) and cetuximab (Erbitux) in combination with platinum-doublet chemotherapy for the treatment of patients with advanced NSCLC.4,5 Therefore, there remains an unmet need for a treatment that can offer patients with advanced NSCLC a greater clinical benefit that is measured by increases in survival and reduction in toxic side effects, with a manageable or improved safety profile.
In separate randomized multicenter Phase II and Phase III clinical trials in NSCLC patients, treatment with BNP7787 in combination with standard chemotherapy (platinum and taxane) resulted in substantial increases in the overall survival of patients with advanced adenocarcinoma of the lung in the first-line treatment setting (Hausheer et al1 and Hausheer et al, unpublished data, 2014). BNP7787 does not interfere with paclitaxel-induced apoptosis or with taxane-, platinum-, vinca alkaloid-, or epothilone-induced cytotoxicity in human cancer cell lines.6 Additionally, studies in animals demonstrated that BNP7787 did not exhibit tumor protection when given with taxane and platinum agents.7,8 Importantly, in Phase I and Phase III clinical trials, BNP7787 did not interfere with the antitumor activity of cisplatin and paclitaxel.1,9 Unlike thiol-containing drugs such as mesna and amifostine, BNP7787 can be administered to patients in large doses (eg, 18.4 g/m2) without notable perturbations in plasma disulfide proportions or in the strongly oxidizing extracellular environment and without any toxic side effects.6 We have previously reported that thiol-containing drugs may dramatically disrupt the normal homeostatic thiol and disulfide balance both in the plasma and inside the cell, and that they are often accompanied by toxic side effects;6 however, for disulfide-containing drugs like BNP7787, there is no obvious mechanism through which they would substantially perturb the ratios of thiols and disulfides in these compartments.6,7,10
Based on computational modeling, we hypothesized that BNP7787 might interact with and modify human anaplastic lymphoma kinase (ALK). ALK belongs to the family of insulin growth factor receptor tyrosine kinases, and fusions of ALK with other genes are common in several diseases and cancers.11 Our focus was on ALK fusions found in NSCLC. At least seven different variants of ALK fusions with the gene encoding the echinoderm microtubule-associated protein-like 4 (EML4) are known to occur in NSCLC; EML4–ALK variants are constitutively active, exhibiting gain of function.12 Additionally, fusions between the tropomyosin receptor kinase fused gene (TFG) and ALK (TFG–ALK), and KIF5B and ALK (KIF5B–ALK), are also known to occur in NSCLC.11,13,14 EML4–ALK fusions are thought to account for approximately 3% of NSCLC cases.11 ALK is coupled to numerous signaling pathways that regulate cell proliferation including Ras–ERK, JAK3–STAT3, and PI3K15 and, therefore, represents an important target for anticancer-drug development. Herein, we characterize the structural and functional consequences of BNP7787 on the kinase domain of human ALK. Specifically, we determine the effect of BNP7787 on ALK activity in the presence and absence of the known ALK inhibitor, crizotinib (PF02341066), and report a high-resolution X-ray crystal structure of the kinase domain of ALK bearing two covalent BNP7787-derived mesna-cysteine adducts.
Materials and methods
Reagents for in vitro kinase assays
N-terminal 6His tagged recombinant human ALK expressed in baculovirus Sf21 was purchased from EMD Millipore (Billerica, MA, USA) (purity ≥60% by sodium dodecyl sulfate polyacrylamide gel electrophoresis) and aliquoted to 1 μL fractions when it was used the first time (to avoid multiple freeze/thaws for subsequent experiments). BNP7787 was prepared by a proprietary method (purity >97%, no mesna was detected by mass spectroscopy). Kinase inhibitor, PF02341066 (crizotinib), was purchased from Selleck Chemicals, LLC. (Houston TX, USA). Polyglutamate-tyrosine (PolyGT) substrate was purchased from Sigma-Aldrich (St. Louis, MO, USA). Kinase assay buffer was prepared and consisted of 20 mM HEPES, 0.1% Brij 96, 10 mM NaF, 1 mM Na3VO4, and 10 mM MnCl2 adjusted to a final pH of 7.5. Half-area 96-well microplates were purchased directly from Corning Incorporated (Corning, NY, USA). ADP-Glo reagents were purchased from Promega (Madison WI, USA) and consisted of ADP, ATP, ADP-Glo, kinase detection reagent buffer, and kinase detection substrate. All other reagents were purchased from Sigma-Aldrich Co (St Louis, MO, USA). A Tecan Ultra microplate reader with XFluor software (V4.51; Tecan [Morrisville, NC, USA]) and RdrOle software (V4.50; Tecan) were used in this study.
ALK kinase activity assay
Kinase activity was evaluated using the ADP-Glo system from Promega Corporation and monitored ADP produced when ALK phosphorylated the PolyGT substrate. Assays typically contained 4 ng/μL of ALK per assay and this concentration typically gave signal to background ratios of 12 or higher (relative to control). Each assay condition was run in triplicate and experiments were repeated independently on different days to ensure reproducibility of results. PolyGT (4:1 ratio) was used as the substrate for phosphorylation and had an average polymer mass ranging from 20,000 to 50,000 g/mol. Typically, 10-μL volume assays in half-area 96-well microtiter plates contained ALK (40 ng total or 4 ng/μL), ATP (100 μM), PolyGT substrate (0.2 μg/μL), and the concentrations of BNP7787 and/or crizotinib as indicated; additionally, kinase assay buffer was added to achieve a final volume of 10 μL per assay. For most assays, a stock of ATP (1 mM) and PolyGT (2 mg/mL) was mixed 1:1 to give an ATP/PolyGT master mix of 0.5 mM ATP and 1 mg/mL PolyGT. Crizotinib was dissolved as a 1 mM stock in dimethyl sulfoxide (DMSO) and then further diluted in kinase assay buffer (DMSO-only controls were run to ensure that DMSO did not interfere with the assay).
Cloning, expression, and purification of the kinase domain of ALK for X-ray crystallographic analyses
Wild type human ALK, consisting of residues 1095–1410, was cloned into a proprietary vector containing a C-terminal 6His tag. Isolated shuttle vector was transformed into DH10Bac cells. Colonies containing bacmid with transposed ALK DNA were picked and grown overnight at 37°C. Bacmid DNA was isolated by DNA isopropanol precipitation and resuspended in 100 μL of sterile water. Bacmid DNA was allowed to resuspend at room temperature for 1 hour prior to transfection. Recombinant bacmid DNA was expressed in SF9 cells at 27°C. The virus generated from a 72-hour infection was stored at 4°C.
Recombinant protein was expressed in SF9 cells at a multiplicity of infection of two in a 48-hour infection at 27°C. The cells were harvested by centrifugation and stored at −80°C. Purification of target protein was done using a two-column system (Ni-NTA and size exclusion). The cell biomass was lysed by sonification in 50 mM Tris-HCl pH 7.8, 500 mM NaCl, 10% glycerol, 20 mM imidazole (buffer A) plus Roche complete protease inhibitor tablets (Roche Diagnostics Corporation, Indianapolis, IN, USA), and 20,000 units benzonase. Target protein was extracted by binding Ni-NTA (Qiagen, Valencia, CA, USA). Protein was eluted with 250–500 mM imidazole pH 7.8. Peak fractions were pooled and aggregated protein was separated from monomeric protein via size exclusion (S200 16/60; GE Healthcare Lifesciences [Piscataway, NJ, USA]) in 50 mM bicine pH 8.4, 150 mM NaCl, 5 mM dithiothreitol (DTT). Monomeric protein was concentrated to ~19 mg/mL.
Preparation of BNP7787-derived mesna adducts on ALK crystals
ALK (19 mg/mL) in 50 mM bicine, pH 8.4, 150 mM NaCl, and 25 mM DTT was incubated at 4°C overnight to fully reduce the protein. DTT was removed by exchanging five times in 50 mM bicine, pH 8.4, 150 mM NaCl using ultrafiltration (10-kDa-cutoff Centricon filters). Fully reduced ALK was incubated with 5 mM BNP7787 and incubated at 4°C overnight. Protein was submitted for mass spectrometry analysis to confirm the presence of at least one BNP7787-derived mesna adduct prior to initiation of protein-crystallization experiments. The predicted mass for ALK was 36,868, and for ALK incubated with BNP7787 a mass of 37,194 was observed. This mass difference of 326 corresponds to two BNP7787-derived mesna moieties on the ALK protein (each mesna moiety has a molecular weight of 164; loss of a proton on the thiol group due to formation of a disulfide with a cysteine residue results in net mass increase of approximately 163 per mesna moiety).
Crystallization of ALK containing BNP7787-derived mesna adducts
Mass spectrometry analysis of C-terminal 6Xhis tag ALK that had been incubated with BNP7787 indicated two likely BNP7787-derived mesna adducts; however, we were unable to obtain crystals from these protein samples. As an alternative, apo-ALK crystals were soaked with BNP7787 and this yielded crystals with two BNP7787-derived mesna adducts. Briefly, the crystal of C-terminal 6Xhis tag ALK was obtained by sitting drop/vapor diffusion method by mixing 2 μL at 17 mg/mL protein (50 mM bicine pH 8.4, 150 mM NaCl, 5 mM DTT) with 2 μL of 0.1 M Tris-HCl pH 8.5, 0.2 M sodium acetate trihydrate, 30% w/v polyethylene glycol 4,000 at 20°C. Diffracting crystals appeared within 5–8 days. Before data collection, the crystals were soaked in 20 mM BNP7787 overnight and transferred into a cryoprotectant solution made up of 20% ethylene glycol v/v in crystallization buffer, after which they were flash-frozen in liquid nitrogen for data collection. Crystals diffracted to 2.1 Å. As previously mentioned, the mass spectrometry analysis of ALK after reaction with BNP7787 (data not shown) suggested two BNP7787-derived mesna adducts consistent with the X-ray structure.
Data collection and processing
Diffraction data were collected at the Advanced Light Source (ALS) (Berkeley, CA). BNP7787-derived mesna adducts were observed on Cys 1156 and Cys 1235. Data were processed using the program package Mosflm as part of the ccp4 program package. Image processing statistics are summarized in Table 1.
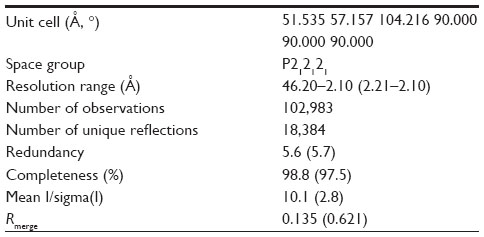 | Table 1 Crystal characteristics and data collection statistics |
Structure solution and refinement
Data were indexed, integrated, scaled, and merged using the program Mosflm. The structure was solved by molecular replacement with Phaser using a monomer from the Protein Data Bank,16 and an internal structure which is similar to 2XP2.17 The structure was consistent with one molecule in the crystal asymmetric unit. The protein model was iteratively refit and refined using MIFit18 and REFMAC5.19 The solved structure is supported by: 1) contiguous electron density for most of the molecule; 2) landmark side chain density features matching the amino acid sequence including cysteines; 3) absence of phi-psi violations; and 4) final R/Rfree values in the normal range. Residual density observed near Cys 1235 and Cys 1156 was modeled as BNP7787-derived mesna adducts. Final statistics are summarized in Table 2. A number of side-chain atoms and protein fragments were not refined. The missing fragments included Gly 1123–Gly 1128 (P-loop), Ser 1136–Pro 1144 (loop connecting β2–β3), Arg 1214–Pro 1218 (loop connecting αD–αE), and Ser 1281–Arg 1284 (part of activation loop or A-loop).
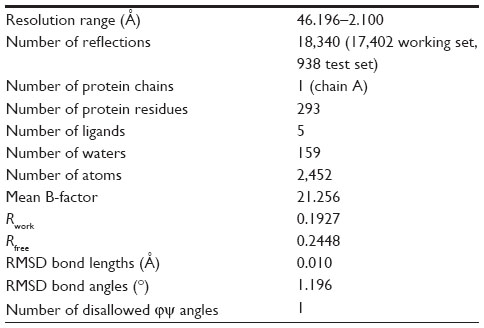 | Table 2 Crystallographic data and refinement statistics |
Results
Crystal structure of ALK bearing BNP7787-derived mesna-cysteine adducts
The crystal structure of ALK in complex with a BNP7787-derived mesna-cysteine adduct was completed at 2.1-Å resolution (Figure 1A; PDB ID code 4TT7). The protein crystallizes as a monomer in the asymmetric unit and BNP7787-derived mesna disulfide bonds were observed with Cys 1235 and Cys 1156. We refer to this process as BNP7787-mediated xenobiotic modification/modulation of protein cysteines. The mesna adduct at Cys 1156 is located in close proximity to the active site and in fact results in substantial disorder of P-loop (or phosphate binding loop) which is highly conserved in protein kinases. This disorder prevented full refinement of the P-loop. While a large fragment of the P-loop is missing from the refined structure, comparison with the P-loop of the apo-ALK suggests that the BNP7787-derived mesna adduct at Cys 1156 interferes with the positioning of Phe 1127 (one of the P-loop residues) into a small pocket now occupied by mesna, resulting in a destabilization of the loop’s binding orientation (Figure 1B). In order to accommodate the BNP7787-derived mesna adduct in the current crystal structure, Cys 1156 flips from a totally solvent exposed orientation (in the wild type ALK) to an inside orientation, thereby modifying the P-loop.
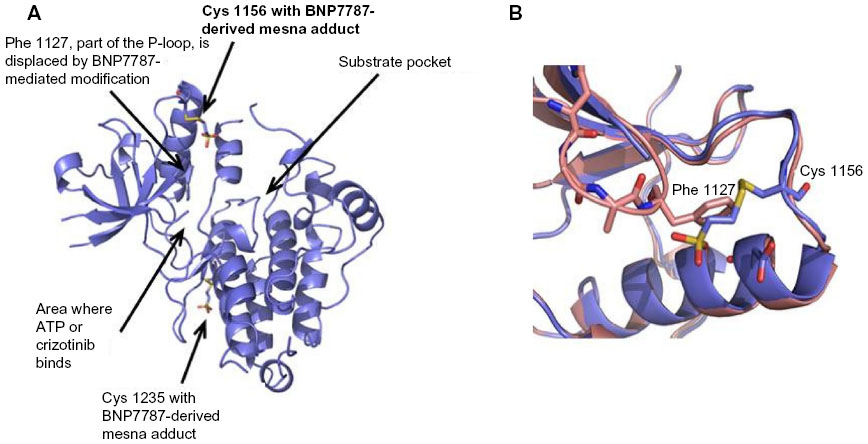 | Figure 1 Ribbon diagrams of ALK with covalently bound BNP7787-derived mesna adducts (PDB IB code 4TT7). |
Ligand binding site
Close-up views of the electron density map at the sites of the BNP7787-derived xenobiotically modified ALK cysteine residues is presented in Figure 2A and B. For both of the BNP7787-derived mesna adducts at Cys 1235 and Cys 1156, a single conformation was observed. Both ligand binding sites are relatively solvent exposed (Figure 2C and D). The BNP7787-derived mesna adduct at Cys 1235 is located in the “back” of the kinase domain relative to the position of the active site. Mesna does not interact with any residues other than Cys 1235 although the sulfonate group is in close proximity to Arg 1231 (Figure 2D).
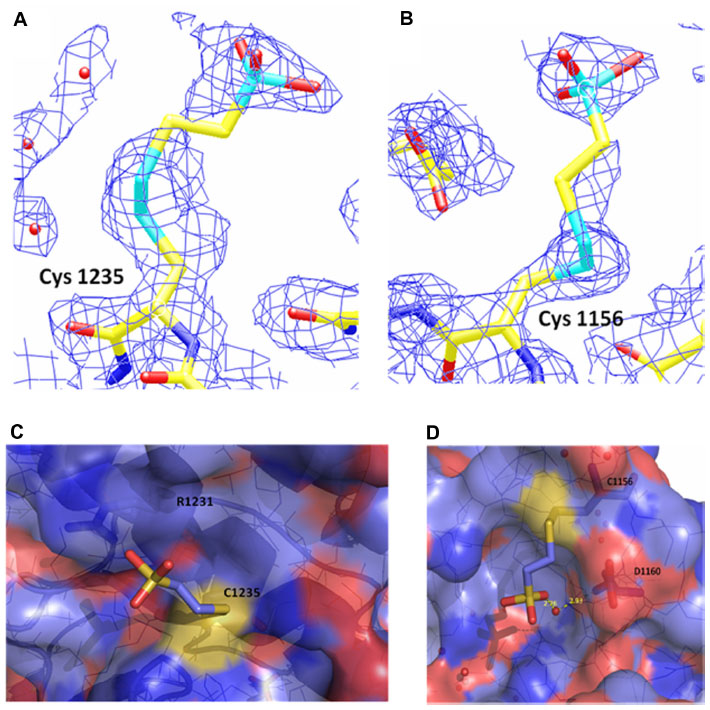 | Figure 2 Electron density and binding site maps showing BNP7787-derived mesna-cysteine adducts on ALK. |
At the Cys 1156 site (which is located in a loop that connects β3 to αC), the mesna sulfonate group makes a water-mediated hydrogen bond with the carbonyl of Asp 1160 (Figure 2D). A fragment of the P-loop (Gly 1123–Gly 1128), another nearby fragment (Ser 1281–Arg 1284, which is part of activation loop or A-loop), and a number of residue side chains (such as Lys 1285) are not in the final refined structure, and interactions between a BNP7787-derived mesna adduct and these missing atoms cannot be ruled out. Additionally, interactions between a BNP7787-derived mesna adduct and Arg 1120 of another protein monomer in the crystal may also be a possibility.
This current structure of ALK with BNP7787-derived mesna adducts at Cys 1156 and Cys 1235 does not have electron density for Tyr 1282, Tyr 1283, and Lys 1285 residues which are part of the ALK activation loop (A-loop); it is not clear if the loss of density for these residues is due to the presence of a BNP7787-derived mesna adduct on Cys 1156 near this A-loop. An overlay of wild type ALK (PDB ID code 3L9P) with the ALK containing BNP7787-derived mesna adduct at Cys 1156 (PDB ID Code 4TT7) indicates that the mesna sulfonate is in potential contact distance with Lys 1285 side chain. However, as a point of reference, these residues (Tyr 1282, Tyr 1283, and Lys 1285) are also disordered in the ALK structure with crizotinib (PDB ID code 2XP2) suggesting that this is an area with inherent disorder.
BNP7787 inhibits ALK activity in vitro
ALK activity assays were run under two ATP concentrations (100 and 500 μM). ATP is often in the millimolar range in vivo,20 and the human body is reported to contain no more than 0.5 moles (250 g) of ATP at any time, but this supply is constantly and efficiently recycled.21 In vivo there are many ATP-dependent enzymes that compete for ATP binding, including kinases, synthetases, helicases, membrane transporters and pumps, chaperones, motor proteins, and large protein complexes like the proteasome;22 therefore, the concentrations of 100 and 500 μM ATP used herein are approximations for ATP concentrations that may be available to ALK in vivo as it competes for ATP with the various other enzymes and proteins that utilize ATP.
BNP7787 inhibited ALK with a half maximal inhibitory concentration (IC50) of 9.16±2.91 mM under assay conditions of 100 μM ATP (Figure 3A) and with an IC50 of 20.80±3.49 mM under assay conditions of 500 μM ATP (Figure 3B). Physiologically, concentrations of BNP7787 as high as 18 mM have been achieved in the clinic.23 BNP7787 has been administered at doses as high as 41 g/m2, and, with concentrations of 18.4 g/m2 used in Phase III trials, maximum serum concentration (Cmax) values in plasma of 10 mM are often observed;23 therefore, the concentrations of BNP7787 required to see an effect on ALK activity in vitro are physiologically relevant. Kinase endpoint assays like the Promega ADP-Glo assay system often classify inhibitors as competitive if their IC50 increases notably as the ATP concentration increases. As the ATP concentration was increased, the IC50 for BNP7787 also increased. From structural work, we observed that BNP7787 covalently modified ALK on Cys 1156 in a loop region of ALK that may subsequently result in partial interference with the phosphate binding site for ALK’s ATP cofactor, and possibly with the A-loop residues. Therefore, while the inhibition of ALK by BNP7787 is not likely classical competitive inhibition – where ATP and BNP7787 have nearly identical or at least significantly overlapping binding sites and only one molecule (either ATP or BNP7787) can occupy that site at a time – it is “competitive-like” based upon the increasing IC50 as the ATP concentration is increased. This conclusion is supported by the X-ray crystallography studies of the ALK structure containing a BNP7787 adduct which indicate that BNP7787 modification of ALK results in a perturbation of the P-loop near where the ATP binding site is located (Figures 1 and 2).
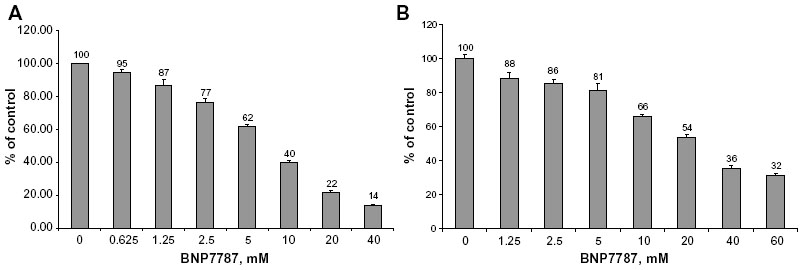 | Figure 3 Concentration-dependent inhibition of ALK activity by BNP7787 in presence of ATP. |
Crizotinib inhibits ALK activity in vitro
Crizotinib is a reported ATP-competitive inhibitor of ALK.24,25 In the in vitro kinase studies reported herein, we observed that crizotinib inhibited ALK with an IC50 of 27.2±1.83 nM (data not shown) under assay concentrations with 100 μM ATP and with an IC50 of 76.3±16.3 nM with 500 μM ATP (data not shown). As mentioned above, crizotinib has previously been characterized as a competitive inhibitor of ALK, with respect to ATP,24,25 and our data are consistent with this previously reported observation (note that in clinical trials where crizotinib was administered orally at doses of 250 mg twice daily, concentrations of crizotinib of 57 nM were reported25).
BNP7787 potentiates the inhibitory effect of crizotinib on ALK activity in vitro (100 and 500 μM ATP conditions)
We evaluated the effect of physiologically achievable concentrations of BNP7787 near the IC25 and IC50 concentrations of crizotinib under assay conditions with either 100 or 500 μM ATP. Concentrations of crizotinib of 57 nM have been reported in clinical trials;25 therefore, concentrations used in these studies were within physiologically relevant ranges. BNP7787 has been administered at doses as high as 41 g/m2 and Cmax values in plasma of 10 mM are typical.23 Under assay conditions with 100 μM ATP (Figure 4A), 5 mM BNP7787 in combination with 15 nM crizotinib (near the IC25 value for crizotinib, when ATP is 100 μM) resulted in 16% greater inhibition than 15 nM crizotinib alone, whereas 10 mM BNP7787 in combination with 15 nM crizotinib resulted in 29% greater inhibition than 25 nM crizotinib alone. Under assay conditions with 100 μM ATP (Figure 4B), 5 mM BNP7787 in combination with 30 nM crizotinib (near the IC50 value of crizotinib) resulted in 10% greater inhibition than 30 nM crizotinib alone, whereas 10 mM BNP7787 in combination with 30 nM crizotinib resulted in 19% greater inhibition than 30 nM crizotinib alone. These assays near the IC50 value for crizotinib (ie, 30 nM, when ATP is 100 μM) have slightly less impressive stimulation compared to 100 μM ATP and 15 nM crizotinib IC25 conditions. For all of the ALK assays that contained 100 μM ATP, we noted that ALK activity levels of less than 10% were not generally observed; this is a technical limitation of the assay. This means that there is only a range of 40% between the IC50 and the assay lower limit, so any additional inhibition attributable to BNP7787 is confined by this limit. Based on this data, BNP7787 notably potentiates the inhibitory effect of crizotinib on ALK at physiologically relevant concentrations of both BNP7787 and crizotinib. As discussed in preceding sections, BNP7787 alone or crizotinib alone were both also effective at inhibiting ALK in vitro.
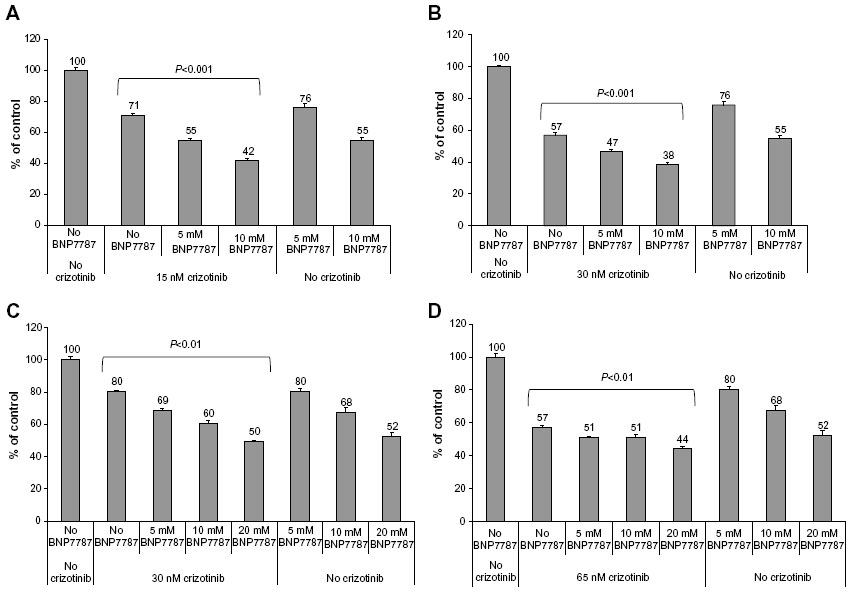 | Figure 4 BNP7787 stimulates crizotinib-mediated inhibition of ALK activity under varying ATP and crizotinib concentration combinations. |
Under assay conditions with 500 μM ATP (Figure 4C), 5 mM BNP7787 in combination with 30 nM crizotinib (near the IC25 value for crizotinib when ATP is 500 μM) resulted in 11% greater inhibition than 30 nM crizotinib alone; 10 mM BNP7787 in combination with 30 nM crizotinib resulted in 20% greater inhibition than 30 nM crizotinib alone; and 20 mM BNP7787 in combination with 30 nM crizotinib resulted in 30% greater inhibition than 30 nM crizotinib alone. Under assay conditions with 500 μM ATP (Figure 4D), both 5 mM and 10 mM BNP7787 in combination with 65 nM crizotinib (near the IC50 value of crizotinib when ATP is 500 μM) resulted in 6% greater inhibition than 65 nM crizotinib alone, whereas 20 mM BNP7787 in combination with 65 nM crizotinib resulted in 13% greater inhibition than 65 nM crizotinib alone.
Discussion
BNP7787 is a multitargeted cysteine-specific agent that covalently modifies the catalytic domain of ALK and inhibits kinase activity
BNP7787 is a novel chemo-enhancing and cytoprotective disulfide agent that has been evaluated in the clinic in patients with NSCLC.1 BNP7787 reacts with and forms mixed disulfides on specific cysteine residues on proteins, yielding specific mesna-cysteine adducts on the target protein. We refer to this process as BNP7787-mediated xenobiotic modulation/modification, and we have observed and characterized this in a variety of proteins important in cell growth and proliferation (data not shown). As an example, we report data herein on the crystal structure of ALK in complex with BNP7787-derived mesna adducts at 2.1-Å resolution. BNP7787-derived mesna adducts were found at Cys 1156 and Cys 1235. Both adducts are relatively solvent exposed, although the adduct at Cys 1156 clearly disrupts the orientations of the P-loop by sterically blocking the typically observed binding site for Phe 1127. Because the P-loop binds the ATP-substrate, this P-loop disruption may or may not alter the kinase activity of ALK or the inhibitory potency of its small molecule inhibitors.
The cysteine-specific, multitargeted nature of BNP7787 is important due to the fact that, except for a few types of cancer (eg, chronic myelogenous leukemia), tumor cells are known to be genomically heterogeneous, and tumors contain subpopulations of cancer cells that often express different tumor-promoting proteins or that have multiple dysregulated, distinct, but key pathways that modulate cell proliferation.26,27 The covalent modification of ALK by BNP7787 results in noteworthy disruptions of the protein’s P-loop region. We hypothesize that BNP7787-mediated cysteine xenobiotic modulation/modification represents a novel mechanism of action for this type of agent, but cysteine-specific modifications, specifically posttranslational modifications of cysteine residues in proteins, are a biological mechanism that may regulate a variety of cellular processes (eg, glutathionylation,28 nitrosylation,29 prenylation,30 and palmitoylation31).
BNP7787 is expected to remain predominantly in the disulfide form in the plasma;6,10,32 however, the intracellular environment and the interstitial space are likely venues for BNP7787 metabolism to mesna, mesna-disulfide heteroconjugates, and free thiols. Any of these species (BNP7787, intracellularly generated BNP7787-derived mesna, or BNP7787-derived mesna-disulfide heteroconjugates) may modify proteins in vivo. The metabolism of BNP7787 to mesna-disulfide heteroconjugates has been observed in in vitro studies and is supported by computational studies on nonenzymatic thiol transfer reactions involving physiological free thiols with BNP7787.6
In summary, BNP7787 has been shown to covalently modify ALK, to inhibit ALK’s kinase activity in vitro, and to potentiate the inhibitory effect of crizotinib (PF02341066) on ALK. BNP7787 is a cysteine-specific, multitargeted modulator of protein function. BNP7787 mediates the nonenzymatic xenobiotic modification of cysteine residues on proteins. BNP7787 is autocatalytic and requires no protein cofactor to xenobiotically modify cysteines, and appears to be specific for cysteine residues located within a particular structural context (ie, not all cysteines in a protein are xenobiotically modified; proprietary computational work by FHH, PYA, and PNP). BNP7787-mediated cysteine xenobiotic modification represents a novel mechanism of action for this type of agent.
Disclosure
Authors ARP, PNP, MZ, PYA, HK, and FHH are or were employed by BioNumerik Pharmaceuticals, Inc. Authors VLN, JB, BC-L, VS, CL are or were employed by Zenobia Therapeutics, Inc. The authors report no other conflicts of interest in this work.
References
Hausheer F, Bain S, Perry M, et al. Comprehensive meta-analysis of survival outcomes from two randomized multicenter trials in first-line advanced non-small cell lung cancer in patients treated with the novel investigational antitumor-enhancing and chemoprotective agent Tavocept. European J Clin Med Oncol. 2010;2(1):7–19. | |
American Cancer Society, Cancer Facts and Figures 2014, American Cancer Society, 1–70, Atlanta, GA. | |
Cancer, Feb 2014, Fact Sheet No 297 [webpage on the Internet], Geneva: World Health Organization; Available from: http://www.who.int/mediacenter/factsheets/fs297/en/index.html. Accessed November 13, 2014. | |
Rosell R, Robinet G, Szczesna A, et al. Randomized phase II study of cetuximab plus cisplatin/vinorelbine compared with cisplatin/vinorelbine alone as first-line therapy in EGFR-expressing advanced non-small-cell lung cancer. Ann Oncol. 2008;19(2):362–369. | |
US Food and Drug Administration. Avastin Label. US Food and Drug Administration; 2004. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/125085s225lbl.pdf. Accessed November 13, 2014. | |
Hausheer FH, Kochat H, Parker AR, et al. New approaches to drug discovery and development: a mechanism-based approach to pharmaceutical research and its application to BNP7787, a novel chemoprotective agent. Cancer Chemother Pharmacol. 2003;52 Suppl 1:S3–S15. | |
Hausheer FH, Kanter P, Cao S, et al. Modulation of platinum-induced toxicities and therapeutic index: mechanistic insights and first- and second-generation protecting agents. Semin Oncol. 1998;25(5):584–599. | |
Boven E, Verschraagen M, Hulscher TM, et al. BNP7787, a novel protector against platinum-related toxicities, does not affect the efficacy of cisplatin or carboplatin in human tumour xenografts. Eur J Cancer. 2002;38(8):1148–1156. | |
Takeda K, Negoro S, Matsui K, et al. Phase I safety and pharmacokinetic trial of BNP7787 in patients receiving cisplatin (CDDP) and paclitaxel (PTX) for advanced non-small cell lung cancer (NSCLC):an Osaka phase 1 study group trial. American Society of Clinical Oncology. 2002; Abstract 453. | |
Pendyala L, Schwartz G, Smith P, Zdanowicz J, Murphy M, Hausheer F. Modulation of plasma thiols and mixed disulfides by BNP7787 in patients receiving paclitaxel/cisplatin therapy. Cancer Chemother Pharmacol. 2003;51(5):376–384. | |
Palmer RH, Vernersson E, Grabbe C, Hallberg B. Anaplastic lymphoma kinase: signalling in development and disease. Biochem J. 2009;420(3):345–361. | |
Horn L, Pao W. EML4-ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol. 2009;27(26):4232–4235. | |
Zhang X, Zhang S, Yang X, et al. Fusion of EML4 and ALK is associated with development of lung adenocarcinomas lacking EGFR and KRAS mutations and is correlated with ALK expression. Mol Cancer. 2010;9:188. | |
Grande E, Bolós MV, Arriola E. Targeting oncogenic ALK: a promising strategy for cancer treatment. Mol Cancer Ther. 2011;10:569–579. | |
Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8(1):11–23. | |
Lee CC, Jia Y, Li N, et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J. 2010;430(3):425–437. | |
Cui JJ, Tran-Dube M, Shen H, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem. 2011;54:6342–6363. | |
MIFit Open Source Project [homepage on the Internet]. 2010. Available from: http://code.google.com/p/mifit/. Accessed. | |
Murshudov GN, Vagin AA, Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. | |
Lu X, Errington J, Chen VJ, Curtin NJ, Boddy AV, Newell DR. Cellular ATP depletion by LY309887 as a predictor of growth inhibition in human tumor cell lines. Clin Cancer Res. 2000;6(1):271–277. | |
University of Leeds. Nature’s batteries’ may have helped power early lifeforms [webpage on the Internet]. Rockville: ScienceDaily; May 25, 2010. Available from: http://www.sciencedaily.com/releases/2010/05/100525094906.htm. Accessed June 1, 2012. | |
Rappas M, Niwa H, Zhang X, Mechanisms of ATPases - a multi-disciplinary approach. Curr Protein Pept Sci. 2004;5(2):89–105. | |
Verschraagen M, Boven E, Zegers I, Hausheer FH, Van der Vijgh WJ. Pharmacokinetics of BNP7787 and its metabolite mesna in plasma and ascites: a case report. Cancer Chemother Pharmacol. 2003;51(6):525–529. | |
Bang YJ. The potential for crizotinib in non-small cell lung cancer: a perspective review. Ther Adv Med Oncol. 2011;3(6):279–291. | |
Ou SH. Crizotinib: a novel and first-in-class multitargeted tyrosine kinase inhibitor for the treatment of anaplastic lymphoma kinase rearranged non-small cell lung cancer and beyond. Drug Des Devel Ther. 2011;5:471–485. | |
Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138(5):822–829. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34(2):85–96. | |
Marino SM, Gladyshev VN. Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J Mol Biol. 2010;395(4):844–859. | |
Hougland JL, Hicks KA, Hartman HL, Kelly RA, Watt TJ, Fierke CA. Identification of novel peptide substrates for protein farnesyltransferase reveals two substrate classes with distinct sequence selectivities. J Mol Biol. 2010;395(1):176–190. | |
Dietrich LE, Ungermann C. On the mechanism of protein palmitoylation. EMBO Rep. 2004;5(11):1053–1057. | |
Parker AR, Petluru PN, Wu M, Zhao M, Kochat H, Hausheer FH. BNP7787-mediated modulation of paclitaxel- and cisplatin-induced aberrant microtubule protein polymerization in vitro. Mol Cancer Ther. 2010;9(9):2558–2567. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.