")
Back to Journals » Journal of Pain Research » Volume 11
Novel agonist of α4β2* neuronal nicotinic receptor with antinociceptive efficacy in rodent models of acute and chronic pain
Authors Sudo RT, Hayashida K, Santos AN, Kawatani M, Monteiro CES, Moreira RD, Trachez MM , Montes GC, Zapata-Sudo G
Received 29 March 2018
Accepted for publication 24 July 2018
Published 30 October 2018 Volume 2018:11 Pages 2453—2462
DOI https://doi.org/10.2147/JPR.S169637
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Katherine Hanlon
Roberto T Sudo,1,2 Kenichiro Hayashida,3 Aluizio N Santos,2 Masahito Kawatani,3 Carlos ES Monteiro1 Roberto D Moreira,4 Margarete M Trachez,1 Guilherme C Montes,1 Gisele Zapata-Sudo1
1Program of Research in Drug Development of Biomedical Science, Institute of Federal University of Rio de Janeiro, Rio de Janeiro, Brazil; 2Post-Graduation Program in Medicine (General Surgery) of Federal University of Rio de Janeiro, Rio de Janeiro, Brazil; 3Department of Neurophysiology, Akita University School of Medicine, Akita, Japan; 4Cristalia Produtos Quimicos e Farmacêuticos Ltda, Itapira, São Paulo, Brazil.
Objective: To demonstrate the antinociceptive and antihypersensitivity mechanisms of Cris-104 (1-{2-[5-(4-fluorophenyl)–1H-pyrazol-4-yl]ethyl}piperidine), a novel selective α4β2* nicotinic acetylcholine receptor (nAChR) agonist, in rodent acute/inflammatory and chronic pain models.
Materials and methods: Hot-plate and formalin tests in mice were used to examine Cris-104-induced antinociceptive effects on thermal/inflammatory pain. Cris-104 effects on hypersensitivity, norepinephrine (NE) release in the spinal dorsal horn, and neuronal activity in the locus coeruleus (LC) were examined in rats with lumbar spinal nerve ligation using behavioral, microdialysis, and extracellular recording methods. Cris-104 effects on spontaneous locomotion were examined in an open-field test.
Results: Cris-104 induced dose-dependent antinociception effects in hot-plate and formalin tests, and these effects were blocked by the general nAChR antagonist mecamylamine, the selective α4β2* nAChR antagonist dihydro-beta-erythroidine, and the α2-adrenoceptor antagonist yohimbine, but not by the α1-adrenoceptor antagonist prazosin. Systemic and spinally perfused Cris-104 increased NE concentrations in microdialysates from the spinal cord in both normal and SNL rats. Systemic Cris-104 increased neuronal activity in the LC of normal rats. Mecamylamine blocked the effects of Cris-104 on spinal NE release and LC neuronal activity. Systemic Cris-104 did not affect locomotor activity significantly.
Conclusion: The α4β2 neuronal nAChR agonist, Cris-104, was effective for treatment of pain via descending noradrenergic inhibition of pain signaling.
Keywords: pain, nicotinic receptor, Cris-104, epibatidine
Introduction
Nicotinic acetylcholine receptors (nAChRs) are widely expressed in the central nervous system (CNS) and in peripheral nervous system, predominantly in neuromuscular junctions. While neuromuscular junction nAChRs are formed by α, β, δ, ε, or ϒ subunits, CNS nAChRs are composed of only α (α2–7, α9–10) and β (β2–4) subunits, with their distribution and function being dependent upon the particular arrangement of these subunits.1–3 In the CNS, α and β subunits form homo- or heteropentameric nAChRs.4,5 Pharmacologically important α4β2-containing nAChRs have two main pentameric arrangements, namely (α4)2(β2)3 and (α4)3(β2)2, which have different affinities to acetylcholine.5
Several studies have identified α4β2* (* denotes additional subunit) nAChRs as important pharmacological targets to treat smoking-cessation symptoms, cognitive impairment (including in Alzheimer’s disease), and pain.6,7 Stimulation of supraspinal and midbrain α4β2* nAChRs activates descending inhibitory mechanisms, thereby reducing pain signal neurotransmission in the spinal cord.1,8,9 Interestingly, many approved treatments for neuropathic pain – including gabapentinoids, norepinephrine (NE) re-uptake inhibitors, and the α2-adrenoceptor agonist clonidine recruit, augment, or mimic the actions of the descending locus coeruleus (LC) – spinal cord NE inhibitory pathway, underscoring the importance of this pathway in neuropathic pain control.10–13
Epibatidine is a natural alkaloid α4β2* αnAChR agonist extracted from the skin of the Epipedobutes tricolor frog; it has an analgesic potency 200-fold higher than morphine and has been considered previously for development into a drug to treat pain and smoking cessation.14 However, in clinical trials, epibatidine and its related compounds were found to produce adverse secondary effects (eg, nausea and vomiting), likely due to interactions with multiple nAChR subtypes.14 We have been working on the development of a new α4β2 nAChR agonist using a medicinal chemical strategy. Our preliminary work led us to select a compound called Cris-104 (Figure 1) as a putative candidate for development based on its efficacy in rats with diabetic neuropathy and its advantageous physical chemistry profile (solubility, stability, and easy synthetic pathway).15
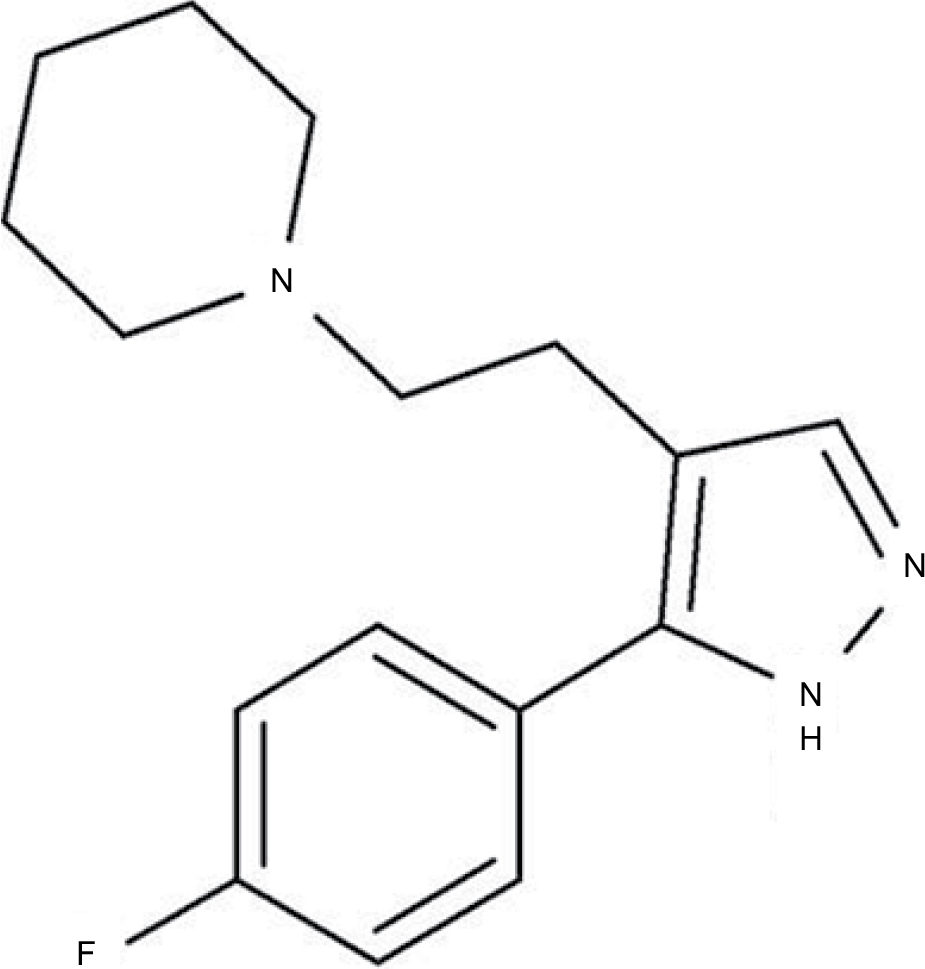 | Figure 1 Chemical structure of Cris-104 (1-{2-[5-(4-fluorophenyl)–1H-pyrazol-4-yl]ethyl}piperidine). |
In the current study, we examined the analgesic effects of Cris-104 on thermal and formalin-induced nociception in mice, as well as the effects of Cris-104 on hypersensitivity in rats after lumbar spinal nerve ligation (SNL) at L5–L6. Additionally, we examined Cris-104’s analgesic potential by way of its effects on descending noradrenergic neurotransmission by testing the effects of systemically or locally administered Cris-104 on NE release in the spinal cord and neuronal activity in the LC, as well as its behavioral effect on locomotion.
Materials and methods
Animals and drugs
Experiments were performed on male Wistar and Sprague Dawley rats (Rattus norvegicus; 180–220 g) and male Swiss mice (Mus musculus; 20–25 g) housed in cages at 22±2°C with a relative humidity of 60%–65%, 12/12-hour light/dark cycles, and free access to food and water. All experimental procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (National Institute of Health) and approved by the Institutional Animal Care and Use Committees at Federal University of Rio de Janeiro, Brazil and Akita University, Akita, Japan.
Cris-104 (chemical name: 1-{2-[5-(4-fluorophenyl)–1H-pyrazol-4-yl]ethyl}piperidine) was synthesized and provided by Cristália Produtos Químicos e Farmacêuticos Ltda (Itapira, SP, Brazil). Acetylsalicylic acid, yohimbine, and prazosin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Mecamylamine and dihydro-beta-erythroidine were purchased from Tocris Bioscience (Minneapolis, MN, USA). All compounds were dissolved in saline and prepared on the day of the experiments in which they were used.
Acute pain model (hot-plate test)
Mice were placed on a heated (52°C) hot-plate surface (Leticia LE 7406).16 The animal reaction (paw raise, jumping, or licking) time (latency) was determined before and after a 5- to 100-minute treatment with orally administered (by gavage) Cris-104 (1, 10, 30, and 100 mg/kg; n=10/dose). In two other groups (n=10/group), animals were pretreated with the nonselective nicotinic receptor antagonist mecamylamine (1 mg/kg, intraperitoneal [i.p.]) or the selective α4β2* nAChR antagonist dihydro-beta-erythroidine (1 mg/kg i.p.), respectively, 15 minutes before Cris-104 administration (30 mg/kg dose was selected based on the results of previous experiments in this study). Antinociceptive efficacy was indexed as a percentage of the maximum possible effect (%MPE) according to the following formula: %MPE = ([postdrug latency] – [predrug latency]/[cutoff] – [predrug latency)]) × 100.16 The cutoff (3-fold the predrug latency) was the maximal time allowed for the animal stand on the hot plate to avoid paw injury.
Neurogenic/inflammatory pain model (formalin test)
Peripheral neurogenic/inflammatory pain was induced in mice by intraplantar injection of formalin (20 µL, 2.5%) into the right hind paw.17 Biphasic behavior characterized the formalin-induced pain response, with licking and/or biting time (reactivity) being noted in the neurogenic phase (0–5 minutes) and inflammatory phase (15–30 minutes). Saline (10 mL/kg), acetylsalicylic acid (150 mg/kg), or Cris-104 (10, 30, and 100 mg/kg) was administered by gavage 15 minutes before formalin injection. Total licking or biting time was then quantified for a 40-minute observation period. Behavior was also compared among animals (n=10/group) pretreated with dihydro-beta-erythroidine (1 mg/kg i.p.), yohimbine (1 mg/kg i.p.), or prazosin (1 mg/kg i.p.), injected immediately before Cris-104 administration.
Chronic nociception model (SNL)
Chronic nociception was induced by SNL as previously described.18 Briefly, Wistar rats were anesthetized with ketamine (100 mg/kg i.p.) and xylazine (5 mg/kg i.p.) (Cristália, SP, Brazil). After making a dorsal midline incision, the paraspinal muscles were separated at spinal levels L4–S2. The right L6 transverse process was removed and the right L5 and L6 spinal nerves were isolated and ligated tightly with 6–0 silk suture. Sham-operated rats underwent the same procedure with the exception of the nerve ligation. Thermal hypernociception was evaluated with a plantar analgesia meter (model 33; ITC, Woodland Hills, CA, USA) by applying a radiant heat source to the plantar surface of the right hind paw.19 For mechanical hyperalgesia measurement, increasing pressure was applied with a filament (1 mm external diameter), connected to a digital analgesimeter (model EFF301; Insight, Ribeirão Preto, Brazil), to the surface of the same hind paw.20 After establishment of thermal and mechanical hypernociception 7 days after SNL, the animals were divided equally into Cris-104 dose groups (as above, n=6/dose). Reaction to thermal and mechanical stimulation was tested on day 1, 3, and 7 of a 7-day Cris-104 treatment period and at the same intervals after suspension of Cris-104 administration.
Microdialysis
Lumbar spinal microdialysis was performed in normal and SNL Sprague Dawley rats as previously described.10 SNL rats (prepared as described above) were used for the experiments 6 weeks after surgery. Anesthesia was induced with 2% isoflurane and then maintained with 1.5% isoflurane. A heating blanket was used to maintain a rectal temperature 36.5±0.5°C. The right femoral vein was cannulated for saline infusion (1 mL/h) and Cris-104 injection, and spinal levels L3–L6 were exposed by T13–L1 laminectomy. A microdialysis probe (CX-I-4–01, outer/inner diameter=0.22/0.20 mm, length=1 mm; EICOM Co., Kyoto, Japan) was inserted into the dorsal horn ipsilateral to the SNL surgery 1 hour prior to the experiment and perfused with Ringer’s solution (1.0 µL/min). Fractions were collected every 30 minutes for 2.5 hours starting 1 hour prior to intravenous injection of Cris-104 (10 mg/kg), local perfusion of Cris-104 (10–100 µM), or local perfusion of epibatidine (3 µM, Sigma-Aldrich Japan, Tokyo, Japan) with or without mecamylamine (100 µM, Sigma-Aldrich Japan) through the microdialysis probe for 90 minutes. NE content in the microdialysates was measured by high-pressure liquid chromatography with electrochemical detection (HTEC-500, EICOM Co.).
Extracellular recording from the LC
Male Sprague Dawley rats were anesthetized with chloral hydrate (400 mg/kg i.p.), and a heating pad was used to maintain their rectal temperatures at 36.5±0.5°C. The right jugular vein was cannulated for injection of anesthetic and test drugs. Supplemental doses of anesthetic were given to maintain anesthesia and prevent nociceptive reactions. Each animal was then placed in a stereotaxic frame with its head oriented at a 15° angle to the horizontal plane (nose down). The skull was exposed, and a 3-mm burr hole was drilled over the cerebellum. The dura was removed carefully to enable insertion of the electrode. Single-unit extracellular recordings from LC neurons were performed as previously reported.21,22 A tungsten electrode (in vitro impedance, 1.0–1.3 MΩ) with a stainless tube for dye injection (Unique Medical Co., LTD, Tokyo, Japan) was inserted into the LC (3.8–3.9 mm posterior and 1.1 mm lateral to lambda, and 5.5–6.5 mm ventral from the brain surface, according to a rat brain atlas).23 Electrode signals were amplified, discriminated, monitored, and recorded with UAS-308S data acquisition system, and the data were analyzed offline in Unique Acquisition 3 (system and software from Unique Medical Co., Ltd). LC neurons were identified by the following standard criteria: spontaneous firing rate, 0.2–5 Hz; a positive–negative action potential shape; and a biphasic excitation–inhibition response to pinch stimulation in the contralateral hindpaw.24 The basal firing rate was recorded at least 1 minute prior to drug administration and only one LC unit per animal was used for analysis.
After the experiment, all animals received an intra-LC injection of methylene blue (50 nL) and were euthanized by an intravenous injection of pentobarbital (150 mg/kg). Each animal’s brain was removed and cut into coronal sections on a microtome, and the placement of the cannula was verified visually. Only data from animals with successful LC electrode placement were included in the analyses.
Locomotor activity
Spontaneous locomotor activity was measured in an automated open-field apparatus (45×45 cm; LE 8811, Panlab, Hollister, MA, USA) in which 16 infrared photocells were positioned at every 2.5 cm.25 Normal Wistar rats were placed individually in the center of the chamber, and beam interruptions were counted as instances of motor activity over a 40-minute observation period divided into eight 5-minute intervals. Locomotor activity was measured before as well as 1, 3, and 7 days after treatment with Cris-104 (100 mg/kg by gavage) and reported in the form of number of movements per minute. The motor activity was performed 24 hours after drug administration. A single dose of locomotion-suppressing diazepam (10 mg/kg by gavage) was used as a positive control.
Statistical analysis
Unless otherwise stated, results are expressed as means ± standard errors of the mean (SEM). One-way ANOVA or two-way repeated measures ANOVA followed by Dunnett’s test or Bonferroni’s tests, respectively, was applied; P<0.05 was considered significant. All tests were performed in GraphPad Prism Version 4.0 (GraphPad, San Diego, CA, USA).
Results
Cris-104-induced thermal antinociception
In the hot-plate test (Figure 2A), we obtained mean %MPE values of 15.9±4.0%, 22.7±2.5%, 34.4±7.1%, and 56.1±5.5% for the 1, 10, 30, and 100 mg/kg doses of orally administered Cris-104, respectively (Figure 2A, inset). The highest %MPE for Cris-104, which was observed with the 30 mg/kg (91.4 µmol/kg) dose, was similar to that of morphine at 10 mg/kg (13.2 µmol/kg). Neither the nonselective nAChR antagonist mecamylamine (1 mg/kg i.p.) nor the selective α4β2 nAChR antagonist dihydro-beta-erythroidine (1 mg/kg i.p.) caused a significant antinociception effect in the hot-plate test alone (Figure 2B), but they reduced the antinociceptive effect of Cris-104 (30 mg/kg) from 34.4±7.1% to 6.3±7.2% (P<0.001) and to 17.2±6.6% (P<0.05), respectively (Figure 2B).
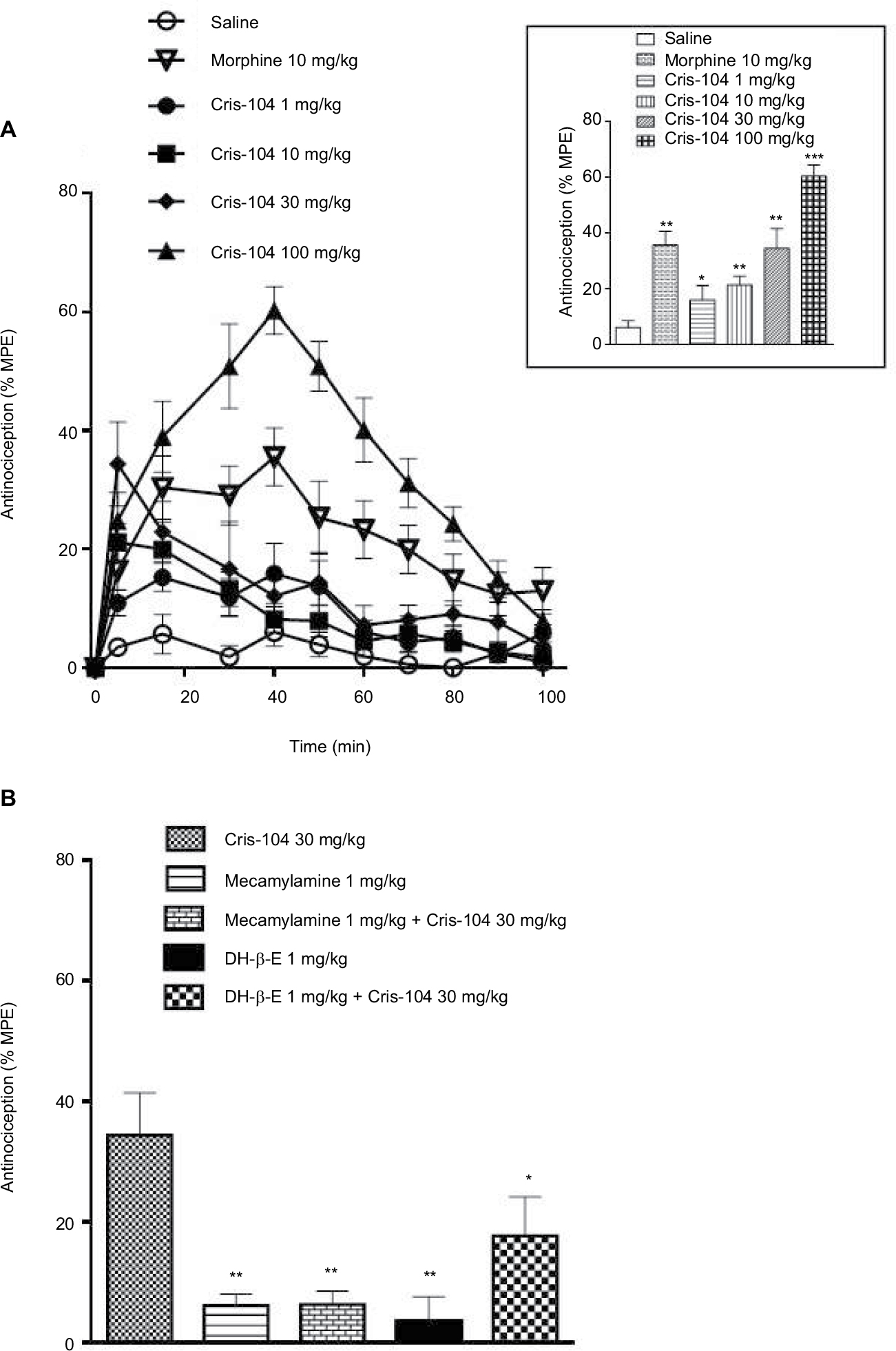 | Figure 2 Antinociceptive effect of Cris-104 on the hot-plate-induced nociception in mice. Notes: (A) Time dependent dose-response curve for Cris-104 (1, 10, 30, and 100 mg/kg p.o.). The upper figure represents the average + SEM (n=10) value extracted from the highest response in the time-course experiments. Morphine (10 mg/kg p.o.) was used as a control drug. (B) Cris-104 (30 mg/kg p.o.) was administered 15 minutes after treatment with mecamylamine (1 mg/kg i.p.) or dihydro-beta-erythroidine (1 mg/kg i.p.). Note absence of antinociceptive effect caused by mecamylamine (1 mg/kg i.p.) or dihydro-beta-erythroidine (1 mg/kg i.p.). Each bar represents mean ± SEM (n=6) measured at highest response in the time-course curve. *P<0.05, **P<0.01, ***P<0.001 vs saline. One-way ANOVA followed by Dunnett’s test. Abbreviations: i.p., intraperitoneal; %MPE, percentage of the maximum possible effect; p.o., peroral; SEM, standard errors of the mean. |
Cris-104 antinociception
Saline (vehicle control) treated mice had postformalin paw-licking times of 65.0±5.7 seconds in the neurogenic phase and 151.4±9.9 seconds in the inflammatory phase. Orally administered acetylsalicylic acid (150 mg/kg), used as a reference drug, did not affect licking time in the neurogenic phase, but reduced licking time significantly in the inflammatory phase. Cris-104 reduced licking time in both phases dose dependently (Figure 3A, B). Dihydro-beta-erythroidine (1 mg/kg) and yohimbine (1 mg/kg) each reversed the antinociceptive effect of Cris-104 (30 mg/kg) in the inflammatory phase, while prazosin (1 mg/kg) did not (Figure 3B). The antinociceptive effect of Cris-104 (30 mg/kg) was not affected by dihydro-beta-erythroidine (1 mg/kg) or yohimbine (1 mg/kg).
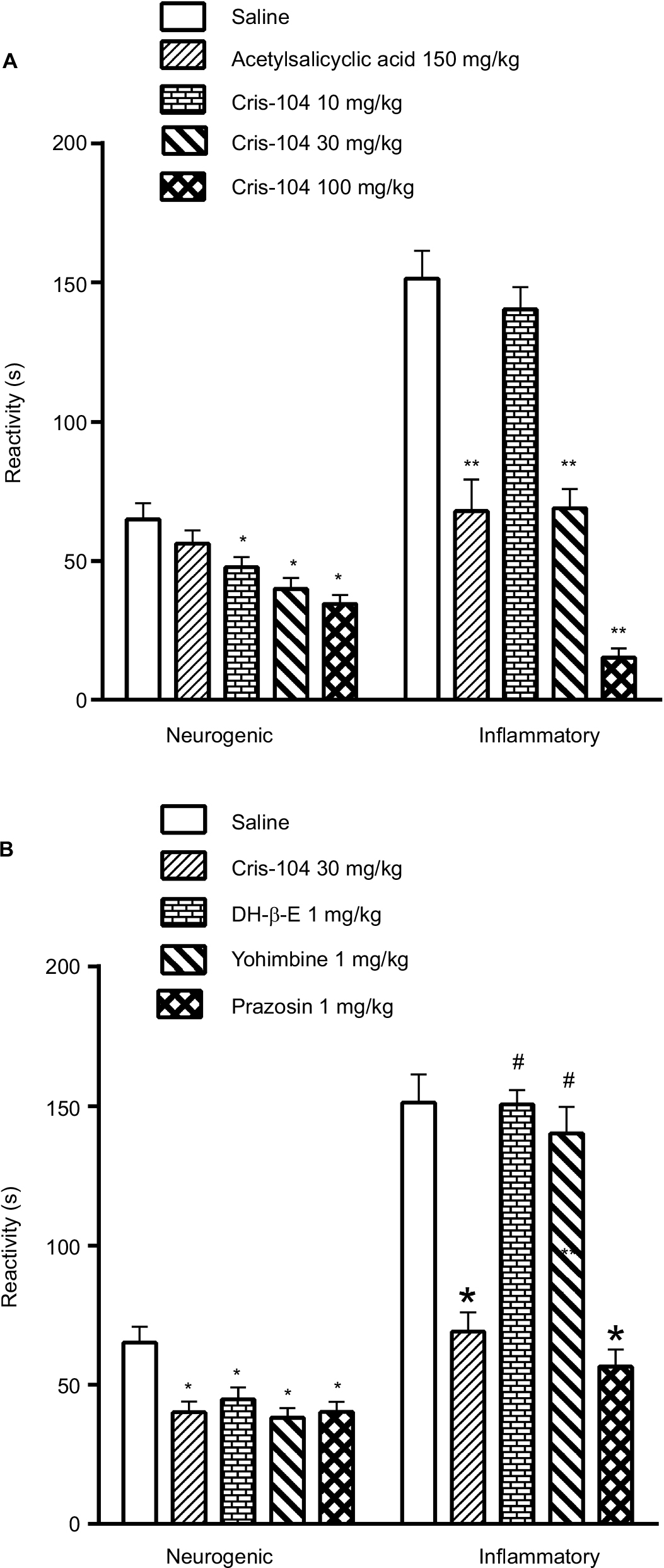 | Figure 3 Antinociceptive effect of Cris-104 on formalin-induced pain-related behavior in mice. Notes: Licking and/or biting time (reactivity) measured 0–5 minutes (neurogenic phase) and 15–30 minutes (inflammatory phase) after intraplantar injection of formalin (20 µL, 2.5%). (A) Mice were treated with saline, acetylsalicylic acid (150 mg/kg p.o.), or Cris-104 (10, 30, or 100 mg/kg p.o.) 15 minutes before formalin injection. (B) Pretreatment with dihydro-beta-erythroidine (1 mg/kg i.p.), yohimbine (1 mg/kg i.p.), or prazosin (1 mg/kg i.p.) 15 minutes before Cris-104 (30 mg/kg p.o.) administration. Each bar represents mean ± SEM (n=10). *P<0.05, **P<0.01, #P<0.05 vs Cris-104. One-way ANOVA followed by Dunnett’s test and two-way ANOVA followed by Bonferroni’s test. Abbreviations: i.p., intraperitoneal; SEM, standard errors of the mean. |
Antihypersensitivity effect of Cris-104 in chronic pain model
Paw withdrawal latency to thermal stimulation (PWL) and paw withdrawal threshold to mechanical stimulation (PWT) were reduced in all SNL groups 7 days after nerve injury compared with the sham group (Figure 4A, B). Repeated oral treatments with Cris-104 (10, 30, and 100 mg/kg/d) for 7 days increased both PWL and PWT dose dependently, and these effects ceased after suspension of the Cris-104 treatment.
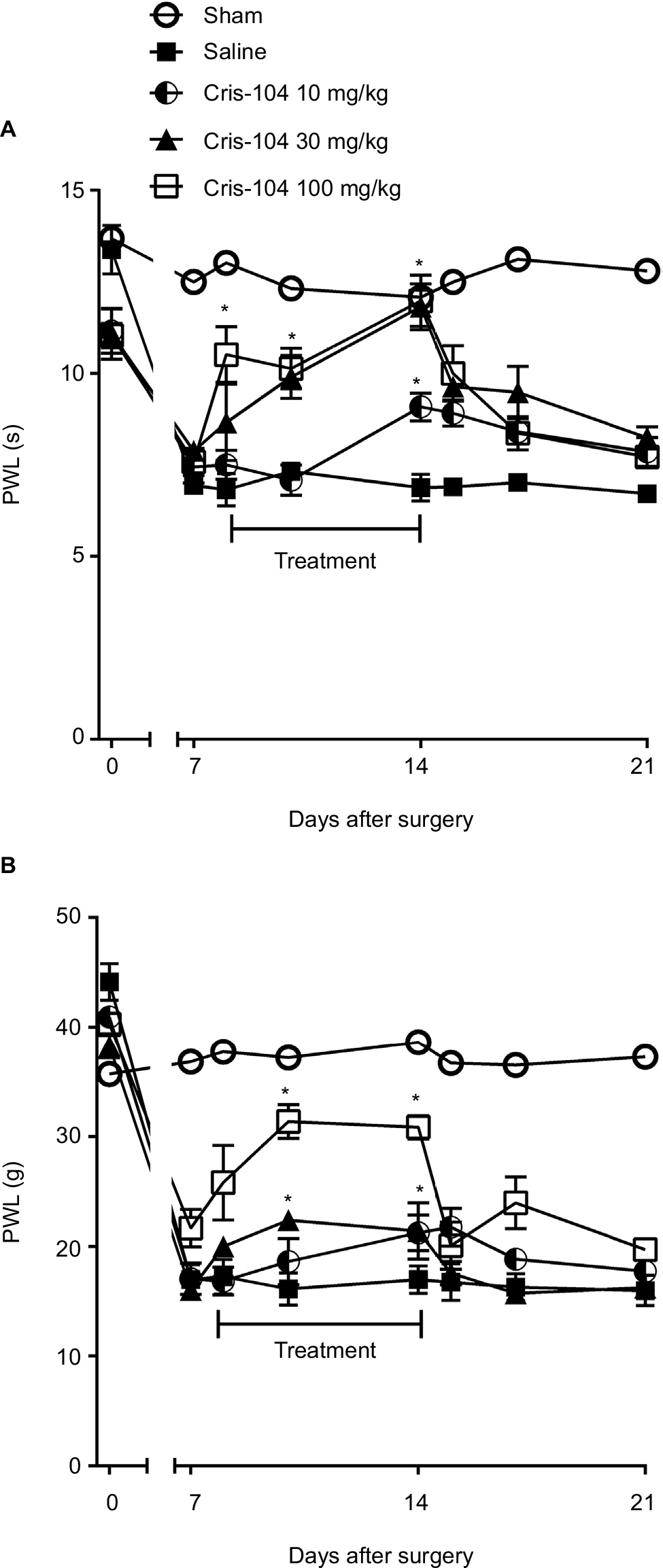 | Figure 4 Effects of Cris-104 on SNL-induced thermal and mechanical hyperalgesia in rats. Notes: Cris-104 (10, 30, and 100 mg/kg p.o.) was administered once a day for 7 days staring from 7 days after SNL surgery and behavior testing was performed prior to the Cris-104 administration. (A) Paw withdrawal latency (PWL) to thermal stimulation. (B) Paw withdrawal threshold (PWT) to mechanical stimulation. Data represent mean ± SEM (n=6). *P<0.05 vs 7 days after SNL. One-way ANOVA followed by Dunnett’s test. Abbreviations: SEM, standard errors of the mean; SNL, spinal nerve ligation. |
Spinal cord NE release
Local perfusion of Cris-104 (10 and 100 µM) or epibatidine (3 µM) through the microdialysis probe or intravenously administered Cris-104 (10 mg/kg) increased NE in microdialysates from the dorsal spinal cord of normal rats (Figure 5A). Simultaneous perfusion with mecamylamine (100 µM) blocked the effect of locally perfused Cris-104 (100 µM) on spinal NE release completely in normal rats. Similarly, locally perfused Cris-104 (10 and 100 µM) or epibatidine (3 µM), or intravenous Cris-104 (10 mg/kg), induced spinal NE release in SNL rats (Figure 5B).
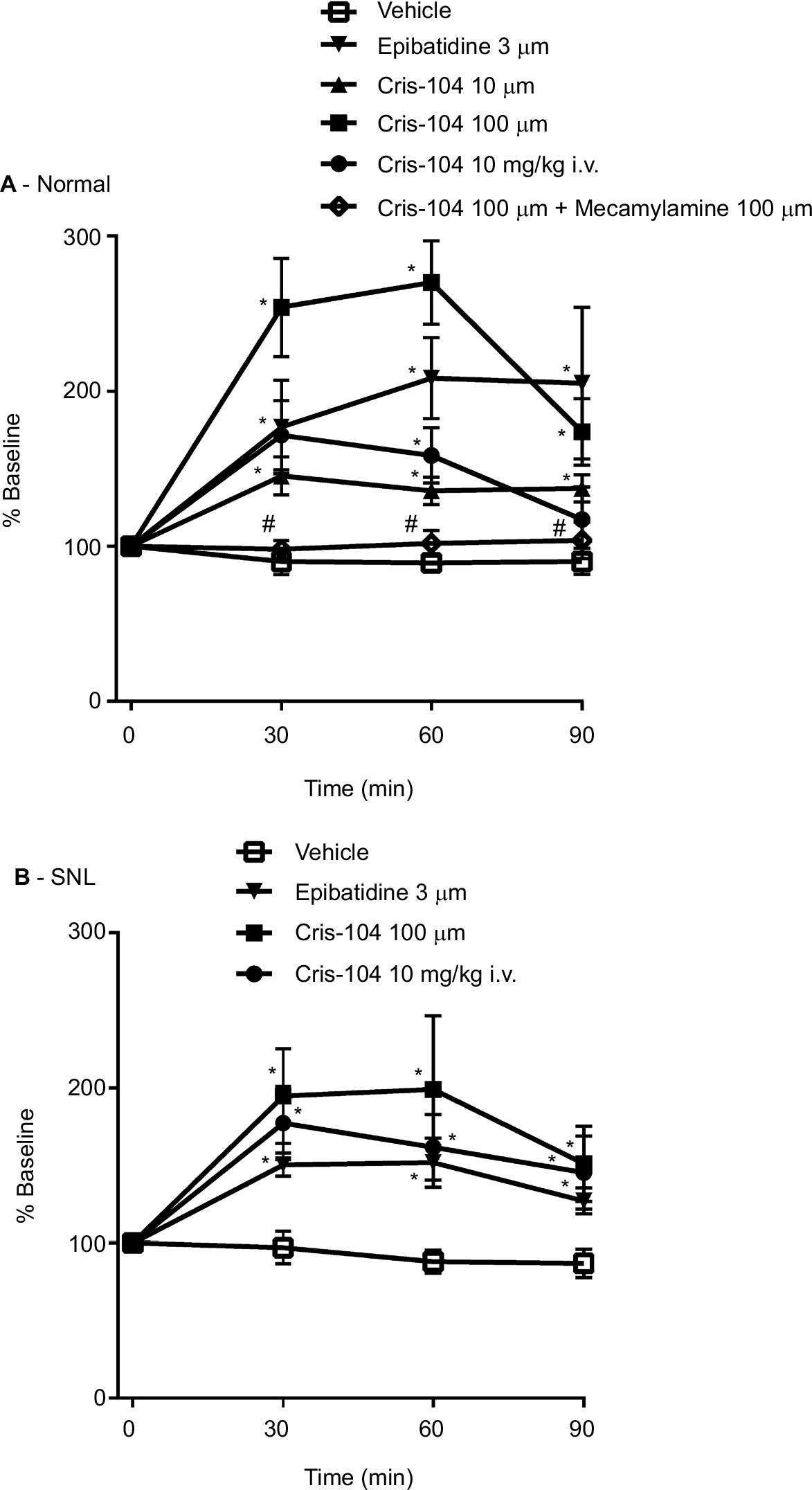 | Figure 5 Changes in noradrenaline concentrations in microdialysates from the spinal dorsal horn of normal (A) and SNL (B) rats were presented over time as percentage of baseline. Vehicle, Cris-104 (10 and 100 µM), Cris-104 (100 µM) + mecamylamine (100 µM), or epibatidine (3 µM) was perfused through the microdialysis probe. Notes: In some animals, Cris-104 (10 mg/kg) was injected through the IV line. Data represent mean ± SEM (n=5–7). *P<0.05 vs saline; #P<0.05 vs Cris-104 (100 µM). One-way ANOVA followed by Dunnett’s test and two-way ANOVA followed by Bonferroni’s test. Abbreviations: SEM, standard errors of the mean; SNL, spinal nerve ligation. |
LC neuronal activity
LC neuronal firing increased substantially after intravenous Cris-104 (10 mg/kg) (representative experiment in Figure 6A). Average baseline firing activity of LC neurons prior to the drug administration did not differ between the Cris-104 alone (0.49±0.14 Hz, N=9) and Cris-104 with mecamylamine (0.59±0.12 Hz, N=9, P=0.598) groups in intact rats. There were significant main effects of treatment (F1,1040=8.48, P=0.010) and time (F65,1040=3.71, P<0.001) on LC neuronal firing, as well as a significant treatment × time interaction (F65,1040=2.38, P<0.001; Figure 6B). Post hoc testing revealed that Cris-104 increased firing activity of LC neurons, relative to baseline, from the 2nd to the 12th postinjection observation (all P<0.05), and this effect was blocked by coadministered mecamylamine.
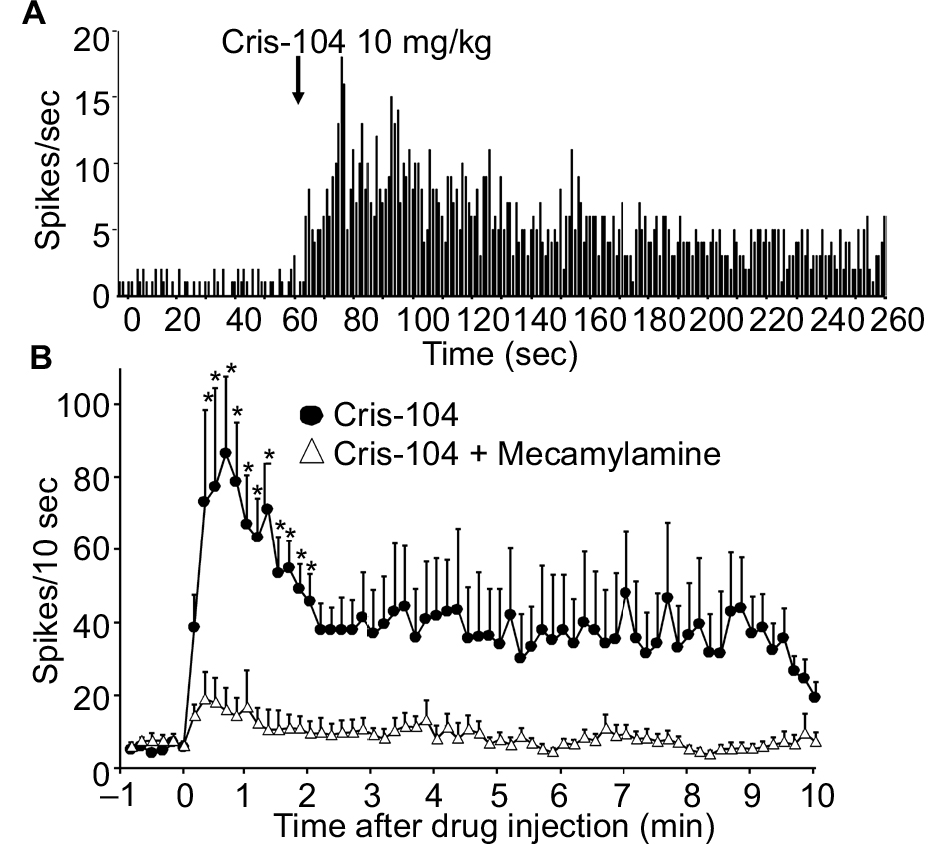 | Figure 6 Normal rats received an intravenous injection of Cris-104 (10 mg/kg) with or without mecamylamine (3 mg/kg). Notes: LC neuronal firing rates are presented over time as spikes per 10 seconds (mean + SEM). (A) Representative LC recording experiment. (B) Pooled data from nine experiments. *P<0.05 vs time 0. Abbreviations: LC, locus coeruleus; SEM, standard errors of the mean. |
As shown in Figure 7, relative to the no-drug control condition (121.3±12.0 movements/min, N=5), locomotor activity was not significantly reduced 1 (90.7±11.3 movements/min, N=5, P=0.10), 3 (89.8±8.1 movements/min, N=5, P=0.06), or 7 days (101.6±17.7 movements/min, N=5, P=0.37) after administration of Cris-104 (100 mg/kg), though locomotor activity at day 3 showed a nonsignificant trend toward inhibition. In contrast, a single dose of diazepam (10 mg/kg) reduced locomotor activity very significantly (50.7±6.4 movements/min, N=4, P=0.002). All measurements were performed 24 hours after oral administration of each tested compound.
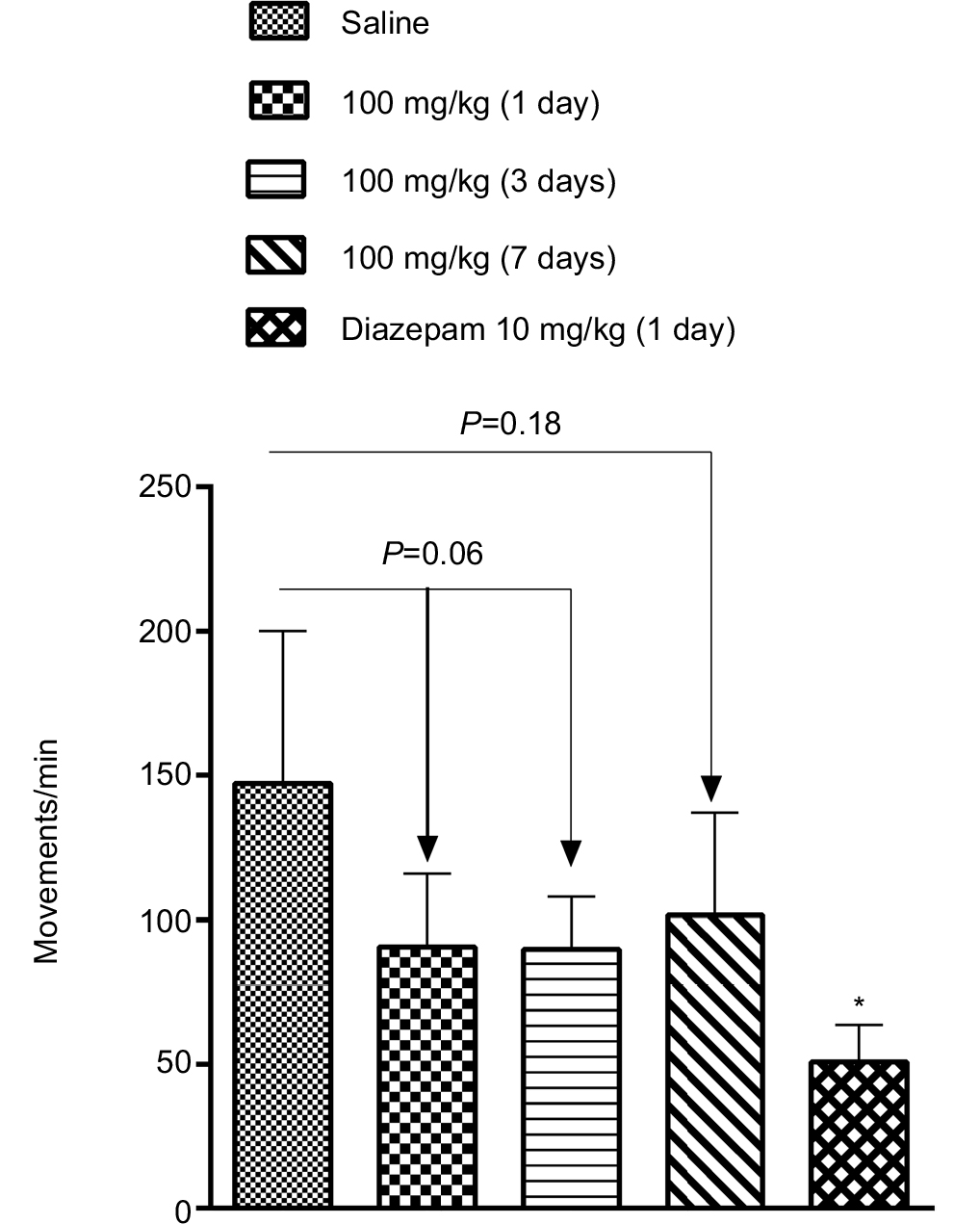 | Figure 7 Normal rats received oral administration of Cris-104 (100 mg/kg) once a day for 7 days and open-field test was performed prior to the Cris-104 administration. Notes: A single oral dose of diazepam (10 mg/kg) or saline was used as positive or negative control, respectively. Total number of movements measured during 45 minutes was normalized and the bars represent average + SEM (n=4–5). *P<0.05 vs saline. Abbreviation: SEM, standard errors of the mean. |
Discussion
In the present study, we found that Cris-104 induced dose-dependent antinociception in hot-plate and formalin tests, and this effect was blocked by the general and α4β2-selective nAChR antagonists and an α2-adrenoceptor antagonist, but not an α1-adrenoceptor antagonist. Systemic and spinally perfused Cris-104 increased NE levels in the spinal cord in both normal and SNL rats. Systemic Cris-104 increased LC neuronal activity in normal rats, without significant effects on locomotion, and this effect was blocked by nAChR antagonism.
Previously, we demonstrated that Cris-104 is not chemically related to epibatidine and that it can reduce thermal and mechanical hyperalgesia in a streptozotocin-induced peripheral diabetic neuropathy animal model.15 Cris-104 and related compounds (Cris-111, Cris-158, Cris-159, and Cris-160; chemical structures not shown) were submitted to screening assays for binding affinity to α7-nAchRs, low-affinity α4β2* nAChRs (cytisine sensitive), high-affinity α4β2 nAChRs, and α1- and α2-adrenoceptors, as well as to 5-HT2/7 serotonin, D2 dopamine, GABAB and GABAA, NMDA glutamate, and H3 histamine receptors by MDS Pharma Services (Taipei, Taiwan). The protocols were based on percentage of displacement induced by a fixed (10 µM) concentration of the compound to a specific agonist for each receptor and >50% displacement was considered relevant to drug-development candidate. Cris-104 displacement was differentiated between different α4β2 nAChR types: 61% displacement occurred with high-affinity (α4)2(β2)3 nAChRs, but only 17% displacement occurred with low-affinity, cytisine-sensitive (α4)3(β2)2 nAChRs. Binding assay and in vitro experiments with nAChR antagonists are in agreement in supporting the hypothesis that effects of Cris-104 on behavior and NE release are likely mediated via α4β2 nAChRs.
In vitro absorption, distribution, metabolism, and excretion studies have shown that Cris-104 binds 53% to plasma proteins; the permeability through parallel artificial membrane permeability assay at pH 7.4 was 13.6±7.1 cm/s, suggesting that Cris-104 should have satisfactory permeability across biological membranes, including the blood–brain barrier.27 Physicochemical properties showed that the logD octanol/PBS values for Cris-104 at pH 5.5 and 7.4 were 0.17±2.9 (n=3) and 1.49±8.6 (n=3), respectively. Optimal range for drug absorption in the intestine is logD7.4 1–3.28 The stability at pH 1.1, 7.4, and 9.4 was 91.3±0.7% (n=3), 98.7±2.9% (n=3), and 91.2±1.7% (n=3), respectively. Cris-104 exposed to rat liver microsomes was found to have a clearance rate of 1.8 mL/min/kg indicating that it appears to be stable across a wide pH range and to have a low metabolic clearance rate (low clearance rate cutoffs set at <5 mL/min/kg).28
Descending noradrenergic pain pathways can be activated by pharmacological stimulation of α4β2* nAChR-expressing neurons in the LC.7 The current study demonstrated that systemic Cris-104 has two interesting effects: (1) increased firing of LC neurons and (2) induction of spinal NE release. That is, nAChR stimulation with Cris-104 affected descending noradrenergic pain pathways both indirectly via supraspinal effects on LC neuronal firing and directly via intraspinal effects on NE release. Moreover, these antinociception effects could be blocked with the α2-adrenoceptor antagonist yohimbine.
Prior attempts to develop nAChR agonist compounds into drugs failed due to dose-dependent adverse secondary effects, including decreased body temperature and locomotor activity, increased blood pressure, dizziness, nausea, and vomiting.14,26,28,29 Those effects were likely due to actions on ganglionic α4β2* nAChRs in the peripheral nervous system. Peripheral side effects were observed on clinical trials with epibatidine, which is a nonselective agonist of α4β2* nACh receptors. However, even a selective agonist such as ABT-594 was unsuccessful in Phase II clinical study,30 indicating that the selectivity is not the main factor responsible for the appearance of side effects. In the present work, side effects were not detected, such as aggressiveness, piloerection, reduced food intake, or impairment of motor activity induced by Cris-104. Therefore, it is not possible to indicate that Cris-104 could be a positive allosteric modulator of α4β2* nACh receptors, which could be a factor to increase the safety of drug discovery.
Because Cris-104 shows selectivity for (α4)2(β2)3 nAChRs, adverse effects mediated by (α4)3(β2)2 nAChRs may be avoided and prostration, aggressiveness, piloerection, and reduced food intake were not induced by Cris-104 in rodent. The absence of significant effects of Cris-104 on locomotion or any signs of gastric malaise (eg, lying flat on belly) with a supraefficacious dose bode well for Cris-104 being better tolerated by patients.
Conclusion
The novel designed α4β2* nAChR agonist Cris-104 is a promising compound with analgesic profile interfering with the descending noradrenergic pathways by stimulating noradrenergic neurons in the LC and stimulating their terminals in the spinal cord.
Acknowledgments
Cristália Produtos Químicos e Farmacêuticos Ltda (Cristalia Chemical and Pharmaceutical Products, Inc.) for providing Cris-104. This work was supported by Nacional Council of Scientific and Tecnology Research (CNPq) with fellowships (RT Sudo and G Zapata-Sudo), Smoking Research Foundation (Tokyo Japan, K Hayashida) and Kaken (16K08985, Tokyo Japan, K Hayashida), Coordination of Superior Level Staff Improvement (CAPES), Carlos Chagas Research Resource Foundation of State of Rio de Janeiro (FAPERJ), and Nacional Science and Tecnology Institute of Drug Development (INCT-INOFAR).
Disclosure
RDM is the director of the Research and Innovation Section at Cristalia Produtos Quimicos e Farmacêuticos Ltda. The authors report no other conflicts of interest in this work.
References
Naser PV, Kuner R, Molecular KR. Molecular, cellular and circuit basis of cholinergic modulation of pain. Neuroscience. 2018;387:135–148. | ||
Sun Y, Yang Y, Galvin VC, Yang S, Arnsten AF, Wang M. Nicotinic α4β2 cholinergic receptor influences on dorsolateral prefrontal cortical neuronal firing during a working memory task. J Neurosci. 2017;37(21):5366–5377. | ||
Nemecz Á, Prevost MS, Menny A, Corringer PJ. Emerging molecular mechanisms of signal transduction in pentameric ligand-gated ion channels. Neuron. 2016;90(3):452–470. | ||
Dani JA. Neuronal nicotinic acetylcholine receptor structure and function and response to nicotine. Int Rev Neurobiol. 2015;124:3–19. | ||
Moroni M, Zwart R, Sher E, Cassels BK, Bermudez I. alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol Pharmacol. 2006;70(2):755–768. | ||
Lloyd GK, Williams M. Neuronal nicotinic acetylcholine receptors as novel drug targets. J Pharmacol Exp Ther. 2000;292(2):461–467. | ||
Cucchiaro G, Chaijale N, Commons KG. The locus coeruleus nucleus as a site of action of the antinociceptive and behavioral effects of the nicotinic receptor agonist, epibatidine. Neuropharmacology. 2006;50(7):769–776. | ||
Li X, Eisenach JC. Nicotinic acetylcholine receptor regulation of spinal norepinephrine release. Anesthesiology. 2002;96(6):1450–1456. | ||
Hayashida K, Parker R, Eisenach JC. Oral gabapentin activates spinal cholinergic circuits to reduce hypersensitivity after peripheral nerve injury and interacts synergistically with oral donepezil. Anesthesiology. 2007;106(6):1213–1219. | ||
Hayashida K, Obata H, Nakajima K, Eisenach JC. Gabapentin acts within the locus coeruleus to alleviate neuropathic pain. Anesthesiology. 2008;109(6):1077–1084. | ||
Obata H, Li X, Eisenach JC. alpha2-Adrenoceptor activation by clonidine enhances stimulation-evoked acetylcholine release from spinal cord tissue after nerve ligation in rats. Anesthesiology. 2005;102(3):657–662. | ||
Pan HL, Chen SR, Eisenach JC. Intrathecal clonidine alleviates allodynia in neuropathic rats: interaction with spinal muscarinic and nicotinic receptors. Anesthesiology. 1999;90(2):509–514. | ||
Sullivan JP, Bannon AW. Epibatidine: pharmacological properties of a novel nicotinic acetylcholine receptor agonist and analgesic agent. CNS Drug Rev. 1996;2(1):21–39. | ||
Nirogi R, Goura V, Abraham R, Jayarajan P. α4β2* neuronal nicotinic receptor ligands (agonist, partial agonist and positive allosteric modulators) as therapeutic prospects for pain. Eur J Pharmacol. 2013;712(1–3):22–29. | ||
Debom R, Trachez MM, Zapata-Sudo G, et al. Novel nicotinic receptor agonist reduces hiperalgesia and allodynia of neuropathic pain in diabetic rats. J Diab Metab. 2014;5:396–400. | ||
Sudo RT, Calasans-Maia JA, Galdino SL, et al. Interaction of morphine with a new alpha2-adrenoceptor agonist in mice. J Pain. 2010;11(1):71–78. | ||
Sudo RT, Neto ML, Monteiro CE, et al. Antinociceptive effects of hydroalcoholic extract from Euterpe oleracea Mart. (Açaí) in a rodent model of acute and neuropathic pain. BMC Complement Altern Med. 2015; 15:208. | ||
Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50(3):355–363. | ||
Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32(1):77–88. | ||
Vivancos GG, Verri WA, Cunha TM, et al. An electronic pressure-meter nociception paw test for rats. Braz J Med Biol Res. 2004;37(3):391–399. | ||
Berrocoso E, Micó JA, Ugedo L. In vivo effect of tramadol on locus coeruleus neurons is mediated by alpha2-adrenoceptors and modulated by serotonin. Neuropharmacology. 2006;51(1):146–153. | ||
Ganesh A, Gonzalez-Sulser A, Chaijale N, Cucchiaro G. Electrophysiologic effects of systemic and locally infused epibatidine on locus coeruleus neurons. Eur J Pharmacol. 2008;584(1):93–99. | ||
Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego: Academic Press; 2007. | ||
Cedarbaum JM, Aghajanian GK. Noradrenergic neurons of the locus coeruleus: inhibition by epinephrine and activation by the alpha-antagonist piperoxane. Brain Res. 1976;112(2):413–419. | ||
Silva GA, Kummerle AE, Antunes F, et al. Impairment of locomotor activity induced by the novel N-acylhydrazone derivatives LASSBio-785 and LASSBio-786 in mice. Braz J Med Biol Res. 2013;46(3):263–269. | ||
Gerzanich V, Peng X, Wang F, et al. Comparative pharmacology of epibatidine: a potent agonist for neuronal nicotinic acetylcholine receptors. Mol Pharmacol. 1995;48(4):774–782. | ||
Yan Z, Caldwell GW. Stable-isotope trapping and high-throughput screenings of reactive metabolites using the isotope MS signature. Anal Chem. 2004;76(23):6835–6847. | ||
Kerns EH, di L, Carter GT. In vitro solubility assays in drug discovery. Curr Drug Metab. 2008;9(9):879–885. | ||
Boyce S, Webb JK, Shepheard SL, Russell MG, Hill RG, Rupniak NM. Analgesic and toxic effects of ABT-594 resemble epibatidine and nicotine in rats. Pain. 2000;85(3):443–450. | ||
Rowbotham MC, Duan WR, Thomas J, Nothaft W, Backonja MM. A randomized, double-blind, placebo-controlled trial evaluating the efficacy and safety of ABT-594 in patients with diabetic peripheral neuropathic pain. Pain. 2009;146(3):245–252. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.