")
Back to Journals » International Journal of Nanomedicine » Volume 16
Novel Approaches of Dysregulating Lysosome Functions in Cancer Cells by Specific Drugs and Its Nanoformulations: A Smart Approach of Modern Therapeutics
Authors Allemailem KS , Almatroudi A , Alrumaihi F , Almatroodi SA, Alkurbi MO, Basfar GT , Rahmani AH, Khan AA
Received 22 May 2021
Accepted for publication 8 July 2021
Published 26 July 2021 Volume 2021:16 Pages 5065—5098
DOI https://doi.org/10.2147/IJN.S321343
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Israel (Rudi) Rubinstein
Khaled S Allemailem,1,2 Ahmad Almatroudi,1 Faris Alrumaihi,1 Saleh A Almatroodi,1 Mohammad O Alkurbi,3 Ghaiyda Talal Basfar,3 Arshad Husain Rahmani,1 Amjad Ali Khan2
1Department of Medical Laboratories, College of Applied Medical Sciences, Qassim University, Buraydah, Saudi Arabia; 2Department of Basic Health Sciences, College of Applied Medical Sciences, Qassim University, Buraydah, Saudi Arabia; 3Department of Laboratory Medicine, Faculty of Applied Medical Sciences, Umm Al-Qura University, Makkah, Saudi Arabia
Correspondence: Amjad Ali Khan
Department of Basic Health Sciences, College of Applied Medical Sciences, Qassim University, P.O. Box 6699, Buraydah, 51452, Saudi Arabia
Tel +966-16-3800050 Ext. 15445
Fax +96616 3801628
Email [email protected]
Abstract: The smart strategy of cancer cells to bypass the caspase-dependent apoptotic pathway has led to the discovery of novel anti-cancer approaches including the targeting of lysosomes. Recent discoveries observed that lysosomes perform far beyond just recycling of cellular waste, as these organelles are metabolically very active and mediate several signalling pathways to sense the cellular metabolic status. These organelles also play a significant role in mediating the immune system functions. Thus, direct or indirect lysosome-targeting with different drugs can be considered a novel therapeutic approach in different disease including cancer. Recently, some anticancer lysosomotropic drugs (eg, nortriptyline, siramesine, desipramine) and their nanoformulations have been engineered to specifically accumulate within these organelles. These drugs can enhance lysosome membrane permeabilization (LMP) or disrupt the activity of resident enzymes and protein complexes, like v-ATPase and mTORC1. Other anticancer drugs like doxorubicin, quinacrine, chloroquine and DQ661 have also been used which act through multi-target points. In addition, autophagy inhibitors, ferroptosis inducers and fluorescent probes have also been used as novel theranostic agents. Several lysosome-specific drug nanoformulations like mixed charge and peptide conjugated gold nanoparticles (AuNPs), Au-ZnO hybrid NPs, TPP-PEG-biotin NPs, octadecyl-rhodamine-B and cationic liposomes, etc. have been synthesized by diverse methods. These nanoformulations can target cathepsins, glucose-regulated protein 78, or other lysosome specific proteins in different cancers. The specific targeting of cancer cell lysosomes with drug nanoformulations is quite recent and faces tremendous challenges like toxicity concerns to normal tissues, which may be resolved in future research. The anticancer applications of these nanoformulations have led them up to various stages of clinical trials. Here in this review article, we present the recent updates about the lysosome ultrastructure, its cross-talk with other organelles, and the novel strategies of targeting this organelle in tumor cells as a recent innovative approach of cancer management.
Keywords: lysosome, cancer, lysosomotropic agents, nanoparticles, lysosome drug targeting
Introduction
Previously, the lysosomes were just thought to be static, enzyme-loaded, membrane-enclosed organelles, mainly involved in the degradation and recycling of different macromolecules.1 This limited view of lysosomes has been dramatically overturned by some recent discoveries. Lysosomes are dynamic organelles which participate in vast number of cellular activities in addition to the degradation of different biomaterials. The different activities of lysosomes include roles in metabolic signalling, gene regulation, plasma membrane repair, cell migration and adhesion.2 The lysosomes also contribute in the regulation of environmental stimuli and immune response, especially the contribution to macrophages in both innate and adaptive immune responses. These organelles are the final destination of engulfed pathogens by macrophages, which also contribute in processing and secretion of inflammatory signals, and produce peptides which bind with major histocompatibility complex (MHC) molecules.3
Multitude of innate and adaptive mechanisms are deployed by the immune system to prevent malignant transformation as well as ward off pathogens (immune surveillance). An apparent tumor represents malignant cell clones having the capability to evade immune recognition (immune evasion).4 The treatment of cancer since last decade has been revolutionized by promoting anti-tumor immune response in patients with different tumor types. The important therapeutics that target T cell inhibitory checkpoint proteins like PD(L)1 and CTLA-4, are effective in different cancers, leading to tumor burden reduction and increased long-term survival of patients. The significant effects of these immunotherapies have encouraged for additional measures that modulate anti-tumor immunity through effects on T cell, myeloid cells and other cell types within tumor microenvironment.5
A typical mammalian cell can have between 50 to 1,000 lysosomes, depending upon its role in the different cellular activities. The numbers, activity and internal composition of lysosomes vary continuously in response to the environmental cues. The lysosomes significantly communicate with other organelles and cellular structures by constantly exchanging their information and contents. The position of lysosomes within the cell is dynamically regulated, so any dysregulation of these activities may result in different diseases. Lysosomal dysfunctions could result in rare lysosome storage diseases (LSD), almost (1:5,000 cases). These LSD include common metabolic and neurodegenerative diseases, in addition to cancer.6
Lysosomes receive cargoes which are destined for degradation or recycling through endocytosis or autophagy. In cancer cells, the routine signalling pathways are dysregulated, which leads to the variation in lysosomal structure and functions.7 So, these activities make the cancer cells more susceptible to lysosome membrane permeabilization (LMP) by different endogenous (oxidative stress, p53 activation) and exogenous (cationic amphiphilic drugs) triggers.
Cancer cells practice a smart strategy of bypassing LMP facilitated lysosomal cell death (LCD) by the caspase-dependent apoptotic pathway. This strategy is believed to be a novel target of apoptosis and drug resistant cancer cells.8 Some important drugs used include antihistamine, antimalarial and anticancer drugs. Furthermore, drugs used to screen LMP like fluphenazine, thioridazine or toremifene, are under clinical trials, as they are known to achieve lysosomal accumulation, thus leading to LMP.9
Drug loaded-nanoparticles/nanoformulations have revolutionized the therapeutic and diagnostic strategies. But these approaches can lead to toxicity concerns, so extra efforts are required to focus on their physicochemical properties and targeting strategies.10 To understand comprehensively the novel direct or indirect lysosome dysregulation target sites within the cancer cells, it is important to understand the recent advances in lysosomal ultrastructure and its role within the cells. In addition, the mechanism of action of lysosome-targeted anticancer drugs and their formulations is important to understand to engineer more specific antitumor drugs and their nanoformulation in future. Here, in this review, we describe in detail the recent updates regarding the lysosome ultrastructure, its signalling and crosstalk with other organelles, the lysosomal dysregulation during cancerous state, and the novel strategies of direct or indirect lysosome targeting in cancer cells with drugs and their nanoformulations.
Lysosome as Eukaryotic Cell Organelle
Lysosomes are housekeeping organelles that perform the enzymatic degradative functions in coordination with cellular metabolism. These organelles are usually 0.2–0.3 µm in diameter and they originate from the Golgi apparatus. Recently, multiple models have been proposed for the biogenesis of lysosomes.11 These organelles can move around the cytoplasm, change their shape and size, and/ or can undergo fission and fusion. The lysosome’s lumen is acidic with pH between 4.5–5.0 which is maintained by the action of proton pumps in their membrane.12 Different types of enzymes which can digest proteins, carbohydrates, nucleic acids and lipids are present in lysosomes. Among those are the cathepsin proteases which represent a diverse enzyme family.13
The lysosome contains lipid bilayer which forms the external limiting membrane containing vast numbers of embedded proteins and intraluminal vesicles (Figure 1). Like other cellular membranes, the lysosomal membrane composition is properly regulated.14 However, unlike other cellular membranes, the proportion of sphingolipids and glycerophospholipids is richer in the lysosomal membrane, besides having a rich amount of bis (monoacylglycerol) phosphate (BMP).15
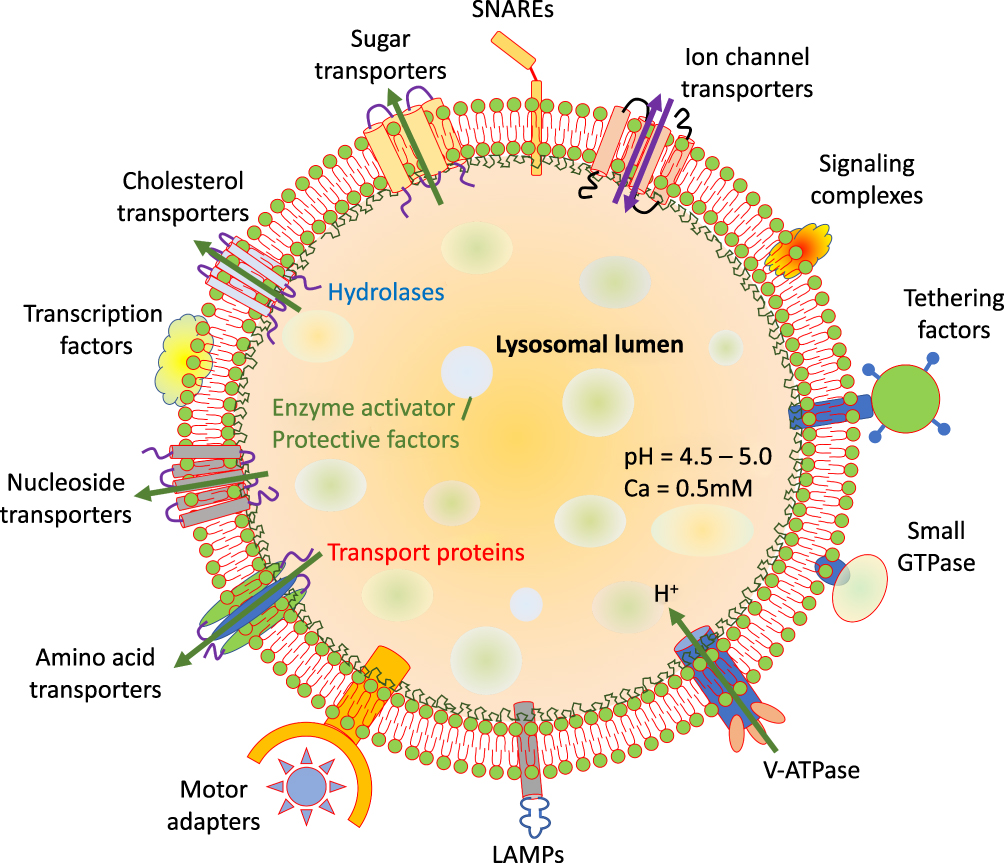 |
Figure 1 General structure and properties of lysosomes. Lysosome lipid bilayer with peripheral and integral membrane proteins with different functions. |
The lysosomes are covered by a lipid bilayer embedded with different types of proteins among which lysosome associated membrane protein 1 and 2 (LAMP-1 and -2) are most abundant. LAMP (-1 and -2) constitute almost half of the lysosomal transmembrane proteins. They are highly glycosylated at luminal N-terminal domain, and are essential for the lysosomal structural integrity.16 The glycosylated tails of these proteins form a sugar coat (glycocalyx), to protect the lysosomal membrane from hydrolase degradation. The LAMPs also serve in lysosomal trafficking, chaperone-facilitated autophagy, exocytosis, autophagosome-lysosome fusion and cholesterol transport.17
Besides the lysosomal integral membrane protein 2 (also identified as SCARB2) and v-ATPase, the lysosome lipid bilayer also contains some other proteins like lysosome integral membrane protein 1 (LIMP1) also known as tetraspanin or CD63 in addition to other less characterized transport proteins.18 The luminal side of these proteins is generally highly glycosylated that gaps the lysosomal membrane from stored digestive enzymes (Figure 1).19
The final step during autophagy results in the fusion of autophagosomes and lysosome and this step is facilitated by the soluble NSF (N‐ethylmaleimide‐sensitive factor) attachment receptor (SNARE) proteins. Among the different SNARE proteins, the lysosome localized SNAREs include vesicle-associated membrane protein 7 or 8 (VAMP7 or VAMP8) and Q-SNAREs are autophagosome-localized which includes STX17 and SNAP29. Recent studies have revealed the role of R-SNARE and YKT6 in lysosome-autophagosome fusion as well.20
Multiple fission and fusion events take place along the endo-lysosomal system for the transportation of proteins and lipids. The mechanism of membrane fusion at endosome and lysosomes takes place by two homologous tethering complexes known as homotypic fusion and vacuole protein sorting (HOPS) and class C core vacuole/endosome tethering (CORVET). Both of these two complexes are hetero-hexamers and interact with Rab GTPases and SNAREs and tether the membranes. CORVET is a Rab5 effector complex while HOPS binds efficiently to late endosomes and lysosomes via Rab7.21
The lysosomes are constantly engaged in functional and physical interactions with other cell organelles either with signal transduction or by membrane contact sites. The master regulators like transcription factor EB (TFEB) and microphthalmia associated transcription factor (MITF) control the lysosomal biogenesis. These proteins sense the signals from the cytoplasm and are translocated to the nucleus to start the induction of lysosomal biogenesis network gene transcription.22 In cytoplasm, mammalian target of rapamycin complex 1 (mTORC1) phosphorylates the MITF and TFEB and are retained there by binding to 14-3-3 proteins.23 During the stressful condition, mTOR pathway is inhibited that leads to the activation of lysosomal biogenesis.
Lysosomal Signalling and Cross-Talk with Other Organelles
Previously the lysosomes were thought to be in relative isolation, but recent years have witnessed that these organelles are fully engaged with all other organelles within the cell (Figure 2). Lysosomes possess a unique position to get information about various biomolecular degradation and recycling events as this organelle can sense the nutritional status of a particular cell. A key factor in this nutrient-regulation mechanistic process is performed by mTORC1, known as a chief regulator of cellular biosynthetic pathways.24 This complex is dynamically associated with lysosomes, which supports the cell growth and anabolism by sensing the growth factors and nutrients. This complex also inhibits the catabolic pathways like autophagy by mediating the phosphorylation of Unc-51-like kinase 1 (ULK1).25 mTORC1 can also regulate the lysosome reformation at the time of autophagy, and helps to restore the organization of mature lysosomes during prolonged starvation.26 The recruitment of mTORC1 on lysosomal surface initiates its activation, which is mediated by resistance associated gene (RAG)-GTPase.27 The lysosomal surface recruitment of mTORC1 is also mediated by cholesterol-binding Niemann-Pick type C1 protein (NPC1). The RAG GTPase also modulates the recruitment of some more nutrient-responsive elements like tuberous sclerosis complex (TSC) and folliculin-interacting protein 1 (FNIP) complex and TFEB. The TFEB is a master modulator for lysosome biogenesis and autophagy (Figure 2).22
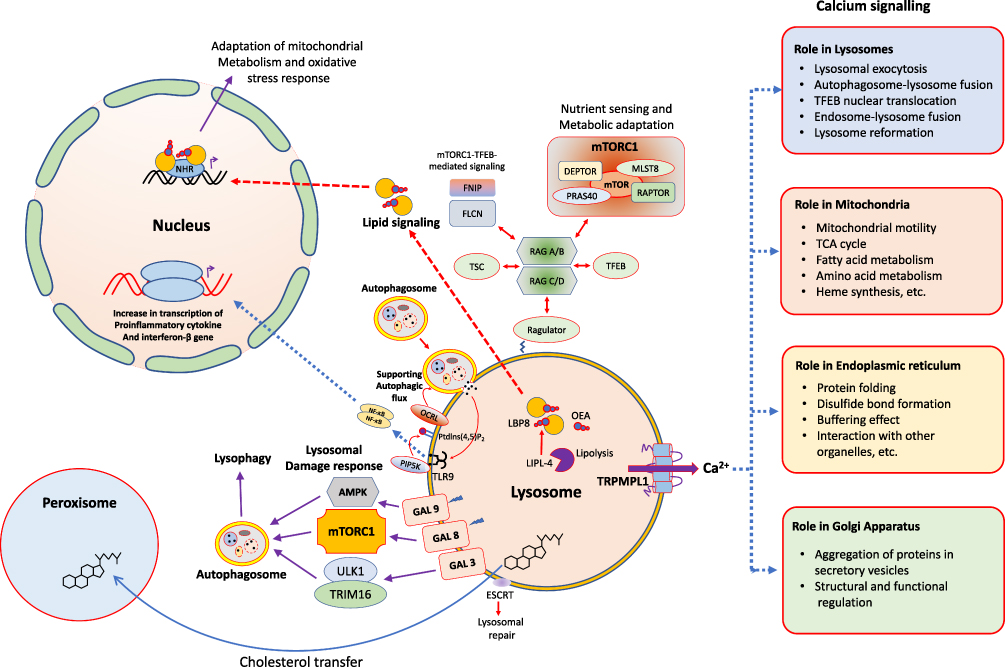 |
Figure 2 The lysosome as an intracellular signaling core. Different types of cellular processes being controlled by signaling pathways and commenced from the surface of lysosomes. |
The lysosome regulates some of its important functions through the release of Ca2+, which includes its fusion with other organelles and structures like plasma membrane, endosomes and autophagosomes (Figure 2).28 There are three main kinds of Ca2+ channels on lysosomal membrane which includes trimeric Ca2+ two transmembrane channel (P2X4), two-pore channels (TPC) and trimeric transient receptor potential cation channel of the mucolipin family (TRPML).28 The Ca2+ mediated interaction results in repair of membrane damage, endocytic membrane trafficking and autophagy. In addition, the Ca2+ release from lysosomes also results in the formation of contact sites with endoplasmic reticulum (ER), which in turn refills back the lysosome with Ca2+.29 The Ca2+ homeostasis is also significant for the proper functioning of other organelles and the acidification within lysosomal lumen, an important requirement for the activity of lysosomal hydrolases (Figure 2).30
The lysosomal Ca2+ channels can respond to different stimuli like pH, stress, nutrient level as well as the level of phospholipids, sphingosine, ATP and NADP. The stimuli of all these molecules with the Ca2+ channels suggests that their activities are differentially modulated, which depends on the overall cell conditions.31 The TRPML1 also called as mucolipin 1, lysosomal Ca2+ channel is the most characterized which mediates the release of Ca2+ from the lysosomes and gets activated by several stimuli like reactive oxygen species (ROS)32 and starvation.33 This channel is also activated by phosphatidyl inositol-3,5-bisphosphate (PI-3,5-bisP), which associates the lysosomal Ca2+ signaling to some intracellular trafficking (Figure 2).34
The TRPML1 also makes a positive-feedback loop with TFEB, where TRPML1 regulates the phosphorylation and subcellular localization of TFEB. In back, TFEB regulates the gene expression of TRPML1.35 The activity of TRPML1 is also linked with some cellular processes in immune cells, which includes large particle phagocytosis.36 In addition, TRPML1 mediates TFEB for the promotion of intracellular clearance of accumulated substrates in LSDs.35 Collectively, all these characteristics of TRPML1 makes it a novel target for some pharmacological modulations in variety of diseases, including cancer. Lysosomes are damaged by its rupture or membrane permeabilization during several circumstances like the severe infection or by the use of lysosomotropic drugs. This damage leads to cathepsin leakage, which promotes to the progression of programmed cell death.37
The lysosomes can fuse with other lysosomes (homotypic fusion) in addition to plasma membrane, autophagosomes, late endosomes, macropinosomes and phagosomes (heterotypic fusion). These fusion activities are mediated by the assembly of trans-SNARE complex, which is composed of one R-SNARE and multiple Q-SNAREs. These fusions are promoted by the Ca2+ release from the lysosomes. Similarly, each organelle can fuse with lysosome through a specific trans-SNARE complex and some precise regulators (Figure 2). Some recent studies have shown that the lysosome and autophagosome fusion is regulated negatively by mTORC1, which promotes the tumor suppressor protein, ultra violet radiation resistance associated gene (UVRAG) phosphorylation. This leads to enhanced Rubicon interaction and diminished HOPS interaction, so inhibiting the fusion events.38
The lysosomal exocytosis leads to the fusion of lysosomes with the plasma membrane and this process is mediated by some novel lysosomal functions that includes plasma membrane repair.39 In addition, the fusion of lysosomes also leads to the formation of cancer cell invasive protrusions40 and the lysosome content secretion into the extracellular space, that occurs usually in bone resorption.41
The discovery of non-fusogenic contacts of lysosomes with other organelles like Golgi complex, endoplasmic reticulum (ER), peroxisomes and mitochondria is quite recent. These contacts require organelle-specific tethering proteins.42 These interactions lead to the maturation of early to late endosomes and then to lysosomes, accompanied by increased contacts with the ER.43 These contacts lead to ER tubular rearrangements and the budding of endosomal tubules.44 The ER-lysosomal contacts also make non-vesicular transfer of lipids between these two organelles as free cholesterol formed in lysosome is exported out of lysosome by the action of NPC (-1, -2). In contrast STARD3 mediates the cholesterol transfer in opposite direction.45
Lysosomes and Cancer Progression
Cancer cells possess relatively fragile and bigger lysosomes as compared to the normal cell counterparts.46 Many types of cancers have altered sphingolipid metabolism, overexpressing sphingosine kinase and downregulating acidic sphingomyelinase.47,48 All these regulations affect the lysosomal membrane function and structure. The cancer cells show increased lysosomal biogenesis which also results in lysosomal enlargement.49 These enlarged lysosomes engulf a significant amount of chemotherapeutic drugs and block it to reach their final destination. In addition, lysosomes arrange a mechanism for the exocytosis of such drugs from cancer cells.49 All these means render a cancer cell drug-resistant, thus emphasizing the lysosomes as a novel strategy as drug targets for cancer therapy. Furthermore, the enhanced activity of phosphatidyl inositol-3 kinase (PI3K), a characteristic feature of cancer cells, confers stability to the tumor cell lysosomes.50 Several cellular processes like size, maturation, and lysosome activity are regulated by PI3K.51 The PI3K inhibition shifts the TNF-facilitated cell death cascade from caspase-dependent mode to cathepsin-dependent way.52
Lysosomes play an important role in cancer biology as this organelle commands to fuel the enhanced requirement of energy sources during the tumor progression.53 Several cancers related to breast, lung, pancreatic, and prostate, etc. have been found to rely mainly on the lysosomal-induced autophagic degradation and the recycling activities, which acts like nutrient-scavenging pathways.53 The lysosomal-autophagic pathways support the proliferation and cancer cell growth like melanoma, renal cell carcinoma, pancreatic adenocarcinoma, etc., as it is associated with the overexpression of microphthalmia-transcription factor E (MiT-TFE) genes. In these cancer types, the mTORC1 induction by MiT-TFE permits the associated hyperactivation of mTORC1-facilitated and autophagy-reconciled nutrient scavenging biosynthetic pathways.
Thus, the unusual catabolic and biosynthetic activation together supports the energy demanding cancer metabolism within the tumor cells.54 The acidic medium of tumor cells leads to lysosome redistribution towards the periphery of the cells,46 and this activity enhances the tissue proliferation by enhanced mTORC1 and mTORC2 signaling.55 This change also leads to the exocytosis of lysosomal hydrolases, matrix metalloproteinases, and integrins that enhance the invasion and metastasis steps.56 The cancer cells possess remarkable proteolytic activity, that assists it to digest the extracellular matrix (ECM). The cancer invasion is associated with increased expression of cathepsin B and other cysteine cathepsins.57 The cathepsins released from the cancer cells degrade the ECM components like laminin, elastin, and fibronectin, which eases the invasion, angiogenesis, and metastasis.58
The cancer cells are efficiently successful to develop chemotherapeutic resistance, which depends upon pro-apoptotic pathways modulation and the modifications of lysosome-facilitated cell death pathways. Cancer cells overexpress cytosolic and lysosomal protease inhibitors which inhibit the LMP.59 In addition, the cytosolic heat shock protein 70 (Hsp70) gets translocated to the lysosomal lumen within the cancer cells. This translocation helps to stabilize the lysosomal membranes by endorsing the acid sphingomyelinase activity.60 This procedure also helps to protect the tumor cells against cytosolic leakage of lysosomal proteases. Any depletion of Hsp70 leads to a lysosome-facilitated cell death program.61 In addition, the cancer cell viability is enhanced by cathepsin inhibitors, with augmented lysosomal activity.62 These findings show that the drug nanoformulations which can target the P13K and the lysosomal Hsp70 accumulation or the cathepsin activity, may work as novel therapeutic agents to make the cancer cells susceptible to lysosome mediated cell death.63
Lysosome Targeting in Cancer Cells
For the purpose of proliferation and adaptation to a new environment, cancer cells adapt with increased lysosomal functions. Different factors like p53, Bcl-2 family members, sphingosine, and oxidative stress are altered during the cancerous state and these alterations can lead to increased LMP.64 Lysosomal disruptions in turn enhance the oxidative stress further, which promotes the lipid peroxidation, mitochondrial dysfunctions, and autophagy. All these alterations lead to cathepsins release, which promotes the degradation of different macromolecules. In addition, these changes can trigger cancer cell death through autosis, apoptosis, or ferroptosis.
Lysosomes perform their role both during catabolic (macropinocytosis and autophagy) and anabolic pathways, as driven forward by mTORC1. All these pathways are potential targets in cancer therapy. Autophagy leads to the delivery of cellular materials to the lysosomes for degradation and it performs multiple functions in cancer progression.65 As autophagy can have both antitumor and protumor effects, the recent efforts of targeting the cancer cell autophagy as a treatment strategy is given a high priority. Autophagy has the ability to promote the tumor growth and facilitates the chemoresistance during cancer therapy.66 Macropinocytosis leads to the delivery of extracellular proteins to lysosomes, so can promote the cancer growth, especially in RAS-driven cancers.67 The inhibition of mTOR functions is widely recognized as anticancer therapy in preclinical trial patients.68
Some antimalarial drugs like quinacrine, chloroquine, and hydroxychloroquine have been found to inhibit the lysosomal functions by inhibiting the autophagy cascade.66 These drugs are used as anticancer agents but have no effect on mTORC1 regulation.69 Chloroquine possess the DNA binding capacity and its dimerization increases its potency as an autophagy inhibitor. Similarly the dimerization of other antimalarial drugs like quinacrine has been found to surpass the tumor growth.
A simple model of drug action by dimeric quinacrine 661 (DQ 661) in cancer cells is represented in Figure 3 as an example to understand the basic mechanism of its action as anticancer activity. DQ661 has been used as a photo-labeling probe to recognize its molecular target as palmitoyl-protein thioesterase 1 (PPT1). This enzyme is used for depalmitoylation of proteins and DQ661 targets the tumor cells by binding and inhibiting PPT1. In addition, this drug possesses multitarget binding activity as it blocks lysosomal activity and some major catabolic functions of macropinocytosis and autophagy and also inhibits mTORC1. So this drug can block the degradative as well as signaling functions of lysosomes. DQ661 inhibits mTOR by disrupting the complex at lysosomal membrane and prevents the amino acid-dependent regulation of mTORC1 kinase activity (Figure 3).
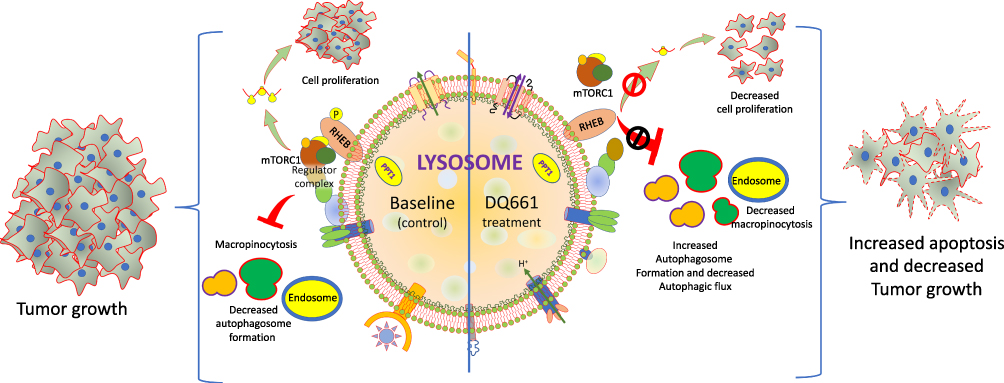 |
Figure 3 The inhibition of PPT1 by DQ661 and the regulation of multiple lysosome-facilitated signaling processes. The left half of the lysosome median line indicates the basal (control) conditions in absence of DQ661. The right side of the lysosome median line shows the effect of DQ661 binding to PPT1 directly within the lysosomal lumen, resulting in decreased macropinocytosis, autophagic flux, proliferation, and decreased tumor growth and enhanced apoptosis. |
All these findings show that DQ661 possess in vivo anticancer activity against different tumor models in both immunodeficient and immunocompetent models. These findings show that lysosome inhibition by these drugs is critical for the anticancer effect. Furthermore, the inhibition of PPT1 also affects the proper localization of lysosomal v-ATPase activity, which is responsible for maintaining lysosome as acidic by the pH gradient. DQ661 will be useful to illustrate exactly the regulation of mTORC1 complex at the lysosomal membrane. Conclusively, the PPT1 identification as a novel therapeutic cancer target suggests that protein palmitoylation needs deeper investigations.70
The cancer cell lysosomes can be targeted at different stages, as discussed here.
Lysosome Membrane Permeabilization
Lysosome membrane permeabilization (LMP) targeting has been fully supported to be a novel therapeutic strategy in different cancers.71 LMP can either be slight or complete and leads to lipid peroxidation and a partial or complete discharge of lysosomal contents. Some of the contents include cathepsins, which cleave and degrade different proteins.37 Cancer cells experience altered metabolism with increased ROS that destabilizes the lysosomes and pushes them for LMP.37 The increased ROS production and the release of lysosomal cathepsins can initiate cell death through mitochondrial dysregulation and ultimately cell membrane permeabilization.72
Cancer cells show an altered sphingolipid metabolism that results in an enhanced sphingosine amount which amplifies the LMP. These findings have also been supported when sphingosine is added to some cell lines, which induces their LMP. Some other agents which induce the LMP are tumor necrosis factor-α (TNF-α), DNA damaging drug mediated p53 phosphorylation, which gets translocated to the lysosomes and induces LMP.22 Cancer cells can smartly regulate this permeabilization through some cellular components like cholesterol, Hsp70, α-tocopherol which minimize the lysosome permeabilization.73
Some cell lines when transformed with oncogenes like Src and Ras have been found to exhibit distorted lysosomal localization and decreased LAMP (-1, -2) expression that primes the cells for LMP.16 Some cancer cells enhance their lysosomal size, biogenesis, and change in Hsp70 expression, thus creating destabilized lysosomes.61 These findings show that cancer cells may be sensitive, if approached for lysosomal cell death. Targeting of LMP is a novel strategy to kill different cancer cell types like breast cancer,35 skin cancer,74 bone cancer, cervical, ovarian, prostate cancer,48 colon cancer,16 lung cancer,75 and acute myeloid leukemia (AML).76
Some of the examples of LMP inducers and their mechanism of action are listed in Table 1.
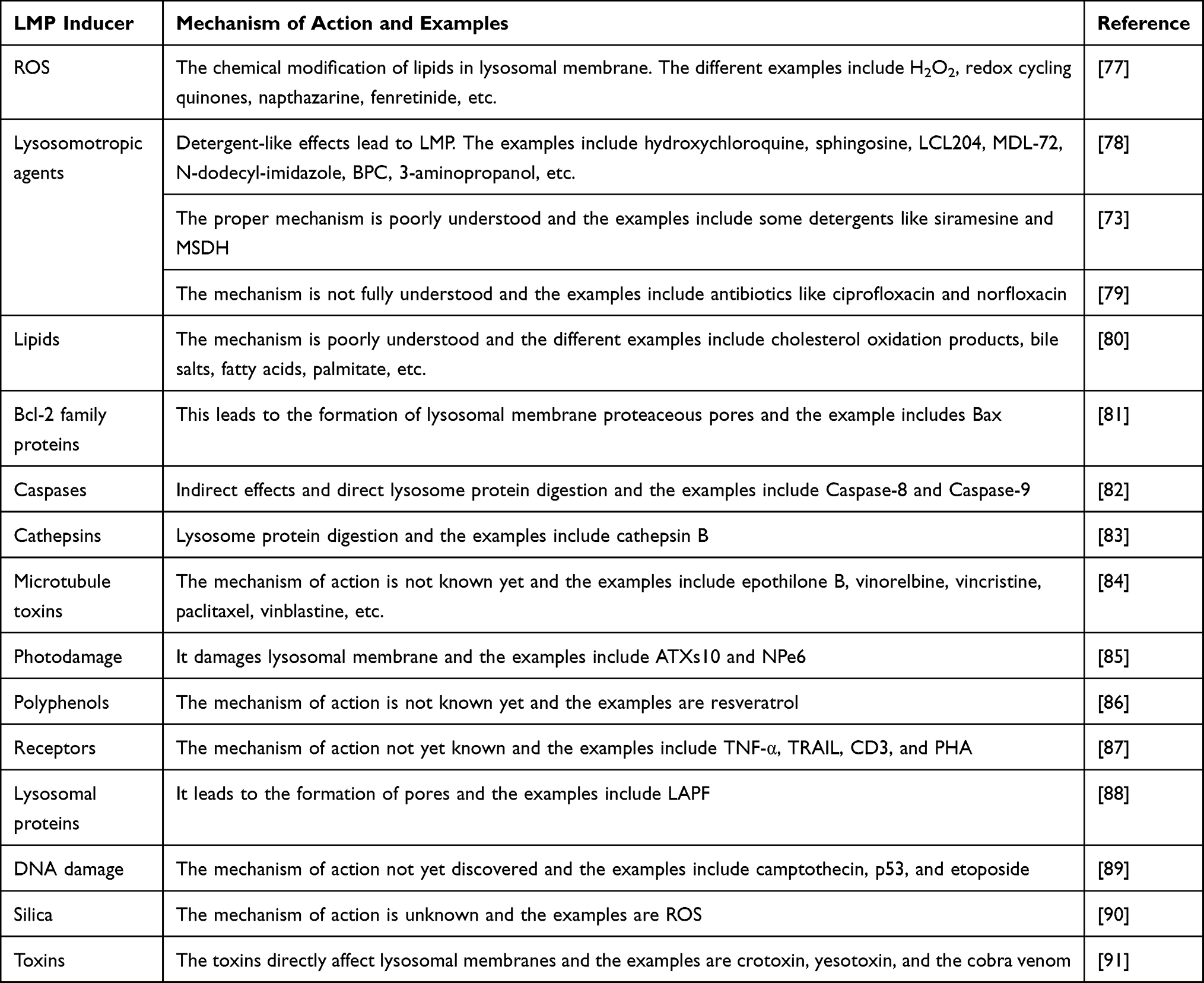 |
Table 1 Different Types of LMP Inducers and Their Mechanisms of Action |
Lysosomes and Apoptosis
Apoptosis is programmed cell death involving both lysosomal and mitochondrial cooperation and the activation of caspases. A novel strategy of treating all types of cancers non-surgically is by targeting apoptosis. Several anticancer drugs target different stages of both intrinsic and extrinsic pathways.92 The two common strategies include inhibition of anti-apoptotic molecules and the stimulation of proapoptotic molecules.93 Some important targets include inhibitors of Bcl-294 ligands for death-receptors,95 X-linked inhibitor of apoptosis protein (XIAP) inhibition,95 and use of alykylphopholipid (APL) analogs which perform like apoptotic signals.96 Some important plant derived compounds which possess apoptosis induction capabilities include aloe-emodin, black cohosh curcumin, epigallocatechin-3-gallate (EGCG), genistein, graviola, juglone, and quercetin.97
The cancer cells meticulously modulate the central control points of apoptotic pathways, including inhibitor of apoptosis (IAP) and FLICE-inhibitory protein (c-FLIP). The cancer cells precisely suppress apoptosis and develop resistance to apoptotic agents by the expression of antiapoptotic proteins like Bcl-2 or by downregulating proapoptotic proteins like BAX.
The release of cathepsins from lysosomes targets BH3 interacting-domain death agonist (BID) which allow its translocation to the mitochondria where it interacts with Bax and Bak.98 The antiapoptotic Bcl-2 family members possess the capacity to prevent LMP in addition to control the mitochondrial regulation.71 It is strongly recommended that there is a link between mitochondrial dysfunctions and the lysosomal disruption. The loss of membrane potential favors the enhanced ROS production which destabilizes the lysosomal membrane through lipid peroxidation and promotes its rupture, which is associated with the activation of caspase 8 and 9.99 Overall, these findings show that lysosomes possess a remarkable role in either initiating or executing the apoptotic pathways.
A novel synthetic retinoid 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalene carboxylic acid (AHPN/CD437) has been confirmed to induce powerful apoptosis in different cancer cell types. The treatment of human leukemia HL-60 cells with CD437 result in rapid apoptosis induction through the caspase activation, mitochondrial functions, and morphological alterations. Treatment with some antioxidants like α-tocopherol acetate effectively inhibits the CD437-mediated apoptosis. In addition, the pretreatment of cells with pepstatin A (cathepsin D inhibitor) blocks the CD437-mediated free radical formation and apoptosis. These observations suggest the role of cathepsin D in initiation of apoptotic cell death. These findings have been confirmed by measuring intracellular distribution of cathepsin D through immunofluorescence, which indicates the release of this enzyme from lysosomes to the cytoplasm. The lysosome labeling with some lysosomotropic agents has confirmed that CD437 induces the lysosomal leakage and apoptosis induction.100
Lysosomes and Autophagy
The fusion of lysosomes with autophagosomes results in the formation of autolysosomes, in which the degradation of intracellular and extracellular materials take place. Autophagy performs a significant role in the adaptation of cancer cells to stress, as it protects these cells from death or any induction of its progress.101 During the normal conditions, the cellular homeostasis is maintained through lysosomes by its biogenesis which occurs through biosynthesis and endocytic pathways. But, during the stressful conditions, the lysosome number gets decreased, as they play a vital role in macromolecular degradation for recycling or removing the damaging organelles. The restoration of lysosomes takes place through autophagic lysosomal reformation (ALR).102
The autophagy also regulates the lysosome cycle by permitting to engulf the damaged lysosomes with autophagosomes which later bind with active lysosomes to remove them from the cells.103 This process leads to the recycling of amino acids and other nutrients to the cell.104 However, if this process is not properly regulated, the destruction of intracellular structures can lead to cellular collapse and autosis, which is totally dependent on lysosomes.105
The alteration of autophagy in cancer cells can prove to be a promising strategy of tumor management. Some drugs are known to target different types of autophagic processes, from its initiation to the degradation step.106 The suppression of autophagy promotes the therapeutic effects of anticancer agents and leads to apoptosis.107 Chloroquine has been used as an autophagy inhibitor which enhances apoptosis and the therapeutic effects of photososan-II-mediated photodynamic therapy (PS-PDT) in colorectal cancer cells.108
Some common autophagy regulators, such as rapamycin and its derivatives, like temsirolimus, everolimus, chloroquine, and hydroxychloroquine, are regularly used in cancer therapy. Temsirolimus and everolimus induce autophagy by the inhibition of mTORC1, and have been approved by the Food and Drug Administration (FDA) for cancer therapy. Chloroquine and hydroxychloroquine directly inhibit autophagy by the alteration of lysosomal pH, inhibition of autophagic degradation, and accumulation of autophagosomes.109
Table 2 lists some common anticancer natural compounds or synthetic drugs which can modulate the autophagy by different mechanisms.
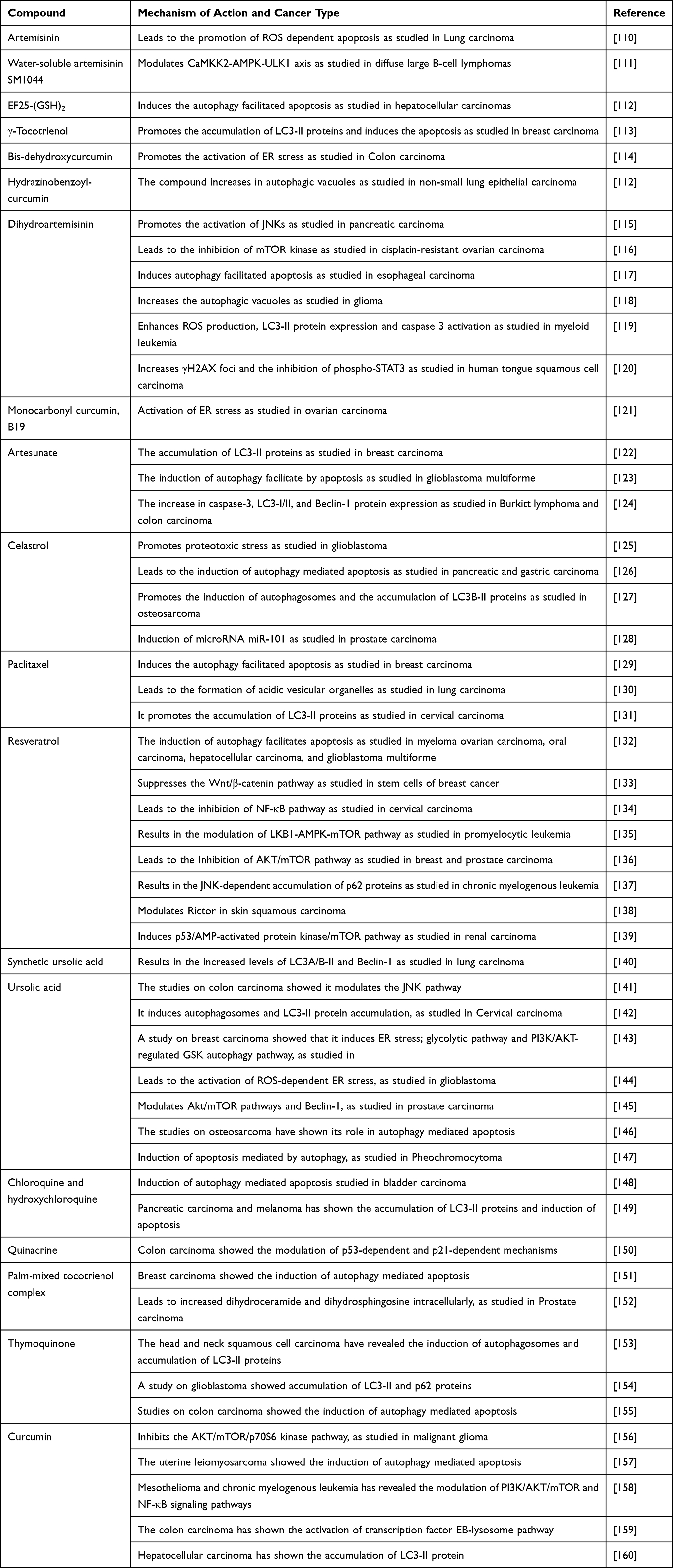 |
Table 2 Examples of Different Compounds Which Act as Modulators of Autophagy Used as Cancer Prevention and Therapy and the Examples of Different Cancer Where Used |
Lysosomes and Ferroptosis
There is an increased metabolic rate and higher turnover of iron-containing proteins in cancer cells, that lead to the accumulation of iron in their lysosomes and the sensitization to ROS-induced LMP.161 These cells have a higher ROS production rate with more cathepsin release from their lysosomes which can induce their cell death.162 This lysosome-facilitated cell death offers a novel option to treat the cancer cells which are resistant to general apoptotic cell death. However, cancer cells meticulously evade the lysosome-sponsored cell death by reorganizing their lysosome metabolism.
Cancer cells exhibit an additional iron demand as compared to the normal non-cancerous cells. So this extra requirement for iron can make such cells susceptible to iron-catalyzed necrosis, known as ferroptosis. Ferroptotic cell death is a distinct type which results from the accumulation of iron-dependent ROS.163 The ferroptosis is regulated by the proteins like ferritin, transferrin, and cysteine antiporter receptors which are responsible for the regulation of iron level.164 Lysosomes are one of the major storage locations of iron and, in the presence of H2O2, the free Fe undergoes Fenton reaction resulting in reactive iron and thus increasing the ROS. A lysosome disrupting compound called siramesine increases the lysosomal pH that results in its leakage which is mediated in part by sphingomyelinase inhibition.48 All this leads to increased reactive iron and ROS, finally mediating cell death.73 The actual role of lysosomes in regulating ferroptosis through increased active iron and ROS is still not fully understood and needs further investigations. The ferroptosis-inducer drugs as approved by the FDA bear high expectations for the potential of tumor management as a new promising way to kill cancer cells. Table 3 lists some important drugs that can either induce or inhibit the iron metabolism facilitated ferroptosis with different types of mechanisms involved.
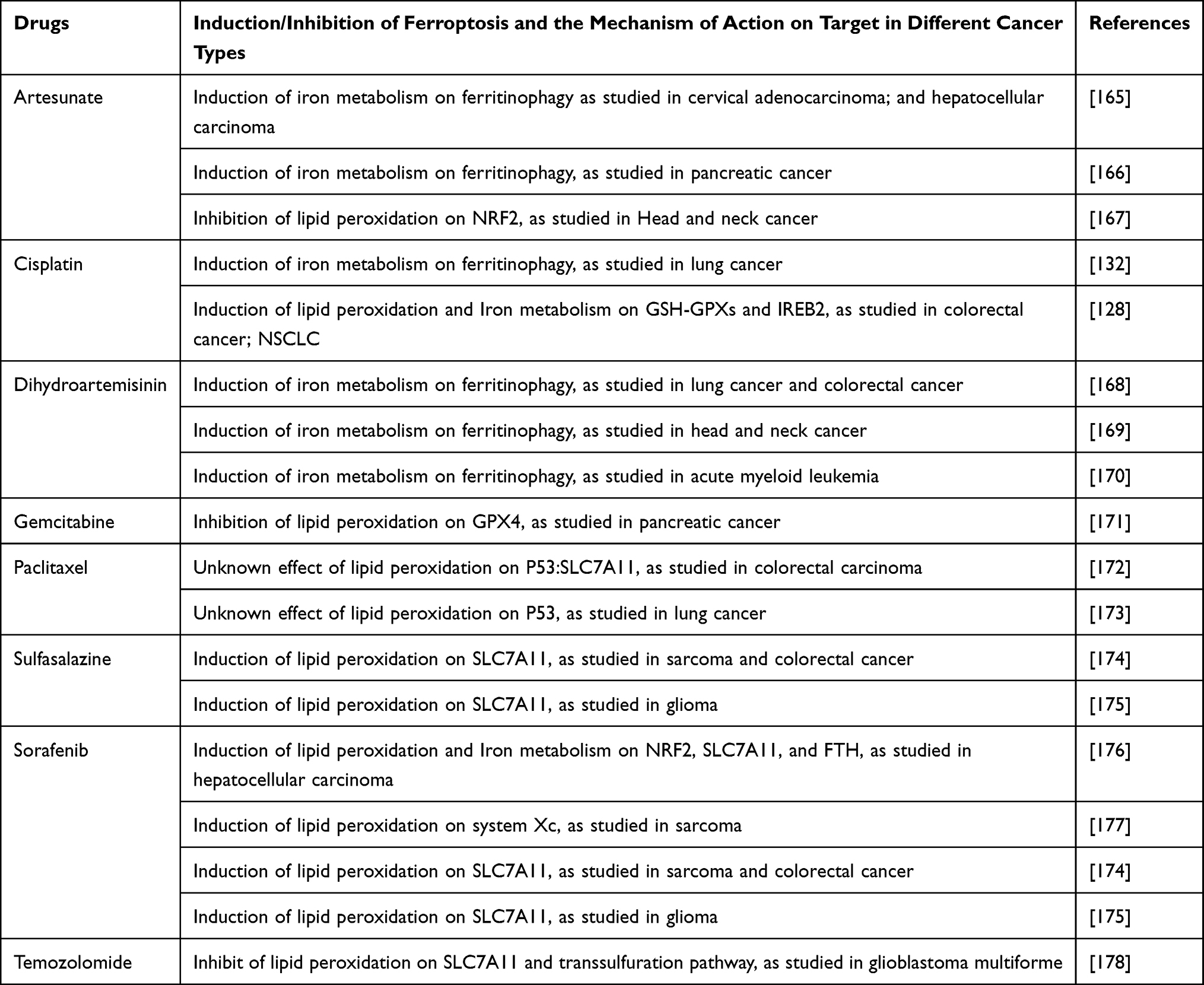 |
Table 3 Examples of Different Drugs and Other Compounds Which Modulate the Iron Metabolism Mediated Ferroptosis with Different Types of Mechanisms |
Lysosomotropic Agents
Lysosomotropic agents consist of weak-base cationic or lipophilic amphiphilic drugs which gets accumulated inside the lysosomes. The lysosomal membrane allows the diffusion of these compounds across and get trapped due to their protonation inside the lysosomes.71 Their excessive accumulation initiates the lysosomal membrane damage and finally causes LMP. Lysosomotropic agents include kinase inhibitors such as ML-9,179 metal nanoparticles (NPs),180 and some pharmaceutically important drugs, which include nortriptyline, siramesine, desipramine, clomipramine, imipramine, etc.48 The chemical structure of some of these compounds is listed in Figure 4.
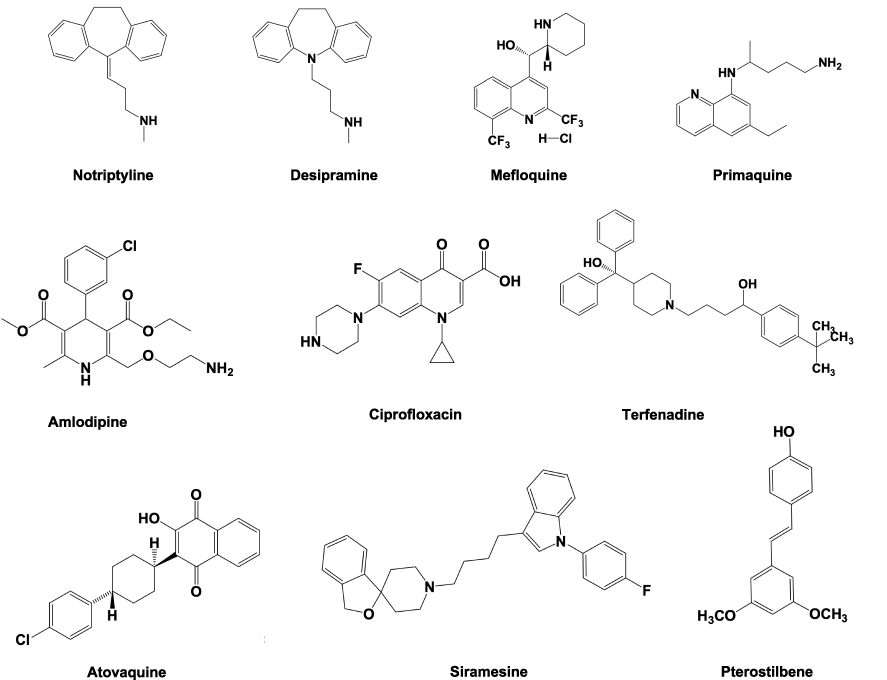 |
Figure 4 Chemical structure of some important lysosomotropic compounds used to induce LMP. |
These compounds have been used in the fight against colon cancer, breast cancer, and CLL cells. In addition, antimalarial drugs like chloroquine and mefloquine have been found to be effective in lymphoma, leukemia, and breast cancer.181 Further, antiallergic drugs like loratadine and terfenadine have been reported to induce breast and lung cancer cell death.48 In addition, the treatment with stilbenoid antioxidant, pterostilbene and antipsychotics, thioridazine, chlorpromazine, and aripiprazole have been found to possess good efficacy in leukemia and breast cancers. Except chloroquine, most of these drugs are FDA-approved and have been properly investigated in clinical trials.181 In future, these lysosomotropic agents will provide a significant foundation to be clinically investigated for their therapeutic role in different types of cancers.
Methods, Strategies, and Characterization Used for the Preparation of NPs
The current advances towards the biomedical application of NPs have established different methods to synthesize these entities from diverse materials like metals, metal oxides, semiconductors, ceramics, polymers, etc. So, on the basis of their origin, NPs possess unique structural, physiochemical, and morphological characteristics, which are important for their wide range of applications like center-point drug targeting, bioimaging, molecular tagging, etc. The structural analogs of NPs include liposomes, dendrimers, quantum dots, polymeric micelles, and each structural analoghas specific applications.182 The synthesis methods of NPs are mainly divided into three categories: physical methods, chemical methods, and bio-assisted methods.
Furthermore, two basic approaches employed for the preparation of NPs include a top-down approach and a bottom-up approach. In the top-down approach, the synthesis of NPs is initialized with a bulk material that leaches out systematically bit-after-bit, resulting in the generation of required NPs. Some commonly used top-down methods include electron beam lithography, photolithography, milling technique, anodization, ion and plasma etching, etc. The bottom-up approach for NP synthesis involves assembling or coalescence of atoms and molecules generating distinct NPs. Some commonly used bottom-up approaches include sol-gel processing, chemical vapor deposition, laser pyrolysis, plasma or flame spraying, bio-assisted synthesis, and chemical or electrochemical nanostructural precipitation.183
The characterization of NPs by different methods is equally important to control their desired in vivo and in vitro behavior. These entities are characterized by their morphology, size, and surface charge by utilizing highly advanced microscopic techniques such as transmission electron microscopy, scanning electron microscopy, and atomic force microscopy. The colloidal stability of NPs is determined through zeta potential, which is an indirect measure of surface charge. The NPs and drug interaction are characterized by using differential scanning calorimetry. In addition, the binding and internalization of targeted nanoformulations against specific cells is determined by cell uptake assays. The biodistribution, intracellular uptake, and subcellular localization of these NPs could be confirmed by confocal microscopy.184
Lysosomal Targeting with Drug Nanoformulations
The practice of using a single unit drug nanoformulation as a therapeutics and diagnostics (theranostics) composite is now well-known as a novel approach of the drug delivery system.185 There are several advantages of using theranostic nanoformulations as compared to the conventional systemic administration of native drugs. Native drugs have the problems of limited solubility, easy inactivation, and fast biodegradation. Some advantages of using drug nanoformulations include extended circulation time, higher concentration at tumor site, multiple synergistic drugs, and diagnostic system delivery.186 Some more advantages include controlled drug release at the tumor site through stimulus-sensitive delivery systems (eg, temperature, pH, enzyme-sensitive nanoformulation, overcoming multidrug resistance and enhanced therapeutic efficacy. The approach of a drug delivery system even up to the organelle level (third level drug targeting) with the aid of different nanoformulations has revolutionized the therapeutic approach for different diseases, including cancer.187
Lysosomes are considered as novel targets for anticancer therapeutics as cancer cells can bypass cell death through the classical caspase-dependent apoptosis pathway. This enables us to focus on targeting apoptosis and drug-resistant cancer cells as a novel therapeutic strategy.188 Cancer cell specific and particularly organelle-directed drug targeting is one of the major challenges in pharmaceutical research. This area requires a multidisciplinary approach for center-point delivery of novel therapeutics without affecting nearby healthy tissues. These drug delivery system nanoformulations are constructed by keeping certain criteria in observations. Several organelle-specific small molecules like heterocyclics, peptide substrates, and oligonucleotides have been widely used.189 Recently, different polymeric carriers like N-(2-hydroxypropyl) methacrylamide (HMPA) and mesoporous silica nanoparticles (NPs) (MSNPs) have been used to target several cancerous tissues.190 These nanoformulations act by passive targeting by taking the benefit of enhanced permeation and retention effect (EPR) of the cancerous tissues. These nanoformulations are hardly selective but get simply distributed by blood circulation. So most of these administered nanoformulations get accumulated within the lungs, spleen, and liver. The researchers have constructed novel drug delivery systems by focusing on different cellular proteases as target sites.191
Among different organelles, lysosome targeting in tumor cells is reported to be one of the potential ways of cancer treatment.192 As lysosomes are rich in proteases, the cathepsin family of enzymes are considered as potential targets of therapeutic strategy in cancer management. In cancer cells, the lysosomal cysteine proteases, including cathepsin B (Cat B), are highly upregulated at mRNA and protein levels.193 The overexpression of Cat B is reported in breast cancer, oesophageal cancer, and other tumors.194 So targeting the Cat B appears to be a promising strategy for novel drug delivery against different types of cancer cells. A tetrapeptide, Gly-Leu-Phe-Gly (GLPG) possesses higher plasma stability and the least hydrolysis by Cat B.195 So the novel targeting of Cat B-enriched cancer cells enhances the efficacy of anticancer drugs, and minimal toxicity to the normal cells.
A synthetic Cat B peptide sequence, GLPG has been engineered by conjugation with a sorbitol core attached with multiple guanidine residues, which targets the cancer cell lysosomes. This strategy mimics the Arg-8-mer or Tat, which shows potential translocation across the blood–brain barrier, cell membrane, and mitochondria.196 There are some advantages of using sorbitol as a delivery carrier, as it possesses the highest density of functionality among other organic compounds having multiple hydroxyl groups. Sorbitol is naturally occurring and devoid of toxicity with the positively charged guanidine groups displaying an association with negatively-charged phospholipids present on cell membrane and other organellar membranes, thus facilitating its entry through the lipid bilayer.197 Doxorubicin, a potential antitumor drug, has been delivered in different cancer cells by using sorbitol as a carrier system conjugated with Cat B cleavable peptide sequence. However, only a few nanoformulations have been tested so far, and these nanoformulations also exhibit little weaker cancer selectivity, but have been found to be effective during combination therapies.188
Lysosomal Targeting with Free or Peptide-Facilitated Gold-Nanoparticles
Recently, different types of NPs, including gold NPs (AuNPs), dendrimers, fullerenes, neodymium oxide, and quantum dots have been shown to be autophagy inducers.198 The AuNPs are one of the most commonly used nanoformulations which initiates the induction and accumulation of autophagosomes. These particles are engulfed by the cancer cells via endocytosis in a size-dependent manner. These NPs ultimately show lysosomal accumulation, which leads to their degradation through the alkalinization of lysosomal pH. Further, it has been observed that AuNPs induce the accumulation of autophagosome and the processing of autophagosome marker protein, microtubule-associated light chain 3 (LC3). However, the degradation of p62, an autophagy substrate, is blocked in AuNP-exposed cells, indicating that the accumulation of autophagosome is the consequence of autophagy flux blockade, rather than autophagy induction. So the AuNPs accumulation of autophagosome clarifies its role on lysosomes. The use of AuNPs and their lysosomotropic-agents tagging can be a novel strategy of targeting specific tumor cells as a cancer therapeutics strategy.199
Gold nanoparticles (AuNPs) with a diameter of about 13 nm tagged with cell membrane penetrating peptide (CPP) and lysosomal sorting peptides (LSPs) have been targeted to lysosomes. The results of this study have confirmed that LSP is quite efficient in transporting AuNPs tagged with CPP up to lysosomes and other lysosome like structures. There are some novel advantages of using LSPs tagged with different types of metal and non-metal based NPs to treat different types of enzyme replacement therapies (ERT) or targeted drug delivery within the lysosomes.200
Co-Targeting of GRP78 and Lysosomes with TPP-PEG-Biotin Self-Assembled NPs
Glucose-regulated protein 78 (GRP78) is a type of Hsp70, a marker protein overexpressed in cancer cells.201 In tumor cells, GRP78 is used to combat the stressful environment and promotes the proliferation, metastasis, survival, and resistance to different anticancer drugs.202 In addition, the lysosomes in cancer cells help in recycling of dysfunctional organelles and other contents by autophagy to reuse the basic components.203 These cells require a continuous supply of functional proteins for their faster cell division, as compared to the normal cells. So, these cells overexpress the molecular chaperones like GRP78 to regulate the overloading of proteins within ER.
A novel strategy of co-targeting GRP78 and lysosomes and destroying them can lead to the accumulation of by-products including unfolded proteins. This significant co-targeting strategy is desperately needed as an anti-cancer therapeutic approach. In this regard, Ruthenium (II, III) complexes have been found to possess strong affinities for thiol containing proteins like glutathione (GSH), transferrin and bovine serum albumin (BSA), etc.204 The strong affinity of ruthenium for transferrin is novel, as it has more specificity for cancer cells as compared to the normal cells due to the highly cancer active metabolism requiring Fe2+ ions.205
A photo-dynamic therapy substance made from tetraphenylporphyrin (TPP)-polyethylene glycol (PEG)-biotin produces ROS when irradiated at 660 nm.206 After the covalent bonding between TPP and PEG, this material undergoes self-assembly making novel photosensitizer-NPs. The TPP-PEG-biotin can be efficiently delivered to different cancer cells, like HepG2 and MCF-7, with overexpressed biotin receptors. The TPP-PEG-biotin formulation has been localized within lysosomes of MCF-7 cells.206 These self-assembly NPs of TPP-PEG-biotin can also encapsulate chemotherapeutic drugs targeting multiple organelles.
The nanoformulation made from TPP-PEG-biotin loaded with Ru-1 has been used to co-target GRP78 and lysosomes as a recent novel anti-tumor therapeutic strategy. In comparison to the previous approaches, based on doxorubicin encapsulation using TPP-PEG-biotin,206 this approach is based on co-targeting strategy. The co-targeting of different locations within the cancer cells adds the damaging sites and proves to be more lethal to them. The co-targeting of lysosomes and GRP78 has been proven to be a very efficient anti-cancer therapeutic strategy.
Lysosome Targeting with Octadecyl-Rhodamine-B Liposomes
The Gaucher disease type 1 (GD1) is recognized by increased incidence of gammopathy and risk of developing multiple myeloma and possibly other hematological malignancies.207 This disease is caused by a deficiency of the lysosomal hydrolase, acid β-glucosidase and results in accumulation of its primary substrate, glucosylceramide (GC), which in the systemic circulation is derived primarily from the turnover of senescent blood cell membranes.208 The liposomes loaded with β-glucosidase from human origin have been found to degrade GM1-ganglioside within feline fibroblast lysosomes and drastically reduced the 70–80% accumulation of its substrate, galactocerebroside.209 However, these liposome-based formulations are not used clinically for ERT, so there is a drastic need to use such nanoformulations, having enhanced enzyme loading efficiency and proper targeting within the lysosomes.
The surface modification of liposomes with novel lysosomotropic octadecyl-rhodamine B has significantly increased the proper delivery of such nanoformulations within the lysosomes of HeLa cells.210 Octadecyl rhodamine B (Rh) has significantly improved the delivery of a model marker fluorescein isothiocyanate (FITC)-dextran within HeLa cell lysosomes.211 These lysosomotropic agent modified liposomes can prove to be a novel therapeutic strategy for the treatment of Gaucher’s fibroblasts and model diseased cells. Keeping this in mind, lysosomotropic Rh tagged liposomes were loaded with velaglucera alpha (VPRIV) and their intracellular and lysosomal delivery has been investigated.
In parallel, some novel liposomal nanoformulations loaded with varied therapeutic enzymes have proved to be a promising strategy of ERT.212 The biodistribution of β-fructofuranosidase containing liposomes has demonstrated up to 50% enzyme activity build-up in liver cell lysosomal fractions.213 In parallel, liposome encapsulated neuraminidase, α-mannosidase, and β-glucosidase have been intravenously administered for their therapeutic purposes.214
Induction of Lysosome-Mediated Cell Necrosis by Cationic Liposomes
Cationic liposomes (CLs) are commonly used as gene delivery vectors, having excellent biocompatibility and biodegradability. The CLs induce enhanced autophagy and promote cell death by significant accumulation of autophagosomes.215 It has been hypothesized that CLs induce LMP through mTOR-independent autophagic flux.216 In addition, the autophagic flux is altered by CLs at early stage and inhibiting at the later stages. This leads to the induction of cellular toxicity by the induction of LMP and the inhibition of autophagic flux. These hypotheses have been supported by the study of CLs on autophagic flux and dysfunction of lysosomes in human liver epithelial cell lines. The results have shown cellular toxicity by the induction of LMP and the inhibition of autophagic flux, with the release of cytoplasmic cathepsin B, enhanced ROS production, and mitochondrial dysfunctions, the key mediators of cellular toxicity.217
Besides this, the cationic liposomes (CLs) have also been used to impair Na+/K+-ATPase in lung cells to induce cell necrosis.218 Furthermore, the CLs can destabilize the plasma membranes by inducing the formation of nonbilayer lipid structures and promotes pronounced cell membrane disruption.219
Carbon Nanotubes-Mediated Autophagy Blockade
Multiwalled carbon nanotubes (MWCNTs) induce an abnormal accumulation of autophagosomes, possibly because of autophagy blockade.220 The autophagy blockage occurs through the modulation of synaptosomal-associated protein (SNAPIN) expression. The chemical nature of NPs and its shape, length, size, crystal phase, and surface properties impact differently on autophagosome accumulation. In an interesting study by Cohignac et al,268 the shape of NPs (CNTs versus spherical carbon or titanium NPs) impacts the induction or blockade of autophagy. Indeed, MWCNTs promote the blockade of autophagic flux, whereas spherical NPs (TiO2 NPs) lead to the activation of functional autophagy.
Furthermore, CNTs have been found to exert cellular toxicity in many cell types through LMP. The LMP leads to enhanced oxidative stress, mitochondrial dysfunctions, and cathepsins release.221 In addition, LMP is a potential mechanism of autophagic flux inhibition, through the blockade of lysosome-autophagosome fusion. This leads to the accumulation of autophagosomes and their substrates like ubiquitinated protein aggregates.215
Lysosomal Targeting by Saposin C Protein Nanovesicles
Saposins or sphingolipid activator proteins (SAPs) are nonenzymatic glycoproteins present in lysosomes. These glycoproteins are usually smaller in size and are essential for the degradation of sphingolipids and membrane digestion. These glycoproteins are comprised of five types as saposin A-D and the GM2 activator protein.222 Saposins play an important role in lipid transport, lipid microdomain assembly, lipid membrane binding capability and reorganization of the biological membranes.223
It has been found that phosphatidylserine is abundantly found in cancer cells,224 so it could be a novel target for saposin C. Several researchers support the idea of a linkage between cellular membrane aberrations and ceramide-facilitated initiation of apoptosis in cancer cells.225 Therefore, some novel agents have been identified which interfere with cancer cell membranes including lysosome membranes and modulate their organization, signal transduction, fluidity, and metabolic activities.226
Saposin C-dioleoyl phosphatidylserine (Sap-C-DPS) nanovesicles with a diameter of almost 190 nm were prepared and these entities presented specific cancer cell targeting. Following the administration of Sap-C-DPS nanoformulation, it got preferentially accumulated in cancer cells in tumor-induced mice. Sap-C-DPS led to the induction of apoptosis preferentially in different cancer cell types and spared the normal cells and tissues. The mechanism of Sap-C-DPS-mediated apoptosis has been found to be through the elevation of intracellular ceramides followed by the activation of caspases. The Sap-C-DPS nanoformulation has been found to significantly inhibit the growth of malignant peripheral nerve sheath tumor and preclinical xenograft. These nanoformulations can prove to be novel cancer-specific agents for the treatment of a broad range of tumors.227
Lysosome Targeting with Mixed-Charged NPs
There are some limitations with unicharged (cationic or anionic) NPs as pure anionic NPs are slowly internalized by target cells while cationic NPs, due to their strong electrostatic attractions with membranes, depolarize the membranes and generate hydrophilic membrane pores promoting their membrane permeabilization.228 Unfortunately, these NPs are non-selectively cytotoxic.229 These issues have been resolved by using mixed charged NPs in different proportions.
The AuNPs functionalized with positively charged N,N,N-trimethyl(11-mercaptoundecyl) ammonium chloride (TMA) and negatively charged 11-mercaptoundecanoic acid (MUA) ligands were prepared to form mixed charged NPs.188 Surprisingly, these mixed charged NPs have been found to possess several intriguing properties, as these are much stable, and can be precipitated or crystallized out at different pH values.230 It is assumed that these pH-dependent mixed charged NPs could prove to be novel for selective targeting of cancerous cells and their lysosomes.
Based on this innovative strategy, lysosomes have been targeted with mixed charged NPs, which gradually disrupt the lysosomal membrane integrity, finally initiating lysosome-facilitated cell death, quite selectively in cancer cells (Figure 5). These NPs cluster at cell surface, followed by the internalization of about 50–100 nm NPs through endocytosis and their gradual accumulation within multivesicular endosomes, which finally leads to shipment to the lysosomes. These mixed charged NPs form superacrystals within the lysosomes. This promotes the lysosomal swelling with a gradual weakening of lysosomal membrane integrity and finally promotes the cell death. In contrast to cancerous cells, in normal cells these mixed charged NPs show limited aggregation, and are excluded through exocytosis, so these cells get least harm. So the use of these mixed charged NPs against different cancers is presumed to be a novel strategy in fighting against cancer (Figure 5).188
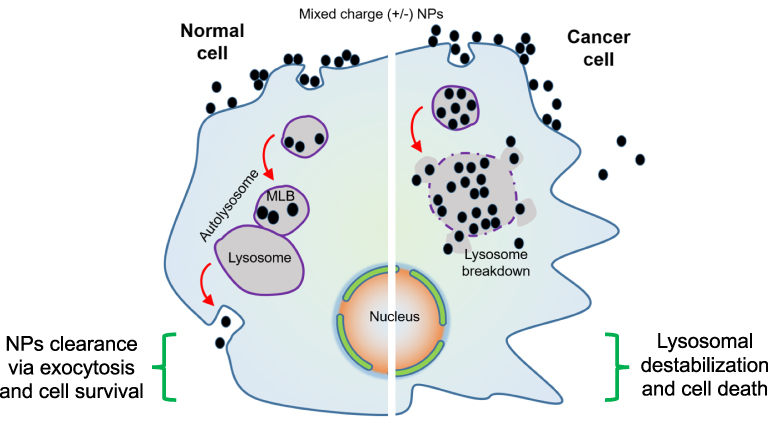 |
Figure 5 The role of mixed charged NPs within normal and cancer cells. Normal cells clear these NPs through proper exocytosis and are least damaged. In cancer cells these NPs form superacrystals and lead to lysosome breakdown resulting in cell death. |
Lysosome Targeting with Au-ZnO Hybrid NPs
Zinc oxide NPs (ZnONPs) have been conjugated with lysosome targeting peptide, to selectively enter the cancer cells through the endocytic pathway. These particles get rapidly accumulated within the lysosomes and initiate ROS generation, thus might propose a new strategy of LMP-dependent apoptotic cell death progression.231 ZnONPs were combined with AuNPs to check the progression of LMP in real-time by fluorescence quenching to understand ROS mediated lysosomal death pathways. These hybrid NPs combine the merits of both ZnO and AuNPs, achieving excellent catalytic activity and fluorescence quenching. Furthermore, these hybrid NPs have been conjugated with FITC-labeled cathepsin B substrate sequence (Arg-Arg, RR)71 and also the αvβ3 integrin-targeting peptide (RGD).232 The resulting FITC-RR-ZnO-Au-RGD NPs bind specifically to integrin αvβ3-rich HepG2 cells, accumulate within their lysosomes, and mediate ROS production and also enable real-time monitoring of LMP-dependent apoptosis in these tumor cells.
Lysosomal Membrane Permeabilization by Targeted Magnetic NPs
Targeted lysosomal dysregulation by magnetic NPs is considered as a novel alternative to overcome cancer resistance. These NPs selectively target cancer cells and enhance its LMP, so these NPs demonstrate as powerful tools in tumor therapeutics. In a novel study, iron oxide magnetic NPs (FeONPs) have been engineered to selectively target epidermal growth factor receptor (EGFR) overexpressed on cancerous cells. These NPs induce LMP under the action of an alternating magnetic field. The enhanced LMP led to the production of excessive ROS that resulted in decreased tumor viability. In these cells, the cytosolic activity of lysosomal protease cathepsin B was confirmed by confocal microscopy. These innovative findings suggest that the lysosomal death pathway can be remotely controlled in cancer cells by triggering their membrane permeabilization through the administration of magnetic FeONPs.233
Mesoporous Silica NPs Can Alter the Lysosomal Exocytosis Rate
Mesoporous Silica NPs (MSNPs) have gained much attention due to some specific properties like large internal pore volume, large surface area, good chemical and thermal stability, and tunable pore size.234 Cancer cells engulf these NPs by energy dependent endocytosis and the majority of these NPs are colocalized within endo/lysosomal compartments.235 It has been reported that MSNPs can be used as efficient imaging vectors and drug delivery vehicles in different types of cancers.236 Different animal studies have demonstrated the role of MSNPs in cancer growth inhibition by targeting lysosomes.237 The ultimate fate of MSNPs after the engulfment by cancer cells has not been fully understood. The cellular mechanism involving the exocytosis of these NPs from the cells also needs to be investigated in detail. The possible mechanisms include co-localization of these NPs with the lysosomes and may enter the Golgi apparatus for excretion or undergo lysosomal exocytosis.
In a previous study, the exocytosis of MSNPs was observed when these entities were not surface modified. But coating the surface of these NPs with 3-(trihydroxysilyl) propyl methylphosphonate improves the dispersibility of these NPs, so can be used to carry targeting moieties, thus improving its cancer therapy.238 In one study, phosphonate-modified MSNPs (P-MSNPs) have been examined to study lysosomal exocytosis. It has been observed that these NPs recover intact after their cellular excretion and this is mediated by the fusion of lysosomes with the plasma membrane. Further, it has been demonstrated that the exocytosis of these P-MSNPs can be regulated by controlling the lysosomal exocytosis. The anticancer drug is released from these NPs via diffusion, so the exposure time of these NPs within the cells could influence the amount of drug release. By decreasing the rate of exocytosis of these NPs, the camptothecin loaded P-MSNPs have been found to improve the cellular effects of drug delivery.
T-Cell Lysosome Targeting for Cancer Immunotherapy
In solid tumors, T-cell immunotherapy faces a great challenge, due to minimal activation, synthesis, and release of lysosome specific therapeutic proteins like granzyme B and perforins. In a novel study, a special type of NPs [mineralized metal-organic framework (MOF)] coupled with CD63 (lysosome targeting aptamer) have been engineered. These NPs target the lysosomes of T-cells and enhance their anticancer potential. The MOF is synthesized from dimethylimidazole and Zn2+ and Calcium carbonate (CaCO3) is used for the mineralization of these NPs. CaCO3 improves the composite material stability for encapsulating the therapeutic proteins and provides calcium ions with synergistic potential. In addition, these NPs are ideal lysosome delivery vectors and possessan efficient protein encapsulation capacity besides having acid sensitivity. Before mineralization, T-cell required therapeutic proteins (granzyme B and perforins) are preloaded with the MOF. The treatment of a specific cancer also involves the T-cell pretreatment with processed tumor-specific antigens to produce or activate memory before reprograming their lysosomes. By using these novel NPs, a significant control of breast cancer enhancement has been achieved.239
Table 4 elaborates some more examples of different types of NPs used to target lysosomes. This table also briefs the NPs size, cellular uptake mechanism, in vitro and in vivo models used, and the imaging and therapy applications (Table 4).
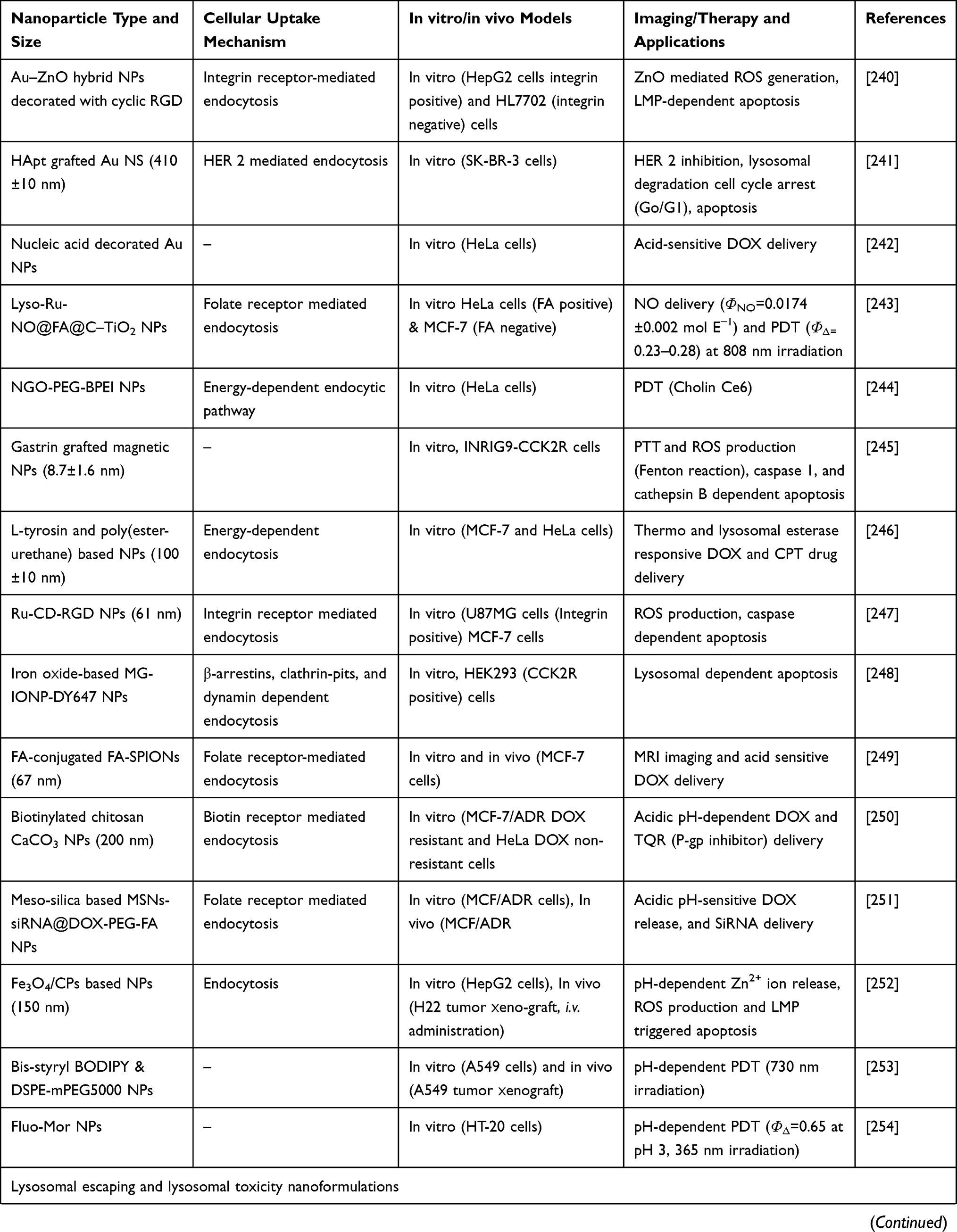 |
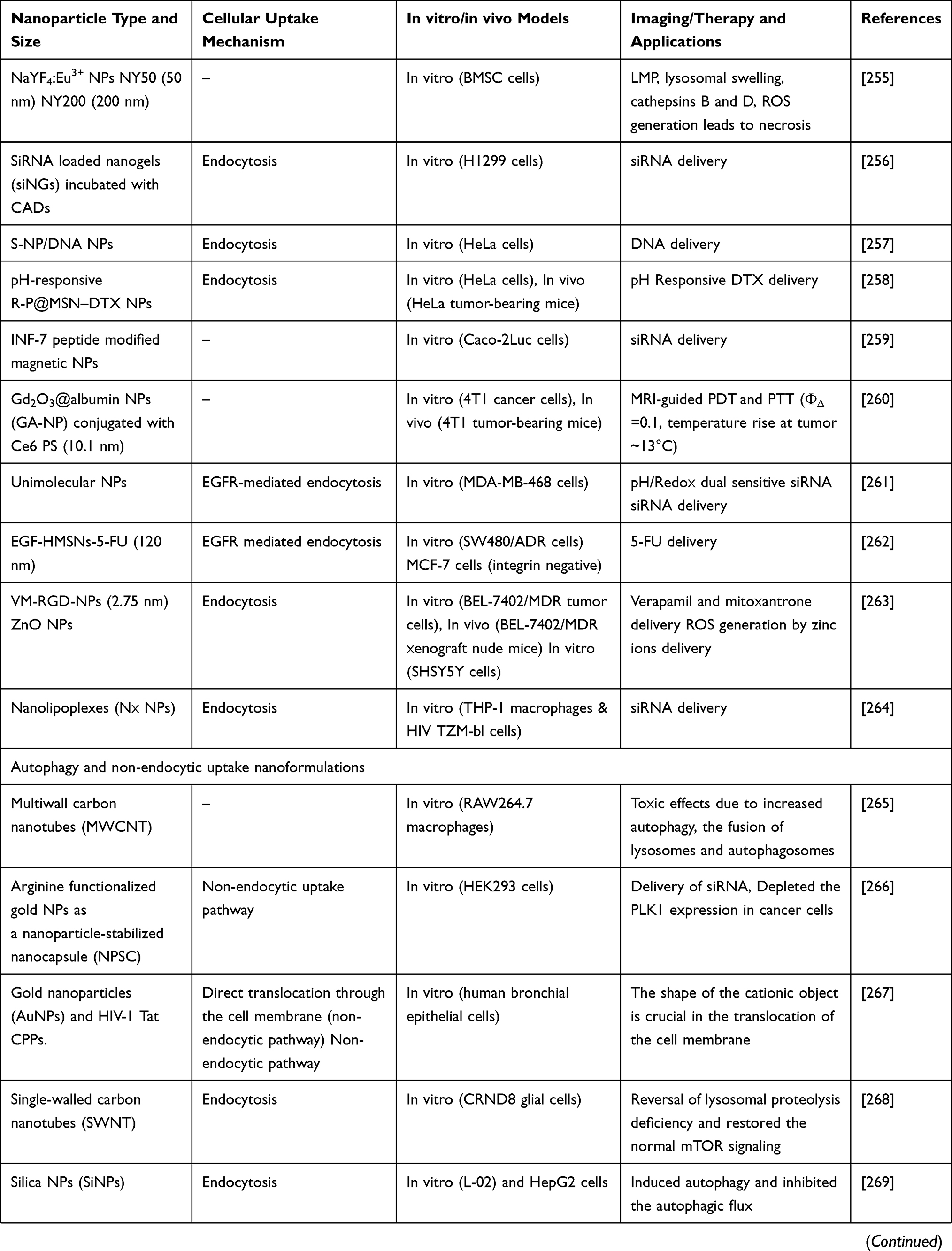 |
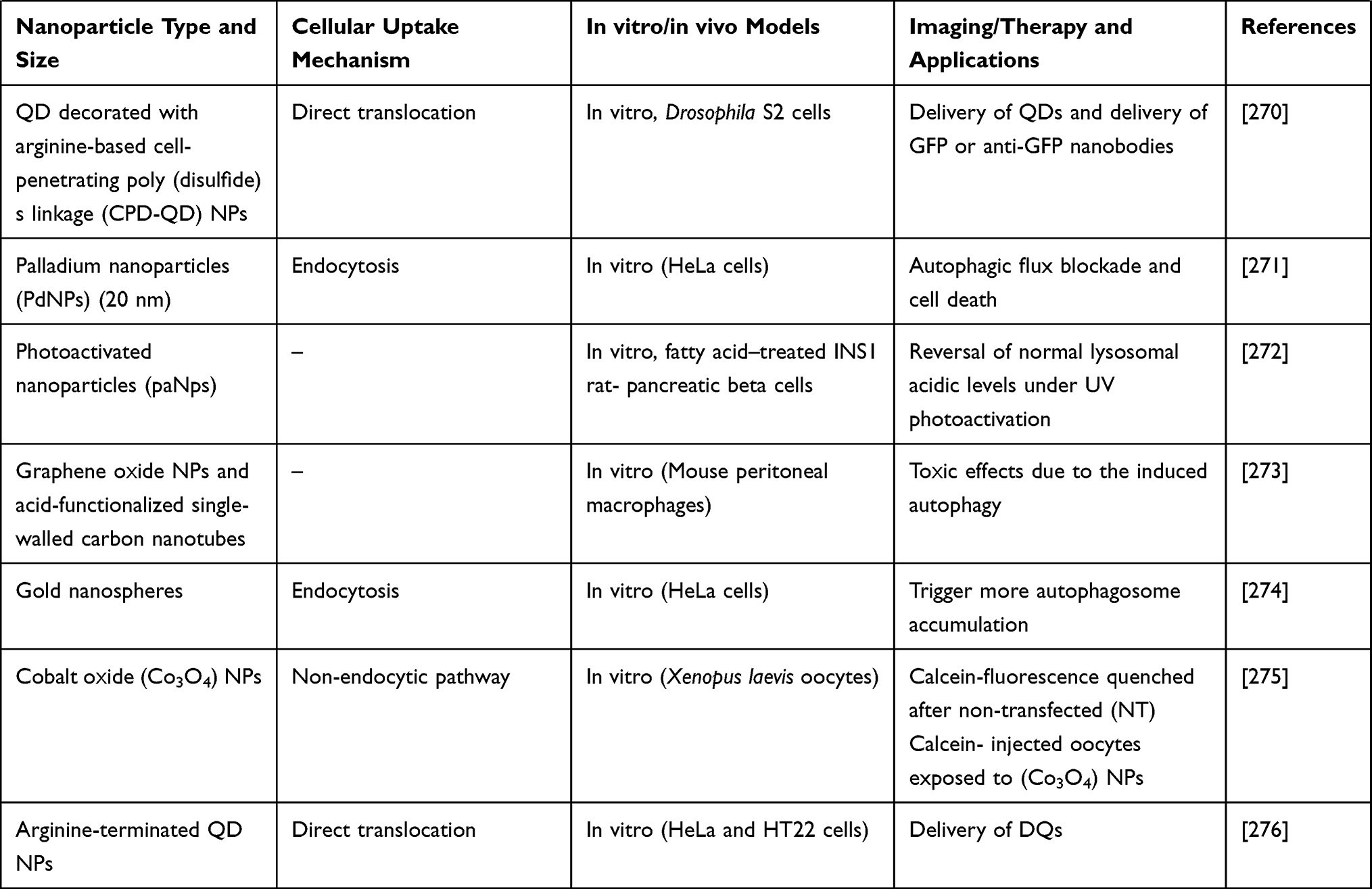 |
Table 4 Different Lysosome-Targeted Nanoformulations Used for Therapy and Imaging Purpose Based on Autophagy and Non-Endocytic Uptake in Different Cancer Cell Types |
Nanoparticles and Immune System
The therapeutic applications of NPs are often challenged by its toxicity concerns involving their interaction with various components of the immune system. It is now well known that NPs size, shape, surface charge, steric effects, and hydrophobicity/hydrophilicity can dictate NPs compatibility with the immune system.277 NPs are constantly engineered to either avoid recognition by the immune system or specifically inhibit or enhance their immune response. NPs can be engineered to modulate the cellular trafficking, thus influencing the immune system. Lysosomes are the principal subcellular catabolic organelles which are meant for degradation and recycling of both intracellular and extracellular materials, which are the final steps in phagocytosis and autophagy. Autophagy and phagocytosis in macrophages play an essential role and serve as a bridge between innate and adaptive immunity. Exposure of macrophage lysosomes with different pathogens leads to distinct alterations in its proteomics, which is closely associated with macrophage immune functions like antigen presentation, toll-like receptor activation, and inflammation.278 In addition, cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells play a vital role in the immune system, as they eliminate both virally infected and tumorigenic cells. Regulated exocytosis of perforins and proapoptotic granzymes from the secretory lysosomes of these cells helps in clearance of target cells.279
NPs elicit the immune response by either direct immuno-stimulation or by direct interaction with antigen presenting cells (APC) or by delivery of antigens to specific cellular compartments.280 Metal drug nanoformulations are well known to possess cytotoxic anticancer potential and can interact with the cancer-immune interface and can reverse immune evasion aspects. Metal drug nanoformulations have the fidelity to induce a long-lasting anticancer immune response.281 After conventional chemotherapy, the anticancer immune response may contribute to control the cancer. In addition, treatment with radiotherapy and some chemotherapeutic drugs, like anthracyclines, can induce specific immune responses leading to immunogenic cancer cell death. The residual cancer cells are eliminated by this anticancer immune response which can also maintain micrometastases in the dormancy stage.282
Within the biological system, different molecules interact with NPs and ultimately lead to the formation of a protein coat around it called the protein corona. This protein corona of NPs plays a significant role in modulating the macrophage behavior.283 NPs are usually first picked up by macrophages which can lead to immunosuppression or immunostimulation, and can promote autoimmune or inflammatory disorders or increase the host’s susceptibility to infection and cancer. The immune cells can inadvertently recognize the NPs as foreign entities which can result in multilevel immune response resulting in toxicity and diminish their therapeutic efficacy. For example, granuloma formation has been reported in the skin and lungs in animals exposed to CNTs.284
When NPs are engineered to behave as self without immune recognition by the host, this is considered the first success in the field of bioimaging or drug delivery. NPs are provided a hydrophilic environment by tagging with polyethylene glycol (PEG) or other polymers, to shield from immune recognition.285 But some data reveals that even after PEGylation some NPs can elicit antibodies. These antibodies promote faster clearance of such NPs from blood and change their pharmacokinetic profile.286 So, NP-specific antibodies can affect the safety and efficacy of their therapeutic potential.
NPs mediated immunosuppression can be either desirable or inadvertent. On one hand immunosuppression can lower the defense against cancer cells and infections, but on the other hand, it may augment the therapeutic advantages of treatment for autoimmune diseases and allergies and can also prevent the transplanted organ rejection. Most studies focus on inflammatory potential of NPs, while few studies have demonstrated the immunosuppression by the inhalation of CNTs suppressing B-cell functions and the production of TGF-β by alveolar macrophages.287 NPs can aid to deliver immunosuppressive drugs and prevent immunosuppressive properties of some other drugs.288 In one study, poly(D,L-lactide-co-glycolide) (PLGA) NPs have been used to deliver glucocorticoids in mouse model inflamed joints as a treatment for arthritis.289
Some breast cancer patients treated with Abraxane (paclitaxel bound to human serum albumin) NPs have demonstrated grade 4 neutropenia (decreased number of neutrophils), a form of myelosuppression.290 Some immunosuppressive agents (eg, corticosteroids, cadmium, tetrachlorodibenzo-p-dioxin) act by impairing the development and functions of T-cells. So the use of quantum dots (cadmium containing NPs) on thymus needs adequate studies to understand their therapeutic potential.291
NPs are also evaluated for their potential to stimulate adaptive and innate immune responses. The activation of a complement system can be damaging if the NPs inadvertently, or by their design, face the systemic circulation, which can lead to anaphylaxis and other hypersensitivity reactions.292 The nanoformulation size is considered as a major factor to determine whether it induces type I (interferon-γ) or type II (Interleukin-4) cytokines, thus contributing to different types of immune response.293
The basics of NPs mediated immune recognition and examples of the allergic reactions due to its exposure in humans and laboratory test animals have been reported somewhere else.294 Some allergic reactions are constantly reported during the occupational hazards as exposure to nanoformulations. For example, toxic epidermal necrolysis-like dermatitis has been reported in workers manufacturing dendrimers.295
NPs loaded with immunotherapy particles can elicit a strong antitumor response. The immune cells have the capacity to proliferate and propagate the response further by activating complementary immune cells. Immunotherapy-loaded NPs can be directly conjugated to the surface of T-cells. This approach is used to release payloads to augment the functions of either T-cells themselves or to release the payload to modify the tumor microenvironment.296 Some standard payloads include cytokines like IL-21,297 and cytotoxic drugs like SN-38. Unlike free NPs, T-cells can concentrate their payloads in tumors by two orders of magnitude. The immunomodulatory NPs can be conjugated to the surface of any leukocyte population, thus facilitating a broad utility for cancer management.
Clinical Trials and Cancer Management
The research over cancer management by lysosome-targeted drugs and its nanoformulation is going on to evaluate the clinical trials and its phases. More than 40 clinical trials have been performed by using hydroxychloroquine (HCQ) on humans and dogs worldwide.298 Six Phase I/II clinical trials have been accomplished in patients having melanoma, glioblastoma, refractory myeloma, and other cancers.299–301 These clinical trials also comprise some combination therapies planned from preclinical investigations.302–304 These trials have demonstrated that cancer management in humans can be efficiently achieved safely through autophagy inhibition. These trials reported the accumulation of autophagic vesicles in peripheral blood mononuclear and cancer cells. The treatment combinations were tolerated even to higher doses without any metabolic dysfunctions, liver damage, or some neurological impairments.298 However, some dose limiting toxicities have also been reported by using HCQ–cancer drug combinations. The Phase II clinical trials have also revealed that some more potent drug formulations are required to have a better outcome as higher doses of HCQ alone did not demonstrate better therapeutic efficacy for previously treated metastatic pancreatic cancer.305 The CQ dimerization led to the formation of Lys05, which is far more potent as a single agent and in combination with B-Raf protooncogene serine/threonine protein kinase (BRAF) inhibitors.306
Future Aspects of Targeting Lysosomes and Cancer Management
The recent findings clearly mention that the degradative functions of lysosomes are closely linked to multiple pathways, which control the overall cellular homeostasis. Now past are the days when lysosomes were considered as isolated organelles with limited functions and little contacts with other organelles and processes. Some recent research has elicited the role of lysosomal activity involving cell trafficking, nutrient sensing, kinase signaling and death signaling. The new studies must report the lysosomal functions in the context of the entire cell and organism.
Despite the recently published massive research about lysosomes, its connection with other organelles and cellular processes, it just represents the tip of an iceberg. The number of new queries build up much faster than the previous doubts are cleared. Some of the new queries to be resolved in the future are to understand the role of hundreds of peripheral and integral lysosomal membrane proteins whose functions have not been discovered yet. In addition, how do ion and nutrient transporters respond to cellular metabolism and connect lysosomes with cellular environment, needs to be solved.
Regarding the cancer cells, the knowledge about lysosomes is still in its infancy. The future tasks include to know the role of lysosomes in a varied cellular environment, and to know if these variations are the same in all cancer cells or change from cell-to-cell or vary at different stages of cancer cells. The approach of direct targeting lysosomes with anticancer drug nanoformulations within the tumor cells will dramatically decrease the drug action at non-specific locations, side-effects, and unwanted higher drug load. The specific pin-point drug targeting tactics are the ultimate goal of future therapeutics in cancer management.
Some novel biomarkers availability for the assessment of drug efficacy is one of the major limitations for clinical trials. The current methods for the visualization of autophagic vesicles accumulation in cancer cells include electron microscopy along with Western blotting and immunohistochemistry. Some recent evidence has encouraged that more potent autophagy inhibitors will be available which can be used synergistically with radiotherapy and conventional chemotherapy. Although in recent years, the knowledge about NPs and their interaction with immune system components have improved, but still many questions require a thorough understanding and deeper investigation. Some more mechanistic studies are needed to investigate the NPs immunomodulatory effects to improve the understanding of physicochemical parameters in relation with the immune system.
The organelle-targeting nanoformulations face tremendous challenges because of wide variations in biological systems. Different nanoformulations like micelles, liposomes, dendrimers, CNTs, etc. tagged/loaded with different lysosomotropic agents and anticancer drugs are being worked out in current research. However, it is of utmost importance to have comprehensive research to comprehend appropriately the safety aspects of these nanoformulations when used in human subjects. Furthermore, it is a very challenging task to prepare perfectly targeted oriented lysosomal drug nanoformulations in cancer cells without any toxicity to nearby normal tissues.
To overcome these challenges, it is very important to know the physico-chemical properties of different nanoformulations like size, shape, charge, drug loading, and its release capacity in addition to specific targeting. So the challenges of cancer management by lysosome targeted drug nanoformulations are quite tough, which are expected to be resolved appropriately by undergoing vigorous research in this area.
Conclusion
The recent updates about the structure and function of lysosomes as well as their role in different diseases including cancer is quite fascinating. The lysosomes are now well recognized as the central communication hub of the major metabolic activities within the cell. The knowledge about the changes within lysosomes in cancer advancement and treatment is still quite young, but some current innovative advances in this area promise speedy progress in the near future. The recent updates about the lysosome ultrastructure, role in different diseases, its cross-talk with other organelles, and the use of some drug nanoformulations, which directly or indirectly target this organelle within the tumor cells, is currently being mentioned to boost the therapeutic strategies. In different cancer cells, the lysosomes have been targeted with different lysosomotropic drug nanoformulations to perturb its signal transduction cascades, pH, Ca2+ homeostasis, membrane permeabilization, and disruption in autophagy and apoptosis.
One of the distinguished hallmarks of cancer is its ability to escape or dodge the immune response. Some more recent scientific advances elucidate the implementation of innovative approaches for immunotherapies to eradicate or treat diverse cancers. NPs can facilitate the location, pharmacokinetics, and co-delivery of special immunomodulatory drugs eliciting the anticancer responses which cannot be achieved by free drugs. The convergence of biotechnology, nanotechnology, cancer immunotherapy, and drug delivery approaches can now be employed to eradicate the cancer menace in the not-too-distant future. Only a few nanoformulations like peptide facilitated-AuNPs, TPP-PEG-biotin, octadecyl-rhodamine B, cationic liposomes, and mixed charged NPs have been engineered and satisfactorily used to target lysosomes in cancer cells. Despite the initial success of some free drugs or drug-conjugated nanoformulations targeted to lysosomes, systematic pre-clinical and clinical surveys are required for their authentic use in final clinical settings.
Abbreviations
AKT, serine-threonine–specific protein kinase; AMP, adenosine monophosphate; AMPK, 5′-adenosine monophosphate-activated protein kinase; CaMKK2, calcium/calmodulin-dependent protein kinase kinase 2; EB, transcription factor EB; EF25-(GSH)2, 3,5-bis(2-hydroxybenzylidine)tetrahydro-4H-pyran-4-1 glutathione conjugate (a water-soluble curcumin analog); ER, endoplasmic reticulum; ESCRT, endosomal sorting complex required for transport; FNIP, folliculin interacting protein; γH2AX, γ-H2a histone family member X; GAL-3,-8,-9, galactins; GSK, glycogen synthase kinase; GTPase, guanosine triphosphatase; JNKs, c-Jun NHκ-terminal kinases; LBP8, lipid-binding protein 8; LC3A/B-II, microtubule-associated protein 1A/1B light chain 3B, modified lipid form; LC3-II, light chain 3 phosphatidylethanolamine conjugate; LKB1, liver kinase B1; miR-101, microRNA 101; mTOR, mechanistic target of rapamycin; mTORC1, mechanistic target of rapamycin complex 1; NF-κB, nuclear factor κ light chain-enhancer of activated B cells; OCRL, oculocerebrorenal syndrome protein; OEA, oleoylethanolamide; p21, cyclin-dependent kinase-interacting protein 1; p53, tumor protein p53; p70S6, ribosomal protein S6 kinase; PI3K, phosphoinositide 3-kinase; PIP5K, phosphatidylinositol 4-phosphate 5-kinases; RAG GTPases, Ragulator GTPase; ROS, ROS proto-oncogene, receptor tyrosine kinase; TFEB, transcription factor EB; TLR9, toll-like receptor 9; TRIM16, tripartite motif-containing protein 16; TSC, tuberous sclerosis complex; ULK1, Unc-51-like kinase 1; Wnt, wingless/integrated.
Funding
Researchers would like to thank the Deanship of Scientific Research, Qassim University for funding publication of this project.
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Hesketh GG, Wartosch L, Davis LJ, Bright NA, Luzio JP. The lysosome and intracellular signalling. Prog Mol Subcell Biol. 2018;57:151–180.
2. Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. 2020;21(2):101–118. doi:10.1038/s41580-019-0185-4
3. Weber K, Schilling JD. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J Biol Chem. 2014;289(13):9158–9171. doi:10.1074/jbc.M113.531202
4. Englinger B, Pirker C, Heffeter P, et al. Metal drugs and the anticancer immune response. Chem Rev. 2018;119(2):1519–1624.
5. Egen JG, Ouyang W, Wu LC. Human anti-tumor immunity: insights from immunotherapy clinical trials. Immunity. 2020;52(1):36–54. doi:10.1016/j.immuni.2019.12.010
6. Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018;6(24):476. doi:10.21037/atm.2018.11.39
7. Maxfield FR, Willard JM, Lu S, editors. Lysosomes: Biology, Diseases, and Therapeutics. John Wiley & Sons; 2016.
8. Domagala A, Fidyt K, Bobrowicz M, Stachura J, Szczygiel K, Firczuk M. Typical and atypical inducers of lysosomal cell death: a promising anticancer strategy. Int J Mol Sci. 2018;19(8):2256. doi:10.3390/ijms19082256
9. Pagliero RJ, D’Astolfo DS, Lelieveld D, et al. Discovery of small molecules that induce lysosomal cell death in cancer cell lines using an image-based screening platform. Assay Drug Dev Technol. 2016;14(8):489–510. doi:10.1089/adt.2016.727
10. Zoroddu MA, Medici S, Ledda A, Nurchi VM, Lachowicz JI, Peana M. Toxicity of nanoparticles. Curr Med Chem. 2014;21(33):3837–3853. doi:10.2174/0929867321666140601162314
11. Trivedi PC, Bartlett JJ, Pulinilkunnil T. Lysosomal biology and function: modern view of cellular debris bin. Cells. 2020;9(5):1131. doi:10.3390/cells9051131
12. Bouche V, Espinosa AP, Leone L, Sardiello M, Ballabio A, Botas J. Drosophila Mitf regulates the V-ATPase and the lysosomal-autophagic pathway. Autophagy. 2016;12(3):484–498. doi:10.1080/15548627.2015.1134081
13. Stoka V, Turk V, Turk B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev. 2016;1(32):22–37. doi:10.1016/j.arr.2016.04.010
14. Liaudet-Coopman E, Beaujouin M, Derocq D, et al. Cathepsin D: newly discovered functions of a long-standing aspartic protease in cancer and apoptosis. Cancer Lett. 2006;237(2):167–179. doi:10.1016/j.canlet.2005.06.007
15. Kolter T, Sandhoff K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010;584(9):1700–1712. doi:10.1016/j.febslet.2009.10.021
16. Fehrenbacher N, Bastholm L, Kirkegaard-Sørensen T, et al. Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res. 2008;68(16):6623–6633. doi:10.1158/0008-5472.CAN-08-0463
17. Schneede A, Schmidt CK, Hölttä‐Vuori M, et al. Role for LAMP‐2 in endosomal cholesterol transport. J Cell Mol Med. 2011;15(2):280–295. doi:10.1111/j.1582-4934.2009.00973.x
18. Eskelinen EL, Tanaka Y, Saftig P. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003;13(3):137–145. doi:10.1016/S0962-8924(03)00005-9
19. Kundra R, Kornfeld S. Asparagine-linked oligosaccharides protect Lamp-1 and Lamp-2 from intracellular proteolysis. J Biol Chem. 1999;274(43):31039–31046. doi:10.1074/jbc.274.43.31039
20. Tian X, Teng J, Chen J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy. 2020;24:1–9.
21. Kleine Balderhaar HJ, Ungermann C. CORVET and HOPS tethering complexes–coordinators of endosome and lysosome fusion. J Cell Sci. 2013;126(6):1307–1316. doi:10.1242/jcs.107805
22. Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14(5):283–296. doi:10.1038/nrm3565
23. Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5(228):ra42. doi:10.1126/scisignal.2002790
24. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. doi:10.1016/j.cell.2017.02.004
25. Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. doi:10.1091/mbc.e08-12-1248
26. Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010a;465(7300):942–946. doi:10.1038/nature09076
27. Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol. 2019;21(2):133–142. doi:10.1038/s41556-018-0244-7
28. Li P, Gu M, Xu H. Lysosomal ion channels as decoders of cellular signals. Trends Biochem Sci. 2019;44(2):110–124. doi:10.1016/j.tibs.2018.10.006
29. Wang ZC, Liu Y, Wang H, Han QK, Lu C. Research on the relationship between artesunate and Raji cell autophagy and apoptosis of Burkitt’s lymphoma and its mechanism. Eur Rev Med Pharmacol Sci. 2017;21(9):2238–2243.
30. Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;17(74):69–86. doi:10.1146/annurev-physiol-012110-142317
31. Cheng X, Shen D, Samie M, Xu H. Mucolipins: intracellular TRPML1–3 channels. FEBS Lett. 2010;584(10):2013–2021. doi:10.1016/j.febslet.2009.12.056
32. Zhang X, Cheng X, Yu L, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016a;7(1):1–2.
33. Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17(3):288–299. doi:10.1038/ncb3114
34. Dong XP, Wang X, Shen D, et al. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat Commun. 2010;1(4):38.
35. Medina DL, Fraldi A, Bouche V, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21(3):421–430. doi:10.1016/j.devcel.2011.07.016
36. Samie M, Wang X, Zhang X, et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell. 2013;26(5):511–524. doi:10.1016/j.devcel.2013.08.003
37. Wang F, Gómez‐Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19(12):918–931. doi:10.1111/tra.12613
38. Kim YM, Jung CH, Seo M, et al. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell. 2015a;57(2):207–218. doi:10.1016/j.molcel.2014.11.013
39. Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell. 2001;106(2):157–169. doi:10.1016/S0092-8674(01)00421-4
40. Naegeli KM, Hastie E, Garde A, et al. Cell invasion in vivo via rapid exocytosis of a transient lysosome-derived membrane domain. Dev Cell. 2017;43(4):403–417. doi:10.1016/j.devcel.2017.10.024
41. Baron R, Neff L, Brown W, Courtoy PJ, Louvard D, Farquhar MG. Polarized secretion of lysosomal enzymes: co-distribution of cation-independent mannose-6-phosphate receptors and lysosomal enzymes along the osteoclast exocytic pathway. J Cell Biol. 1988;106(6):1863–1872. doi:10.1083/jcb.106.6.1863
42. Wu H, Carvalho P, Voeltz GK. Here, there, and everywhere: the importance of ER membrane contact sites. Science. 2018;361:6401. doi:10.1126/science.aan5835
43. Friedman JR, DiBenedetto JR, West M, Rowland AA, Voeltz GK, Hegde RS. Endoplasmic reticulum–endosome contact increases as endosomes traffic and mature. Mol Biol Cell. 2013;24(7):1030–1040. doi:10.1091/mbc.e12-10-0733
44. Dong R, Saheki Y, Swarup S, Lucast L, Harper JW, De Camilli P. Endosome-ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell. 2016;166(2):408–423. doi:10.1016/j.cell.2016.06.037
45. Wilhelm LP, Wendling C, Védie B, et al. STARD 3 mediates endoplasmic reticulum‐to‐endosome cholesterol transport at membrane contact sites. EMBO J. 2017;36(10):1412–1433. doi:10.15252/embj.201695917
46. Glunde K, Guggino SE, Solaiyappan M, Pathak AP, Ichikawa Y, Bhujwalla ZM. Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia. 2003;5(6):533–545. doi:10.1016/S1476-5586(03)80037-4
47. Don AS, Hsiao JH, Bleasel JM, Couttas TA, Halliday GM, Kim WS. Altered lipid levels provide evidence for myelin dysfunction in multiple system atrophy. Acta Neuropathol Commun. 2014;2(1):1–4. doi:10.1186/s40478-014-0150-6
48. Petersen NH, Olsen OD, Groth-Pedersen L, et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. 2013;24(3):379–393. doi:10.1016/j.ccr.2013.08.003
49. Zhitomirsky B, Assaraf YG. Lysosomes as mediators of drug resistance in cancer. Drug Resist Updat. 2016;24:23–33. doi:10.1016/j.drup.2015.11.004
50. Jamora M, Fuchs E, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003;112(4):535–548. doi:10.1016/S0092-8674(03)00108-9
51. Mousavi SA, Brech A, Berg T, Kjeken R. Phosphoinositide 3-kinase regulates maturation of lysosomes in rat hepatocytes. Biochem J. 2003;372(3):861–869. doi:10.1042/bj20021136
52. Madge LA, Li JH, Choi J, Pober JS. Inhibition of phosphatidylinositol 3-kinase sensitizes vascular endothelial cells to cytokine-initiated cathepsin-dependent apoptosis. J Biol Chem. 2003;278(23):21295–21306. doi:10.1074/jbc.M212837200
53. Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25(5):1037–1043. doi:10.1016/j.cmet.2017.04.004
54. Di Malta C, Siciliano D, Calcagni A, et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. 2017;356(6343):1188–1192. doi:10.1126/science.aag2553
55. Li X, Rydzewski N, Hider A, et al. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat Cell Biol. 2016;18(4):404–417. doi:10.1038/ncb3324
56. Bian B, Mongrain S, Cagnol S, et al. Cathepsin B promotes colorectal tumorigenesis, cell invasion, and metastasis. Mol Carcinog. 2016;55(5):671–687. doi:10.1002/mc.22312
57. Ibrahim SA, El-Ghonaimy EA, Hassan H, et al. Hormonal-receptor positive breast cancer: IL-6 augments invasion and lymph node metastasis via stimulating cathepsin B expression. J Adv Res. 2016;7(5):661–670. doi:10.1016/j.jare.2016.06.007
58. Small DM, Burden RE, Jaworski J, et al. Cathepsin S from both tumor and tumor‐associated cells promote cancer growth and neovascularization. Int J Cancer Res. 2013;133(9):2102–2112. doi:10.1002/ijc.28238
59. Silverman GA, Bartuski AJ, Çataltepe S, et al. SCCA1 and SCCA2 are proteinase inhibitors that map to the serpin cluster at 18q21.3. Tumor Biol. 1998;19(6):480–487. doi:10.1159/000030041
60. Kirkegaard T, Roth AG, Petersen NH, et al. Hsp70 stabilizes lysosomes and reverts Niemann–Pick disease-associated lysosomal pathology. Nature. 2010;463(7280):549–553. doi:10.1038/nature08710
61. Gyrd-Hansen M, Nylandsted J, Jattela M. Heat shock protein 70 promotes cancer cell viability by safeguarding lysosomal integrity. Cell Cycle. 2004;3(12):1484–1485. doi:10.4161/cc.3.12.1287
62. Liu N, Raja SM, Zazzeroni F, et al. NF‐κB protects from the lysosomal pathway of cell death. EMBO J. 2003;22(19):5313–5322. doi:10.1093/emboj/cdg510
63. Halaby R. Role of lysosomes in cancer therapy. Res Rep Biol. 2015;28(6):147–155. doi:10.2147/RRB.S83999
64. Dielschneider RF, Henson ES, Gibson SB. Lysosomes as oxidative targets for cancer therapy. In: Oxidative Medicine and Cellular Longevity. 2017:2017.
65. Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30(17):1913–1930. doi:10.1101/gad.287524.116
66. Levy JM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–542. doi:10.1038/nrc.2017.53
67. Commisso C, Davidson SM, Soydaner-Azeloglu RG, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497(7451):633–637. doi:10.1038/nature12138
68. Xie J, Wang X, Proud CG. mTOR inhibitors in cancer therapy. F1000Research. 2016;5:2078. doi:10.12688/f1000research.9207.1
69. Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8(2):200–212. doi:10.4161/auto.8.2.18554
70. Towers CG, Thorburn A. Targeting the lysosome for cancer therapy. Cancer Discov. 2017;7(11):1218–1220. doi:10.1158/2159-8290.CD-17-0996
71. Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27(50):6434–6451. doi:10.1038/onc.2008.310
72. Yu F, Chen Z, Wang B, et al. The role of lysosome in cell death regulation. Tumor Biol. 2016;37(2):1427–1436. doi:10.1007/s13277-015-4516-6
73. Ostenfeld MS, Fehrenbacher N, Høyer-Hansen M, Thomsen C, Farkas T, Jäättelä M. Effective tumor cell death by σ-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 2005;65(19):8975–8983. doi:10.1158/0008-5472.CAN-05-0269
74. Mena S, Rodriguez ML, Ponsoda X, Estrela JM, Jäättela M, Ortega AL. Pterostilbene-induced tumor cytotoxicity: a lysosomal membrane permeabilization-dependent mechanism. PLoS One. 2012;7(9):e44524. doi:10.1371/journal.pone.0044524
75. Chen QY, Shi JG, Yao QH, et al. Lysosomal membrane permeabilization is involved in curcumin-induced apoptosis of A549 lung carcinoma cells. Mol Cell Biochem. 2012;359(1):389–398. doi:10.1007/s11010-011-1033-9
76. Sukhai MA, Prabha S, Hurren R, et al. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. J Clin Invest. 2012;123(1):315–328.
77. Terman A, Kurz T, Gustafsson B, Brunk UT. Lysosomal labilization. IUBMB Life. 2006;58(9):531–539. doi:10.1080/15216540600904885
78. Boya P, Gonzalez-Polo RA, Poncet D, et al. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene. 2003b;22(25):3927–3936. doi:10.1038/sj.onc.1206622
79. Boya P, Andreau K, Poncet D, et al. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med. 2003a;197(10):1323–1334. doi:10.1084/jem.20021952
80. Paris C, Bertoglio J, Breard J. Lysosomal and mitochondrial pathways in miltefosine-induced apoptosis in U937 cells. Apoptosis. 2007;12(7):1257–1267. doi:10.1007/s10495-007-0052-1
81. Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol. 2006;290(6):G1339–46. doi:10.1152/ajpgi.00509.2005
82. Gyrd-Hansen M, Farkas T, Fehrenbacher N, et al. Apoptosome-independent activation of the lysosomal cell death pathway by caspase-9. Mol Cell Biol. 2006;26(21):7880–7891. doi:10.1128/MCB.00716-06
83. Werneburg NW, Guicciardi ME, Bronk SF, Gores GJ. Tumor necrosis factor-α-associated lysosomal permeabilization is cathepsin B dependent. Am J Physiol. 2002;283(4):G947–56. doi:10.1152/ajpgi.00151.2002
84. Groth-Pedersen L, Ostenfeld MS, Høyer-Hansen M, Nylandsted J, Jäättelä M. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome-destabilizing siramesine. Cancer Res. 2007;67(5):2217–2225. doi:10.1158/0008-5472.CAN-06-3520
85. Ichinose S, Usuda J, Hirata T, et al. Lysosomal cathepsin initiates apoptosis, which is regulated by photodamage to Bcl-2 at mitochondria in photodynamic therapy using a novel photosensitizer, ATX-s10 (Na). Int J Oncol. 2006;29(2):349–355.
86. Trincheri NF, Nicotra G, Follo C, Castino R, Isidoro C. Resveratrol induces cell death in colorectal cancer cells by a novel pathway involving lysosomal cathepsin D. Carcinogenesis. 2007;28(5):922–931. doi:10.1093/carcin/bgl223
87. Heinrich M, Neumeyer J, Jakob M, et al. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and-3 activation. Cell Death Differ. 2004;11(5):550–563. doi:10.1038/sj.cdd.4401382
88. Li N, Zheng Y, Chen W, et al. Adaptor protein LAPF recruits phosphorylated p53 to lysosomes and triggers lysosomal destabilization in apoptosis. Cancer Res. 2007;67(23):11176–11185. doi:10.1158/0008-5472.CAN-07-2333
89. Paquet C, Sane AT, Beauchemin M, Bertrand R. Caspase-and mitochondrial dysfunction-dependent mechanisms of lysosomal leakage and cathepsin B activation in DNA damage-induced apoptosis. Leukemia. 2005;19(5):784–791. doi:10.1038/sj.leu.2403717
90. Hamilton RF
91. Yan CH, Liang ZQ, Gu ZL, Yang YP, Reid P, Qin ZH. Contributions of autophagic and apoptotic mechanisms to CrTX-induced death of K562 cells. Toxicon. 2006;47(5):521–530. doi:10.1016/j.toxicon.2006.01.010
92. Bao H, Zhang Q, Zhu Z, et al. BHX, a novel pyrazoline derivative, inhibits breast cancer cell invasion by reversing the epithelial-mesenchymal transition and down-regulating Wnt/-catenin signaling. Sci Rep. 2017;7:9153. doi:10.1038/s41598-017-09655-7
93. O'Brien MA, Kirby R. Apoptosis: A review of pro‐apoptotic and anti‐apoptotic pathways and dysregulation in disease. J Vet Emerg Crit Care (San Antonio). 2008;18(6):572–585.
94. Zaman S, Wang R, Gandhi V. Targeting the apoptosis pathway in hematologic malignancies. Leuk Lymphoma. 2014;55:1980–1992. doi:10.3109/10428194.2013.855307
95. Lopez J, Tait SWG. Mitochondrial apoptosis: killing cancer using the enemy within. Br J Cancer. 2015;112(6):957–962. doi:10.1038/bjc.2015.85
96. Villa-Pulgarín JA, Gajate C, Botet J, et al. Mitochondria and lipid raft-located FoF1-ATP synthase as major therapeutic targets in the antileishmanial and anticancer activities of ether lipid edelfosine. PLoS Negl Trop Dis. 2017;11(8):e0005805. doi:10.1371/journal.pntd.0005805
97. Pfeffer CM, Singh AT. Apoptosis: a target for anticancer therapy. Int J Mol Sci. 2018;19(2):448.
98. Oberle C, Huai J, Reinheckel T, et al. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 2010;17(7):1167–1178. doi:10.1038/cdd.2009.214
99. Dielschneider RF, Eisenstat H, Mi S, et al. Lysosomotropic agents selectively target chronic lymphocytic leukemia cells due to altered sphingolipid metabolism. Leukemia. 2016;30(6):1290–1300. doi:10.1038/leu.2016.4
100. Zang Y, Beard RL, Chandraratna RA, Kang JX. Evidence of a lysosomal pathway for apoptosis induced by the synthetic retinoid CD437 in human leukemia HL-60 cells. Cell Death Differ. 2001;8(5):477–485. doi:10.1038/sj.cdd.4400843
101. Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11(4):777–790. doi:10.1089/ars.2008.2270
102. Chen Y, Henson ES, Xiao W, et al. Tyrosine kinase receptor EGFR regulates the switch in cancer cells between cell survival and cell death induced by autophagy in hypoxia. Autophagy. 2016;12(6):1029–1046. doi:10.1080/15548627.2016.1164357
103. Hasegawa J, Maejima I, Iwamoto R, Yoshimori T. Selective autophagy: lysophagy. Methods. 2015;15(75):128–132. doi:10.1016/j.ymeth.2014.12.014
104. Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2021;20:1–382.
105. Liu Y, Shoji-Kawata S, Sumpter RM, et al. Autosis is a Na+, K+-ATPase–regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia–ischemia. Proc Natl Acad Sci U S A. 2013;110(51):20364–20371. doi:10.1073/pnas.1319661110
106. Morel E, Mehrpour M, Botti J, et al. Autophagy: a Druggable Process. Annu Rev Pharmacol Toxicol. 2017;57(1):375–398. doi:10.1146/annurev-pharmtox-010716-104936
107. Jin F, Wang Y, Li M, et al. MiR-26 enhances chemosensitivity and promotes apoptosis of hepatocellular carcinoma cells through inhibiting autophagy. Cell Death Dis. 2017;8(1):e2540. doi:10.1038/cddis.2016.461
108. Xiong L, Liu Z, Ouyang G, et al. Autophagy inhibition enhances photocytotoxicity of Photosan-II in human colorectal cancer cells. Oncotarget. 2017;8(4):6419–6432. doi:10.18632/oncotarget.14117
109. Redmann M, Benavides GA, Berryhill TF, et al. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2017;11:73–81. doi:10.1016/j.redox.2016.11.004
110. Ganguli A, Choudhury D, Datta S, Bhattacharya S, Chakrabarti G. Inhibition of autophagy by chloroquine potentiates synergistically anti-cancer property of artemisinin by promoting ROS dependent apoptosis. Biochimie. 2014;1(107):338–349. doi:10.1016/j.biochi.2014.10.001
111. Cheng C, Wang T, Song Z, et al. Induction of autophagy and autophagy‐dependent apoptosis in diffuse large B‐cell lymphoma by a new antimalarial artemisinin derivative, SM 1044. Cancer Med. 2018;7(2):380–396. doi:10.1002/cam4.1276
112. Zhou GZ, Zhang SN, Zhang L, Sun GC, Chen XB. A synthetic curcumin derivative hydrazinobenzoylcurcumin induces autophagy in A549 lung cancer cells. Pharm Biol. 2014;52(1):111–116. doi:10.3109/13880209.2013.816971
113. Tiwari RV, Parajuli P, Sylvester PW. Synergistic anticancer effects of combined γ-tocotrienol and oridonin treatment is associated with the induction of autophagy. Mol Cell Biochem. 2015;408(1):123–137. doi:10.1007/s11010-015-2488-x
114. Basile V, Belluti S, Ferrari E, et al. bis-Dehydroxy-Curcumin triggers mitochondrial-associated cell death in human colon cancer cells through ER-stress induced autophagy. PLoS One. 2013;8(1):e53664. doi:10.1371/journal.pone.0053664
115. Jia G, Kong R, Ma ZB, et al. The activation of c-Jun NH 2-terminal kinase is required for dihydroartemisinin-induced autophagy in pancreatic cancer cells. J Exp Clin Cancer Res. 2014;33(1):1. doi:10.1186/1756-9966-33-8
116. Feng X, Li L, Jiang H, Jiang K, Jin Y, Zheng J. Dihydroartemisinin potentiates the anticancer effect of cisplatin via mTOR inhibition in cisplatin-resistant ovarian cancer cells: involvement of apoptosis and autophagy. Biochem Biophys Res Commun. 2014;444(3):376–381. doi:10.1016/j.bbrc.2014.01.053
117. Du XX, Li YJ, Wu CL, et al. Initiation of apoptosis, cell cycle arrest and autophagy of esophageal cancer cells by dihydroartemisinin. Biomed Pharmacother. 2013;67(5):417–424. doi:10.1016/j.biopha.2013.01.013
118. Zhang ZS, Wang J, Shen YB, et al. Dihydroartemisinin increases temozolomide efficacy in glioma cells by inducing autophagy. Oncol Lett. 2015a;10(1):379–383. doi:10.3892/ol.2015.3183
119. Wang Z, Hu W, Zhang JL, Wu XH, Zhou HJ. Dihydroartemisinin induces autophagy and inhibits the growth of iron-loaded human myeloid leukemia K562 cells via ROS toxicity. FEBS Open Bio. 2012;2(1):103–112. doi:10.1016/j.fob.2012.05.002
120. Shi X, Wang L, Li X, et al. Dihydroartemisinin induces autophagy-dependent death in human tongue squamous cell carcinoma cells through DNA double-strand break-mediated oxidative stress. Oncotarget. 2017;8(28):45981. doi:10.18632/oncotarget.17520
121. Qu W, Xiao J, Zhang H, et al. B19, a novel monocarbonyl analogue of curcumin, induces human ovarian cancer cell apoptosis via activation of endoplasmic reticulum stress and the autophagy signaling pathway. Int J Biol Sci. 2013;9(8):766. doi:10.7150/ijbs.5711
122. Chen K, Shou LM, Lin F, et al. Artesunate induces G2/M cell cycle arrest through autophagy induction in breast cancer cells. Anticancer Drugs. 2014;25(6):652–662. doi:10.1097/CAD.0000000000000089
123. Berte N, Lokan S, Eich M, Kim E, Kaina B. Artesunate enhances the therapeutic response of glioma cells to temozolomide by inhibition of homologous recombination and senescence. Oncotarget. 2016;7(41):67235. doi:10.18632/oncotarget.11972
124. Jiang F, Zhou JY, Zhang D, Liu MH, Chen YG. Artesunate induces apoptosis and autophagy in HCT116 colon cancer cells, and autophagy inhibition enhances the artesunate-induced apoptosis. Int J Mol Med. 2018;42(3):1295–1304.
125. Boridy S, Le PU, Petrecca K, Maysinger D. Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells. Cell Death Dis. 2014;5(5):e1216. doi:10.1038/cddis.2014.182
126. Lee HW, Jang KS, Choi HJ, Jo A, Cheong JH, Chun KH. Celastrol inhibits gastric cancer growth by induction of apoptosis and autophagy. BMB Rep. 2014a;47(12):697. doi:10.5483/BMBRep.2014.47.12.069
127. Li HY, Zhang J, Sun LL, et al. Celastrol induces apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells: an in vitro and in vivo study. Cell Death Dis. 2015;6(1):e1604. doi:10.1038/cddis.2014.543
128. Guo J, Xu B, Han Q, et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat. 2018;50(2):445. doi:10.4143/crt.2016.572
129. Veldhoen RA, Banman SL, Hemmerling DR, et al. The chemotherapeutic agent paclitaxel inhibits autophagy through two distinct mechanisms that regulate apoptosis. Oncogene. 2013;32(6):736–746. doi:10.1038/onc.2012.92
130. Xi G, Hu X, Wu B, et al. Autophagy inhibition promotes paclitaxel-induced apoptosis in cancer cells. Cancer Lett. 2011;307(2):141–148. doi:10.1016/j.canlet.2011.03.026
131. Zou SH, Du X, Lin H, Wang PC, Li M. Paclitaxel inhibits the progression of cervical cancer by inhibiting autophagy via lncRNARP11-381N20. 2. Eur Rev Med Pharmacol Sci. 2018;22(10):3010–3017.
132. Tomas‐Hernández S, Blanco J, Rojas C, et al. Resveratrol potently counteracts quercetin starvation‐induced autophagy and sensitizes HepG2 cancer cells to apoptosis. Mol Nutr Food Res. 2018;62(5):1700610. doi:10.1002/mnfr.201700610
133. Fu Y, Chang H, Peng X, et al. Resveratrol inhibits breast cancer stem-like cells and induces autophagy via suppressing Wnt/β-catenin signaling pathway. PLoS One. 2014;9(7):e102535. doi:10.1371/journal.pone.0102535
134. Garcia-Zepeda SP, García-Villa E, Díaz-Chávez J, Hernández-Pando R, Gariglio P. Resveratrol induces cell death in cervical cancer cells through apoptosis and autophagy. Eur J Cancer Prev. 2013;22(6):577–584. doi:10.1097/CEJ.0b013e328360345f
135. Fan Y, Chiu JF, Liu J, et al. Resveratrol induces autophagy-dependent apoptosis in HL-60 cells. BMC Cancer. 2018;18(1):1. doi:10.1186/s12885-018-4504-5
136. Selvaraj S, Sun Y, Sukumaran P, Singh BB. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol Carcinog. 2016;55(5):818–831. doi:10.1002/mc.22324
137. Puissant A, Robert G, Fenouille N, et al. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010;70(3):1042–1052. doi:10.1158/0008-5472.CAN-09-3537
138. Back JH, Zhu Y, Calabro A, et al. Resveratrol‐mediated downregulation of Rictor attenuates autophagic process and suppresses UV‐induced skin carcinogenesis. Photochem Photobiol. 2012;88(5):1165–1172. doi:10.1111/j.1751-1097.2012.01097.x
139. Liu Q, Fang Q, Ji S, Han Z, Cheng W, Zhang H. Resveratrol-mediated apoptosis in renal cell carcinoma via the p53/AMP-activated protein kinase/mammalian target of rapamycin autophagy signaling pathway. Mol Med Rep. 2018;17(1):502–508.
140. Mendes VI, Bartholomeusz GA, Ayres M, Gandhi V, Salvador JA. Synthesis and cytotoxic activity of novel A-ring cleaved ursolic acid derivatives in human non-small cell lung cancer cells. Eur J Med Chem. 2016;10(123):317–331. doi:10.1016/j.ejmech.2016.07.045
141. Xavier CP, Lima CF, Pedro DF, Wilson JM, Kristiansen K, Pereira-Wilson C. Ursolic acid induces cell death and modulates autophagy through JNK pathway in apoptosis-resistant colorectal cancer cells. J Nutr Biochem. 2013;24(4):706–712. doi:10.1016/j.jnutbio.2012.04.004
142. Leng S, Hao Y, Du D, et al. Ursolic acid promotes cancer cell death by inducing Atg5‐dependent autophagy. Int J Cancer Res. 2013;133(12):2781–2790.
143. Lewinska A, Adamczyk-Grochala J, Kwasniewicz E, Deregowska A, Wnuk M. Ursolic acid-mediated changes in glycolytic pathway promote cytotoxic autophagy and apoptosis in phenotypically different breast cancer cells. Apoptosis. 2017;22(6):800–815. doi:10.1007/s10495-017-1353-7
144. Shen S, Zhang Y, Zhang R, Tu X, Gong X. Ursolic acid induces autophagy in U87MG cells via ROS-dependent endoplasmic reticulum stress. Chem Biol Interact. 2014;218:28–41. doi:10.1016/j.cbi.2014.04.017
145. Shin SW, Kim SY, Park JW. Autophagy inhibition enhances ursolic acid-induced apoptosis in PC3 cells. Biochim Biophys Acta Mol Cell Res. 2012;1823(2):451–457. doi:10.1016/j.bbamcr.2011.10.014
146. Wu CC, Huang YF, Hsieh CP, Chueh PJ, Chen YL. Combined use of zoledronic acid augments ursolic acid-induced apoptosis in human osteosarcoma cells through enhanced oxidative stress and autophagy. Molecules. 2016;21(12):1640. doi:10.3390/molecules21121640
147. Jung J, Seo J, Kim J, Kim JH. Ursolic acid causes cell death in PC-12 cells by inducing apoptosis and impairing autophagy. Anticancer Res. 2018;38(2):847–853.
148. Lin YC, Lin JF, Wen SI, et al. Chloroquine and hydroxychloroquine inhibit bladder cancer cell growth by targeting basal autophagy and enhancing apoptosis. Kaohsiung J Med Sci. 2017;33(5):215–223. doi:10.1016/j.kjms.2017.01.004
149. Frieboes HB, Huang JS, Yin WC, McNally LR. Chloroquine-mediated cell death in metastatic pancreatic adenocarcinoma through inhibition of autophagy. JOP J Pancreas. 2014;15(2):189–197.
150. Wang T, Goodall ML, Gonzales P, et al. Synthesis of improved lysomotropic autophagy inhibitors. J Med Chem. 2015;58(7):3025–3035. doi:10.1021/jm501586m
151. Tran AT, Ramalinga M, Kedir H, Clarke R, Kumar D. Autophagy inhibitor 3-methyladenine potentiates apoptosis induced by dietary tocotrienols in breast cancer cells. Eur J Nutr. 2015;54(2):265–272. doi:10.1007/s00394-014-0707-y
152. Jang Y, Rao X, Jiang Q. Gamma-tocotrienol profoundly alters sphingolipids in cancer cells by inhibition of dihydroceramide desaturase and possibly activation of sphingolipid hydrolysis during prolonged treatment. J Nutr Biochem. 2017;1(46):49–56. doi:10.1016/j.jnutbio.2017.04.003
153. Chu SC, Hsieh YS, Yu CC, Lai YY, Chen PN. Thymoquinone induces cell death in human squamous carcinoma cells via caspase activation-dependent apoptosis and LC3-II activation-dependent autophagy. PLoS One. 2014;9(7):e101579. doi:10.1371/journal.pone.0101579
154. Pazhouhi M, Sariri R, Rabzia A, Khazaei M. Thymoquinone synergistically potentiates temozolomide cytotoxicity through the inhibition of autophagy in U87MG cell line. Iran J Basic Med Sci. 2016;19(8):890.
155. Chen MC, Lee NH, Hsu HH, et al. Thymoquinone induces caspase-independent, autophagic cell death in CPT-11-resistant lovo colon cancer via mitochondrial dysfunction and activation of JNK and p38. J Agric Food Chem. 2015a;63(5):1540–1546. doi:10.1021/jf5054063
156. Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol. 2007;72(1):29–39. doi:10.1124/mol.106.033167
157. Li B, Takeda T, Tsuiji K, et al. Curcumin induces cross-regulation between autophagy and apoptosis in uterine leiomyosarcoma cells. Int J Gynecol Cancer. 2013;23(5):803–808. doi:10.1097/IGC.0b013e31828c9581
158. Masuelli L, Benvenuto M, Di Stefano E, et al. Curcumin blocks autophagy and activates apoptosis of malignant mesothelioma cell lines and increases the survival of mice intraperitoneally transplanted with a malignant mesothelioma cell line. Oncotarget. 2017;8(21):34405. doi:10.18632/oncotarget.14907
159. Zhang J, Wang J, Xu J, et al. Curcumin targets the TFEB-lysosome pathway for induction of autophagy. Oncotarget. 2016c;7(46):75659. doi:10.18632/oncotarget.12318
160. Rainey N, Motte L, Aggarwal BB, Petit PX. Curcumin hormesis mediates a cross-talk between autophagy and cell death. Cell Death Dis. 2015;6(12):e2003. doi:10.1038/cddis.2015.343
161. Eaton JW, Qian M. Molecular bases of cellular iron toxicity. Free Radic Biol Med. 2002;32(9):833–840. doi:10.1016/S0891-5849(02)00772-4
162. Windelborn JA, Lipton P. Lysosomal release of cathepsins causes ischemic damage in the rat hippocampal slice and depends on NMDA‐mediated calcium influx, arachidonic acid metabolism, and free radical production. J Neurochem. 2008;106(1):56–69. doi:10.1111/j.1471-4159.2008.05349.x
163. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
164. Schonberg DL, Miller TE, Wu Q, et al. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell. 2015;28(4):441–455. doi:10.1016/j.ccell.2015.09.002
165. Tang M, Chen Z, Wu D, Chen L. Ferritinophagy/ferroptosis: iron‐related newcomers in human diseases. J Cell Physiol. 2018;233(12):9179–9190. doi:10.1002/jcp.26954
166. Kang R, Xie Y, Zeh HJ, Klionsky DJ, Tang D. Mitochondrial quality control mediated by PINK1 and PRKN: links to iron metabolism and tumor immunity. Autophagy. 2019;15(1):172–173. doi:10.1080/15548627.2018.1526611
167. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi:10.1016/j.redox.2019.101107
168. Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ. 2020;27(1):242–254. doi:10.1038/s41418-019-0352-3
169. Lin R, Zhang Z, Chen L, et al. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 2016;381(1):165–175. doi:10.1016/j.canlet.2016.07.033
170. Du J, Wang T, Li Y, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. 2019;1(131):356–369. doi:10.1016/j.freeradbiomed.2018.12.011
171. Zhu S, Zhang Q, Sun X, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017;77(8):2064–2077. doi:10.1158/0008-5472.CAN-16-1979
172. Lv C, Qu H, Zhu W, et al. Low-dose paclitaxel inhibits tumor cell growth by regulating glutaminolysis in colorectal carcinoma cells. Front Pharmacol. 2017;4(8):244. doi:10.3389/fphar.2017.00244
173. Giannakakou P, Robey R, Fojo T, Blagosklonny MV. Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene. 2001;20(29):3806–3813. doi:10.1038/sj.onc.1204487
174. Song X, Zhu S, Chen P, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc–activity. Curr Biol. 2018;28(15):2388–2399. doi:10.1016/j.cub.2018.05.094
175. Sehm T, Fan Z, Ghoochani A, et al. Sulfasalazine impacts on ferroptotic cell death and alleviates the tumor microenvironment and glioma-induced brain edema. Oncotarget. 2016;7(24):36021. doi:10.18632/oncotarget.8651
176. Sun X, Ou Z, Chen R, et al. Activation of the p62‐Keap1‐NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–184. doi:10.1002/hep.28251
177. Dixon SJ, Patel DN, Welsch M, et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;20(3):e02523. doi:10.7554/eLife.02523
178. Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol Rep. 2015b;33(3):1465–1474. doi:10.3892/or.2015.3712
179. Kondratskyi A, Yassine M, Slomianny C, et al. Identification of ML-9 as a lysosomotropic agent targeting autophagy and cell death. Cell Death Dis. 2014;5(4):e1193. doi:10.1038/cddis.2014.156
180. Sabella S, Carney RP, Brunetti V, et al. A general mechanism for intracellular toxicity of metal-containing nanoparticles. Nanoscale. 2014;6(12):7052–7061. doi:10.1039/c4nr01234h
181. Choi AR, Kim JH, Woo YH, Kim HS, Yoon S. Anti-malarial drugs primaquine and chloroquine have different sensitization effects with anti-mitotic drugs in resistant cancer cells. Anticancer Res. 2016;36(4):1641–1648. doi:10.21873/anticanres.11171
182. de Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomed. 2008;3:133–149. doi:10.2147/IJN.S596
183. Daraio C, Jin S. Synthesis and patterning methods for nanostructures useful for biological applications. In: Daraio C, Jin S, Silva GA, Parpura V, editors. Nanotechnology for Biology and Medicine. New York: Springer; 2012:27–44.
184. Jain AK, Thareja S. In vitro and in vivo characterization of pharmaceutical nanocarriers used for drug delivery. Artif Cells Nanomed Biotechnol. 2019;47(1):524–539. doi:10.1080/21691401.2018.1561457
185. Chen F, Ehlerding EB, Cai W. Theranostic nanoparticles. J Nucl Med. 2014;55(12):1919–1922. doi:10.2967/jnumed.114.146019
186. Khan AA, Allemailem KS, Almatroudi A, Almatroodi SA, Alsahli MA, Rahmani AH. Novel strategies of third level (organelle-specific) drug targeting: an innovative approach of modern therapeutics. J Drug Deliv Sci Technol. 2020;29:102315.
187. Allemailem KS, Almatroudi A, Alsahli MA, et al. Novel strategies for disrupting cancer-cell functions with mitochondria-targeted antitumor drug–loaded nanoformulations. Int J Nanomedicine. 2021;16:3907. doi:10.2147/IJN.S303832
188. Borkowska M, Siek M, Kolygina DV, et al. Targeted crystallization of mixed-charge nanoparticles in lysosomes induces selective death of cancer cells. Nat Nanotechnol. 2020;15(4):331–341. doi:10.1038/s41565-020-0643-3
189. Nasrollahi SA, Taghibiglou C, Azizi E, Farboud ES. Cell‐penetrating peptides as a novel transdermal drug delivery system. Chem Biol Drug Des. 2012;80(5):639–646. doi:10.1111/cbdd.12008
190. Zhong YJ, Shao LH, Li YA. Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy. Int J Oncol. 2013;42(2):373–383. doi:10.3892/ijo.2012.1754
191. Cudic M, Fields GB. Extracellular proteases as targets for drug development. Curr Protein Pept Sci. 2009;10(4):297–307. doi:10.2174/138920309788922207
192. Zhang L, Sheng R, Qin Z. The lysosome and neurodegenerative diseases. Acta Biochim Biophys Sin (Shanghai). 2009a;41(6):437–445. doi:10.1093/abbs/gmp031
193. Berquin IM, Sloane BF. Cathepsin B expression in human tumors. Adv Exp Med Biol. 1996;389:281–294.
194. Reinheckel T, Peters C, Krüger A, Turk B, Vasiljeva O. Differential impact of cysteine cathepsins on genetic mouse models of de novo carcinogenesis: cathepsin B as emerging therapeutic target. Front Pharmacol. 2012;11(3):133.
195. Maiti KK, Jeon OY, Lee WS, et al. Design, synthesis, and membrane‐translocation studies of inositol‐based transporters. Angew Chem Int Ed. 2006;45(18):2907–2912. doi:10.1002/anie.200600312
196. Maiti KK, Lee WS, Takeuchi T, et al. Guanidine‐containing molecular transporters: sorbitol‐based transporters show high intracellular selectivity toward mitochondria. Angew Chem. 2007;119(31):5984–5988. doi:10.1002/ange.200701346
197. Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. The design of guanidinium-rich transporters and their internalization mechanisms. Adv Drug Deliv Rev. 2008;60(4–5):452–472. doi:10.1016/j.addr.2007.10.016
198. Wei P, Zhang L, Lu Y, Man N, Wen L. C60 (Nd) nanoparticles enhance chemotherapeutic susceptibility of cancer cells by modulation of autophagy. Nanotechnology. 2010;21(49):495101. doi:10.1088/0957-4484/21/49/495101
199. Ma X, Wu Y, Jin S, et al. Gold nanoparticles induce autophagosome accumulation through size-dependent nanoparticle uptake and lysosome impairment. ACS Nano. 2011;5(11):8629–8639. doi:10.1021/nn202155y
200. Dekiwadia CD, Lawrie AC, Fecondo JV. Peptide‐mediated cell penetration and targeted delivery of gold nanoparticles into lysosomes. J Pept Sci. 2012;18(8):527–534. doi:10.1002/psc.2430
201. Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014b;14(4):263–276. doi:10.1038/nrc3701
202. Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 2007;67(8):3496–3499. doi:10.1158/0008-5472.CAN-07-0325
203. Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining” clean” cells. Autophagy. 2005;1(3):131–140. doi:10.4161/auto.1.3.2017
204. Nesic M, Popović I, Leskovac A, Petković M. Biological activity and binding properties of [Ru (II)(dcbpy) 2 Cl 2] complex to bovine serum albumin, phospholipase A 2 and glutathione. Biometals. 2016;29(5):921–933. doi:10.1007/s10534-016-9964-y
205. Purushothaman B, Arumugam P, Ju H, Kulsi G, Samson AA, Song JM. Novel ruthenium (II) triazine complex [Ru (bdpta)(tpy)] 2+ co-targeting drug resistant GRP78 and subcellular organelles in cancer stem cells. Eur J Med Chem. 2018;156:747–759. doi:10.1016/j.ejmech.2018.07.048
206. Purushothaman B, Choi J, Park S, et al. Biotin-conjugated PEGylated porphyrin self-assembled nanoparticles co-targeting mitochondria and lysosomes for advanced chemo-photodynamic combination therapy. J Mater Chem B. 2019;7(1):65–79. doi:10.1039/C8TB01923A
207. Zabner J, Fasbender AJ, Moninger T, Poellinger K, Welsh MJ. J. Biol Chem. 1995;270:18997–19007. doi:10.1074/jbc.270.32.18997
208. Wattiaux R, Wattiaux-de Coninck S. Biology of the Lysosome. Lloyd and Mason, editors. New York: Plenum Press; 1996:387.
209. Kornfeld S. Trafficking of lysosomal enzymes in normal and disease states. J Clin Invest. 1986;77(1):1–6. doi:10.1172/JCI112262
210. Kornfeld S, Reitman ML, Varki A, Goldberg D, Gabel CA. Steps in the phosphorylation of the high mannose oligosaccharides of lysosomal enzymes. Ciba Found Symp. 1982;92(92):138–156.
211. Izbicki T, Mazur J, Izbicka E. Epidemiology of neuroblastoma: analysis of a single institution. Anticancer Res. 2003;23(2C):1933–1938.
212. Schwake M, Schröder B, Saftig P. Lysosomal membrane proteins and their central role in physiology. Traffic. 2013;14(7):739–748. doi:10.1111/tra.12056
213. Biederbick A, Rose S, Elsasser HP. A human intracellular apyrase-like protein, LALP70, localizes to lysosomal/autophagic vacuoles. J Cell Sci. 1999;112(15):2473–2484. doi:10.1242/jcs.112.15.2473
214. Lloyd-Evans E, Platt FM. Lysosomal Ca2+ homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium. 2011;50(2):200–205. doi:10.1016/j.ceca.2011.03.010
215. Stern ST, Adiseshaiah PP, Crist RM. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol. 2012;9(1):1–7. doi:10.1186/1743-8977-9-20
216. Man N, Chen Y, Zheng F, Zhou W, Wen LP. Induction of genuine autophagy by cationic lipids in mammalian cells. Autophagy. 2010;6(4):449–454. doi:10.4161/auto.6.4.11612
217. Yang K, Lu Y, Xie F, et al. Cationic liposomes induce cell necrosis through lysosomal dysfunction and late-stage autophagic flux inhibition. Nanomedicine. 2016;12(23):3117–3137. doi:10.2217/nnm-2016-0289
218. Andreichenko K, Shelyuk O, Prylutska S, et al. Effect of multi‐walled iron‐filled carbon nanotubes on ATPase activity and superprecipitation of natural actomyosin. Materwiss Werksttech. 2013;44(2–3):103–106. doi:10.1002/mawe.201300103
219. Capco DG, Chen Y. Preface. In: Nanomaterial Impacts on Cell Biology and Medicine. NY, USA: Springer; 2014.
220. Yu KN, Kim JE, Seo HW, et al. Differential toxic responses between pristine and functionalized multiwall nanotubes involve induction of autophagy accumulation in murine lung. J Toxicol Environ Health A. 2013;76(23):1282–1292. doi:10.1080/15287394.2013.850137
221. Sohaebuddin SK, Thevenot PT, Baker D, Eaton JW, Tang L. Nanomaterial cytotoxicity is composition, size, and cell type dependent. Part Fibre Toxicol. 2010;7(1):1–7. doi:10.1186/1743-8977-7-22
222. Sandhoff K, Kolter T, Harzer K. Sphingolipid activator proteins. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease.
223. Wang Y, Grabowski GA, Qi X. Phospholipid vesicle fusion induced by saposin C. Arch Biochem Biophys. 2003;415(1):43–53. doi:10.1016/S0003-9861(03)00219-4
224. Ran S, Thorpe PE. Phosphatidylserine is a marker of tumor vasculature and a potential target for cancer imaging and therapy. Int J Radiat Oncol Biol Phys. 2002;54(5):1479–1484. doi:10.1016/S0360-3016(02)03928-7
225. Ditaranto-Desimone K, Saito M, Tekirian TL, et al. Neuronal endosomal/lysosomal membrane destabilization activates caspases and induces abnormal accumulation of the lipid secondary messenger ceramide. Brain Res Bull. 2003;59(6):523–531. doi:10.1016/S0361-9230(02)00948-6
226. Hendrich AB, Michalak K. Lipids as a target for drugs modulating multidrug resistance of cancer cells. Curr Drug Targets. 2003;4(1):23–30. doi:10.2174/1389450033347172
227. Thekkedath R, Koshkaryev A, Torchilin VP. Lysosome-targeted octadecyl-rhodamine B-liposomes enhance lysosomal accumulation of glucocerebrosidase in Gaucher’s cells in vitro. Nanomedicine. 2013;8(7):1055–1065. doi:10.2217/nnm.12.138
228. Jiang Y, Huo S, Mizuhara T, et al. The interplay of size and surface functionality on the cellular uptake of sub-10 nm gold nanoparticles. ACS Nano. 2015;9(10):9986–9993. doi:10.1021/acsnano.5b03521
229. Kim B, Han G, Toley BJ, Kim CK, Rotello VM, Forbes NS. Tuning payload delivery in tumour cylindroids using gold nanoparticles. Nat Nanotechnol. 2010;5(6):465–472. doi:10.1038/nnano.2010.58
230. Pillai PP, Kowalczyk B, Pudlo WJ, Grzybowski BA. Electrostatic titrations reveal surface compositions of mixed, on-nanoparticle monolayers comprising positively and negatively charged ligands. J Phys Chem C. 2016;120(7):4139–4144. doi:10.1021/acs.jpcc.5b12599
231. Zhang ZY, Xu YD, Ma YY, et al. Biodegradable ZnO@ polymer core–shell nanocarriers: pH‐triggered release of doxorubicin in vitro. Angew Chem Int Ed. 2013;52:4127. doi:10.1002/anie.201300431
232. Chu JJ, Ng ML. Interaction of West Nile virus with αvβ3 integrin mediates virus entry into cells. J Biol Chem. 2004;2004(52):54533. doi:10.1074/jbc.M410208200
233. Domenech M, Marrero-Berrios I, Torres-Lugo M, Rinaldi C. Lysosomal membrane permeabilization by targeted magnetic nanoparticles in alternating magnetic fields. ACS Nano. 2013;7(6):5091–5101. doi:10.1021/nn4007048
234. Yanes RE, Tamanoi F. Development of mesoporous silica nanomaterials as a vehicle for anticancer drug delivery. Ther Deliv. 2012;3(3):389–404. doi:10.4155/tde.12.9
235. Meng H, Xue M, Xia T, et al. Use of size and a copolymer design feature to improve the biodistribution and the enhanced permeability and retention effect of doxorubicin-loaded mesoporous silica nanoparticles in a murine xenograft tumor model. ACS Nano. 2011;5(5):4131–4144. doi:10.1021/nn200809t
236. Zhu CL, Lu CH, Song XY, Yang HH, Wang XR. Bioresponsive controlled release using mesoporous silica nanoparticles capped with aptamer-based molecular gate. J Am Chem Soc. 2011;133(5):1278–1281. doi:10.1021/ja110094g
237. Lu J, Li Z, Zink JI, Tamanoi F. In vivo tumor suppression efficacy of mesoporous silica nanoparticles-based drug-delivery system: enhanced efficacy by folate modification. Nanomedicine. 2012;8(2):212–220. doi:10.1016/j.nano.2011.06.002
238. Mamaeva V, Rosenholm JM, Bate-Eya LT, et al. Mesoporous silica nanoparticles as drug delivery systems for targeted inhibition of Notch signaling in cancer. Mol Ther. 2011;19(8):1538–1546. doi:10.1038/mt.2011.105
239. Zhao Q, Gong Z, Li Z, et al. Target reprogramming lysosomes of CD8+ T cells by a mineralized metal–organic framework for cancer immunotherapy. Adv Mater. 2021;33(17):2100616. doi:10.1002/adma.202100616
240. Jaattela M. Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene. 2004;23(16):2746–2756. doi:10.1038/sj.onc.1207513
241. Huang Y, Mao K, Zhang B, Zhao Y. Superparamagnetic iron oxide nanoparticles conjugated with folic acid for dual target-specific drug delivery and MRI in cancer theranostics. Mater Sci Eng C. 2017;70:763–771. doi:10.1016/j.msec.2016.09.052
242. Lee H, Dam DHM, Ha JW, Yue J, Odom TW. Enhanced human epidermal growth factor receptor 2 degradation in breast cancer cells by lysosome-targeting gold nanoconstructs. ACS Nano. 2015;9(10):9859–9867. doi:10.1021/acsnano.5b05138
243. Zeng Y, Yang Z, Luo S, et al. Fast and facile preparation of PEGylated graphene from graphene oxide by lysosome targeting delivery of photosensitizer to efficiently enhance photodynamic therapy. RSC Adv. 2015;5(71):57725–57734. doi:10.1039/C5RA07535A
244. Yuan X, Xiao F, Zhao H, et al. High-Yield method to fabricate and functionalize DNA nanoparticles from the products of rolling circle amplification. ACS Appl Bio Mater. 2018;1(2):511–519. doi:10.1021/acsabm.8b00238
245. Zeng L, Gupta P, Chen Y, et al. The development of anticancer ruthenium(ii) complexes: from single molecule compounds to nanomaterials. Chem Soc Rev. 2017;46(19):5771–5804. doi:10.1039/C7CS00195A
246. Cheng W, Liang C, Wang X, et al. A drug-self-gated and tumor microenvironment-responsive mesoporous silica vehicle:“four-in-one” versatile nanomedicine for targeted multidrug-resistant cancer therapy. Nanoscale. 2017;9(43):17063–17073. doi:10.1039/C7NR05450E
247. Gao W, Cao W, Zhang H, Li P, Xu K, Tang B. Targeting lysosomal membrane permeabilization to induce and image apoptosis in cancer cells by multifunctional Au-ZnO hybrid nanoparticles. Chem Commun. 2014;50(60):8117–8120. doi:10.1039/c4cc03793f
248. Xue S, Tan C, Chen M, et al. Tumor-targeted supramolecular nanoparticles self-assembled from a ruthenium-β-cyclodextrin complex and an adamantane-functionalized peptide. Chem Commun. 2017;53:842–845. doi:10.1039/C6CC08296C
249. Ju E, Chen Z, Li W, et al. Embedding magnetic nanoparticles into coordination polymers to mimic zinc ion transporters for targeted tumor therapy. Chem Commun. 2016;52(85):12598–12601. doi:10.1039/C6CC06467A
250. Sharma A, Lee M-G, Shi H, et al. Overcoming drug resistance by targeting cancer bioenergetics with an activatable prodrug. Chem. 2018;4(10):2370–2383. doi:10.1016/j.chempr.2018.08.002
251. Clerc P, Jeanjean P, Hallali N, et al. Targeted magnetic intra-lysosomal hyperthermia produces lysosomal reactive oxygen species and causes caspase-1 dependent cell death. J Control Release. 2018;270:120–134. doi:10.1016/j.jconrel.2017.11.050
252. Leanza L, Biasutto L, Manago A, Gulbins E, Zoratti M, Szabo I. Intracellular ion channels and cancer. Front Physiol. 2013;4:227–233. doi:10.3389/fphys.2013.00227
253. Gangopadhyay M, Mukhopadhyay SK, Gayathri S, et al. Fluorene–morpholine-based organic nanoparticles: lysosome-targeted pH-triggered two-photon photodynamic therapy with fluorescence switch on-off. J Mater Chem B. 2016;4(10):1862–1868. doi:10.1039/C5TB02563J
254. Sharma A, Arambula JF, Koo S, et al. Hypoxia-targeted drug delivery. Chem Soc Rev. 2019;48:771–813.
255. Mirshafiee V, Sun B, Chang CH, et al. Toxicological profiling of metal oxide nanoparticles in liver context reveals pyroptosis in kupffer cells and macrophages versus apoptosis in hepatocytes. ACS Nano. 2018;12(4):3836–3852. doi:10.1021/acsnano.8b01086
256. Cerda MB, Batalla M, Anton M, et al. Enhancement of nucleic acid delivery to hard-totransfect human colorectal cancer cells by magnetofection at laminin coated substrates and promotion of the endosomal/lysosomal escape. RSC Adv. 2015;5(72):58345–58354. doi:10.1039/C5RA06562C
257. Joris F, De Backer L, Van de Vyver T, Bastiancich C, De Smedt SC, Raemdonck K. Repurposing cationic amphiphilic drugs as adjuvants to induce lysosomal siRNA escape in nanogel transfected cells. J Control Release. 2018;269:266–276. doi:10.1016/j.jconrel.2017.11.019
258. Zhou L, Yang T, Wang J, et al. Size-tunable Gd2O3@Albumin nanoparticles conjugating chlorin e6 for magnetic resonance imaging-guided photo-induced therapy. Theranostics. 2017;7(3):764–774. doi:10.7150/thno.15757
259. Berger E, Breznan D, Stals S, et al. Cytotoxicity assessment, inflammatory properties, and cellular uptake of neutraplex lipid-based nanoparticles in THP-1 monocyte-derived macrophages. Nanobiomedicine. 2017;4:1–14. doi:10.1177/1849543517746259
260. Chen L, She X, Wang T, et al. Overcoming acquired drug resistance in colorectal cancer cells by targeted delivery of 5-FU with EGF grafted hollow mesoporous silica nanoparticles. Nanoscale. 2015;7(33):14080–14092. doi:10.1039/C5NR03527A
261. Guo S, Huang L. Nanoparticles escaping RES and endosome: challenges for siRNA delivery for cancer therapy. J Nanomater. 2011;2011:Article ID 742895.
262. Yuan Y, Zhang C, Liu B. A photoactivatable AIE polymer for light-controlled gene delivery: concurrent endo/lysosomal escape and DNA unpacking. Angew Chem Int Ed. 2015;54(39):11419–11423. doi:10.1002/anie.201503640
263. Li L, Sun W, Li L, et al. A pHresponsive sequential-disassembly nanohybrid for mitochondrial targeting. Nanoscale. 2017;9(1):314–325. doi:10.1039/C6NR07004C
264. Chen G, Wang Y, Xie R, Gong S. Tumor-targeted pH/redox dual-sensitive unimolecular nanoparticles for efficient siRNA delivery. J Control Release. 2017;259:105–114. doi:10.1016/j.jconrel.2017.01.042
265. Wan B, Wang Z, Lv Q, et al. Single-walled carbon nanotubes and graphene oxides induce autophagosome accumulation and lysosome impairment in primarily cultured murine peritoneal macrophages. Toxicol Lett. 2013;221(2):118–127. doi:10.1016/j.toxlet.2013.06.208
266. Ghosh S, Panja P, Dalal C, Jana NR. Arginine-Terminated, chemically designed nanoparticle for direct cell translocation. ACS Appl Bio Mater. 2019;2(1):339–348. doi:10.1021/acsabm.8b00617
267. Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium rich peptides into cells. J Am Chem Soc. 2004;126(31):9506–9507. doi:10.1021/ja0482536
268. Cohignac V, Landry MJ, Ridoux A, et al. Carbon nanotubes, but not spherical nanoparticles, block autophagy by a shape-related targeting of lysosomes in murine macrophages. Autophagy. 2018;14(8):1323–1334.
269. Duan J, Yu Y, Li Y, et al. Silica nanoparticles induce autophagy and endothelial dysfunction via the PI3K/Akt/ mTOR signaling pathway. Int J Nanomed. 2014;9:5131–5141. doi:10.2147/IJN.S71074
270. Bossi E, Zanella D, Gornati R, Bernardini G. Cobalt oxide nanoparticles can enter inside the cells by crossing plasma membranes. Sci Rep. 2016;6(1):22254–22263. doi:10.1038/srep22254
271. Wang J, Yu Y, Lu K, et al. Silica nanoparticles induce autophagy dysfunction via lysosomal impairment and inhibition of autophagosome degradation in hepatocytes. Int J Nanomed. 2017;12:809–825. doi:10.2147/IJN.S123596
272. Zhang L, Chen X, Wu J, et al. Palladium nanoparticles induces autophagy and autophagic flux blockade in HeLa cells. RSC Adv. 2018;8(8):4130–4141. doi:10.1039/C7RA11400A
273. Nakamura S, Yoshimori T. New insights into autophagosome–lysosome fusion. J Cell Sci. 2017;130:1209–1216.
274. Xue X, Wang L, Sato Y, et al. Single single walled carbon nanotubes alleviate autophagic/lysosomal defects in primary glia from a mouse model of Alzheimer’s disease. Nano Lett. 2014;14(9):5110–5117. doi:10.1021/nl501839q
275. Lin J, Alexander-Katz A. Cell membranes open “Doors” for cationic nanoparticles/ biomolecules: insights into uptake kinetics. ASC Nano. 2013;7(12):10799–10808. doi:10.1021/nn4040553
276. Derivery E, Bartolami E, Matile S, Gonzalez-Gaitan M. Efficient delivery of quantum dots into the cytosol of cells using cell-penetrating poly(disulfide)s. J Am Chem Sci. 2017;139(30):10172–10175. doi:10.1021/jacs.7b02952
277. Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv Drug Deliv Rev. 2009;61(6):428–437. doi:10.1016/j.addr.2009.03.009
278. Gao Y, Chen Y, Zhan S, Zhang W, Xiong F, Ge W. Comprehensive proteome analysis of lysosomes reveals the diverse function of macrophages in immune responses. Oncotarget. 2017;8(5):7420. doi:10.18632/oncotarget.14558
279. Casey TM, Meade JL, Hewitt EW. Organelle proteomics: identification of the exocytic machinery associated with the natural killer cell secretory lysosome. Mol Cell Proteomics. 2007;6(5):767–780. doi:10.1074/mcp.M600365-MCP200
280. Jenkins SJ, Ruckerl D, Cook PC, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of Th2 inflammation. Science. 2011;332(6035):1284–1288. doi:10.1126/science.1204351
281. Terenzi A, Pirker C, Keppler BK, Berger W. Anticancer metal drugs and immunogenic cell death. J Inorg Biochem. 2016;1(165):71–79. doi:10.1016/j.jinorgbio.2016.06.021
282. Zitvogel L, Apetoh L, Ghiringhelli F, André F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? J Clin Invest. 2008;118(6):1991–2001. doi:10.1172/JCI35180
283. Fedeli C, Segat D, Tavano R, et al. Variations of the corona HDL:albumin ratio determine distinct effects of amorphous SiO2 nanoparticles on monocytes and macrophages in serum. Nanomedicine. 2014;9(16):2481–2497. doi:10.2217/nnm.14.22
284. Poland CA, Duffin R, Kinloch I, et al. Carbon nanotubes introduced into the abdominal cavity of mice show asbestoslike pathogenicity in a pilot study. Nat Nanotechnol. 2008;3(7):423–428. doi:10.1038/nnano.2008.111
285. Moghimi SM. Chemical camouflage of nanospheres with a poorly reactive surface: towards development of stealth and targetspecific nanocarriers. Biochim Biophys Acta. 2002;1590(1–3):131–139. doi:10.1016/S0167-4889(02)00204-5
286. Ishida T, Kiwada H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int J Pharm. 2008;354(1–2):56–62. doi:10.1016/j.ijpharm.2007.11.005
287. Mitchell LA, Lauer FT, Burchiel SW, McDonald JD. Mechanisms for how inhaled multiwalled carbon nanotubes suppress systemic immune function in mice. Nat Nanotechnol. 2009;4(7):451–456. doi:10.1038/nnano.2009.151
288. Stinchcombe TE, Socinski MA, Walko CM, et al. Phase I and pharmacokinetic trial of carboplatin and albumin-bound paclitaxel, ABI-007 (Abraxane) on three treatment schedules in patients with solid tumors. Cancer Chemother Pharmacol. 2007;60(5):759–766. doi:10.1007/s00280-007-0423-x
289. Higaki M, Ishihara T, Izumo N, Takatsu M, Mizushima Y. Treatment of experimental arthritis with poly(D, L-lactic/glycolic acid) nanoparticles encapsulating betamethasone sodium phosphate. Ann Rheum Dis. 2005;64(8):1132–1136. doi:10.1136/ard.2004.030759
290. Blum JL, Savin MA, Edelman G, et al. Phase II study of weekly albumin-bound paclitaxel for patients with metastatic breast cancer heavily pretreated with taxanes. Clin Breast Cancer. 2007;7(11):850–856. doi:10.3816/CBC.2007.n.049
291. Lafuente A, Gonza´lez-Carracedo A, Romero A, Esquifino AI. Effect ofcadmiumon lymphocyte subsets distribution in thymus and spleen. J Physiol Biochem. 2003;59(1):43–48. doi:10.1007/BF03179867
292. Szebeni J, Alving CR, Rosivall L. Animal models of complement-mediated hypersensitivity reactions to liposomes and other lipid-based nanoparticles. J Liposome Res. 2007;17(2):107–117. doi:10.1080/08982100701375118
293. Mottram PL, Leong D, Crimeen-Irwin B, et al. Type 1 and 2 immunity following vaccination is influenced by nanoparticle size: formulation of a model vaccine for respiratory syncytial virus. Mol Pharm. 2007;4(1):73–84. doi:10.1021/mp060096p
294. Nygaard UC, Hansen JS, Samuelsen M, Alberg T, Marioara CD, Løvik M. Single-walled and multi-walled carbon nanotubes promote allergic immune responses in mice. Toxicol Sci. 2009;109(1):113–123. doi:10.1093/toxsci/kfp057
295. Toyama T, Matsuda H, Ishida I, et al. A case of toxic epidermal necrolysis-like dermatitis evolving from contact dermatitis of the hands associated with exposure to dendrimers. Contact Dermatitis. 2008;59(2):122–123. doi:10.1111/j.1600-0536.2008.01340.x
296. Huang B, Abraham WD, Zheng Y, et al. Active targeting of chemotherapy to disseminated tumors using nanoparticle- carrying Tcells. Sci Transl Med. 2015;7(291):291ra294. doi:10.1126/scitranslmed.aaa5447
297. Cheung AS, Zhang DKY, Koshy ST, Mooney DJ. Scaffolds that mimic antigen- presenting cells enable ex vivo expansion of primary T cells. Nat Biotechnol. 2018;36(2):160–169. doi:10.1038/nbt.4047
298. Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2016;35(1):1–11. doi:10.1038/onc.2015.99
299. Mahalingam D, Mita M, Sarantopoulos J, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy. 2014;10(8):1403–1414. doi:10.4161/auto.29231
300. Rangwala R, Chang YC, Hu J, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10(8):1391–1402. doi:10.4161/auto.29119
301. Barnard RA, Wittenburg LA, Amaravadi RK, Gustafson DL, Thorburn A, Thamm DH. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxy-chloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy. 2014;10(8):1415–1425. doi:10.4161/auto.29165
302. Amaravadi RK, Yu D, Lum JJ, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117(2):326–336. doi:10.1172/JCI28833
303. Bray K, Mathew R, Lau A, et al. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS One. 2012;7(7):e41831. doi:10.1371/journal.pone.0041831
304. Xie X, White EP, Mehnert JM, Fimia GM. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS One. 2013;8(1):e55096. doi:10.1371/journal.pone.0055096
305. Wolpin BM, Rubinson DA, Wang X, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist. 2014;19(6):637–638. doi:10.1634/theoncologist.2014-0086
306. Amaravadi RKWJ, Winkler JD. Lys05: a new lysosomal autophagy inhibitor. Autophagy. 2012;8(9):1383–1384. doi:10.4161/auto.20958
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.